

Abstract
Edaravone, a novel antioxidant, acts by trapping hydroxyl radicals, quenching active oxygen and so on. Its cardioprotective activity against experimental autoimmune myocarditis (EAM) was reported. Nevertheless, it remains to be determined whether edaravone protects against cardiac remodelling in dilated cardiomyopathy (DCM). The present study was undertaken to assess whether edaravone attenuates myocardial fibrosis, and examine the effect of edaravone on cardiac function in rats with DCM after EAM. Rat model of EAM was prepared by injection with porcine cardiac myosin 28 days after immunization, we administered edaravone intraperitoneally at 3 and 10 mg/kg/day to rats for 28 days. The results were compared with vehicle-treated rats with DCM. Cardiac function, by haemodynamic and echocardiographic study and histopathology were performed. Left ventricular (LV) expression of NADPH oxidase subunits (p47phox, p67phox, gp91phox and Nox4), fibrosis markers (TGF-β1 and OPN), endoplasmic reticulum (ER) stress markers (GRP78 and GADD 153) and apoptosis markers (cytochrome C and caspase-3) were measured by Western blotting. Edaravone-treated DCM rats showed better cardiac function compared with those of the vehicle-treated rats. In addition, LV expressions of NADPH oxidase subunits levels were significantly down-regulated in edaravone-treated rats. Furthermore, the number of collagen-III positive cells in the myocardium of edaravone-treated rats was lower compared with those of the vehicle-treated rats. Our results suggest that edaravone ameliorated the progression of DCM by modulating oxidative and ER stress-mediated myocardial apoptosis and fibrosis.
Keywords: edaravone, oxidative stress, endoplasmic reticulum stress, apoptosis, fibrosis, dilated cardiomyopathy
Introduction
Inflammation and autoimmunity are involved in many cardiac diseases among which myocarditis, an inflammatory heart disease, causes both acute and chronic heart failure as a result [1]. A model of rat experimental autoimmune myocarditis (EAM) resembles human giant-cell myocarditis, and the recurrent form of EAM leads to dilated cardiomyopathy (DCM) [2]. Neurohumoral factors such as cytokines and chemokines, and myocardial remodelling including myocardial apoptosis, play important roles in the progression of EAM [3, 4]. Excessive production of reactive oxygen species (ROS) such as superoxide induced by inflammatory stimuli is an important observation in failing hearts and produces myocardial injury in autoimmune-mediated heart failure [5] and it has also been reported that the patients with heart failure were found with excess levels of ROS in their plasma [6]. Myocardial fibrosis probably plays an important role in both diastolic and systolic dysfunctions and has adverse clinical consequences that result in increases in mortality because of progressive heart failure [7]. Many kinds of cytokines, such as basic fibroblast growth factor, Angiotensin (Ang)-II, transforming growth factor (TGF)-β1, and collagen-III, have been suggested to play an important role in structural remodelling of the non-myocyte compartment of the myocardium after heart failure [8].
Edaravone, a novel free radical scavenger, may be an effective agent for myocardial inflammation [9] by combating oxidative stress. It was reported by various studies about the protective effect of edaravone against myocardial injury. Edaravone significantly reduced myocardial infarct size and improved cardiac function and left ventricular (LV) remodelling 14 days after infarction [10]. It was also reported that edaravone significantly attenuates pressure overload-induced cardiac hypertrophy mediated through its antioxidative function [11]. We have also reported the protective effects of edaravone against EAM induced by cardiac myosin, focusing on oxidative stress, endoplasmic reticulum (ER) stress, inflammatory cytokines and myocardial apoptosis [12]. But it is now of interest to identify whether treatment with edaravone can inhibit the progression of EAM into DCM.
Thus, the purpose of the present study was to test the effect of edaravone treatment against porcine cardiac myosin-induced chronic heart failure model, focusing on its inhibitory effects on oxidative stress, myocardial apoptosis and fibrosis.
Materials and methods
Materials
Edaravone was obtained from Mitsubishi Research, Japan and Lewis rats (male, 8 weeks old) were purchased from Charles River Japan Inc. (Kanagawa, Japan). All the chemicals and reagents were purchased from Sigma Aldrich, Tokyo, Japan unless otherwise mentioned.
Experimental design
All experiments were carried out using 8-week-old male Lewis rats and were performed in accordance with the guidelines of our institute.
Lewis rats (n = 30) were injected in the footpads with antigen-adjuvant emulsion in accordance with a procedure described previously [13]. In brief, porcine cardiac myosin was dissolved in phosphate-buffered saline at 5 mg/ml and emulsified with an equal volume of complete Freund's adjuvant with 11 mg/ml Mycobacterium tuberculosis H37RA (Difco Laboratories, Detroit, MI, USA). Experimental autoimmune myocarditis in rats was induced by immunization with 0.1 ml of emulsion once by subcutaneous injection into the rear footpads (0.1 ml to each footpad). The morbidity of EAM was 100% in rats immunized by this procedure. Twenty-eight days after immunization, the surviving Lewis rats were divided into three groups and received an intraperitoneal injection of edaravone [3 and 10 mg/kg/day; Group Ed 3 (n = 9) and Group Ed 10 (n = 9)] or vehicle [Group V (n = 8)] for 28 days. These doses were selected as per the previous report suggesting its cardioprotective effects [11]. Age-matched Lewis rats without immunization were used as normal controls [Group N (n = 6)].
Haemodynamic study
Eight weeks after immunization, cardiac function of each rat was measured by haemodynamic study, and cardiac pressure changes were recorded as described previously [14]. The rats were anaesthetized with 2% halothane in O2 during the surgical procedures preceding haemodynamic measurements; this concentration was then reduced to 0.5% to minimize the anaesthetic effect.
Transthoracic echocardiographic analysis
Echocardiographic studies were carried out with a 7.5-MHz transducer (Aloka Inc., Tokyo, Japan) under 0.5% halothane anaesthesia, which has produced no effect on measurement. The LV dimensions in diastole (LVDd) and systole (LVDs), percentage ejection fraction (%EF) and percentage fractional shortening (%FS) were estimated using M-mode measurements. Doppler analysis was carried out to estimate the E/A ratio [15].
Histopathology
The body weight (bw) of rats was noted just before the surgical procedure. After the echocardiographic analysis, the rats were killed, and the wet heart was isolated and weighed to calculate the ratio of heart weight (HW) to bw. The excised myocardium was kept in formalin and the mid-ventricle sections were then embedded with paraffin.
Analysis of cardiac apoptosis by terminal transferase-mediated dUTP nick-end labelling (TUNEL) assays
The TUNEL assay was performed as specified in the instructions for the in situ apoptosis detection kit (Takara Bio Inc., Shiga, Japan). Sections embedded in paraffin were mounted and examined using light microscopy. Digital photomicrographs were obtained by using a colour image analyser (CIA-102; Olympus, Tokyo, Japan) at 200× magnification, and 25 random fields from each heart were chosen and the number of TUNEL-positive nuclei was quantified in a blinded manner. For each group, three sections were scored for apoptotic nuclei. Only nuclei that were clearly located in cardiac myocytes were considered [16].
Azan-Mallory staining for myocardial fibrosis
The area of myocardial fibrosis in LV tissue sections stained with Azan-Mallory was quantified by using a colour image analyser (CIA-102; Olympus) and the blue fibrotic areas were measured as opposed to the red myocardium at 200x magnification. The results were presented as the ratio of the fibrotic area to the whole area of the myocardium [17, 18].
Immunohistochemical assay
Immunohistochemical staining was performed [19] with the formalin-fixed, paraffin-embedded cardiac tissue sections of the rats from different groups. After deparaffinization and hydration, the slides were washed in Tris-buffered saline (TBS; 10 mmol/l Tris HCl, 0.85% NaCl, pH 7.5) containing 0.1% bovine serum albumin. Endogenous peroxidase activity was quenched by incubating the slides in 0.6% H2O2 in methanol. To perform antigen retrieval, the sections were pre-treated with trypsin for 15 min. at 37°C. After overnight incubation with the goat polyclonal anti-collagen type III antibody (diluted 1:100) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 4°C, the slides were washed in TBS and horseradish peroxidase-conjugated secondary antibody was then added and the slides were further incubated at room temperature for 45 min. The slides were washed in TBS and incubated with diaminobenzidine tetrahydrochloride as the substrate, and counterstained with haematoxylin. A negative control without primary antibody was included in the experiment to verify the antibody specificity. Measurement of myocardial immunoreactivity for collagen-III was performed in 100 randomly selected fields in heart sections in 200x magnification by light microscopy.
In situ detection of superoxide production in hearts
Superoxide generation was estimated by dihydroethidium (DHE) staining as previously described [20]. To evaluate in situ superoxide production from hearts, unfixed frozen cross-sections of the specimens were stained with DHE (Molecular Probes, Eugene, OR, USA) according to the previously validated method. In the presence of superoxide, DHE is converted to the fluorescent molecule ethidium, which can then label nuclei by intercalating with DNA. Briefly, the unfixed frozen LV tissues were cut into 10-μm-thick sections and incubated with DHE 10 μM at 37°C for 30 min. in a light-protected humidified chamber. Fluorescence images (200x magnification) were obtained by using a fluorescence microscope equipped with a rhodamine filter.
Western immunoblotting analysis
This analysis was carried out as per the earlier method by Thandavarayan et al. [21]. The myocardial tissue samples obtained from different groups were homogenized with lysis buffer. Protein concentrations in these homogenized samples were measured by the bicinchoninic acid method. For Western blots, proteins were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and identified with the following polyclonal antibodies to quantify their myocardial levels: rabbit polyclonal anti-p47phox, goat polyclonal anti-p67phox, goat polyclonal anti-gp91phox, goat polyclonal anti-nox4, mouse polyclonal anti-osteopontin (OPN), rabbit polyclonal anti-TGF-β1, goat polyclonal anti-glucose regulated protein (GRP)78, mouse polyclonal anti-growth arrest and DNA damage inducible gene (GADD)153 and rabbit polyclonal anti-cleaved caspase-3 antibodies and rabbit polyclonal anti-glyceraldehyde 3 phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Biotechnology or Cell Signaling) (diluted 1:100). The cytosolic fraction was separated from the homogenate as per the previous method [22]. Briefly, the tissues were minced and added to ice-cold homogenizing buffer (250 mM sucrose, 20 mM Hepes-KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 1 mg/ml aprotinin, 1 mg/ml leupeptin). After 30 min. incubation on ice, cells were homogenized with polytron homogenizer (single stroke for 20 sec.). The homogenates obtained were then subjected to a series of centrifugations at 100,000×g for 60 min. to collect the cytosolic fractions for the estimation of cytochrome C level. According to the molecular weight of the primary antibody, we have used 7.5%, 10% and 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA), and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk or 5% bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA) in TBS-T (20 mmol/l Tris, pH 7.6, 137 mmol/l, NaCl, and 0.05% Tween).
After incubation with primary antibody, the bound antibody was visualized with respective horseradish peroxidase-coupled secondary antibody (Santa Cruz Biotechnology) and chemiluminescence-developing agents (Amersham Biosciences, Buckinghamshire, UK). The level of GAPDH was estimated in every sample. Films were scanned and band densities were quantified with densitometric analysis by using Scion Image program (Epson GT-X700, Tokyo, Japan). Finally, Western blot data were normalized with cardiac GAPDH.
Statistical analysis
All the values are expressed as means ± S.E.M. Statistical analysis of differences between the groups was performed by one-way analysis of variance, followed by Dunnett's or Tukey's test. A value of P < 0.05 was considered statistically significant.
Results
Effect of edaravone on myocardial dimensions
Although heart rate was not different among the four groups of rats, transthoracic echocardiographic studies of group V rats showed evidence for the cardiac remodelling with increased LVDs (7.42 ± 0.67 versus 3.44 ± 0.19 mm, P < 0.01), reduced %FS (15 ± 0.68 versus 49.2 ± 1.9%, P < 0.01) and %EF (28.8 ± 3.71 versus 85.1 ± 1.6%, P < 0.01) and reduced E/A ratio. These results indicate the impairment of cardiac function in the DCM rats of group V compared with that in group N. Treatment with edaravone (3 or 10 mg/kg) significantly prevented all of these changes compared with those in group V (Table 1 and Fig. 1).
Table 1.
Changes in haemodynamic and echocardiographic parameters in rats with dilated cardiomyopathy (DCM) after treatment with edaravone
| Parameter | Group N | Group V | Group Ed 3 | Group Ed 10 |
|---|---|---|---|---|
| Histopathology | ||||
| bw (g) | 412 ± 8 | 329 ± 5** | 338 ± 7** | 337 ± 8** |
| HW (g) | 0.96 ± 0.01 | 1.24 ± 0.03** | 1.06 ± 0.03 | 1.08 ± 0.01*# |
| HW/bw (g/kg) | 2.32 ± 0.03 | 3.62 ± 0.14** | 3.20 ± 0.11** | 3.27 ± 0.12** |
| Haemodynamic data | ||||
| HR (beats/min) | 378 ± 19 | 329 ± 8 | 349 ± 14 | 364 ± 7 |
| Mean BP (mmHg) | 99 ± 6 | 82 ± 4* | 75 ± 2** | 75 ± 3** |
| CVP (mmHg) | 1.0 ± 0.4 | 4.2 ± 0.4* | 3.3 ± 0.9 | 3.2 ± 0.8 |
| LVP (mmHg) | 128 ± 5 | 101 ± 4** | 118 ± 5# | 106 ± 1** |
| LVEDP (mmHg) | 19.7 ± 1.4 | 40.4 ± 2.3** | 33.8 ± 3.7* | 35.9 ± 4.4* |
| +dP/dt (mmHg/min) | 6446 ± 881 | 4205 ± 77* | 4769 ± 211 | 4530 ± 404 |
| −dP/dt (mmHg/min) | 7291 ± 1124 | 3933 ± 413** | 4054 ± 191** | 3730 ± 274** |
| Echocardiographic data | ||||
| LVDd (mm) | 6.7 ± 0.31 | 7.62 ± 0.32 | 7.56 ± 0.22 | 7.5 ± 0.1 |
| LVDs (mm) | 3.44 ± 0.19 | 7.42 ± 0.67** | 5.44 ± 0.12**,## | 5.48 ± 0.11**,## |
| FS (%) | 49.2 ± 1.9 | 15 ± 0.68** | 32.7 ± 1.49**,## | 25.8 ± 1.4**,##,† |
| EF (%) | 85.1 ± 1.6 | 28.8 ± 3.71** | 64.4 ± 1.58**,## | 56.3 ± 2.29**,## |
| E/A ratio | 2.94 ± 0.05 | 0.32 ± 0.01** | 1.73 ± 0.10**,## | 2.05 ± 0.16**,## |
Group N: age-matched intact rats; Group V: rats with DCM treated with vehicle; Group Ed3: rats with DCM treated with edaravone (3 mg/kg); Group Ed10: rats with DCM treated with edaravone (10 mg/kg); bw: body weight; HW: heart weight; CVP: central venous pressure; LVP: left ventricular pressure; LVEDP: left ventricular end-diastolic pressure; dP/dt: rate of intra-ventricular pressure rise and decline; LVDd: left ventricular dimension in diastole; LVDs: left ventricular dimension in systole; FS: fractional shortening; EF: ejection fraction. **P < 0.01 and *P < 0.05 versus Group N, ##P < 0.01 and ##P < 0.05 versus Group V, †P < 0.05 versus Group E3. Values are mean ± S.E.M. (one-way anova followed by Tukey's multiple comparison test).
Fig 1.

Echocardiographic data. Group N, age-matched intact rats; Group V, rats with dilated cardiomyopathy (DCM) treated with vehicle; Group Ed 3, rats with DCM treated with edaravone (3 mg/kg); Group Ed 10, rats with DCM treated with edaravone (10 mg/kg).
Effect of edaravone on cardiac hypertrophy
Dilated cardiomyopathy is a condition where there will be increased HW with respect to bw, indicating the cardiac hypertrophy and dilatation of the cardiac chambers. Here, in the present study, myocarditis was confirmed with the increased HW of the group V rats. The ratio (HW/bw) was found to be significantly higher (3.62 ± 0.14, P < 0.01) when compared with group N (2.32 ± 0.03). In the edaravone-treated rats, this value was less than that of group V rats (Table 1).
Effect of edaravone on myocardial fibrosis and its marker molecules
Progression of EAM to DCM involves myocardial remodelling in the form of extensive replacement fibrosis, which can be identified by the measurement of its marker proteins. The hearts from group V DCM rats showed massive fibrosis and increased expressions of OPN and TGF-β1 compared with those from group N. Azan-Mallory staining of the myocardial tissue sections revealed the extensive fibrosis in the myocardium of group V rats, whereas the edaravone-treated (3 or 10 mg/kg) rats were significantly protected from these changes (Fig. 2A). This antifibrotic effect has also been confirmed by immunohistochemical studies for deposition of collagen-III in the myocardial tissue sections of edaravone-treated rats (Fig. 2B). In addition, treatment with edaravone significantly reduced the myocardial expression of OPN and TGF- β1 than in group V DCM rats (Fig. 3A and B).
Fig 2.
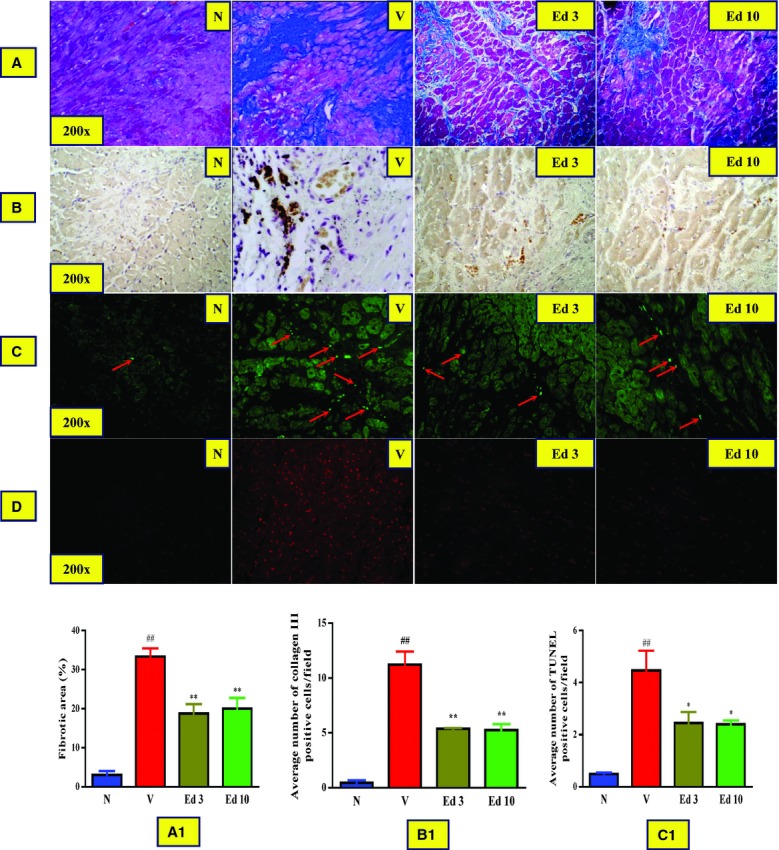
(A and A1) Azan-Mallory staining for fibrosis of the cross-sectional tissue slices of hearts (Fibrosis is indicated by blue area as opposed to the red myocardium and bar graph showing the percentage fibrotic area, respectively). (B and B1) Immunohistochemical stained myocardial tissue sections for collagen III and bar graph showing the average number of collagen-III positive cells/field, respectively. (C and C1) TUNEL stained myocardial tissue sections for apoptotic nuclei and bar graph showing the average number of TUNEL positive apoptotic nuclei/field, respectively. (D) Dihydroethidium-stained myocardial tissue sections for superoxide production. Group N: age-matched intact rats; Group V: rats with dilated cardiomyopathy (DCM) treated with vehicle; Group Ed 3: rats with DCM treated with edaravone (3 mg/kg); Group Ed 10: rats with DCM treated with edaravone (10 mg/kg); ##P < 0.01 versus Group N, *P < 0.05 and **P < 0.01 versus Group V (one-way ANOVA followed by Dunnett's test). Values are expressed as mean ± S.E.M., n = 3.
Fig 3.

Myocardial expressions of osteopontin (OPN), transforming growth factor-β1 (TGF-β1) and cytochrome (cyt)-C. (A–C) Densitometric data of protein analysis. The mean density values of OPN, TGF-β1 and cyt-C (Expressed as a ratio relative to that of GAPDH) with representative Western blots showing specific bands for OPN, TGF-β1 and cyt-c. GAPDH was used as an internal control. Group N: age-matched intact rats; Group V: rats with dilated cardiomyopathy (DCM) treated with vehicle; Group Ed 3: rats with DCM treated with edaravone (3 mg/kg); Group Ed 10: rats with DCM treated with edaravone (10 mg/kg). ##P < 0.01 versus Group N, *P < 0.05 and **P < 0.01 versus Group V (one-way ANOVA followed by Dunnett's test). Values are expressed as mean ± S.E.M., n = 3.
Effect of edaravone on myocardial apoptosis
Apoptotic cell death plays a major role in several cardiovascular disorders and is also an important myocardial remodelling process leading to cardiac dysfunction via cardiomyocyte loss. TUNEL staining reveals the number of apoptotic cells present in the stained area, and increased apoptotic cell count indicates the disease progression. In the present study, the number of TUNEL positive nuclei was significantly higher in myocardial tissue sections from group V rats than in group N. However, treatment with edaravone (3 or 10 mg/kg) significantly decreased their number compared with that in group V (Fig. 2C). In addition, the cytosolic level of cytochrome C, an apoptosis marker, was significantly elevated in the group V rats, whereas treatment with edaravone (3 or 10 mg/kg) significantly prevented this change (Fig. 3C).
Effect of edaravone on oxidative stress
Oxidative stress is involved in the pathogenesis of various cardiovascular disorders, which has also been reported to be involved in the progression of EAM to DCM. In the present study, the rats of group V suffered from the excess oxidative stress as identified by DHE staining and Western blotting for its marker molecules. Involvement of superoxides in the progression of EAM has been confirmed by the DHE staining of the myocardial tissue sections of group V rats compared with the group N rats, whereas the treatment with edaravone (3 or 10 mg/kg) significantly protected from these changes (Fig. 2D). The cardiac expressions of NADP(H) oxidase subunits like p47phox, p67phox, gp91phox and Nox4 were significantly higher in group V rats compared with that in group N, and these changes were significantly reversed by the treatment with edaravone (Fig. 4A and D).
Fig 4.

Myocardial expressions of p47phox, p67phox, gp91phox and Nox4. (A–D) Densitometric data of protein analysis. The mean density values of p47phox, p67phox, gp91phox and Nox4 (Expressed as a ratio relative to that of GAPDH) with representative Western blots showing specific bands for p47phox, p67phox, gp91phox and Nox4. GAPDH was used as an internal control. An equal amount of protein sample obtained from whole ventricular homogenate was applied in each lane. These bands are representative of three separate experiments. Group N: age-matched intact rats; Group V: rats with dilated cardiomyopathy (DCM) treated with vehicle; Group Ed 3: rats with DCM treated with edaravone (3 mg/kg); Group Ed 10: rats with DCM treated with edaravone (10 mg/kg). ##P < 0.01 versus Group N, *P < 0.05 and **P < 0.01 versus Group V (one-way ANOVA followed by Dunnett's test). Values are expressed as mean ± S.E.M., n = 3.
Effect of edaravone on ER stress marker proteins
Various cellular stresses are reported to be increased during myocardial injury, of which ER stress is the one which directly leads to myocardial cell loss via apoptosis involving caspases. The elevated levels of various ER stress markers were identified in the DCM hearts of group V rats. The myocardial expressions of GRP78 and GADD153 were significantly increased in the group V rats. However, these changes were significantly attenuated by the treatment with edaravone (3 or 10 mg/kg) as demonstrated by the decreased levels of these above-mentioned markers (Fig. 5A and B). Prolonged ER stress stimulates apoptotic signalling via caspases, and the present study with edaravone has provided significant suppression in the myocardial levels of caspase-3, which is the downstream of the caspase-mediated apoptotic signalling when compared with the group V rats (Fig. 5C).
Fig 5.

Myocardial expressions of GRP78, GADD153, and caspase-3. (A–D) Densitometric data of protein analysis. The mean density values of GRP78, GADD153 and caspase-3 (expressed as a ratio relative to that of GAPDH) with representative Western blots showing specific bands for GRP78, GADD 153, Cyt C and caspase-3. GAPDH was used as an internal control. Group N: age-matched intact rats; Group V: rats with DCM treated with vehicle; Group Ed 3: rats with dilated cardiomyopathy (DCM) treated with edaravone (3 mg/kg); Group Ed 10: rats with DCM treated with edaravone (10 mg/kg). ##P < 0.01 versus Group N, *P < 0.05 and **P < 0.01 versus Group V (one-way ANOVA followed by Dunnett's test). Values are expressed as mean ± S.E.M., n = 3.
Discussion
The present findings clearly suggest that edaravone, a novel free radical scavenger, reduced the severity of EAM and also prevented its progression to DCM. The cardioprotection offered by edaravone treatment may be partly as a result of the suppression of oxidative and ER stresses.
Edaravone treatment has protected the hearts from functional deterioration as demonstrated by haemodynamic study. There was a significant improvement in the cardiac haemodynamics of the edaravone-treated rats, as the LV pressure parameters were significantly improved when compared with those in the group V. Similarly, echocardiography also confirmed the improved cardiac performance in the edaravone-treated rats as their %FS and %EF values were improved significantly when compared with the group V rats. No significant change in the HR was observed; however, the HW/bw ratio of the edaravone-treated rats was less when compared with the group V rats. Thus, our present study confirmed the effect of edaravone in maintaining the cardiac function against DCM derived from EAM induced by immunization with porcine cardiac myosin.
Oxidative stress and inflammation are thought to play important roles in the progression of cardiovascular diseases. An increase in the level of oxidative stress is implicated in the pathogenesis of heart failure and various autoimmune disorders including DCM mediated by cardiac myosin [23]. Investigations suggest that free radicals may be important contributors to the deterioration of the decompensating myocardium [24]. Treatments that reduce the levels of oxidative stress or inflammation have thus been found to improve myocardial function in patients with advanced heart failure as well as in animal models of this condition. The NADPH oxidase system, present in cardiac and vascular tissues, is also a candidate for the source of the superoxide [25]. The NADPH oxidase complex is a cluster of proteins that promote donation of an electron from NADPH to molecular oxygen to produce superoxide. Activation of electron transfer from NADPH to molecular oxygen requires recruitment of the cytosolic subunits p47phox, p67phox and other subunits [26, 27]. Thus, estimation of these proteins is an important mode for the identification of oxidative stress during various disorders. Our study has confirmed that the DCM rats suffer from oxidative stress as demonstrated by the increased levels of myocardial NADPH oxidase subunits like p47phox, p67phox, gp91phox and Nox4. The results from edaravone treatment showed that the treated rats have been protected from myocardial oxidative stress as observed with their reduced myocardial levels of the above-mentioned NADPH oxidase subunits. This effect was expected as edaravone has been reported to provide protection against oxidative stress in various animal models. Zhang et al. [28] reported the protective effect of edaravone against acute myocardial ischaemia/reperfusion injury where they have suggested its antioxidative role in reducing the infarct size. It has also been reported that edaravone at 10 mg/kg dose provided significant cardioprotection and reduced the ROS production in pressure overload-induced cardiac hypertrophy [11]. Consistent with these reports and from the present results, we can suggest that edaravone treatment can provide significant cardioprotection in rats with DCM, at least in part via the inhibition of oxidative stress.
Oxidative stress is known to induce cardiomyocyte apoptosis, an important contributor to hypertrophic remodelling and cell dysfunction [29] in a variety of cell types by activating intracellular cell death signalling cascades [30]. Apoptosis is the key contributor to cell loss during heart failure causing the loss of contractile muscle mass, which is largely irreplaceable [31]. There are various reports regarding the involvement of apoptosis during myocardial injury and damage. Most of them suggest that activation of caspases is the main finding during this process and blocking these caspases was reported to be effective against apoptosis-mediated myocardial functional loss [32, 33]. Activation of these caspases is mainly via the functional stress in the ER. The ER is classically characterized as an organelle that participates in the folding of membrane and secretory proteins. Stimuli such as ischaemia, hypoxia, heat shock, genetic mutation, oxidative stress and increased protein synthesis that cause ER dysfunction are collectively known as ER stress [34]. Any disturbance in its function causes ER stress, leading to up-regulation of ER chaperones such as GRP78. GRP78 serves as the master modulator of unfolded protein response network by binding to various ER stress sensors. Increased GRP78 was reported in ER stress-associated apoptosis of cardiocytes in the heart failure [35]. In our present study, the protein levels of GRP78 and GADD153 were markedly up-regulated in the group V rats confirming the ER stress, whereas the treatment with edaravone significantly attenuated this change, suggesting its protective effect against ER stress. Similarly, the change in the cytosolic level of cytochrome C was also reversed by the treatment with edaravone. When ER stress is excessive and/or prolonged, however, apoptotic signals are initiated by the ER, proceed through various routes involving caspases and finally induce apoptosis by activation of caspase-3 [34, 36]. Similarly, we have also observed the elevated myocardial level of cleaved caspase-3, which has confirmed the activation of apoptosis in the group V rats that could have been mediated via oxidative stress and/or ER stress. Interestingly, the rats treated with edaravone showed significant suppression of caspase-3, suggesting its antiapoptotic role in DCM. Apart from this, the antiapoptotic role of edaravone is also confirmed by the reduction in the number of TUNEL positive apoptotic cells in the myocardial sections of the DCM rats treated with edaravone. From these results, we can suggest that treatment with edaravone is effective in preventing myocardial apoptosis possibly via modulation of oxidative and ER stresses.
Cardiac adaptation in response to intrinsic or external stress involves a complex process of chamber remodelling and myocyte molecular modifications and growing evidence highlights oxidative stress as an important mechanism for this maladaptation [37]. ROS generated by NADPH oxidase plays a key role in cardiac remodelling [38]. Myocardial fibrosis, the replacement of fibrous tissue in place of damaged myocardium is the hallmark of DCM, was observed in DCM hearts as measured by Azan–Mallory staining and increased concentrations of its marker molecules (TGF-β1 and OPN). Excessive expressions of TGF-β1 and collagen-III are involved in the myocardial fibrosis, which promote the synthesis of extracellular matrix constituents such as proteoglycans and fibronectin. In addition, they suppress the degradation of extracellular matrix. It was reported that, in a rat model of dimethylnitrosamine-induced liver cirrhosis, treatment with 10 mg/kg of edaravone produced significant reduction in the fibrotic area [39]. Similarly, in a pressure overload-induced LV hypertrophic rat model, cardiac fibrosis was significantly attenuated by the treatment with the same dose of edaravone [11]. Consistent with those reports, in the present study, treatment with edaravone has reduced the percentage area of fibrosis and reduced myocardial levels of TGF-β1, OPN and collagen III. From these results, we can confirm that edaravone, a novel antioxidant is effective in preventing the myocardial fibrosis in the rats with DCM.
Conclusion
There are several reports regarding the protective role of edaravone against cardiac impairment in various immune-mediated cardiac inflammatory conditions [40, 41] and ischaemia-reperfusion injury [42] including the one which we have published earlier regarding its protective effect against EAM in rats [12], but its effect on progression of EAM to chronic cardiac complications like DCM is not available. The present study has provided lines of evidence for the protective effects of edaravone against the deterioration of cardiac architecture during EAM-mediated DCM.
Acknowledgments
This research was supported by a Yujin Memorial Grant, Ministry of Education, Culture, Sports, Science and Technology, Japan, and by a grant from the Promotion and Mutual Aid Corporation for Private Schools, Japan.
Conflict of interest
The authors declare that there are no conflicts of interest with this work.
References
- 1.Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death. Circulation. 2001;34:199–204. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- 2.Kodama M, Hanawa H, Saeki M, et al. Rat dilated cardiomyopathy after autoimmune giant cell myocarditis. Circ Res. 1994;75:278–84. doi: 10.1161/01.res.75.2.278. [DOI] [PubMed] [Google Scholar]
- 3.Okura Y, Yamamoto T, Goto S, et al. Characterization of cytokine and iNOS mRNA expression in situ during the course of experimental autoimmune myocarditis in rats. J Mol Cell Cardiol. 1997;29:491–502. doi: 10.1006/jmcc.1996.0293. [DOI] [PubMed] [Google Scholar]
- 4.Ishiyama S, Hiroe M, Nishikawa T, et al. The Fas/Fas ligand system is involved in the pathogenesis of autoimmune myocarditis in rats. J Immunol. 1998;161:4695–701. [PubMed] [Google Scholar]
- 5.Ishiyama S, Hiroe M, Nishikawa T, et al. Nitric oxide contributes the progression of myocardial damage in experimental autoimmune myocarditis in rats. Circulation. 1997;95:489–96. doi: 10.1161/01.cir.95.2.489. [DOI] [PubMed] [Google Scholar]
- 6.Belch JJ, Bridges AB, Scott N, et al. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–8. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi K. Effects of water deprivation on immunoreactive angiotensin II levels in plasma, cerebroventricular perfusate and hypothalamus of the rat. Acta Endocrinol. 1981;97:137–44. doi: 10.1530/acta.0.0970137. [DOI] [PubMed] [Google Scholar]
- 8.Schelling P, Fischer H, Ganten D. Angiotensin and cell growth: a link to cardiovascular hypertrophy? J Hypertens. 1991;9:3–15. [PubMed] [Google Scholar]
- 9.Tada S, Nakamoto N, Kameyama K, et al. Clinical usefulness of edaravone for acute liver injury. J Gastroenterol Hepatol. 2003;18:851–7. doi: 10.1046/j.1440-1746.2003.03064.x. [DOI] [PubMed] [Google Scholar]
- 10.Onogi H, Minatoguchi S, Chen XH, et al. Edaravone reduces myocardial infarct size and improves cardiac function and remodelling in rabbits. Clin Exp Pharmacol Physiol. 2006;33:1035–41. doi: 10.1111/j.1440-1681.2006.04483.x. [DOI] [PubMed] [Google Scholar]
- 11.Tsujimoto I, Hikoso S, Yamaguchi O, et al. The antioxidant edaravone attenuates pressure overload-induced left ventricular hypertrophy. Hypertension. 2005;45:921–6. doi: 10.1161/01.HYP.0000163461.71943.e9. [DOI] [PubMed] [Google Scholar]
- 12.Shimazaki H, Watanabe K, Veeraveedu PT, et al. The antioxidant edaravone attenuates ER-stress-mediated cardiac apoptosis and dysfunction in rats with autoimmune myocarditis. Free Radic Res. 2010;44:1082–90. doi: 10.3109/10715762.2010.499904. [DOI] [PubMed] [Google Scholar]
- 13.Kodama M, Matsumoto Y, Fujiwara M, et al. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250–62. doi: 10.1016/0090-1229(90)90039-s. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe K, Nakazawa M, Fuse K, et al. Protection against autoimmune myocarditis by gene transfer of interleukin-10 by electroporation. Circulation. 2001;104:1098–100. doi: 10.1161/hc3501.096190. [DOI] [PubMed] [Google Scholar]
- 15.Thandavarayan RA, Watanabe K, Ma M, et al. Dominant-negative p38alpha mitogen activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. Am J Physiol Heart Circ Physiol. 2009;297:H911–9. doi: 10.1152/ajpheart.00124.2009. [DOI] [PubMed] [Google Scholar]
- 16.Thandavarayan RA, Watanabe K, Sari FR, et al. Modulation of doxorubicin-induced cardiac dysfunction in dominant-negative p38α mitogen-activated protein kinase mice. Free Radic Biol Med. 2010;49:1422–31. doi: 10.1016/j.freeradbiomed.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Sukumaran V, Watanabe K, Veeraveedu PT, et al. Beneficial effects of olmesartan, an angiotensin II receptor type 1 antagonist, in rats with dilated cardiomyopathy. Exp Biol Med. 2010;235:1338–46. doi: 10.1258/ebm.2010.010016. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe K, Sukumaran V, Veeraveedu PT, et al. Regulation of inflammation and myocardial fibrosis in experimental autoimmune myocarditis. Inflamm Allerg Drug Targets. 2011;10:218–25. doi: 10.2174/187152811795564091. [DOI] [PubMed] [Google Scholar]
- 19.Arozal W, Watanabe K, Veeraveedu PT, et al. Beneficial effects of angiotensin II receptor blocker, olmesartan, in limiting the cardiotoxic effect of daunorubicin in rats. Free Radic Res. 2010;44:1369–77. doi: 10.3109/10715762.2010.509399. [DOI] [PubMed] [Google Scholar]
- 20.Miller FJ, Jr, Gutterman DD, Rios CD, et al. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 21.Thandavarayan RA, Watanabe K, Ma M, et al. 14-3-3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy. Biochem Pharmacol. 2008;75:1797–806. doi: 10.1016/j.bcp.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Kwon KB, Kim EK, Han MJ, et al. Induction of apoptosis by Radix Paeoniae Alba extract through cytochrome c release and the activations of caspase-9 and caspase-3 in HL-60 cells. Biol Pharm Bull. 2006;29:1082–6. doi: 10.1248/bpb.29.1082. [DOI] [PubMed] [Google Scholar]
- 23.Sukumaran V, Watanabe K, Veeraveedu PT, et al. Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int J Biol Sci. 2011;7:154–67. doi: 10.7150/ijbs.7.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prasad K, Gupta JB, Kalra J, et al. Oxidative stress as a mechanism of cardiac failure in chronic volume overload in canine model. J Mol Cell Cardiol. 1996;28:375–85. doi: 10.1006/jmcc.1996.0035. [DOI] [PubMed] [Google Scholar]
- 25.Muthalif MM, Karzoun NA, Gaber L, et al. Angiotensin II-induced hypertension: contribution of Ras GTPase/Mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension. 2000;36:604–9. doi: 10.1161/01.hyp.36.4.604. [DOI] [PubMed] [Google Scholar]
- 26.Ming W, Li S, Billadeau DD, et al. The Rac effector p67phox regulates phagocyte NADPH oxidase by stimulating Vav1 guanine nucleotide exchange activity. Mol Cell Biol. 2007;27:312–23. doi: 10.1128/MCB.00985-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyaw M, Yoshizumi M, Tsuchiya K, et al. Atheroprotective effects of antioxidants through inhibition of mitogen-activated protein kinases. Acta Pharmacol Sin. 2004;25:977–85. [PubMed] [Google Scholar]
- 28.Zhang YM, Wang Y, Liu XH, et al. Cardioprotective effect of edaravone pharmacological postconditioning on acute myocardial ischemia/reperfusion injury: experiment with rats. Zhonghua Yi Xue Za Zhi. 2008;88:2558–61. [PubMed] [Google Scholar]
- 29.Cesselli D, Jakoniuk I, Barlucchi L, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–86. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 30.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 31.Chandrashekhar Y, Narula J. Death hath a thousand doors to let out life. Circ Res. 2003;92:710–4. doi: 10.1161/01.RES.0000069364.24820.8B. [DOI] [PubMed] [Google Scholar]
- 32.Juhasz B, Der P, Szodoray P, et al. Adrenocorticotrope hormone fragment (4-10) attenuates the ischemia/reperfusion-induced cardiac injury in isolated rat hearts. Antioxid Redox Signal. 2007;9:1851–61. doi: 10.1089/ars.2006.1535. [DOI] [PubMed] [Google Scholar]
- 33.Kovacs P, Bak I, Szendrei L, et al. Non-specific caspase inhibition reduces infarct size and improves post-ischaemic recovery in isolated ischaemic/reperfused rat hearts. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:501–7. doi: 10.1007/s002100100483. [DOI] [PubMed] [Google Scholar]
- 34.Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–45. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- 35.Bhimji S, Godin DV, McNeill JH. Myocardial ultrastructural changes in alloxan-induced diabetes in rabbits. Acta Anat. 1986;125:195–200. doi: 10.1159/000146161. [DOI] [PubMed] [Google Scholar]
- 36.Mao W, Fukuoka S, Iwai C, et al. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol. 2007;293:H1636–45. doi: 10.1152/ajpheart.01377.2006. [DOI] [PubMed] [Google Scholar]
- 37.Takimoto E, Kass DA. Role of oxidativestress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–8. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 38.Bendall JK, Rinze R, Adlam D, et al. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res. 2007;100:1016–25. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka H, Ueda H, Fukuchi H, et al. Antifibrotic effect of edaravone in rat liver cirrhosis induced by dimethylnitrosamine. Clin Exp Med. 2009;9:229–33. doi: 10.1007/s10238-009-0034-4. [DOI] [PubMed] [Google Scholar]
- 40.Nimata M, Okabe TA, Hattori M, et al. MCI-186 (edaravone), a novel free radical scavenger, protects against acute autoimmune myocarditis in rats. Am J Physiol Heart Circ Physiol. 2005;289:H2514–8. doi: 10.1152/ajpheart.00661.2005. [DOI] [PubMed] [Google Scholar]
- 41.Okabe TA, Kishimoto C, Hattori M, et al. Cardioprotective effects of 3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186), a novel free radical scavenger, on acute autoimmune myocarditis in rats. Exp Clin Cardiol. 2004;9:177–80. [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe K, Ma M, Wen J, et al. Effects of edaravone in heart of aged rats after cerebral ischemia-reperfusion injury. Biol Pharm Bull. 2007;30:460–4. doi: 10.1248/bpb.30.460. [DOI] [PubMed] [Google Scholar]