

Abstract
Endometriosis is a disease characterized by the localization of endometrial tissue outside the uterine cavity. The differences observed in migration of human endometrial stromal cells (hESC) obtained from patients with endometriosis versus healthy controls were proposed to correlate with the abnormal activation of Raf-1/ROCKII signalling pathway. To evaluate the mechanism by which Raf-1 regulates cytoskeleton reorganization and motility, we used primary eutopic (Eu-, n = 16) and ectopic (Ec-, n = 8; isolated from ovarian cysts) hESC of patients with endometriosis and endometriosis-free controls (Co-hESC, n = 14). Raf-1 siRNA knockdown in Co- and Eu-hESC resulted in contraction and decreased migration versus siRNA controls. This phenotype was reversed following the re-expression of Raf-1 in these cells. Lowest Raf-1 levels in Ec-hESC were associated with hyperactivated ROCKII and ezrin/radixin/moesin (E/R/M), impaired migration and a contracted phenotype similar to Raf-1 knockdown in Co- and Eu-hESC. We further show that the mechanism by which Raf-1 mediates migration in hESC includes direct myosin light chain phosphatase (MYPT1) phosphorylation and regulation of the levels of E/R/M, paxillin, MYPT1 and myosin light chain (MLC) phosphorylation indirectly via the hyperactivation of ROCKII kinase. Furthermore, we suggest that in contrast to Co-and Eu-hESC, where the cellular Raf-1 levels regulate the rate of migration, the low cellular Raf-1 content in Ec-hESC, might ensure their restricted migration by preserving the contracted cellular phenotype. In conclusion, our findings suggest that cellular levels of Raf-1 adjust the threshold of hESC migration in endometriosis.
Keywords: endometriosis, endometrial stromal cells, Raf-1/ROCKII signalling pathway, actin cytoskeleton and cellular migration, reproductive endocrinology, cell signalling and endometriosis
Introduction
Endometriosis is defined by the presence of endometrial glands and stroma outside the uterine cavity. It is regarded as an oestrogen driven disease, as it affects women mainly during their reproductive age [1, 2]. Different hypotheses on the pathogenesis of this disease exist [3]. The most widely favoured hypothesis encompasses a phenomenon called retrograde menstruation [3]. It is believed that viable endometrial cells and tissue can reach the abdomen through the Fallopian tubes at the time of menstruation. They adhere to the peritoneal wall, invade the mesothelial cell layer and basement membrane through enzymatic degradation and provoke angiogenesis. This process is associated with deregulated remodelling, proliferation, migration and adhesion of endometrial cells [4, 5]. Although retrograde menstruation is a common phenomenon occurring in more than 80% of women [6], only 10–20% develop endometriosis, suggesting that further conditions must be in place to support the endometriotic growth. Genetic, endocrine, immune and environmental factors have been suggested in the pathogenesis of endometriosis. In particular, the intrinsic molecular aberrations in pelvic endometriotic implants were proposed to contribute significantly to the establishment of the disease. These include aberrant expression of cytokines [7], matrix-metalloproteinases [8], resistance to the protective action of progesterone [9] and aberrant expression of aromatase [10]. Alterations in the expression pattern of different adhesion regulatory proteins such as Focal Adhesion Kinase (FAK) [11, 12], integrins [13] and cadherins [14, 15] have also been observed in both eutopic and ectopic cells from women with endometriosis. These changes were associated with a more invasive phenotype of eutopic endometrial cells of patients with endometriosis [16]. One of the steps of invasion is cell migration.
Cell migration is a complex, multi-step process involving changes in the cytoskeleton, cell-substrate adhesion and the extracellular matrix. Coordinated cell movement can theoretically be divided into four mechanistically separate steps: lamellipodium extension, formation of new adhesion foci, cell body translocation and detachment of the tail [17]. All of these processes are linked to cytoskeleton remodelling. The key regulators of cytoskeleton dynamics and therefore of cell migration are the small Rho-GTPase family proteins Rho, Rac and Cdc42. Rho induces assembly of stable focal adhesions, leading to decreased motility. It promotes actomyosin contractility, necessary for cell body translocation, whereas Rac and Cdc42 are both required at the leading edge of migrating cells [18]. One of the downstream targets of Rho is ROCKII (also known as Rho-kinase). ROCKII regulates cell body contraction during migration by acting on actomyosin contractility. In particular, ROCKII affects MLC phosphorylation, both by inhibiting MLC phosphatase (MYPT1) and by phosphorylation of MLC [19]. It also affects changes of actin cytoskeleton relevant to cell migration through its downstream targets ezrin/radixin/moesin (E/R/M) protein family [20]. Ezrin was proposed to be involved in endometrial pathologies such as uterine adenocarcinomas [21] and endometriosis [22].
During the last decade, growing experimental evidence showed that ovarian steroids differentially affect the migration and invasion properties of normal (Co-hESC), eutopic (Eu-hESC) and ectopic (Ec-hESC) human endometrial stromal cells derived from women with and without endometriosis [23, 24]. However, whether these cells behave different compared with each other without any steroid stimulation remains to be clarified. Little is known about whether a difference in basal, i.e. unstimulated migration behaviour of these three types of cells exists. It is also still not well understood how the molecular differences between Co-, Eu- and Ec-hESC are implicated in the regulation of their basal migration. In an attempt to provide answers to this question, we recently showed that the increased migratory potential of Eu-hESC versus Co-hESC is due to the abnormal activation of the Raf-1/RhoA/ROCKII signalling pathway [25]. In particular, we demonstrated that Raf-1 operates as a negative regulator of ROCKII activity and that the protein levels of Raf-1 are lower in Eu-hESC, compared with Co-hESC, ensuring the abnormal activation of this pathway in Eu- versus Co-hESC. Moreover, it became obvious that endometriosis shares some characteristics with malignancy, i.e. development of local and distant foci, attachment to and invasion of other tissues and subsequent damage to affected organs [26]. However, it is still not obvious why ectopic lesions are unable to further invade other tissues, thereby resulting in malignant disease [27]. We hypothesize that Ec-hESC have a restricted ability to migrate compared with Co- and Eu-hESC due to a significant loss of Raf-1 expression resulting in hyperactivation of the Raf-1/RhoA/ROCKII signalling pathway. Despite the evidence that abnormal expression of RhoA-GTPase, ROCKII-kinases and their downstream cytoskeleton-regulatory effectors might be associated with the establishment of endometriosis [28], little is known at present about the mechanisms by which Raf-1/ROCKII regulates hESC migration. Thus, our present study characterizes the differences in basal cell migration and the molecular mechanisms by which Raf-1/ROCKII signalling pathway regulates cytoskeleton remodelling and motility in Co-, Eu- and Ec-hESC in the absence of additional steroid hormone stimuli.
Materials and methods
Patients and tissue collection
Endometrial samples (n = 38) were obtained from patients (age 18–42) who underwent laparoscopy and additional curettage for diagnosis and/or treatment of endometriosis. Patients clinically presented with ovarian cysts, chronic pelvic pain, dysmenorrhoea and/or infertility. The presence or absence of endometriosis was confirmed visually using laparoscopy and additional histological analysis. In particular, eutopic and ectopic (ovarian endometrioma) endometrium samples of patients with endometriosis (n = 16 and n = 8, respectively) and endometrium samples of age-matched endometriosis-free controls (n = 14) in the proliferative phase of the menstrual cycle were used for primary cell culture preparation as described in [25] and Table S1. None of the donors were taking medications or had received hormonal therapy for at least 6 months prior to surgery. All samples were obtained following Institutional Review Board approval (IRB number 075/2005 General Hospital of Vienna, Vienna, Austria) as well as written informed consent permission from every participating woman. Immediately after surgery, the tissue was transferred in D-MEM + Ham's F12 medium on ice and processed for immunohistochemistry and primary cell culture preparation. Of all prepared cultures, different subsets of randomly chosen samples, indicated with numbers in the respective figure legends, were used for the experiments.
Cell culture
Endometrial tissue was minced into small pieces and incubated with collagenase (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 10 min., followed by filtration, as previously described [29, 30]. This method produces 95–99% pure stromal fibroblasts. The purity of the hESC at passage two was evaluated by immunofluorescence analysis using antibodies against vimentin (stromal cell marker), cytokeratin7 (epithelial cell marker) and CD45 (leucocyte marker) (data not shown). All cultures were 98–99% pure stromal fibroblasts. All cells were then cultured as previously described [25]. Briefly, the cells were cultured on fibronectin-collagen (Gibco, Grand Island, NY, USA) coated dishes in DMEM-F12 without phenol red (Gibco) supplemented with 10% foetal bovine serum (FBS) (Gibco), 2 mM l-glutamine (Gibco) and 1% antibiotics–antimycotics (Gibco) up to passage 4–6. To exclude influences of the serum derived steroid hormones, two passages before experiments, the cells were grown in culture medium containing 10% charcoal stripped-foetal bovine serum (CS-FBS; Gibco). When indicated, specific Raf-1 kinase inhibitors (1 μM GW5074, 1 μM ZM336372; Tocris, Eching, Germany) or ROCKII inhibitor (10 μM Y-27632; Sigma-Aldrich) were added to the growing medium for 24 hrs.
siRNA knockdowns, Western blot analysis and co-immunoprecipitation
Ten micromolar ROCKII siGENOME SMART pool (Thermo Scientific, Dharmacon, Lafayette, CO, USA) and siCONTROL non-targeting (Thermo Scientific) or 10 nM Raf-1 (Cat.No: VHS40462; Invitrogen, Carlsbad, CA, USA) and low-density GCs negative universal control siRNA (Cat.No: 46-2002; Invitrogen) were transfected with siLentFect™ (Bio-Rad Laboratories, Hercules, CA, USA) transfection reagent according to the manufacturer's protocol. The cells were lysed and analysed 48 hrs after transfection using Western blot as described in [25]. Briefly, 20 μg of lysates were used for Western blot and 500 μg for immunoprecipitation. The following antibodies were used for immunoblotting: ROCKII, Raf-1, MYPT1 (BD Transduction Laboratories, Danvers, MA, USA); pMLC (Thr18/Ser19); pE/R/M (Thr567/Thr564/Thr558); pPaxillin (Tyr118), E/R/M and GAPDH (Cell Signalling Technology Inc., Danvers, MA, USA), paxillin (Abcam, Cambrige, UK), p MYPT1 (Thr696) (Millipore, Temecula, CA, USA) and α-tubulin (Sigma-Aldrich) prior to incubation with peroxidase (PE)-conjugated secondary antibodies (Pierce Chemical Co., La Jolla, CA, USA). For all primary antibodies, 1:1000 dilutions (in 1× TBS-0.1% Tween 20 buffer, Promega, San Luis Obispo, CA, USA) supplemented with 1% Bovine Serum Albumin (BSA) and incubated over night at 4°C were used. The secondary antibodies were diluted 1:25,000 in 1× TBS-0.1% Tween 20 buffer and incubated for 1 hr at room temperature. Bound antibodies were detected and quantified as previously described [25].
Immunofluorescence
Cells plated on fibronectin (Roche, Mannheim, Germany) were fixed in 4% Paraformaldehyde (PFA), permeabilized (0.02% Triton-X-100) and blocked with 0.2% gelatine before incubation with the primary antibodies: vimentin (rabbit; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Raf-1 (mouse monoclonal; BD Transduction Laboratories) and staining with appropriate Alexa Fluor-conjugated secondary antibody. Alexa Fluor 596-conjugated phalloidin (Invitrogen) was used to visualize F-actin filaments. Epifluorescence was performed with an Olympus BX50 microscope equipped with soft imaging system-F-View camera and Cell∧P imaging software (Olympus Austria Ges.m.b.H, Vienna, Austria).
Cell migration
Cells were analysed for migration using a Boyden chamber assay with polycarbonate membranes. An equal number of cells (5 × 104) was re-suspended in complete cell media, plated on top of fibronectin-collagen coated chambers (8 μm-pores; BD Biosciences, Bedford, MA, USA) and allowed to migrate for 12 hrs. In case of siRNA knockdown, the cells were used for migration analysis 48 hrs after their initial transfection. The cells on the underside of the membrane were fixed, stained with 4′, 6-diamidino-2-phenylindol (DAPI) and counted (5 random fields/membrane) by two independent investigators.
ROCKII activity assay
ROCKII kinase activity was analysed by an enzyme immunoassay, using Cell Biolabs'96-well ROCKII activity assay kit (Cell Biolabs Inc., San Diego, CA, USA). Experiments were performed according to the manufacturer's protocol, using 10 μl of the protein lysates. Total starting protein concentration for every sample was 1 mg/ml.
Statistical analysis
Data were subjected either to statistical analysis using unpaired Student's t-tests or to anova followed by Post-hoc statistical analysis.
Results
Effects of Raf-1 knockdown on hESC cell morphology
Immunofluorescence analysis of actin cytoskeleton showed that in Co- and Eu-hESC, Raf-1 knockdown leads to a change of cellular phenotype to a more symmetric, contracted morphology (Figs 1A and S1). In Raf-1 knockdown cells, the actin cytoskeleton was organized in tight cortical bundles, and vimentin filaments were forming perinuclear aggregates (Figs 1A and S1). In Co- and Eu-hESC with Raf-1 knockdown, the number of contracted cells was 47% (P < 0.005) and 78% (P < 0.005) higher compared with respective siRNA controls (Fig. 1B). In contrast to the Co- and Eu-hESC, Raf-1 knockdown in Ec-hESC did not affect their morphology (Fig. 1A, lower panel). However, Ec-hESC treated with control siRNA had a more contracted cellular morphology similar to that seen in the Raf-1 knockdown Co- and Eu-hESC (Fig. 1A).
Fig 1.
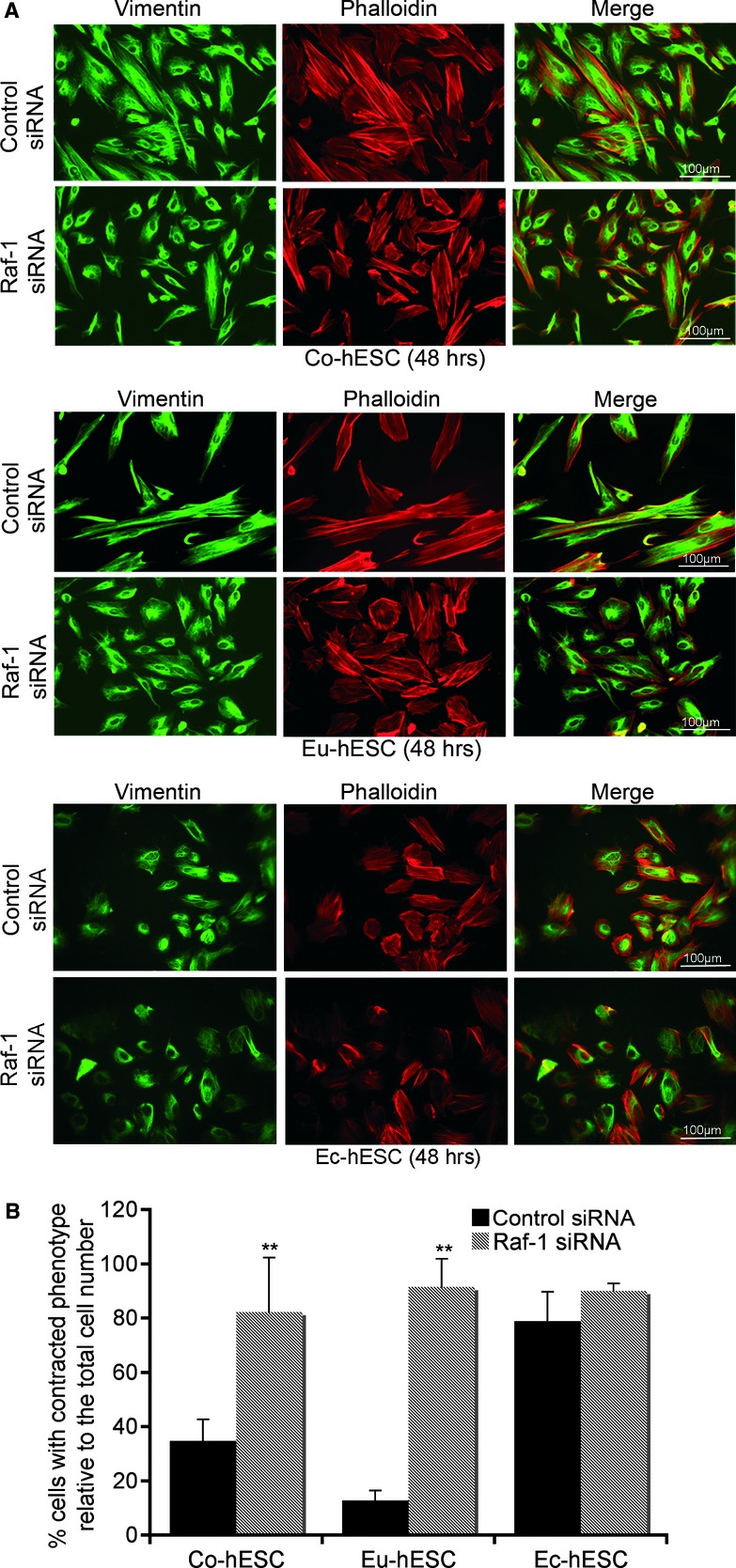
Effects of Raf-1 knockdown on hESC cell morphology. (A) Immunofluorescence analysis of cytoskeleton by vimentin (green), phalloidin (red) and merge (green and red), 48 hrs after transfection of Co-hESC, Eu-hESC and Ec-hESC cells with siRNA are shown, scale bar line = 100 μm. (B) Average number of cells with contracted phenotype (estimated by two independent investigators) out of three biological replicates are given as% of cells with contracted phenotype relative to the total cell number (set to 100%), **P < 0.005.
The re-expression of Raf-1 protein in knockdown Eu- and Co-hESC, following continuous cultivation of knockdown hESC for 6 days after Raf-1 siRNA transfection, rescued the defects seen in cell morphology (Figs 2A and S2A, left panels). After this time span, the number of contracted Eu-hESC was 17% (P < 0.005) and for Co-hESC 34% (P < 0.005), respectively, compared with total cell number (Figs 2A and S2A, right panels). These values were similar to the number of contracted cells observed 48 hrs after control siRNA transfection (Figs 1B, 2A and S2A). The contracted phenotype of Raf-1 knockdown Eu-hESC was associated with a 70% lower migration rate (P < 0.005) compared with the siRNA controls (Fig. 2B). Similar results were obtained for Raf-1 knockdown Co-hESC, where the contracted phenotype was accompanied by a reduction in migration rate to 43% (P < 0.005), compared with the controls (Fig. S2B). The re-expression of Raf-1 restored the migratory phenotype of both Eu- and Co-hESC (Figs 2B and S2B). In contrast to Co- and Eu-hESC, Raf-1 knockdown in Ec-hESC did not significantly change their phenotype (Fig. 1A, lower panel) or migration (Fig. 2C).
Fig 2.
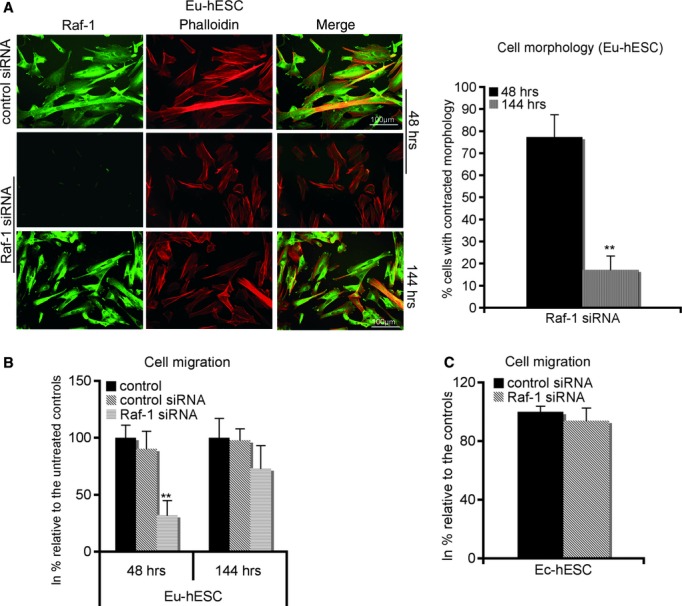
Raf-1 regulates hESC morphology and migration. (A) Immunofluorescence analysis of cytoskeleton by phalloidin (red), Raf-1 cellular levels (green) and merge (green and red) 48 and 144 hrs after transfection of Eu-hESC cells with siRNA are shown on the left. Average number of cells with contracted phenotype (estimated by two independent investigators) out of three biological triplicates are given on the right as % of cells with contracted phenotype relative to the total cell number (set to 100%, ± S.D.). **P < 0.005; scale bar line = 100 μm. (B) Effects of Raf-1 knockdown on cell migration in Eu-hESC 48 and 144 hrs after initial transfection with either control (transfection reagent only) or control siRNA or Raf-1 siRNA are given. Average values of biological triplicates are shown as percentage of migrating cells relative to the controls (mean value set to 100%), **P < 0.005. (C) Raf-1 knockdown does not change the migration rate of Ec-hESC. A migration assay, 48 hrs after Raf-1 knockdown in Ec-hESC, was performed. Average values of biological triplicates are shown as percentage of migrating cells relative to the siRNA controls (mean value set to 100%).
Cellular Raf-1 levels determine hESC motility and morphology
Western blot analysis of Raf-1 revealed that Raf-1 levels in Ec-hESC were 65% (P < 0.005) lower compared with respective levels in Eu-hESC (Fig. 3A). In Eu- and Ec-hESC, the gradual down-regulation of Raf-1 levels compared with the Co-hESC was associated with an increased E/R/M phosphorylation (Fig. 3B, left panel). However, the levels of E/R/M in Eu- and Ec-hESC were not significantly different when compared with Co-hESC (Fig. 3B, right panel). The difference in E/R/M phosphorylation between Eu- and Ec-hESC was also associated with ROCKII hyperactivation in Ec-hESC versus Co- and Eu-hESC (Fig. 3C). In Ec-hESC, ROCKII activity was 25% (P < 0.05) higher and the migration rate was 75% (P < 0.005) lower versus Eu-hESC (Fig. 3C and D). Thus, Raf-1 levels might determine the morphology of hESC.
Fig 3.
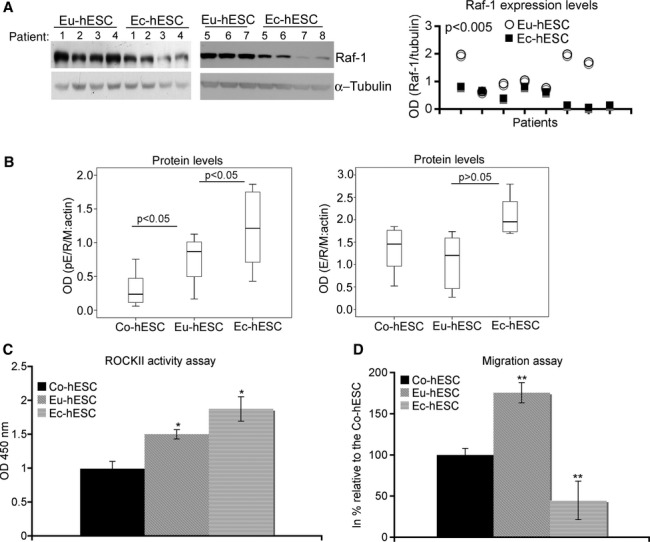
Cellular Raf-1 levels determine hESC motility. (A) Western blot analysis of Raf-1 expression levels in individual hESC cultures (numbered at the top of the immunoblot and corresponding to the sample ID number given in Table S1 (for Ec-hESC; n = 7) and in [25] for Eu-hESC; n = 7) is shown on the left and the graphical representation of the analysis on the right. The level of the protein is shown as optical density (OD) of the Western blot lanes normalized to the OD of α-tubulin. (B) Box plots of the data obtained using Western blot analysis pE/R/M (left panel; n = 8 per group) and total E/R/M levels (right panel; n = 8 per group) in Co-, Eu- and Ec-hESC are given. The levels of the proteins are shown as OD of the Western blot lanes normalized to the OD of β-actin. The corresponding P-values obtained after anova and Post hoc analysis are additionally inserted on the top of the box plots. (C) The ROCK II activity in Co-, Eu- and Ec-hESC is shown as average values of absorbance (OD 450 nm) determined in six biological replicates (mean ± S.D.) and normalized to the absorbance in Co-hESC. The statistical analysis of the data was performed with anova followed by Post-hoc test, *P < 0.05. (D) The analysis of cell migration in Co-, Eu- and Ec-hESC is given in per cent as average values of migrating cells from biological triplicates relative to that in Co-hESC (mean value set to 100%).
The downstream Raf-1/ROCKII effectors E/R/M, paxillin, MYPT1 and MLC regulate shape and migration of hESC
Immunoblot analysis of the downstream Raf-1/ROCKII effectors E/R/M, paxillin and MYPT1 showed increased phosphorylation of these proteins in Raf-1 knockdown Co- and Eu-hESC (Fig. 4A). Knockdown of Raf-1 to 10% (P < 0.005) in Co- and to 20% (P < 0.005) in Eu-hESC resulted in 1.53-fold in Co-hESC (P < 0.005) and 2.93-fold in Eu-hESC (P < 0.005) increase of E/R/M phosphorylation versus respective controls. In Raf-1 knockdown Co- and Eu-hESC, the phosphorylation levels of paxillin were 2-fold higher (P < 0.05 for Co-hESC and P < 0.005 for Eu-hESC) compared with the controls. MYPT1 is a direct Raf-1 target protein implicated in the regulation of cell migration. In particular, it is known that Raf-1 mediated phosphorylation results in MYPT1 inactivation via a mechanism involving direct Raf-1/MYPT1 interaction [31]. To evaluate the role of Raf-1 in the regulation of MYPT1, we performed co-immunoprecipitation analysis. We showed that Raf-1 directly interacts with MYPT1 in Co-, Eu- and Ec-hESC (Fig. S3 and data not shown). However, in Raf-1 knockdown Co- and Eu-hESC, the levels of phosphorylated (inactivated) MYPT1 protein were 6.3-fold and 4.7-fold higher in Co- and Eu-hESC, respectively (P < 0.005), compared with the levels in siRNA controls (Fig. 4A, left and lower right panels). These data suggest that either Raf-1 is not a direct regulator of MYPT1 phosphorylation or that other mechanisms could compensate for the loss of Raf-1 in these cells. In Ec-hESC, knockdown of Raf-1 to 32% (P < 0.005) did not yield to a significant change in either E/R/M or paxillin phosphorylation compared with siRNA controls, but caused a reduction of MYPT1 phosphorylation (Fig. 4B). In these cells, phosphorylated MYPT1 protein was 60% lower (P < 0.005) compared with respective controls (Fig. 4B), and ROCKII activity was 1.3-fold (P < 0.05) higher compared with respective controls (Fig. 4C).
Fig 4.
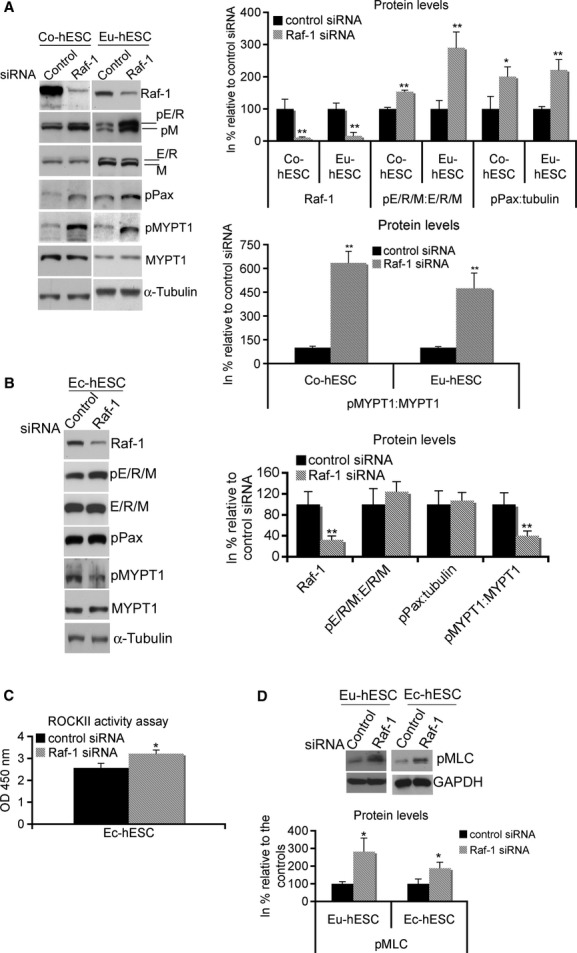
Raf-1 knockdown affects phosphorylation levels of downstream target proteins. (A) Total cell lysates (TCL) from Co- and Eu-hESC cells were analysed for Raf-1, pE/R/M, phospho-paxillin (pPax), E/R/M, pMYPT1, MYPT1 and α-tubulin, 48 hrs after their transfection with either Raf-1 or control siRNA. The Raf-1 and pPax levels were normalized by total α-tubulin. The pE/R/M and pMYPT1 levels were normalized by E/R/M and MYPT1 respectively. Representative blots from biological triplicates (left panel) and graphical representation of Raf-1, pE/R/M, pPax and pMYPT1 (right panel) are shown as protein levels in% (mean value ± S.D.) relative to their normalized levels (mean value set to 100%) in cells transfected with control siRNA, **P < 0.0001. (B) TCLs from Ec-hESC were analysed for Raf-1, pE/R/M, E/R/M, pPax, pMYPT1, MYPT1 and α-tubulin 48 hrs after their transfection with Raf-1 siRNA or control siRNA. The levels of the analysed proteins were normalized as described in (A). Representative blots from biological triplicates and graphical representation of Raf-1, pE/R/M, pMYPT1 and pPax as described in (A) are shown. **P < 0.0001. (C) ROCK II activity in Raf-1 knockdown Ec-hESC is shown as average values of absorbance (OD 450 nm) determined in four biological replicates (mean ± S.D.) and normalized to the absorbance in siRNA controls (mean value set to 1), *P < 0.05. (D) TCL from Eu- and Ec-hESC were analysed for pMLC, 48 hrs after their transfection with Raf-1 siRNA or control siRNA. The pMLC levels were normalized by GAPDH. Representative blots from biological triplicates and graphical representation of pMLC are shown as protein levels in% (mean value ± S.D.) relative to their normalized levels (mean value set to 100%) in cells transfected with control siRNA, *P < 0.05.
The knockdown of Raf-1 in hESC caused an increase of MLC phosphorylation levels, which were 2.8-fold (P < 0.05) and 1.9-fold (P < 0.05) higher in Eu- and Ec-hESC, respectively, compared with the levels in siRNA controls (Fig. 4D and data not shown).
Kinase active Raf-1 is required for MYPT1 inactivation but not for ROCKII activation
To further analyse whether Raf-1 activity is required for MYPT1 inactivation, Eu-hESC were treated with Raf-1 kinase inhibitors ZM336372 and GW5074 (1 μM) for 24 hrs. The inhibition of Raf-1 reduced MYPT1 phosphorylation to approximately half (P < 0.05) of the levels of untreated cells (Fig. 5, upper panel), showing that Raf-1 kinase activity is required for the direct regulation of MYPT1 phosphorylation. Similar results were obtained for Co- and Ec-hESC under GW5074 administration (Fig. 5, lower panel).
Fig 5.
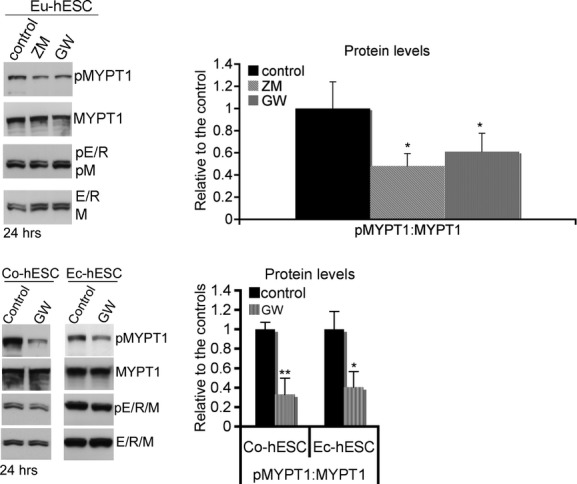
Western blot analysis of MYPT1 and E/R/M phosphorylation analysed 24 hrs after treatment of hESC with specific Raf-1 inhibitors (ZM336372 and GW5074 – 1 μM) is shown on the left. Representative blots from three independent experiments are given. Graphical representation of pMYPT1 (right panels) is shown as protein levels in% (mean value ± S.D.) relative to their normalized levels (mean value set to 1) in the non-treated controls, *P < 0.05.
The E/R/M proteins are known effectors for ROCKII and MYPT1 [20, 32]. The inhibition of Raf-1 kinase activity in Eu-hESC was not able to affect the levels of E/R/M phosphorylation (Fig. 5), thereby excluding MYPT1 as a regulator of E/R/M phosphorylation. Collectively, these data also suggest that in Co- and Eu-hESC, the kinase activity of Raf-1 is required for MYPT1 inactivation, but not for the activation of ROCKII signalling. ROCKII regulates cell body contraction during migration by acting on actomyosin contractility and directly affecting MLC phosphorylation, both by inhibiting MYPT1 and by phosphorylation of MLC [19]. To evaluate whether ROCKII is able to regulate these cytoskeleton proteins and to compensate the loss of Raf-1 in knockdown Co- and Eu-hESC, we performed ROCKII siRNA knockdown.
ROCKII knockdown in hESC
In contrast to the Raf-1 inhibition, ROCKII siRNA knockdown caused a significant reduction of E/R/M, MYPT1, MLC and paxillin phosphorylation in Co- and Eu-hESC (Fig. 6A) versus controls, suggesting that these cytoskeleton proteins are direct effectors for ROCKII. Similar to Co- and Eu-hESC, in Ec-hESC ROCKII knockdown led to reduction of MYPT1 and MLC phosphorylation, but did not affect the activity of E/R/M (Fig. 6B, left panel). In addition, ROCKII knockdown in Ec-hESC led to an increase in paxillin phosphorylation. In these cells, phosphorylated paxillin levels were 2-fold (P < 0.005) higher versus respective controls (Fig. 6B, left and middle panels). Surprisingly, this effect was associated with an increase (22%; P < 0.02) in the migration rate of the ROCKII knockdown Ec-hESC versus controls (Fig. 6B, right panel) on the background of reduced ROCKII activity (Fig. 6C, left panel). However, these cells did not show a significant change in their phenotype. Similar to Raf-1 knockdown Ec-hESC, ROCKII siRNA knockdown cells had preserved their contracted cell morphology (Fig. 6C, right panel).
Fig 6.
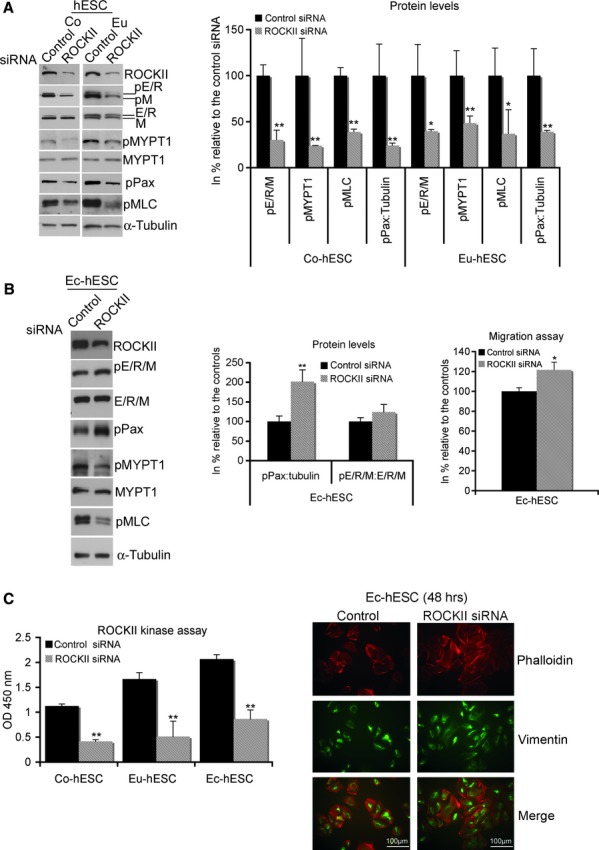
(A) Western blot analysis of ROCKII knockdown in Co- and Eu-hESC, showing down-regulation of E/R/M, MYPT1, paxillin and MLC phosphorylation in knockdown cells versus respective siRNA controls. Representative blots of six independent experiments (left panel) and graphical representation of the levels of phosphorylated proteins (right panel) normalized by α-tubulin are shown as protein level in % (mean ± SD) relative to their normalized levels (set to 100%) in cells transfected with control siRNA, *P < 0.05 and **P < 0.0001. (B) Western blot analysis of ROCKII knockdown in Ec-hESC showing up-regulation of pPax and down-regulation of both pMYPT1 and pMLC in the knockdown cells versus siRNA controls. Representative blots of four independent experiments (left panel) and graphical representation of the level of pPax and pE/R/M (middle panel) normalized either by α-tubulin or E/R/M are shown. The levels are presented in% (mean ± S.D.) relative to their normalized level (set to 100%) in cells transfected with control siRNA, **P < 0.005. The analysis of cell migration in ROCKII knockdown Ec-hESC is given on the right. Average values of migrating cells from four biological replicates are shown in% relative to the values of siRNA controls (mean value set to 100), *P < 0.02. (C) The activity of ROCKII in Co-, Eu- and Ec-hESC is shown as average values of absorbance (OD 450 nm) determined in biological triplicates (mean ± S.D.) and normalized to the absorbance measured in control siRNA treated Co-hESC (mean value set to 1), **P < 0.005 (left panel). Immunofluorescence analysis of the cytoskeleton in Ec-hESC, 48 hrs after their transfection with either ROCKII siRNA or control siRNA and visualized by phalloidin (red), vimentin (green) and merge (red and green) is shown on the right. Scale bar line = 100 μm.
Effects of ROCKII inhibition on MYPT1 phosphorylation
To get an insight into the mechanism of MYPT1 regulation in hESC, we determined the effects of ROCKII inhibition on MYPT1 phosphorylation under Raf-1 knockdown in Eu- and Ec-hESC. Western blot analysis showed that 48 hrs after Raf-1 knockdown, the administration of 10 μM Y-27632 for additional 24 hrs caused significant down-regulation of MYPT1 phosphorylation in Raf-1 knockdown Eu-hESC, but not in Ec-hESC (Fig. S4). The inhibition of ROCKII alone did not affect MYPT1 phosphorylation levels in both cell types. In summary, these data suggest that ROCKII is able to compensate for the loss of Raf-1 function on MYPT1 phosphorylation in knockdown Eu-hESC, but not in Ec-hESC.
Discussion
In the present study, we aimed to elucidate the differences in basal cell migration and the mechanism by which the Raf-1/ROCKII signalling pathway regulates motility of human endometrial stromal cells obtained from tissue samples of patients with and without endometriosis. In contrast to the previous observations [33], we show that Co-, Eu- and Ec-hESC exhibit different basal migratory potentials in the absence of additional steroid hormone stimulation, i.e. increased migration in Eu- and decreased migration in Ec-hESC versus Co-hESC. Together with our previous observations [25] that the levels of Raf-1 protein gradually decrease from Co- to Eu-hESC, we demonstrate that in Ec-hESC Raf-1 levels are even lower than in Eu-hESC. This reduction in Raf-1 levels was associated with a more contracted cellular morphology, impaired cell migration and hyperactive ROCKII signalling. The mechanism by which Raf-1 levels determine the differences in Co- and Eu-hESC migration involves direct action on MYPT1 phosphorylation and indirect regulation of the levels of E/R/M, paxillin, MYPT1 and MLC phosphorylation at the level of ROCKII. The inhibition of Raf-1 did not cause a significant change in the levels of E/R/M phosphorylation, showing that in Co- and Eu-hESC Raf-1 activity is dispensable for the activation of ROCKII and its downstream effectors. Such a mechanism of regulation was previously published in other cell types [34, 35]. We were able to show that the hyperactivation of ROCKII in Ec-hESC is associated with differences in Raf-1 dependent regulation of cell migration, compared with Co- and Eu-hESC. First, the knockdown of Raf-1 in Ec-hESC was neither able to mediate changes in the phenotype nor in the levels of E/R/M and paxillin phosphorylation compared with Co- and Eu-hESC. These data suggest that in contrast to Co-and Eu-hESC, where the cellular Raf-1 levels regulate the rate of migration, the low cellular Raf-1 content in Ec-hESC might ensure their restricted migration by preserving the contracted cellular phenotype. This is in line with the observation that Ec-hESC exhibit an enhanced contractile profile compared with Co-hESC [28]. Second, we further show that ROCKII directly regulates E/R/M phosphorylation in Co- and Eu-hESC. However, in Ec-hESC, ROCKII knockdown did not affect E/R/M activity, arguing for a ROCKII independent mechanism of E/R/M activation. Such a mechanism was previously shown in different cell types [36, 37].
E/R/M phosphorylation enables E/R/M to cross link actin filaments to the plasma membrane [38]. ROCKII induces paxillin and FAK hyperphosphorylation [39] thereby promoting stable focal adhesion formation and reduced cell motility. ROCKII is also able to phosphorylate vimentin, which leads to collapse of the vimentin filaments into perinuclear aggregates [40], a feature that we demonstrated here in Raf-1 knockdown Eu-hESC. This explains the contracted appearance and the compact cortical actin localization in Co- and Eu-hESC lacking Raf-1 protein and in untreated Ec-hESC.
It was recently shown that an in vitro-induced decidualization of Eu- and Ec-hESC could inhibit cellular contractility via suppression of ROCK-mediated signalling [41]. Our observations suggest that Eu-hESC could be more resistant to decidualization signals compared with Co-hESC based upon an increased activation of Raf-1/ROCKII signalling. Impaired decidualization during the menstrual cycle was already seen in endometrium of patients with endometriosis [42].
In hESC, Raf-1 directly phosphorylates and inactivates MYPT1, and this process requires kinase active Raf-1 protein. We show that in Raf-1 knockdown Co- and Eu-hESC MYPT1 is hyperphosphorylated. As MYPT1 dephosphorylates MLC, MYPT1 inactivation increases cell contractility, a phenotype seen in Raf-1 knockdown cells. However, the inactivation of MYPT1 seems to be entirely inconsistent with the levels of Raf-1 protein. We observed that in Co- and Eu-hESC, the lack of Raf-1 in this context is compensated by the hyperactivation of ROCKII, which itself is able to inhibit MYPT1 by phosphorylation [43] and to phosphorylate myosin B light chain [44]. In Ec-hESC, however, the lack of Raf-1 leads to a reduction of MYPT1 inactivation and an increased MLC phosphorylation versus respective controls. In these cells, reduced Raf-1 levels were not compensated by the activation of ROCKII as seen in Co- and Eu-hESC. We speculate here that the differences in the regulation of MYPT1 phosphorylation between Co-, Eu- and Ec-hESC under Raf-1 knockdown are associated with the preservation of Ec-hESC morphology.
Proteins that serve as membrane-cytoskeleton linkers, such as E/R/M, have been identified as substrates of MYPT1 [20, 32]. The phosphorylation of these proteins needs to be sustained to extend filopodia formation [45]. Thus, downstream of Raf-1, ROCKII and MYPT1 are supposed to collectively control the activation of a subset of signalling pathways to regulate cytoskeleton organization in vivo. In Co-hESC, the balance in the functional pathway ensures normal cell motility. We recently showed that Raf-1 levels in untreated Eu-hESC are accompanied with an increased ROCKII activity and enhanced motility versus Co-hESC [25]. Together with our observations that ROCKII regulates E/R/M and paxillin phosphorylation and inactivates MYPT1, we suggest that in untreated Eu-hESC, increased basal cell migration is due to activation of ROCKII signalling leading to enhanced levels of basal E/R/M, paxillin and MYPT1 phosphorylation. In the context of MYPT1 regulation, this is in line with the previously shown function of MYPT1 Tyr696 phosphorylation that allows de-adhesion necessary for cell movement at the rear of the cells [46, 47]. In contrast, further reduction of Raf-1 levels by siRNA knockdown in Co- and Eu-hESC and natively in Ec-hESC contracts hESC and dramatically reduces basal cell migration.
High levels of E/R/M phosphorylation are associated with a more invasive Ec-hESC phenotype [22]. De novo synthesis of ezrin is involved in the acquisition of metastatic potential in endometrial cancer cells [21]. However, in our in vitro cell system, the high E/R/M phosphorylation in Ec-hESC was associated with increased cellular contractility and reduced migratory potential. These data argue against the proposed invasive Ec-hESC phenotype [22] and suggest that increased levels of E/R/M phosphorylation are most probably needed to restrict the ectopic lesions at the place of initial implantation, thereby preventing metastatic disease. We further show that ROCKII knockdown in Ec-hESC leads to up-regulation of paxillin phosphorylation and to activation of cellular motility compared with respective controls. Ezrin is able to trigger FAK activation in signalling events that are not elicited by cell–matrix adhesion [48]. FAK-mediated phosphorylation of paxillin can initiate an autoregulatory loop, which is independent from Raf-1/ROCKII-mediated FAK and paxillin regulation, and involves MAPK family member-ERK-2 [49, 50]. Although there are other possible scenarios (see [51] for review), we speculate that there might be a regulatory cycle in which FAK activation and signalling to ERK-2 can first function to promote FAK release from existing focal contacts and then, through ERK-2 mediated phosphorylation of paxillin, to promote FAK re-binding and activation at new focal contacts in migrating cells. In ROCKII knockdown Ec-hESC increased paxillin phosphorylation might be associated with an increased migration of these cells.
In conclusion, we provide insight into the mechanism of Raf-1/ROCKII-mediated reorganization of the cytoskeleton and the regulation of basal cell migration in hESC with emphasis on the differences between Co-, Eu- and Ec-hESC. In particular, our findings suggest that cellular levels of Raf-1 adjust the threshold of hESC migration in the pathogenesis of endometriosis. This might have the potential to influence the future therapeutic handling of the disease.
Acknowledgments
This study was supported by FWF grant P19327-B02 (to Prof. Walter Tschugguel). We thank the staff members of the Department of Obstetrics and Gynaecology, Medical University of Vienna, for their cooperation and support in collecting the tissue samples needed for this study, and especially Erika Marton for her kind support.
Conflict of interest
The authors confirm that there are no conflicts of interest. Iveta Yotova and Ping Quan contributed equally to this work.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Effects of Raf-1 knockdown on Eu-hESC cell morphology. Immunofluorescence analysis of cytoskeleton by vimentin (green), phalloidin (red), DAPI (blue) and merge (green, red and blue) 48 hrs after transfection of Eu-hESC cells with siRNA are shown. The cells with both cortical F-actin (red) localization and collapsed vimentin (green) filaments are indicated with arrows, scale bar line = 50 µm.
Figure S2. Effects of Raf-1 knockdown on Co-hESC morphology. (A) Immunofluorescence analysis of the cytoskeleton by Raf-1 cellular levels (green), phalloidin (red) and merge (green and red) 48 and 144 hrs after their transfection with Raf-1 siRNA are shown on the left, scale bar line = 100 µm. Average number of cells with contracted phenotype (estimated by two independent investigators) out of three biological replicates are given on the right as % of cells with contracted phenotype relative to the total cell number (set to 100%, ± S.D.),**P < 0.005. (B) Effects of Raf-1 knockdown on cell migration in Co-hESC 48 and 144 hrs after initial transfection with either control (transfection reagent only) or control siRNA or Raf-1 siRNA are given. Averaged values of biological triplicates are shown as percentage of migrating cells relative to the untreated controls (mean value set to 100%), **P < 0.005.
Figure S3. Total cell lysates from two Eu-hESC (numbered in brackets on the top of the immunoblot) and one Ec-hESC were collected and used for immunoprecipitation with Raf-1 antibody and co-immunoprecipitation with MYPT1 antibody. Raf-1/MYPT1 complex formation is visualized by immunoblotting. 500 µg of lysates were used for immunoprecipitation with Raf-1. After blotting, the membrane was cut into two separate pieces at the level of 95 kD marker (indicated with black arrowed line on the blots) and the upper part was incubated with MYPT1 (mouse; BD Transduction Laboratories, MA, USA) to visualize the complex formation between Raf-1 and MYPT1. The down part was incubated with Raf-1 (mouse; BD Transduction Laboratories) to visualize the amount of bound Raf-1. The whole membrane was than subjected to an incubation with peroxidase (PE)-conjugated secondary anti-mouse antibody (Pierce Chemical Co., CA, USA) and the bound antibodies were detected and quantified as previously described [1]; *MYPT1/Raf-1 complex in Ec-hESC.
Figure S4. Effects of ROCKII inhibition on MYPT1 phosphorylation under Raf-1 knockdown in Eu- and Ec-hESC. Western blot analysis of MYPT1 phosphorylation analysed 24 hrs after treatment of either control or Raf-1 siRNA transfected Eu- and Ec-hESC is shown on the left. Representative blots from four independent experiments are given. Graphical representation of pMYPT1 (right panel) is shown as protein levels in % (mean ± S.D.) relative to their normalized levels (mean value set to 100%) in the non-treated controls, **P < 0.005.
Table S1. Subject characteristics. Ectopic endometrium obtained from ovarian endometrioma cysts, used for primary culture preparation and analysis.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Berkley KJ, Rapkin AJ, Papka RE. The pains of endometriosis. Science. 2005;308:1587–9. doi: 10.1126/science.1111445. [DOI] [PubMed] [Google Scholar]
- 2.Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–99. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 3.Dunselman GA, Groothuis PG. Etiology of endometriosis: hypotheses and facts. Gynecol Obstet Invest. 2004;57:42–3. [PubMed] [Google Scholar]
- 4.Klemmt PA, Carver JG, Koninckx P, et al. Endometrial cells from women with endometriosis have increased adhesion and proliferative capacity in response to extracellular matrix components: towards a mechanistic model for endometriosis progression. Hum Reprod. 2007;22:3139–47. doi: 10.1093/humrep/dem262. [DOI] [PubMed] [Google Scholar]
- 5.Flamini MI, Sanchez AM, Genazzani AR, et al. Estrogen regulates endometrial cell cytoskeletal remodeling and motility via focal adhesion kinase. Fertil Steril. 2011;95:722–6. doi: 10.1016/j.fertnstert.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Halme J, Hammond MG, Hulka JF, et al. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet Gynecol. 1984;64:151–4. [PubMed] [Google Scholar]
- 7.Taylor RR, Guerrieri JP, Nash JD, et al. Atypical cervical cytology. Colposcopic follow-up using the Bethesda System. J Reprod Med. 1993;38:443–7. [PubMed] [Google Scholar]
- 8.Osteen KG, Bruner KL, Sharpe-Timms KL. Steroid and growth factor regulation of matrix metalloproteinase expression and endometriosis. Semin Reprod Endocrinol. 1996;14:247–55. doi: 10.1055/s-2007-1016334. [DOI] [PubMed] [Google Scholar]
- 9.Bruner KL, Rodgers WH, Gold LI, et al. Transforming growth factor beta mediates the progesterone suppression of an epithelial metalloproteinase by adjacent stroma in the human endometrium. Proc Natl Acad Sci USA. 1995;92:7362–6. doi: 10.1073/pnas.92.16.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble LS, Simpson ER, Johns A, et al. Aromatase expression in endometriosis. J Clin Endocrinol Metab. 1996;81:174–9. doi: 10.1210/jcem.81.1.8550748. [DOI] [PubMed] [Google Scholar]
- 11.Mu L, Zheng W, Wang L, et al. Focal adhesion kinase expression in ovarian endometriosis. Int J Gynaecol Obstet. 2008;101:161–5. doi: 10.1016/j.ijgo.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Mu L, Zheng W, Wang L, et al. Alteration of focal adhesion kinase expression in eutopic endometrium of women with endometriosis. Fertil Steril. 2008;89:529–37. doi: 10.1016/j.fertnstert.2007.03.060. [DOI] [PubMed] [Google Scholar]
- 13.Lessey BA, Damjanovich L, Coutifaris C, et al. Integrin adhesion molecules in the human endometrium. Correlation with the normal and abnormal menstrual cycle. J Clin Invest. 1992;90:188–95. doi: 10.1172/JCI115835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starzinski-Powitz A, Handrow-Metzmacher H, Kotzian S. The putative role of cell adhesion molecules in endometriosis: can we learn from tumour metastasis? Mol Med Today. 1999;5:304–9. doi: 10.1016/s1357-4310(99)01497-5. [DOI] [PubMed] [Google Scholar]
- 15.Gaetje R, Kotzian S, Herrmann G, et al. Nonmalignant epithelial cells, potentially invasive in human endometriosis, lack the tumour suppressor molecule E-cadherin. Am J Pathol. 1997;150:461–7. [PMC free article] [PubMed] [Google Scholar]
- 16.Martelli M, Campana A, Bischof P. Secretion of matrix metalloproteinases by human endometrial cells in vitro. J Reprod Fertil. 1993;98:67–76. doi: 10.1530/jrf.0.0980067. [DOI] [PubMed] [Google Scholar]
- 17.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 18.Macara IG. Parsing the polarity code. Nat Rev Mol Cell Biol. 2004;5:220–31. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- 19.Kawano Y, Fukata Y, Oshiro N, et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–38. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsui T, Maeda M, Doi Y, et al. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–57. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohtani K, Sakamoto H, Rutherford T, et al. Ezrin, a membrane-cytoskeletal linking protein, is highly expressed in atypical endometrial hyperplasia and uterine endometrioid adenocarcinoma. Cancer Lett. 2002;179:79–86. doi: 10.1016/s0304-3835(01)00857-6. [DOI] [PubMed] [Google Scholar]
- 22.Ornek T, Fadiel A, Tan O, et al. Regulation and activation of ezrin protein in endometriosis. Hum Reprod. 2008;23:2104–12. doi: 10.1093/humrep/den215. [DOI] [PubMed] [Google Scholar]
- 23.Gentilini D, Busacca M, Di Francesco S, et al. PI3K/Akt and ERK1/2 signalling pathways are involved in endometrial cell migration induced by 17beta-estradiol and growth factors. Mol Hum Reprod. 2007;13:317–22. doi: 10.1093/molehr/gam001. [DOI] [PubMed] [Google Scholar]
- 24.Flamini MI, Sanchez AM, Goglia L, et al. Differential actions of estrogen and SERMs in regulation of the actin cytoskeleton of endometrial cells. Mol Hum Reprod. 2009;15:675–85. doi: 10.1093/molehr/gap045. [DOI] [PubMed] [Google Scholar]
- 25.Yotova IY, Quan P, Leditznig N, et al. Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum Reprod. 2011;26:885–97. doi: 10.1093/humrep/der010. [DOI] [PubMed] [Google Scholar]
- 26.Somigliana E, Vigano P, Parazzini F, et al. Association between endometriosis and cancer: a comprehensive review and a critical analysis of clinical and epidemiological evidence. Gynecol Oncol. 2006;101:331–41. doi: 10.1016/j.ygyno.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 27.Borghese B, Mondon F, Noel JC, et al. Gene expression profile for ectopic versus eutopic endometrium provides new insights into endometriosis oncogenic potential. Mol Endocrinol. 2008;22:2557–62. doi: 10.1210/me.2008-0322. [DOI] [PubMed] [Google Scholar]
- 28.Yuge A, Nasu K, Matsumoto H, et al. Collagen gel contractility is enhanced in human endometriotic stromal cells: a possible mechanism underlying the pathogenesis of endometriosis-associated fibrosis. Hum Reprod. 2007;22:938–44. doi: 10.1093/humrep/del485. [DOI] [PubMed] [Google Scholar]
- 29.Aghajanova L, Hamilton A, Kwintkiewicz J, et al. Steroidogenic enzyme and key decidualization marker dysregulation in endometrial stromal cells from women with versus without endometriosis. Biol Reprod. 2009;80:105–14. doi: 10.1095/biolreprod.108.070300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tulac S, Overgaard MT, Hamilton AE, et al. Dickkopf-1, an inhibitor of Wnt signaling, is regulated by progesterone in human endometrial stromal cells. J Clin Endocrinol Metab. 2006;91:1453–61. doi: 10.1210/jc.2005-0769. [DOI] [PubMed] [Google Scholar]
- 31.Broustas CG, Grammatikakis N, Eto M, et al. Phosphorylation of the myosin-binding subunit of myosin phosphatase by Raf-1 and inhibition of phosphatase activity. J Biol Chem. 2002;277:3053–9. doi: 10.1074/jbc.M106343200. [DOI] [PubMed] [Google Scholar]
- 32.Tsukita S, Yonemura S. Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J Biol Chem. 1999;274:34507–10. doi: 10.1074/jbc.274.49.34507. [DOI] [PubMed] [Google Scholar]
- 33.Gentilini D, Vigano P, Somigliana E, et al. Endometrial stromal cells from women with endometriosis reveal peculiar migratory behavior in response to ovarian steroids. Fertil Steril. 2010;93:706–15. doi: 10.1016/j.fertnstert.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Niault T, Sobczak I, Meissl K, et al. From autoinhibition to inhibition in trans: the Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J Cell Biol. 2009;187:335–42. doi: 10.1083/jcb.200906178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrenreiter K, Piazzolla D, Velamoor V, et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168:955–64. doi: 10.1083/jcb.200409162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsui T, Yonemura S, Tsukita S, et al. Activation of ERM proteins in vivo by Rho involves phosphatidyl-inositol 4-phosphate 5-kinase and not ROCK kinases. Curr Biol. 1999;9:1259–62. doi: 10.1016/s0960-9822(99)80508-9. [DOI] [PubMed] [Google Scholar]
- 37.Yonemura S, Matsui T, Tsukita S, et al. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J Cell Sci. 2002;115:2569–80. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- 38.Tsukita S, Yonemura S, Tsukita S. ERM proteins: head-to-tail regulation of actin-plasma membrane interaction. Trends Biochem Sci. 1997;22:53–8. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- 39.Sinnett-Smith J, Lunn JA, Leopoldt D, et al. Y-27632, an inhibitor of Rho-associated kinases, prevents tyrosine phosphorylation of focal adhesion kinase and paxillin induced by bombesin: dissociation from tyrosine phosphorylation of p130(CAS) Exp Cell Res. 2001;266:292–302. doi: 10.1006/excr.2001.5219. [DOI] [PubMed] [Google Scholar]
- 40.Sin WC, Chen XQ, Leung T, et al. RhoA-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol Cell Biol. 1998;18:6325–39. doi: 10.1128/mcb.18.11.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuno A, Nasu K, Yuge A, et al. Decidualization attenuates the contractility of eutopic and ectopic endometrial stromal cells: implications for hormone therapy of endometriosis. J Clin Endocrinol Metab. 2009;94:2516–23. doi: 10.1210/jc.2009-0207. [DOI] [PubMed] [Google Scholar]
- 42.Velarde MC, Aghajanova L, Nezhat CR, et al. Increased mitogen-activated protein kinase kinase/extracellularly regulated kinase activity in human endometrial stromal fibroblasts of women with endometriosis reduces 3′,5′-cyclic adenosine 5′-monophosphate inhibition of cyclin D1. Endocrinology. 2009;150:4701–12. doi: 10.1210/en.2009-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 44.Amano M, Ito M, Kimura K, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura N, Oshiro N, Fukata Y, et al. Phosphorylation of ERM proteins at filopodia induced by Cdc42. Genes Cells. 2000;5:571–81. doi: 10.1046/j.1365-2443.2000.00348.x. [DOI] [PubMed] [Google Scholar]
- 46.Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–38. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen DH, Catling AD, Webb DJ, et al. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J Cell Biol. 1999;146:149–64. doi: 10.1083/jcb.146.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poullet P, Gautreau A, Kadare G, et al. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J Biol Chem. 2001;276:37686–91. doi: 10.1074/jbc.M106175200. [DOI] [PubMed] [Google Scholar]
- 49.Liu ZX, Yu CF, Nickel C, et al. Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J Biol Chem. 2002;277:10452–8. doi: 10.1074/jbc.M107551200. [DOI] [PubMed] [Google Scholar]
- 50.Ishibe S, Joly D, Zhu X, et al. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell. 2003;12:1275–85. doi: 10.1016/s1097-2765(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 51.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.