

Abstract
Cardiac fibrosis after myocardial infarction (MI) has been identified as a key factor in the development of heart failure. Although dysregulation of microRNA (miRNA) is involved in various pathophysiological processes in the heart, the role of miRNA in fibrosis regulation after MI is not clear. Previously we observed the correlation between fibrosis and the miR-24 expression in hypertrophic hearts, herein we assessed how miR-24 regulates fibrosis after MI. Using qRT-PCR, we showed that miR-24 was down-regulated in the MI heart; the change in miR-24 expression was closely related to extracellular matrix (ECM) remodelling. In vivo, miR-24 could improve heart function and attenuate fibrosis in the infarct border zone of the heart two weeks after MI through intramyocardial injection of Lentiviruses. Moreover, in vitro experiments suggested that up-regulation of miR-24 by synthetic miR-24 precursors could reduce fibrosis and also decrease the differentiation and migration of cardiac fibroblasts (CFs). TGF-β (a pathological mediator of fibrotic disease) increased miR-24 expression, overexpression of miR-24 reduced TGF-β secretion and Smad2/3 phosphorylation in CFs. By performing microarray analyses and bioinformatics analyses, we found furin to be a potential target for miR-24 in fibrosis (furin is a protease which controls latent TGF-β activation processing). Finally, we demonstrated that protein and mRNA levels of furin were regulated by miR-24 in CFs. These findings suggest that miR-24 has a critical role in CF function and cardiac fibrosis after MI through a furin–TGF-β pathway. Thus, miR-24 may be used as a target for treatment of MI and other fibrotic heart diseases.
Keywords: myocardial infarction, cardiac fibrosis, microRNA-24, furin, TGF-β, telocyte
Introduction
Cardiac fibrosis after myocardial infarction (MI) plays a key part in regulating heart function in the development of heart failure [1]. It can determine the size, shape and wall thickness of ventricles. Excessive fibrosis leads to ventricular dilation, infarct expansion, and heart failure [2]. During cardiac remodelling, fibroblasts differentiate into myofibroblasts. These are fast-proliferating, α-smooth muscle cell actin (α-SMA)-positive cells with pronounced contractile and secretory properties [3]. Myofibroblasts can be induced by transforming growth factor (TGF)-β and lead to excessive matrix accumulation [1, 4].
Micro-ribonucleic acid (miRNA) molecules are short RNA sequences of 20–23 nucleotides. They act as negative regulators of gene expression by inhibiting mRNA translation or promoting mRNA degradation [5]. It is now apparent that miRNAs are the major players involved in all aspects of remodelling, growth, fibrosis, cell death, vascularization and contraction of cardiac tissue [6]. In the past few years, several miRNAs were found in cardiac interstitial cells such as fibroblasts or telocytes [7] and to regulate cardiac fibrosis. For example, miR-21 is known to regulate fibroblast proliferation and fibrosis [8], and miR-29 has been found to reduce collagen expression [9]. MiR-30 and miR-133 have been identified as regulators of connective tissue growth factor, and to further decrease production of collagen [10]. The role of miRNA in fibrosis in MI has only just started to unravel. Therefore, determining the function of the specific miRNAs involved in the pathogenesis of cardiac fibrosis may provide important new treatments for MI and other types of fibrotic heart disease.
The miR-24 cluster, which includes miR-23a, miR-24 and miR-27a, is abundant in the heart [11]; miR-23a has been demonstrated to regulate cardiac hypertrophy through NFATc3 [12]; miR-27, which is also mentioned as angiogenic miRNA was found in interstitial cells like telocytes [13]. Moreover, we previously found that expression of the miR-24 cluster was considerably changed and was associated with the degree of fibrosis in hypertrophic hearts [14, 15]. Herein we hypothesize that miR-24 may regulate cardiac fibrosis after MI. We used a MI model in mice and primary cardiac fibroblasts (CFs) to test this hypothesis.
Materials and methods
Mouse model of MI, echocardiography, lentiviral expression constructs and transfer in vivo, measurement of the area at risk (AAR) and triphenyltetrazolium chloride (TTC) stain MI areas, immunohistochemical analyses of mouse hearts and CFs, western blot analyses, quantitative real-time polymerase chain reaction (qRT-PCR), CFs culture and miR-24 transfection, transwell migration assay, ELISA test, fluorescence-activated cell sorting (FACS) analyses and statistical analyses are described in the ‘Materials and methods’ section in Appendix S1.
Results
MiR-24 expression is down-regulated after MI
We previously monitored miRNA expression and cardiac fibrosis in remodelling and reverse remodelling of the heart, and found that changes in the expression of miR-24 were associated with fibrosis. To ascertain if miR-24 is dysregulated during MI, we measured expression of the miR-24 at various time-points and different sections in a mouse model of MI. Expression of the miR-24 transcript was down-regulated after MI; expression peaked 1 week after MI, then gradually attenuated in infarct and remote areas over time, and was finally restored to normal levels at 28 days after MI (Fig. 1A). In the same way, expression of collagen-1, fibronectin and TGF-β1 in different areas in the MI model was detected at different times (Fig. 1B–D), a significant correlation was found between expression of miR-24 and fibrosis level.
Fig 1.
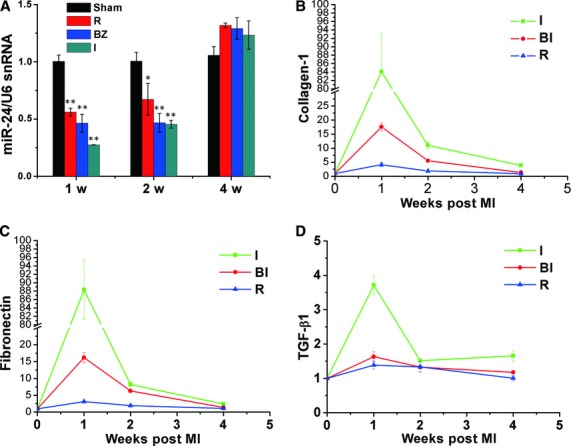
MiR-24 is down-regulated in different areas of the heart after MI. (A) miRNAs were isolated in different areas of the infracted heart and sham-opened hearts 1, 2, and 4 weeks after MI. qRT-PCR was done to determine levels of miR-24 in MI areas and in control groups. Compared with sham-operated hearts, miR-24 expression was significantly decreased in MI areas. (B–D) qRT-PCR was done to determine expression of collagen-1 (B), fibronectin (C), and TGF-β1 (D) in the same samples as (A) (I: infarct zone; BZ: border zone; R: remote zone; n = 3 for each time-point; mean ± S.E.M. *P < 0.05, **P < 0.01 compared with sham groups).
In vivo delivery of lentivirus-miR-24 improves heart function after MI
To examine if miR-24 delivery can improve cardiac function after MI, we injected lentiviral vectors (LV) intramyocardially to modify expression of miR-24 (n = 35 per group). Lentiviral vectors were previously shown to modify expression of miRNAs in vivo [16]. The CFs and a subset of MI tissue samples were detected by GFP-labelled lentiviral vectors 3–7 days after transfection, and the fluorescence of GFP in the CFs and around the injection area indicated an efficient degree of infection (Fig. S1A and B). The expression of GFP was low in the first 2 days, and increased after the third day (Fig. S1E). qRT-PCR revealed the miR-24 expression in injected and border areas was increased threefold by LV-miR-24 transfection 1 week after treatment (Fig. S1C).
After infection, post-MI lethality was not significantly different between the two groups (Fig. 2B). Infarct sizes were significantly reduced in LV-miR-24-treated mice. MiR-24 transfection resulted in 36.9% reduction in scar size quantified by performing masson trichrome staining (Fig. 2A and C). Echocardiography was done before and 2 weeks after left anterior descending artery (LAD) ligation. At baseline, left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) were similar in all groups. After LAD ligation, miR-24 improved LVFS and LVEF in the treatment group compared with the control group (Fig. 2D). Reduction in infarct size could have been caused by the decrease of apoptosis of healthy cardiac tissue or by a reduction in fibrosis. To address this issue, we undertook Evans blue/TTC staining to estimate the AAR and infarct size 24 hrs after MI. The extent of endangered myocardium INF/AAR between the two groups was similar (Fig. 2E). Furthermore, we detected the expression of miR-24 in the border zone (BZ) 24 hrs after MI, and found that the level of miR-24 did not change during the first 24 hrs after transfection (Fig. S1F). This may be the reason why TTC area was not changed after miR-24 treatment.
Fig 2.

Lentivirus-mediated miR-24 transfer in vivo results in reduced scar formation after MI. (A) Whole images of representative hearts two weeks after MI and Masson's trichrome staining of sections from two representative hearts. miR-24 reduced scar size and built a more compact region in the infarct zone (LV: left ventricle; RV: right ventricle). (B) The number of living and dead mice were counted two weeks after MI: there was no significant difference between LV-miR-24- and LV-GFP-infected groups. (LV-GFP: lentivirus-GFP; LV-miR-24: lentivirus-miR-24). (C) Quantification of infarct size as a percentage of epicardial and endocardial circumference occupied by the infarct on all heart slices of the left ventricle 2 weeks after MI (n = 6); measurements were repeated three times (technical triplicates). MiR-24 transfection resulted in 36.9% reduction in scar size. (D) M-mode echocardiography of representative hearts two weeks after MI. Ejection fraction (EF), and fractional shortening (FS) were measured by using high-resolution echocardiography two weeks after MI. *P < 0.05, **P < 0.01 (n = 9). miR-24 could improve heart function after MI. (E) Representative Evans blue/TTC stains on four continuous slices of left ventricle 24 hrs after MI of control mimic or miR-24 transfections. Blinded quantification of AAR size and infarct size (n = 3). Measurements were repeated thrice (technical triplicates). Bars: (A, E) 1 mm. There was no significant difference between the two groups.
In vivo delivery of miR-24 reduces cardiac fibrosis after MI
To examine if miR-24 can regulate cardiac fibrosis after MI, we isolated tissue from the infarct BZ and the entire heart 2 weeks after MI. By comparing LV-miR-24 with LV-GFP, it was observed by western blotting that collagen-1 protein expression decreased in the infarct BZ due to miR-24 treatment and that α-SMA (marker of myofibroblasts) was also decreased by miR-24 treatment (Fig. 3A and B). qRT-PCR revealed that up-regulation of the mRNA level of Col-1a2, Col-3, fibronectin and α-SMA in the BZ (Fig. 3C) or entire heart (Fig. 3D) of MI mice was attenuated in the group treated with miR-24. To confirm that miR-24 treatment could affect collagen synthesis, we stained ventricular sections for interstitial expression of collagen-1, fibronectin and α-SMA. Results indicated that miR-24 transfection could reduce the expression of collagen-1, fibronectin, and α-SMA in the infarct BZ (Figs 3E and S2). These findings suggest that miR-24 may decrease extracellular matrix (ECM) synthesis and prevent transformation of fibroblasts in the infarct BZ of the heart 2 weeks after MI.
Fig 3.
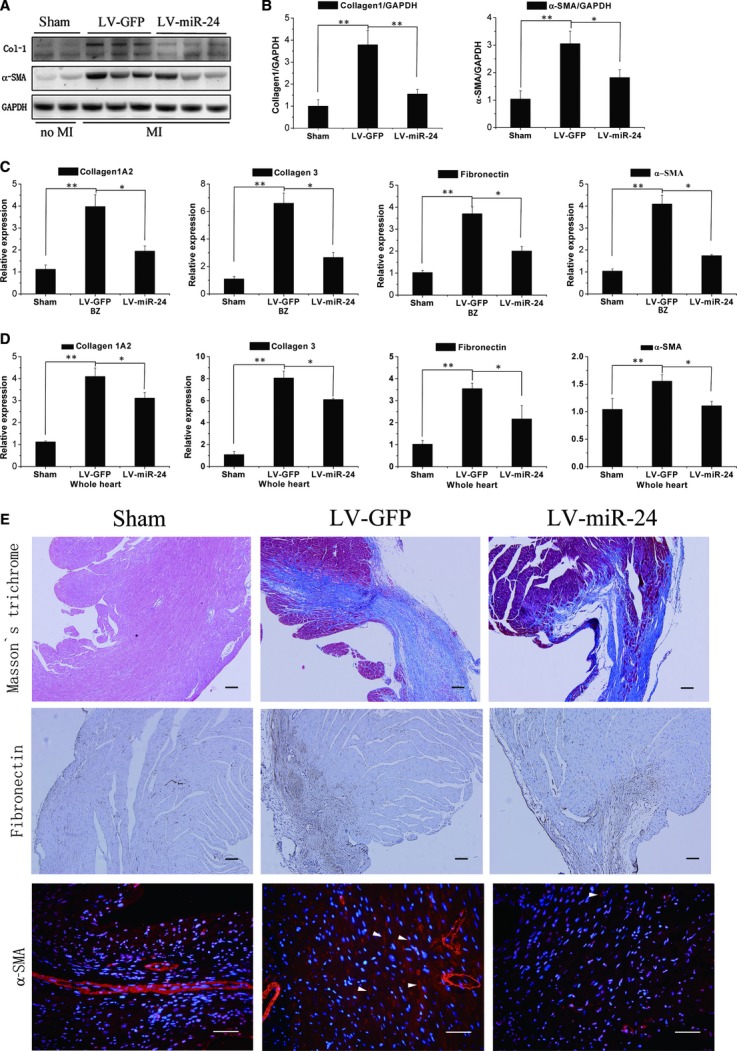
MiR-24 regulates myocardial fibrosis 2 weeks after MI (A, B) Mice receiving control lentiviral vector-GFP (LV-GFP) or lentiviral vector-miR-24 (LV-miR-24) were killed 2 weeks after MI. Protein levels of Col-1, α-SMA and GAPDH in the border zone were determined by western blotting, and densitometric quantification of protein expression (n = 6). (C, D) qRT-PCR was done to determine mRNA levels of Col-1A2, Col-3, fibronectin, and α-SMA in the border zone (C) or whole heart (D) of MI mice. Mean ± S.E.M., *P < 0.05, **P < 0.01 (n = 6). (E) Experiments were done as described in (B). Masson's trichrome blue was used to stain fibrosis, fibronectin was stained to detect ECM remodelling; bars: 100 μm; and α-SMA was stained to detect CF differentiation (arrows denote SMA-positive spindle-shaped myofibroblasts); bars: 50 μm.
MiR-24 down-regulates CF function in vitro
To verify the anti-fibrotic effects of miR-24 treatment observed in vivo, we investigated the effect of miR-24 on various functional aspects of CFs by transfecting oligonucleotide mimics and inhibitors of miR-24 into CFs. qRT-PCR assays confirmed that the expression levels of miR-24 clearly increased (>15-fold) and decreased (<20%) after transfection with mimics and inhibitors of miR-24, respectively (Fig. S1D). Overexpression of miR-24 was shown to correlate with the reduction in collagen-1 protein (by ≍ 40%) and mRNA synthesis. Inhibition miR-24 led to a twofold increase in the mRNA level of collagen-1 and a slight increase in the amount of collagen-1 protein (Fig. 4A–D). Overexpression of miR-24 correlated with a 60% decrease in the myofibroblast transformation marker α-SMA at protein and mRNA levels (Fig. 4A–D), and inhibition of miR-24 could increase α-SMA expression.
Fig 4.
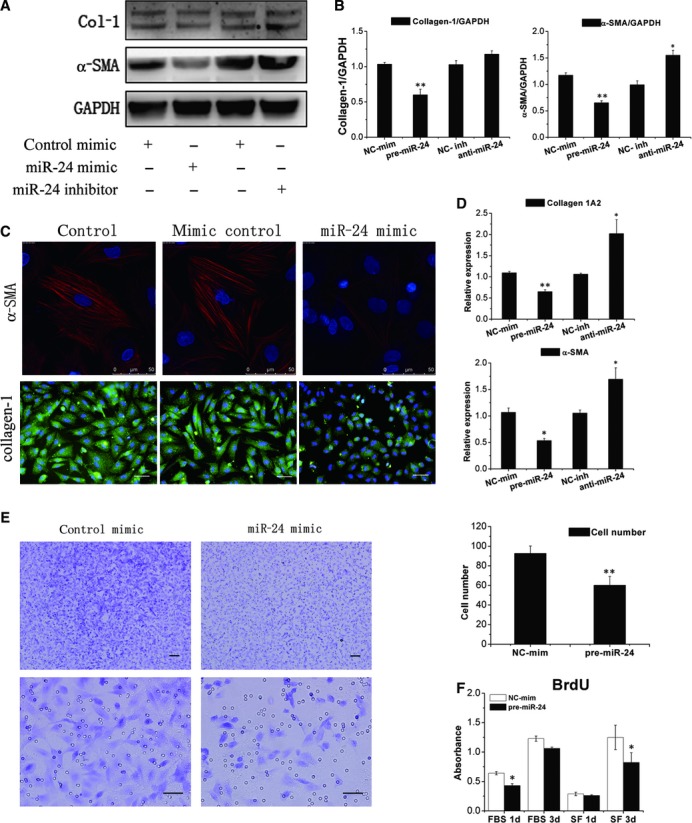
MiR-24 down-regulates CF fibrosis through regulation of the differentiation, and migration of CFs. (A, B) CFs were transfected with a pre-miR-24 or anti-miR-24. Expression of collagen-1 and α-SMA was determined by western blotting followed by densitometric quantification of protein expression (n = 6). (NC-mim, negative control mimic; NC-inh, negative control inhibitor). (C) Immunofluorescence showed that up-regulated miR-24 results in a decrease in the expression of α-SMA and collagen-1 protein in CFs compared with control. Bars: 50 μm. (D) Experiments were done as described in (A), and α-SMA and Col-1A2 mRNA levels in CFs were detected by qRT-PCR (n = 6). (E) A transwell migration assay was used to detect the role of miR-24 in CF migration. Representative images of CFs transfected with a control mimic and pre-miR-24 mimic migrated towards 1% serum media. Quantitative analyses demonstrated the difference in CF migration. Mean ± S.E.M., *P < 0.05, **P < 0.01 (n = 3). Bars: 100 μm up two figures, 50 μm down two figures. (F) BrdU incorporation in CFs grown in serum-free (SF) or with 10% foetal bovine serum (FBS) cultured for 24 and 72 hrs after transfection with a pre-miR-24 mimic (n = 3). Notable miR-24 could decrease proliferation of CFs.
The phenotype switch from CFs to myofibroblasts after MI is known to improve migratory ability and subsequent migration to the infarct zone [17]. To further detect if miR-24 could regulate the migration of CFs, transwell inserts were used. Upon stimulation by serum, miR-24-transfected CFs exhibited significantly reduced migration activity compared with control transfected cells (Fig. 4E). Furthermore, miR-24 has been reported to regulate apoptosis in cardiomyocytes and endothelial cells [18, 19]. However, through loss-of-function and gain-of-function experiments, we could not detect a significant difference of apoptosis in CFs (Fig. S3). To determine if miR-24 affects CF proliferation, we used BrdU incorporation assay to detect the proliferation level in CFs. Overexpression of miR-24 could down-regulate CFs proliferation independent of stimulation by FBS (Fig. 4F).
MiR-24 interaction with the TGF-β1 pathway
The TGF-β1 signalling pathway has been shown to regulate fibroblast function [1, 4]. We next assessed if the role of miR-24 in the activation of CFs is mediated by the TGF-β1 pathway. TGF-β1 up-regulates miR-24 expression in a time- and dose-dependent manner in CFs, and miR-24 expression is down-regulated by high doses of TGF-β1 (Fig. 5A and B). Phosphorylation of Smad2 and Smad3 is downstream of TGF-β signalling, and is necessary for most of the functions of TGF-β. Transfecting CFs with miR-24 precursors could attenuate Smad2/3 phosphorylation, but this regulation was attenuated in response to TGF-β1 stimulation (Fig. 5C). In addition, by performing western blotting and ELISA, we found that TGF-β protein in CFs and supernatants were down-regulated by overexpression of miR-24, and were up-regulated by a miR-24 inhibitor (Fig. 5D and E). Furthermore, we found that miR-24 could regulate the function of exogenously added angiotensin II induced collagen-1 changes in CFs, but could not affect TGF-β1 function (Fig. 5F). These data suggested that miR-24 may regulate the signalling events related to the upstream genes of TGF-β1.
Fig 5.
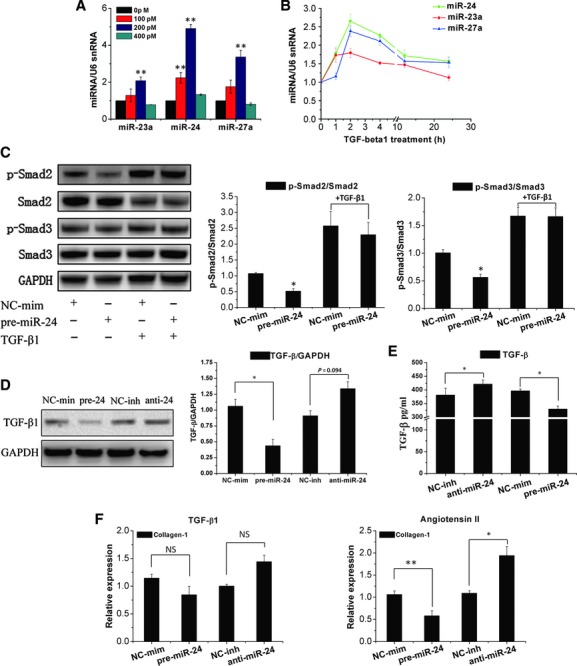
MiR-24 is induced by TGF-β1 and regulates the activities of TGF-β1. (A, B) CFs were treated with TGF-β1 at indicated concentrations (100, 200, 400 pM) for 4 hrs, or with 200 pM TGF-β1 for the indicated time. miR-24 cluster levels were determined by qRT-PCR (n = 3). (C) Smad2/3 phosphorylation were detected by western blotting in cultured CFs treated for 24 hrs with a pre-miR-24 mimic and for an additional 24 hrs with TGF-β1 (200 pM), and quantification of Smad2 or Smad3 phosphorylation normalization to total Smad2 and Smad3 (n = 3). (D) Expression of TGF-β protein was detected by western blotting and normalization to GAPDH (n = 3). (E) The TGF-β expression in culture supernatants was measured by ELISA. miR-24 can significantly inhibit TGF-β release compared with control group, and miR-24 inhibitor could induce TGF-β secretion (n = 3). (F) qRT-PCR determined Col-1A2 mRNA expression in CFs transfected with a miR-24 mimic or inhibitor for 24 hrs, and CFs were then treated with TGF-β1 (200 pM) or angiotensin II (100 nM) for an additional 24 hrs. miR-24 can regulate the function of angiotensin II (but not TGF-β1) in CFs. Mean ± S.E.M., *P < 0.05, **P < 0.01 (n = 3).
Targets of miR-24 function in CFs
Global changes in the transcriptional profile of CFs over-expressing miR-24 were assessed by microarray (data not shown). By using the TargetScan and Sanger database and bioinformatics analyses, we identified furin as a highly likely miR-24 target to regulate the TGF-β1 pathway. Furin is a protease and has been reported to regulate TGF-β maturation. Through target prediction tools we identified one miR-24-conserved binding site in furin mRNA (Fig. 6C). Loss-of-function and gain-of-function experiments indicated that the transfected miR-24 precursors in CFs significantly down-regulated furin expression at protein and mRNA levels, whereas inhibition of miR-24 up-regulated furin expression (Fig. 6A and B). To detect the function of furin in CFs, we used siRNA of furin to inhibit it in CFs and detected the expression of furin, collagen-1 and α-SMA by qRT-PCR and western blotting. The results showed that furin was reduced significantly after transfection, and the expression of collagen-1 and α-SMA was also downregulated in protein and mRNA level (Fig. 6D and E). This means that furin may regulate the fibrosis level in CFs.
Fig 6.
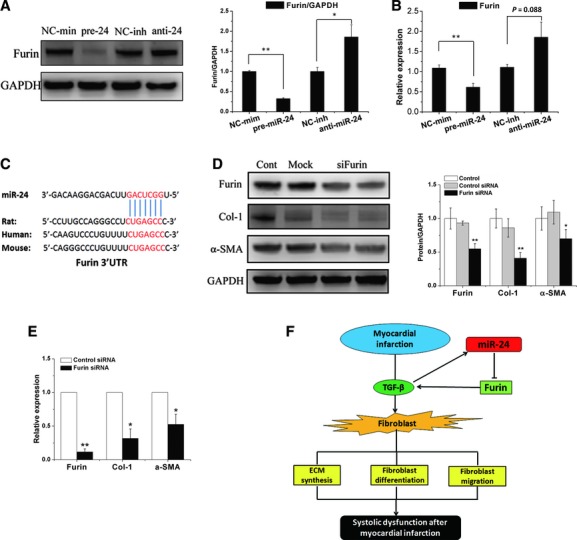
Target of miR-24 and proposed model of miR-24 function. (A) Expression of furin protein was detected by western blotting and normalization to GAPDH. (B) qRT-PCR revealed furin mRNA expression in CFs transfected with a pre-miR-24 mimic or anti-miR-24 inhibitor. Mean ± S.E.M., *P < 0.05, **P < 0.01 (n = 3). (C) Furin putative miR-24 binding sites (potential complementary residues shown in red). (D) CFs were transfected with siRNA of furin, and expression of furin, Collagen-1 and α-SMA was determined by western blotting and quantitative determination (n = 3). (E) qRT-PCR analysis for Furin, Collagen-1 and α-SMA mRNA expression. Mean ± S.E.M. *P < 0.05. **P < 0.01 compared with control siRNA group (n = 4). (F) Proposed model for the regulation of miR-24 in fibrosis after myocardial infarction.
Discussion
After MI, cardiac fibrosis plays a key part in regulating heart function [17]. Cardiac fibrosis remodelling after MI is a dynamic process. Collagen fibres are morphologically evident 1 week after MI, and an organized assembly of these fibres in the form of scar tissue becomes evident at week 2[20]. During the early-phase, reparative deposition of the ECM in the infarct region can maintain the structural integrity of the heart [21]. During later phases, down-regulation of fibrosis (especially in the infarct BZ) may benefit heart function after MI [1, 22, 23]. In this report, we demonstrated the functional involvement of miR-24 in MI fibrosis remodelling through regulation of the TGF-β1 pathway. First, we showed that miR-24 was down regulated after MI, and change in miR-24 expression was closely associated with fibrosis remodelling in the MI heart. Second, we found that miR-24 could improve heart function 2 weeks after MI by shortening the infarct segment through regulating the metabolism of fibrosis. Third, we demonstrated that miR-24 could decrease the differentiation of fibroblasts in the infarct BZ. In vitro experiments showed that miR-24 could decrease the synthesis of ECM proteins, reduce the conversion of fibroblasts to myofibroblasts and attenuate the serum-mediated migration of CFs. Through microarray analyses and bioinformatic analyses we demonstrated that furin may be the major target of miR-24 for regulation of the TGF-β1 pathway. This is the first evidence to suggest a novel role for miR-24 in regulating heart function after MI through regulation of cardiac fibrosis.
The TGF-β has an important role in modulating fibroblast phenotype fibrosis gene expression, and in promoting ECM deposition in the infarct region [4]. The miR-24 cluster has been reported to change TGF-β signalling through several pathways [24, 25]. TGF-β signalling in fibroblasts is based upon Smads [26]. Our data suggest that miR-24 can decrease smad2/3 phosphorylation, and reduce TGF-β secretion in CFs. These findings indicate that miR-24 may target a gene correlate with TGF-β. We used microarray analyses for in vitro identification of miR-24 target genes in the CFs of miR-24-transfect cells. Furin was the gene predicted to connect with TGF-β and with the conserved miR-24 binding site. This finding is consistent with earlier observations that miR-24 may target regulate the furin protein in human trabecular meshwork cells [27]. Furin is a protease, and has been reported to regulate angiotensin II-induced TGF-β activation [28], and to control latent TGF-β activation processing [29, 30]. Elevated furin level could increase collagen production in fibrosis [31]. In our experiment we also found target inhibition of furin could reduce the expression of col-1 and α-SMA. Hence, miR-24 may function in CFs through depressing furin function and then reducing TGF-β activation. A proposed model to explain the role of miR-24 in this process is outlined in Figure 6F.
During the duration of the present study and the writing of this manuscript, two studies demonstrated that miR-24 has different functions in MI. One study reported that overexpression of miR-24 inhibited apoptosis in cardiomyocytes, and was beneficial for MI [19]. Another investigation found that inhibiting miR-24 expression in endothelial cells could reduce apoptosis and limit infarct size in mice [18]. In the present study, we found another mechanism of miR-24 that improves the function of the MI heart through regulation of cardiac fibrosis. MiRNAs usually have multiple targets, and may have different functions in the same organ. For example, miR-21 and miR-29 have been found to have important roles in the growth and apoptosis of cardiac cells [32, 33], and in cardiac fibrosis remodelling [8, 9].
In this article we demonstrate that miR-24 overexpression results in reduction of infracted area through regulation of cardiac fibrosis. In addition we did not find the difference in infarct size 24 hrs after MI between the treatment and control groups, in view of the low expression of miRNA during the first day after transfection, we think it may be caused by different transfection methods. The previous studies have identified many kinds of vectors to carry miRNA, such as minicircle, adenovirus, antagomir, oligonucleotide and Lentivirus [8, 19, 32, 34–36]. These vectors could transfect miRNA via catheter-based injection [8], osmotic minipumps pumping [12], aorta arteries infusion [32] and direct intramyocardial injection [19, 35] and consequently modulate the expression of miRNA in vivo. Lentivirus has been demonstrated to stably express miRNA and generate long-term phenotypes for individual miRNAs [34, 37, 38]. In addition, transfected lentivirus-mediated gene directly into the heart of mice has been shown well tolerated based on low inflammation and immune response level to lentivirus [37, 39, 40].
However, our study does not exclude that miR-24 contribute to infarct reduction also by other mechanisms. In vivo study, we found that miR-24 could reduce cell apoptosis in the infarct region (Fig. S4A). Furthermore, in vitro study we found that overexpression of miR-24 in cardiomyocytes could reduce the apoptosis level induced by hypoxia/serum deprivation (SD) for 48 hrs detected by Caspase-Glo™ 3/7 Assay (Fig. S4B). To detect the function of miR-24 in endothelial cells we detected the expression of factor 8, which is the marker of endothelial cell [41] by histological staining, and we found that there was no significant difference between the miR-24 treatment and control of angiogenesis in infarct zone 1 week after MI (Fig. S5). Furthermore investigations are needed to elucidate the underlying mechanism of miR-24 in affecting the function of MI hearts.
In summary, anti-fibrosis therapy such as anti-TGF-β1 has been shown to benefit infarct healing by shortening and thickening the infracted segment [42]. We found that miR-24 can inhibit furin and interact with TGF-β1 to attenuate fibrosis in the infarct BZ to improve heart function. Fibrosis is a pathological feature common to numerous forms of heart disease, including: MI, ischaemic, dilated and hypertrophic cardiomyopathies and heart failure [43]. Effective therapies to prevent or treat cardiac fibrosis are not available [26]. The present study identified a novel approach of using miRNA therapeutics to regulate the fibrosis of CFs, which may consequently improve heart function and reduce scar size in MI patients.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of China (grant numbers 30872538 and 81170130) and a grant from the Major State Basic Research Development Program of China (973 Program; 2010CB529500).
We thank Professor Xiaodu Wang from the University of Texas (San Antonio, TX, USA) for his excellent language editing. We thank Jun Li, Bianmei Han, Chuangjue Cui, Xuewen Liu, Jianqing Jiao and Dr Jianfeng Hou, Jingzou Chen and Jinghai Chen for excellent technical assistance. This article has been edited by International Science Editing company.
Conflict of interest
All authors have no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 Materials and methods.
Fig. S1 Infection rate in vivo and in vitro (A) The up two figures show that transfected CFs with GFP-labeled lentiviral vectors for three days, and detected infect rate by fluorescence expression, the expression of green fluorescence indicated over 80% of CFs were infected. The below two images shows transfected cardiac fibroblasts with FAM™ dye-labeled miRNA for three days. And the green fluorescence in CFs showed the high transfection rate of cells appear infected. Bars: 100 μm. (B) MI tissue were transfected with GFP-labeled lentiviral vectors and isolated seven days after transfection. Bars: 50 μm. Green: GFP; blue: DAPI; white arrows denote infected areas. (C) LV-miR-24 was delivered into the mouse heart using a local delivery method. Seven days later, the heart was isolated and assayed by qRT-PCR. Expression in the border zone and remote zone were detected. Mean ± S.E.M., *P < 0.05, **P < 0.01 (n = 5). (D) Pre-miR-24 can up-regulate miR-24 by >15-fold. An anti-miR-24 inhibitor can down-regulate miR-24 to < 20% in CFs. (E) LV-GFP were transfected into CFs, the expression of GFP was low in the first 2 days, and increased after the third day. (F) LV-miR-24 and LV-GFP were delivered into mouse heart, and the expression of miR-24 was detected 24 hrs after transfection by qRT-PCR (n = 3).
Fig. S2 MiR-24 regulates α-SMA expression in BZ 2 weeks after MI The border zone of miR-24-treatmed MI hearts was stained with α-SMA (brown). miR-24 attenuated expression of α-SMA in the border zone induced by MI. Bars: up figures, 100 μm; down figures, 50 μm.
Fig. S3 miR-24 does not affect CFs apoptosis CFs were transfected with miR-24, and cultured in 2% FBS media for an additional 48 hrs. Cells undergoing the early stages of apoptosis were detected by FACS analyses. miR-24 treatment did not affect early apoptosis in CFs. Mean ± S.E.M. (n = 6).
Fig. S4 Effect of miR-24 on post-MI cell apoptosis. (A) Cell apoptosis was detected by TUNEL staining histological sections of LV-GFP and LV-miR-24 transfect hearts 1 week post-MI. Nuclei were visualized using Dapi. Bars: 200 μm. Green: TUNEL positive cells; blue: Dapi; white arrows denote TUNEL positive areas, miR-24 treatment could decrease the TUNEL level in MI heart (n = 3). (B) Cardiomyocytes transfected with pre-miR-24 mimic or anti-miR-24 inhibitor were treated with hypoxia/serum deprivation for 48 hrs. Caspase-3 and -7 activity was assayed by Caspase-Glo luminescence assay. Mean ± S.E.M., *P < 0.05, **P < 0.01 compared to the control group (n = 9).
Fig. S5 Effect of miR-24 on post-MI angiogenesis activity. The cardiac microvessel density was measured by Factor VIII-related antigen staining in the border zone 1 week after MI. And the microvessel density was quantified (right panel). Bars: 200 μm (up two panels), 50 μm (down two panels). Mean ± S.E.M., *P < 0.05, **P < 0.01 compared to the sham group (n = 3–6). There was no significant difference between the control and miR-24 transfected groups.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.van den Borne SW, Diez J, Blankesteijn WM, et al. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 2.Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol. 2010;48:558–63. doi: 10.1016/j.yjmcc.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 4.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 6.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419–29. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 7.Cismasiu VB, Radu E, Popescu LM. MiR-193 expression differentiates telocytes from other stromal cells. J Cell Mol Med. 2011;15:1071–4. doi: 10.1111/j.1582-4934.2011.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 9.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–32. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duisters RF, Tijsen AJ, Schroen B, et al. MiR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–8. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- 11.Cheng Y, Ji R, Yue J, et al. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Z, Murtaza I, Wang K, et al. MiR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:12103–8. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manole CG, Cismasiu V, Gherghiceanu M, et al. Experimental acute myocardial infarction: telocytes involvement in neo-angiogenesis. J Cell Mol Med. 2011;15:2284–96. doi: 10.1111/j.1582-4934.2011.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Xu R, Lin F, et al. MicroRNA: novel regulators involved in the remodeling and reverse remodeling of the heart. Cardiology. 2009;113:81–8. doi: 10.1159/000172616. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Zheng Z, Lin FQ, et al. [MiRNA changes in the reverse remodeling heart of rats] Zhonghua Xin Xue Guan Bing Za Zhi. 2010;38:745–50. [PubMed] [Google Scholar]
- 16.Gentner B, Schira G, Giustacchini A, et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods. 2009;6:63–6. doi: 10.1038/nmeth.1277. [DOI] [PubMed] [Google Scholar]
- 17.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Fiedler J, Jazbutyte V, Kirchmaier BC, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–30. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 19.Qian L, Van Laake LW, Huang Y, et al. MiR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–60. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Y, Cleutjens JP, Diaz-Arias AA, et al. Cardiac angiotensin converting enzyme and myocardial fibrosis in the rat. Cardiovasc Res. 1994;28:1423–32. doi: 10.1093/cvr/28.9.1423. [DOI] [PubMed] [Google Scholar]
- 21.Weber KT, Brilla CG. Factors associated with reactive and reparative fibrosis of the myocardium. Basic Res Cardiol. 1992;87:291–301. doi: 10.1007/978-3-642-72474-9_25. [DOI] [PubMed] [Google Scholar]
- 22.Small EM, Thatcher JE, Sutherland LB, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeuchi M, Tsutsui H, Shiomi T, et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64:526–35. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Sun Q, Zhang Y, Yang G, et al. Transforming growth factor-beta-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008;36:2690–9. doi: 10.1093/nar/gkn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan MC, Hilyard AC, Wu C, et al. Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J. 2010;29:559–73. doi: 10.1038/emboj.2009.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–12. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Luna C, Li G, Qiu J, et al. MicroRNA-24 regulates the processing of latent TGFbeta1 during cyclic mechanical stress in human trabecular meshwork cells through direct targeting of FURIN. J Cell Physiol. 2011;226:1407–14. doi: 10.1002/jcp.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stawowy P, Margeta C, Kallisch H, et al. Regulation of matrix metalloproteinase MT1-MMP/MMP-2 in cardiac fibroblasts by TGF-beta1 involves furin-convertase. Cardiovasc Res. 2004;63:87–97. doi: 10.1016/j.cardiores.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Blakytny R, Ludlow A, Martin GE, et al. Latent TGF-beta1 activation by platelets. J Cell Physiol. 2004;199:67–76. doi: 10.1002/jcp.10454. [DOI] [PubMed] [Google Scholar]
- 30.Dogar AM, Towbin H, Hall J. Suppression of latent transforming growth factor (TGF)-beta1 restores growth inhibitory TGF-beta signaling through microRNAs. J Biol Chem. 2011;286:16447–58. doi: 10.1074/jbc.M110.208652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ornatowski W, Poschet JF, Perkett E, et al. Elevated furin levels in human cystic fibrosis cells result in hypersusceptibility to exotoxin A-induced cytotoxicity. J Clin Invest. 2007;117:3489–97. doi: 10.1172/JCI31499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong S, Cheng Y, Yang J, et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–25. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye Y, Hu Z, Lin Y, et al. Downregulation of microRNA-29 by antisense inhibitors and a PPAR-{gamma} agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2010;87:535–44. doi: 10.1093/cvr/cvq053. [DOI] [PubMed] [Google Scholar]
- 34.Surdziel E, Eder M, Scherr M. Lentivirus-mediated antagomir expression. Methods Mol Biol. 2010;667:237–48. doi: 10.1007/978-1-60761-811-9_16. [DOI] [PubMed] [Google Scholar]
- 35.Hu S, Huang M, Li Z, et al. MicroRNA-210 as a novel therapy for treatment of ischaemic heart disease. Circulation. 2010;122:S124–31. doi: 10.1161/CIRCULATIONAHA.109.928424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scherr M, Venturini L, Battmer K, et al. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshimitsu M, Sato T, Tao K, et al. Bioluminescent imaging of a marking transgene and correction of Fabry mice by neonatal injection of recombinant lentiviral vectors. Proc Natl Acad Sci USA. 2004;101:16909–14. doi: 10.1073/pnas.0407572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshimitsu M, Higuchi K, Ramsubir S, et al. Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 2007;14:256–65. doi: 10.1038/sj.gt.3302839. [DOI] [PubMed] [Google Scholar]
- 39.Shai E, Falk H, Honigman A, et al. Gene transfer mediated by different viral vectors following direct cannulation of mouse submandibular salivary glands. Eur J Oral Sci. 2002;110:254–60. doi: 10.1034/j.1600-0722.2002.21200.x. [DOI] [PubMed] [Google Scholar]
- 40.Yoshimitsu M, Higuchi K, Dawood F, et al. Correction of cardiac abnormalities in fabry mice by direct intraventricular injection of a recombinant lentiviral vector that engineers expression of alpha-galactosidase A. Circ J. 2006;70:1503–8. doi: 10.1253/circj.70.1503. [DOI] [PubMed] [Google Scholar]
- 41.Sehested M, Hou-Jensen K. Factor VII related antigen as an endothelial cell marker in benign and malignant diseases. Virchows Arch A Pathol Anat Histol. 1981;391:217–25. doi: 10.1007/BF00437598. [DOI] [PubMed] [Google Scholar]
- 42.Okada H, Takemura G, Kosai K, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–7. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 43.van Rooij E, Olson EN. Searching for miR-acles in cardiac fibrosis. Circ Res. 2009;104:138–40. doi: 10.1161/CIRCRESAHA.108.192492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.