

Abstract
Heparin and low molecular weight heparins have been demonstrated to reduce myocardial ischaemia/reperfusion (I/R) injury, although their use is hampered by the risk of haemorrhagic and thrombotic complications. Chemical and enzymatic modifications of K5 polysaccharide have shown the possibility of producing heparin-like compounds with low anticoagulant activity and strong anti-inflammatory effects. Using a rat model of regional myocardial I/R, we investigated the effects of an epimerized N-,O-sulphated K5 polysaccharide derivative, K5-N,OSepi, on infarct size and histological signs of myocardial injury caused by 30 min. ligature of the left anterior descending coronary artery followed by 1 or 24 h reperfusion. K5-N,OSepi (0.1–1 mg/kg given i.v. 15 min. before reperfusion) significantly reduced the extent of myocardial damage in a dose-dependent manner. Furthermore, we investigated the potential mechanism(s) of the cardioprotective effect(s) afforded by K5-N,OSepi. In left ventricular samples, I/R induced mast cell degranulation and a robust increase in lipid peroxidation, free radical-induced DNA damage and calcium overload. Markers of neutrophil infiltration and activation were also induced by I/R in rat hearts, specifically myeloperoxidase activity, intercellular-adhesion-molecule-1 expression, prostaglandin-E2 and tumour-necrosis-factor-α production. The robust increase in oxidative stress and inflammatory markers was blunted by K5-N,OSepi, in a dose-dependent manner, with maximum at 1 mg/kg. Furthermore, K5-N,OSepi administration attenuated the increase in caspase 3 activity, Bid and Bax activation and ameliorated the decrease in expression of Bcl-2 within the ischaemic myocardium. In conclusion, we demonstrate that the cardioprotective effect of the non-anticoagulant K5 derivative K5-N,OSepi is secondary to a combination of anti-apoptotic and anti-inflammatory effects.
Keywords: heparin-like derivative, K5-N, OSepi, myocardial ischaemia/reperfusion, inflammation, apoptosis
Introduction
Ischaemia of the heart evokes a complex and inter-related sequence of events resulting in myocardial dysfunction and damage. Although reperfusion following transient ischaemia leads to restoration of blood flow, there is compelling evidence to support the notion that reperfusion may exacerbate the injury initially caused by ischaemia, producing a so-called myocardial I/R injury [1]. Many lines of evidence have convincingly shown that inflammatory response plays a major role in determining the extension of myocardial damages evoked by I/R [2]. After the restoration of blood flow, leucocytes infiltrate into the myocardium and release cytokines, proteolytic enzymes and reactive oxygen species (ROS) that significantly contribute to the development of injuries [3]. Moreover, resident mast cells also contribute to the myocardial dysfunction through the release of superoxide [4] and other mediators, such as histamine and leukotrienes, which favour neutrophil adhesion to the endothelium [5]. The combination of multiple triggers, such as free radicals production and calcium overload, has been reported to markedly affect the outcomes of myocardial I/R injury [6]. Apoptosis has also been implicated as a possible mechanism in the development of myocardial infarction [7], and a significant increase in myocardial apoptosis have been documented in both animal models and patients with ischaemic cardiomyopathy [8, 9]. Recently, a close relationship between the temporal and spatial distribution of cytokines in the infarcted myocardium and the detection of apoptosis has been described in patients with myocardial infarction [10]. Therefore, interventions aimed at suppressing both post-ischaemic inflammatory reaction and myocyte apoptosis may represent effective therapeutic strategies in the context of myocardial I/R injury.
Heparin and low molecular weight heparins (LMWH) have been consistently demonstrated to reduce myocardial I/R injury [11, 12]. However, the major drawbacks of both heparin and LMWH still remain their haemorrhagic and thrombotic complications [13]. Although heparin's mechanism of action for myocardial protection has been thought to be dependent on its anticoagulant property, most recent findings suggest that an important contributor to the cardioprotective effect of heparin involves suppression of the post-ischaemic inflammatory response [14]. On this basis, considerable efforts have been made to develop safer heparin derivatives with a low anticoagulant activity and retaining strong anti-inflammatory properties. In this study, we have investigated the effects of a semisynthetic, heparin-like derivative, K5-N,OSepi, obtained by combined chemical and enzymatic modification of the Escherichia coli capsular polysaccharide, in an in vivo experimental model of regional myocardial I/R injury. The capsular polysaccharide of E. coli K5 bacteria can be subjected to various degrees of chemical N-deacetylation/N-sulphation as well as O-sulphation, to yield different heparin-like semisynthetic polysaccharides [15]. Depending on their extent and pattern of sulphation, chemically sulphated K5 derivatives have been shown to possess different degree of antithrombotic/anticoagulant activity. As previously described [16], the semi-synthetic glycosaminoglycan K5 polysaccharide derivative K5-N,OSepi was obtained by N-desacetylation/N, O-sulphation of K5 polysaccharide and epimerization of K5 with the enzyme glucuronyl C5 epimerase. This compound has been demonstrated to be endowed with anti-inflammatory and anti-adhesive effects, but it is devoid of any anti-coagulant activity [16–18]. To date, the effects of this non-anticoagulant heparin-like derivative on myocardial I/R injury have not yet been investigated. Hence, this study was undertaken to extend the investigation on K5-N,OSepi efficacy to conditions associate with myocardial injury.
Materials and methods
Animals
A total of 108 male albino rats, Wistar strain, weighing 250–300 g (Morini, Reggio Emilia, Italy) were quarantined for 7 days with laboratory chow (Rodentia, Bergamo, Italy) and water ad libitum. The experimental protocol conformed to either the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health and the European Commission Directive 86/609/EEC and it was approved by the Animal Care Committee of the University of Florence (Italy).
Preparation and biochemical characterization of the N-, O-sulphated K5 polysaccharide
The precursor of the compound K5-N,OSepi is the capsular K5 polysaccharide obtained from E. coli strain 010:K5:H4, a polymer with the structure [-4)-GlcA β1-4 GlcNAc-(1-]n in which the disaccharidic unit formed by D-glucuronic acid and N-acetylglucosamine is linked by a α 1→4 bond. This structure is similar to N-acetylheparosan, the natural precursory polymer of heparin. The purified K5 polysaccharide was prepared as described by Urbinati et al. [19]. N, O-sulphated K5 polysaccharide was obtained by chemical N- and O- sulphation of the K5 polysaccharide, followed by the enzymatic epimerization with glucuronyl C5 epimerase, according to the method described by Gori et al. [16]. The epimerized product was purified by ultrafiltration, precipitated with ethanol and passed through a cation-exchange resin. The O-sulphation was performed by treating the product with tetrabutylamonium hydroxyde in N-N-dimetylformamide and with pyridine sulphur trioxide at 50°C for 24 hrs. The product was finally depolymerized to the desired molecular weight by controlled nitrous acid deamination. The structural profile of the compound was characterized by nuclear magnetic resonance (1H-NMR and 13C-NMR) analysis; the sulphate content was obtained by sulphate/carboxyl ratio analysis and molecular weight determination was performed by HPLC analysis as described [20]. The compound used in these experiments had an average molecular weight of 6000 Da, a sulphate/carboxyl ratio of 4.0 and an iduronic acid/glucuronic acid ratio of 0.8. K5-N,OSepi was also tested for its antithrombotic/anticoagulant activity, showing 8.96 ± 0.8 U/mg anti-factor Xa activity by using a commercial photometric kit (COATEST Heparin; Cromogenix, Bedford, MA, USA) and by following the protocol provided by the manufacturer.
Surgical procedure and treatments
The rats were anaesthetized by intraperitoneal injection of sodium thiopental (50 mg/kg body weight; Abbott, Latina, Italy) and monitored for body temperature, respiration pattern, loss of righting reflex, unresponsiveness and corneal reflexes. A cannula was inserted into the trachea and the animals were ventilated with air by using a Palmer pump (U. Basile, Comerio, Italy). Subcutaneous peripheral limb electrodes were inserted and electrocardiogram (ECG) was continuously recorded for the entire duration of the experiment. ECGs were analysed to determine the incidence and duration of ventricular fibrillation (VF) and ventricular tachycardia (VT). According to Gelvan et al. [21], VT was recognized as three or more consecutive premature ventricular contractions and VF was recognized as irregular modulating baseline. All rats underwent thoracotomy at the fifth left intercostal space, the pericardium was opened and a loose 00 braided silk suture was placed around the left coronary artery (LCA). Then, the chest was closed by a silk suture to minimize heart displacement. Rats were allowed to equilibrate for 20 min. to enable ECG values to stabilize. Ischaemia was induced by tightening the threads of the coronary suture and was maintained for 30 min. Reperfusion was obtained by reopening the chest and cutting the ligature around the coronary artery. The duration of reperfusion was pre-determined to 1 hr or 24 hrs.
Animals were randomly allocated into different groups (n = 12 per group): sham group (rats underwent the same surgical procedures as above, but without the tightening of the coronary sutures); I/R groups; K5-N,OSepi groups (dose-range 0.1–1 mg/kg 15 min. before reperfusion) and B4/100 group (1 mg/kg 15 min. before reperfusion). B4/100 is a biologically inactive non-sulphated polysaccharide compound used as negative control molecule.
Determination of area at risk and infarct size
At the end of reperfusion (1 hr or 24 hrs), LCA was re-tightened with a 00 braided silk suture in the same place of the previous ligature and 2 ml of Evans Blue (Sigma-Aldrich, St. Louis. MO, USA) was retrogradely injected with a thin catheter inserted into carotid artery to delineate the in vivo area at risk (AAR) [22]. The chest was re-opened and the hearts of the anaesthetized rats were quickly removed. To distinguish between viable ischaemic and infracted tissue, the p-nitro blue tetrazolium (NBT) dye exclusion method was used. On removal, the hearts were attached to a Langendorff's apparatus through a cannula introduced into the aorta and perfused with 10 ml of 1% NBT dissolved in a modified Tyrode solution, at a constant pressure of 40 cm of water at 37°C for 20 min. Following this treatment, the normal myocardium shows an intense blue staining reaction because of the presence of dehydrogenase enzymes, whereas ischaemia-reperfusion-injured lesions remain unstained. Thus, the latter regions appear as clearly delineated, unstained zones. The hearts were detached from the cannula, weighed, fixed in buffered 4% formaldehyde for 12 hrs, and the ventricles sectioned in 1-mm transverse slices from the apex to the ligature. In each slice, the bound areas of the unstained area on the upside surface were traced onto a superimposed acetate sheet and the encircled area was measured by computer-assisted morphometry, as described below. The left ventricular area, AAR, and the area of infarction for each slide were then determined as previously described [22]. In each slice, the volume of the damaged myocardium was calculated by multiplying the unstained surface area for the thickness of the slice. The total volume of the damaged myocardium was calculated as the sum of the partial values of the different slices. To allow a comparison of the extension of myocardial injury between hearts of different sizes, the total volume of the damaged myocardium was divided by the heart weight (grams). All measurements and calculations were performed by a single individual (M.R.), who was blinded to treatment status.
Ultrastructural examination and tissue injury scoring
Electron microscopic examination was carried out on ultrathin sections of heart tissue fragments stained with uranyl acetate and alkaline bismuth subnitrate and examined under a JEM 1010 electron microscope (Jeol, Tokyo, Japan) at 80 kV. In each fragment, two series of six to eight ultrathin sections cut at two different levels were examined and photographed. Myocyte and microvascular endothelium injury was quantified from electron-micrographs (final magnifications range ×10,000–×20,000) as previously reported [23]. The criteria used are reported in Table 1. Each animal was assigned a separate score for myocyte and endothelial injury from two independent observers (D.B. & A.P.), blinded to the experimental groups, and the values were then averaged.
Table 1.
Scoring method of myocyte and endothelial injury
| Injury | Score | Degree of injury | Description |
|---|---|---|---|
| Myocyte | 0 | Normal myocyte | |
| 1 | Slight | Mild intracellular oedema | |
| Mild mitochondrial swelling | |||
| 2 | Moderate | Mild intracellular oedema | |
| Contracture of myofibrils | |||
| Marked mitochondrial swelling with clearing of matrix | |||
| Occasional focal clumping of mitochondrial cristae | |||
| Mild nuclear chromatin clumping | |||
| 3 | Severe | Severe mitochondrial swelling with loss of cristae | |
| Presence of intramitochondrial dense granules | |||
| Disarrangement of myofibrils | |||
| Plasma membrane rupture | |||
| Nuclear degeneration (apoptosis or karyolysis) | |||
| Endothelial | 0 | Normal endothelium | |
| 1 | Slight | Mild-to-moderate endothelial swelling | |
| 2 | Moderate | Marked endothelial swelling | |
| Decreased pinocytotic vesicles | |||
| Mitochondrial swelling | |||
| 3 | Severe | Severe mitochondrial swelling with loss of cristae | |
| Plasma membrane rupture | |||
| Nuclear degeneration (apoptosis or karyolysis) | |||
| Neutrophil adhesion and extravasation |
Evaluation of myeloperoxidase activity
MPO activity, which can be used as a marker for neutrophil accumulation in tissues, was determined as previously described [23]. MPO activity was defined as the quantity of enzyme degrading 1 μmol of peroxide per min. at 37°C and was expressed in mU/g of wet tissue.
Determination of prostaglandin E2 tumour necrosis factor-α production
Prostaglandin E2 (PGE2) and tumour necrosis factor (TNF)-α were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits (Cayman Chemical, Ann Arbor, MI, USA), following the protocol provided by the manufacturer and results are expressed as pg/ml.
Determination of thiobarbituric acid-reactive substances
Thiobarbituric acid-reactive substances (TBARS) are end-products of cell membrane lipid peroxidation by ROS and are considered reliable marker of myocardial cell damage. TBARS were determined by measurement of the chromogen obtained from the reaction of MDA with 2-thiobarbituric acid according to Aruoma et al. [24].
Evaluation of calcium content
The calcium content of the myocardial tissue was measured as described previously [23] by using an atomic absorption spectrophotometer (Perkin-Elmer, Uberlingen, Germany) at a 422-nm wavelength. The relevant values were determined by comparison with a standard curve obtained with increasing concentrations of CaCl2 and expressed as ng of calcium/mg of tissue (dry weight).
Computer-assisted morphometry
The surface areas of the I/R-injured myocardium on slices of hearts stained with nitroblue tetrazoliurn were measured on the corresponding profiles reported on the acetate sheets. These profiles were registered through a CCTV television camera (Sony, Tokyo, Japan) interfaced with an Apple Macintosh LC III personal computer through a Videospigot card (Supermac, Sunnyvale, CA, USA). The software (Image Analysis Program 1.49, National Institute of Health, Bethesda, MD, USA) enables the areas encircled by each profile to be measured. Evaluation of light transmittance across mast cells was performed as previously described [23].
Determination of 8-hydroxy-2′-deoxyguanosine
DNA isolation from cardiac tissue homogenates was performed according to Masini et al. [25]. Samples of DNA extract were used for 8-hydroxy-2′-deoxyguanosine (8-OHdG) determination using a Bioxytech enzyme immunoassay kit (Oxis, Portland, OR, USA), following the instructions provided by the manufacturer. The values are expressed as ng of 8OHdG per μg proteins.
Measurement of caspase-3 activity
The enzymatic activity of caspase-3 was determined using the Ac-Asp-Glu-Val-Asp-AMC (Ac-DEVD-AMC; Bachem AG, Bubendorf, Switzerland) fluorescent substrate [26]. Samples of myocardial tissues were homogenized and then centrifuged at 10,000 g for 10 min. The supernatants were incubated with 40 μM of AC-DEVD-AMC for 60 min. at 37°C. Substrate cleavage was monitored fluorimetrically (Spectrofluo JY3 D; Jobin Yvon, Paris, France) at 380 nm excitation and 460 nm emission wave lengths. Data are expressed as arbitrary units/mg proteins. One unit of enzyme activity is defined as the amount of enzyme required to liberate 40 μmol of Ac-DEVD-AMC upon 60 min. at 37°C.
Western blot analysis
Western blots were carried out as previously described [27]. Proteins were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to polyvinyldenedifluoride membranes. The membranes were then incubated with primary and then secondary antibodies and developed by using the ECL detection system. The immunoreactive bands were visualized by autoradiography and the density of the bands was evaluated densitometrically by using Gel Pro#x00AE;Analyser 4.5, 2000 software (Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
Data are expressed as mean ± SEM. One-way anova followed by Student–Newman–Keuls multiple comparison test or by Student's t-test for unpaired values was performed by using GraphPad Prism 4.02 (GraphPad Software, San Diego, CA, USA); P < 0.05 was considered significant.
Materials
Unless stated otherwise, all compounds were purchased from the Sigma-Aldrich Company Ltd. K5-N,OSepi was kindly provided by INALCO RSM S.p.A (Montale, Pistoia, Italy). Goat polyclonal antibody against ICAM-1, rabbit polyclonal antibodies against Bcl-2, Bid and Bax and horseradish peroxidase-conjugated donkey anti-goat and anti-rabbit IgG were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Luminol ECL detection reagents were from Amersham (Buckinghamshire, UK).
Results
Effect of K5-N,OSepi on the infarct size caused by regional myocardial I/R
The results of the computer-assisted morphometry on the I/R hearts stained with nitroblue tetrazolium showed that administration of K5-N,OSepi 15 min. before reperfusion induced a significant reduction in the extension of myocardial damage in comparison with the non-treated rats at both 1 hr and 24 hrs (Fig. 1A). The mean size of ischaemic lesions in the rats treated with K5-N,OSepi was reduced in a dose-dependent manner at 1 hr reperfusion, with a maximum effect at 1 mg/kg (50% reduction). The highest dose of K5-N,OSepi evoked a similar 50% reduction in infarct volume when measured after 24 hrs reperfusion.
Fig 1.

Effects of K5-N,OSepi on myocardial infarct size evoked by regional I/R. (A) Dose–response effects of K5-N,OSepi (0.1–1 mg/kg) on the extension of left ventricular myocardium with I/R-induced injury, as evaluated on hearts stained with nitro-blue tetrazolium and (B) quantification of infarct size after I/R injury, expressed as per cent of area at risk (AAR), in the absence (vehicle) or presence of K5-N,OSepi (0.3–1 mg/kg) or B4/100 (1 mg/kg). Data are means ± SEM of six animals/group. *P < 0.01 versus vehicle (1 hr reperfusion) and •P < 0.01 versus vehicle (24 hrs reperfusion).
The mean values for the area at risk (AAR) per left ventricle (LV) ranged from 52 ± 6% to 59 ± 8% and were similar in all animal groups, not modified by drug treatment (P > 0.05, data not shown). When compared with sham-operated animals, 30-min. ischaemia followed by 1- or 24-hr reperfusion caused a significant increase in infarct size, expressed as per cent of AAR (57 ± 8% and 61 ± 3%, respectively) (Fig. 1B). The portion of the AAR-rendered necrotic was significantly reduced in the animals treated with K5-N,OSepi (0.1–1 mg/kg) in a dose-dependent manner, reaching a maximum at 1 mg/kg. At this dose, infarct size was 20 ± 5% of the area at risk after 1 hr reperfusion and 24 ± 4% of the area at risk after 24 hrs reperfusion, approximately a 60% reduction in infarct size. In contrast, no tissue protection was found when rats were treated with the polysaccharide B4/100 (1 mg/kg), used as negative control molecule.
Effect of regional myocardial I/R and K5-N,OSepi on haemodynamic parameters and ECG analysis
The mean baseline values of MAP in all groups of animals ranged from 117 ± 6 to 129 ± 7 mmHg, and were not significantly different between groups (P > 0.05, Table 2). In sham-operated rats, infusion of vehicle for K5-N,OSepi, 1 mg/kg, had no significant haemodynamic effects. In rats subjected to coronary I/R, mean values for MAP significantly decreased throughout the experimental period. When compared with I/R-animals treated with vehicle, I/R-animals treated with K5-N,OSepi or B4/100 did not exhibit any significant changes in MAP (P > 0.05, Table 2). The mean baseline values for HR in all groups of animals ranged from 427 ± 17 to 456 ± 11 bpm. In rats subjected to coronary occlusion and reperfusion, there was no significant alteration in HR in comparison with sham-operated animals and drug treatment did not modify the mean values of HR. Examination of ECG recordings showed no ventricular arrhythmias in the sham-operated animals, whereas 33% of the control rats subjected to myocardial I/R injury had ventricular arrhythmias, which appeared within the first 30 min. of reperfusion. K5-N,OSepi administration evoked a significant dose-dependent recovery of myocardial function: VT and VF occurred in 25% of the animals treated with the lowest dose of K5-N,OSepi (0.1 mg/kg) and in none of the animals treated with the highest dose (1 mg/kg) (P < 0.01). Of note, K5-N,OSepi (1 mg/kg) given to normal rats had no effects on HR (data not shown).
Table 2.
Effect of vehicle, K5-N,OSepi or B4/100 on mean arterial pressure (MAP) and heart rate (HR) in sham-operated rats or ischaemic reperfused rats
| Occlusion (min.) | Reperfusion (min.) | ||||
|---|---|---|---|---|---|
| Group | Baseline | 15 | 30 | 30 | 60 |
| Sham (n = 12) | |||||
| MAP | 128 ± 7 | 126 ± 4 | 119 ± 3 | 110 ± 6 | 112 ± 4 |
| HR | 427 ± 14 | 429 ± 16 | 426 ± 12 | 434 ± 16 | 429 ± 7 |
| I/R Vehicle (n = 12) | |||||
| MAP | 129 ± 7 | 122 ± 5 | 115 ± 6 | 94 ± 7 | 95 ± 8 |
| HR | 454 ± 15 | 438 ± 13 | 438 ± 15 | 430 ± 2 | 434 ± 5 |
| I/R + K5-N,OSepi 1 mg/kg (n = 12) | |||||
| MAP | 117 ± 6 | 109 ± 9 | 106 ± 10 | 97 ± 6 | 98 ± 9 |
| HR | 435 ± 39 | 440 ± 37 | 442 ± 38 | 422 ± 13 | 425 ± 24 |
| I/R + B4/100 1 mg/kg (n = 12) | |||||
| MAP | 128 ± 8 | 120 ± 5 | 94 ± 8 | 88 ± 6 | 85 ± 7 |
| HR | 456 ± 11 | 432 ± 11 | 438 ± 12 | 430 ± 8 | 429 ± 13 |
All data are mean ± S.E.M. of n observations.
K5-N,OSepi reduces the histological signs of injury caused by I/R
When compared with normal left ventricular morphology of the sham-operated rats (cardiomyocyte and endothelial cells, Fig. 2A and E, respectively), the animals undergoing regional myocardial ischaemia followed by 1 hr reperfusion showed the typical features of myocardial and endothelial injury related to severe oxygen starvation and dysfunction. In particular, the cardiomyocytes showed ultrastructural alterations, which ranged from intracellular oedema, mitochondrial swelling and myofibril hypercontraction to plasma membrane rupture and myofibril disarrangement (Fig. 2B and F). The microvascular endothelium showed narrowed lumina (arrow) and signs of cell injury, including cytoplasmic and mitochondrial swelling and, in some instances, cell detachment and interstitial haemorrhage (data not shown). In contrast, the left ventricular myocardium from the rats subjected to I/R and treated with K5-N,OSepi (1 mg/kg) showed a marked reduction in the ultrastructural signs of injury, the most prominent changes being intracellular oedema and myofibril hypercontraction in some cardiomyocytes and moderate cytoplasmic swelling of the microvascular endothelium (Fig. 2C and G). Of note, substitution of K5-N,OSepi with inactive B4/100 resulted in ultrastructural signs of myocyte and endothelial injury similar to the untreated I/R rats (Fig. 2E and H). Semi-quantitative scoring performed on cardiomyocyte and endothelial injury confirmed the visual observations and showed that K5-N,OSepi (1 mg/kg), but not B4/100, significantly blunts cardiac cell damage (Fig. 3).
Fig 2.
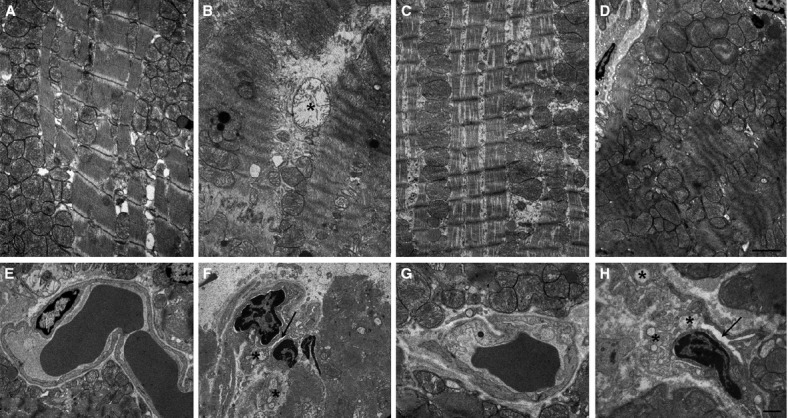
Histopathological examination of biopsies of left ventricular tissues. Representative electron micrographs of cardiomyocytes (upper panels) and blood capillaries (lower panels) from: sham-operated rats (A, E), showing normal mitochondria and myofibrils and microvessels with open lumina and normal organelle complement; rats undergoing 30 min. ischaemia followed by 60 min. reperfusion (B, F), showing mitochondrial swelling (asterisk), severe myofibril hypercontraction and constriction of microvascular lumen (arrow); rats subjected to I/R and treated with 1 mg/kg K5-N,OSepi (C, G), showing cytoplasmic oedema, moderate myofibril hypercontraction and nearly normal microvessels; rats subjected to I/R and treated with 1 mg/kg B4/100 (D, H), showing similar myocyte and endothelial alteration as in the untreated I/R rats. Bars = 1 μm.
Fig 3.
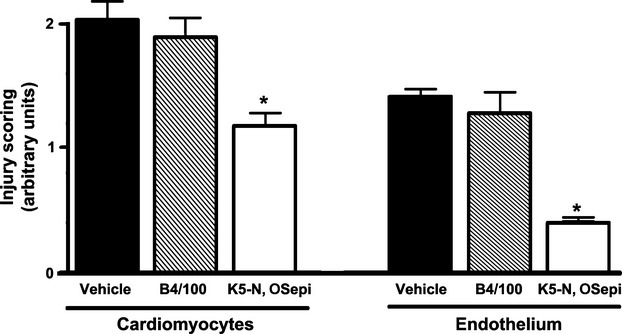
Semi-quantitative scoring of cardiomyocyte and endothelial cell injury in the different groups. Injury scoring was measured subsequent to I/R (30/60 min.) in the absence (vehicle) or presence of B4/100 (1 mg/kg) or K5-N,OSepi (1 mg/kg) treatment. *P < 0.01 versus vehicle and B4/100.
Effects of K5-N,OSepi on oxidative stress and calcium overload induced by I/R injury
Rats that had undergone I/R exhibited a massive increase in tissue markers of oxidative stress, such as TBARS production, an index of peroxidation of cell membrane lipids and 8-OHdG, a marker of free radical-induced DNA damage (Fig. 4A and B, respectively). The robust increase in TBARS and 8-OHdG levels was blunted by K5-N,OSepi, in a dose-dependent manner, with maximum effect at 1 mg/kg. Similarly, I/R caused a 4-fold increase in calcium content in heart homogenates compared with sham-operated animals and this increase was dose-dependently reduced by K5-N,OSepi (Fig. 4C). On the other hand, administration of the inactive compound B4/100 (1 mg/kg) to rats exposed to I/R had no significant effect on any of the measured markers as compared with control rats that underwent I/R.
Fig 4.

Dose–response effects of K5-N,OSepi on lipid peroxidation, free-radical-induced DNA damage and calcium release in left ventricular tissue. TBARS production (A), 8-OHdG levels (B) and calcium content (C) were measured subsequent to sham operation (Sham) or I/R (30/60 min.) in the absence (vehicle) or presence of K5-N,OSepi (0.1–1 mg/kg) or B4/100 (1 mg/kg) treatment. Data are means ± S.E.M. of eight animals/group. *P < 0.01 versus vehicle.
Effects of K5-N,OSepi on leucocyte infiltration and activation
The improvement in the outcome of I/R injury was associated with a reduced neutrophil infiltration measured in reperfused left ventricular samples at both 1 hr and 24 hrs. As shown in Figure 5A, regional myocardial I/R caused a robust increase in MPO activity, a specific marker of local neutrophil activity, at 1 hr reperfusion, which further increased after 24 hrs reperfusion. In K5-N,OSepi-treated animals, the MPO activity was significantly reduced in a dose-dependent manner, with maximum at 1 mg/kg in both the experimental conditions. The adhesion molecule ICAM-1, which is the endothelial ligand for the neutrophil receptor CD11b/CD18, was scarcely detectable in the hearts from sham-operated animals and its expression was strongly induced by 1 hr and 24 hrs reperfusion (Fig. 5B). Administration of K5-N,OSepi drastically reduced the I/R-induced increase in ICAM-1 expression, without any significant differences between 1 hr and 24 hrs reperfusion. PGE2 and TNF-α, typical inflammatory prostanoid and cytokine released by activated leucocytes, were significantly increased in left ventricular tissue of I/R rats, as compared with the sham-operated animals (Fig. 5C and D, respectively). In contrast, administration of K5-N,OSepi, but not B4/100, prevented the I/R-induced rise in PGE2 and TNF-α levels in a dose-dependent manner. The maximal inhibitory effect recorded at 1 hr reperfusion was similar to that recorded after 24 hrs reperfusion.
Fig 5.

Effects of K5-N,OSepi on neutrophil infiltration and cytokines production induced by regional I/R. Myeloperoxidase (MPO) activity (A), ICAM-1 expression (B), PGE2 (C) and TNF-α (D) levels were measured in myocardial homogenates of sham-operated rats (Sham) and rats that underwent 30 min. ischaemia followed by 1 or 24 hr reperfusion (vehicle). K5-N,OSepi (0.1–1 mg/kg in panels (A, C and D) and 1 mg/kg in panel (B) or B4/100 (1 mg/kg) were administered 15 min. before reperfusion. Densitometric analysis of the bands is expressed as relative optical density (O.D.) corrected for the corresponding β-actin contents and normalized using the related sham-operated band. Data are means ± S.E.M. of eight animals/group for MPO, PGE2 and TNF-α and three separate experiments for Western blot analysis. *P < 0.01 versus vehicle (1h reperfusion) and •P < 0.01 versus vehicle (24 hrs reperfusion).
Effects of K5-N,OSepi on cardiac mast cells
Morphometric analysis of cardiac mast cells in the left ventricle showed that light transmittance across mast cells, which is inversely related to the amount of secretory granules, was significantly higher in I/R hearts in comparison with the sham-operated ones (Fig. 6). Conversely, in the rats treated with K5-N,OSepi, but not with B4/100, the light transmittance across cardiac mast cells was reduced in a dose-dependent manner, reaching levels similar to those recorded in sham-operated animals at the highest dose (1 mg/kg).
Fig 6.
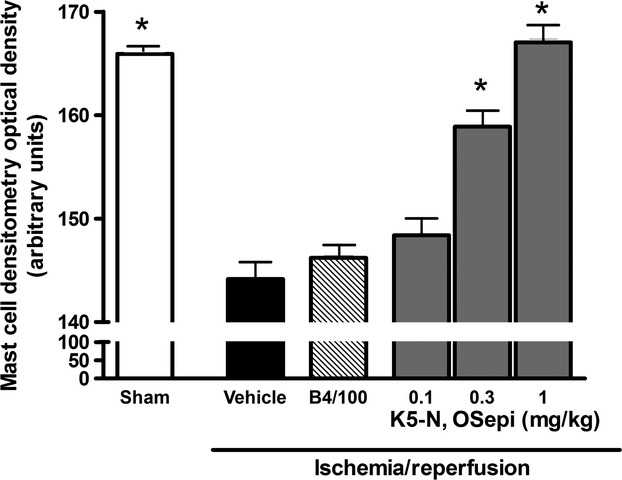
Mast cell densitometry evaluated as light transmittance across left ventricular mast cells. Compared with sham-operated heart (Sham), the I/R hearts (vehicle) show a significant increase in light transmittance, indicating a decrease in intracellularly secretory granules, evaluated at 1 hr reperfusion. This effect is dose-dependently reverted by K5-N,OSepi (0.1–1 mg/kg) treatment, but not by B4/100 (1 mg/kg). Data are means ± S.E.M. of eight animals/group. *P < 0.01 versus vehicle.
Effects of K5-N,OSepi on markers of apoptosis
When compared with sham-operated animals, there was a significant increase in caspase-3 activity within the AAR obtained from rat hearts subjected to myocardial I/R, which was significantly prevented by K5-N,OSepi administration (Fig. 7A). Western blot analysis revealed a significant reduction in the expression of the intact form of the pro-apoptotic protein Bid in the rat hearts subjected to myocardial I/R, when compared with sham-operated rats (Fig. 7B), thus demonstrating its activation by cleavage of intact Bid into truncated forms of Bid. The administration of K5-N,OSepi prevented the I/R-induced activation of Bid, when compared with control rats. Accordingly, the levels of the mitochondrial pro-apoptotic protein Bax were significantly increased by I/R and reduced by K5-N,OSepi administration (Fig. 7C). Interestingly, the basal expression of Bcl-2 protein, a well-known suppressor of apoptosis, was significantly reduced in animals subjected to I/R in comparison with sham-operated rats and this effect was attenuated by treatment with K5-N,OSepi (Fig. 7D). On the contrary, all the above parameters were not changed by B4/100 treatment.
Fig 7.

Effects of K5-N,OSepi on markers of apoptosis in the hearts of rats that underwent myocardial I/R injury. Caspase 3 activity (A) and expression of the apoptotic markers Bid (B), Bax (C) and Bcl-2 (D) were measured in the hearts from sham-operated rats (Sham), rats subjected to 30-min. myocardial ischaemia followed by 1-hr reperfusion (vehicle) or rats subjected to myocardial I/R and treated with K5-N,OSepi (1 mg/kg) or B4/100 (1 mg/kg) treatment. Each immunoblot is from a single experiment and is representative of three separate experiments. Densitometric analysis of the bands is expressed as relative optical density (O.D.) corrected for the corresponding β-actin contents and normalized using the related sham-operated band. Data are means ± S.E.M. of four animals/group for caspase-3 activity and three separate experiments for Western blot analysis. *P < 0.05 versus vehicle.
Overall, these results demonstrate that K5-N,OSepi beneficial effects appear to involve a combination of anti-inflammatory and anti-apoptotic mechanisms. Thus, targeting anti-inflammatory and anti-apoptotic cascades, but not anticoagulant pathways, may hold pharmacological potential for the therapeutic use of heparin-like derivatives in the clinical setting of acute ischaemic events, with significant reduction in the well-known haemorrhagic complications.
Discussion
At doses greatly exceeding those needed for anticoagulation, heparin and low-molecular weight heparins substantially reduce myocardial infarction [11, 12]. However, their potential for causing and haemorrhagic and thrombotic complications continues to arouse caution [13, 28]. Previous studies have revealed that heparin protection against coronary occlusion is independent of the activity of heparin as an anticoagulant and has been attributed largely to inhibition of complement [12] and neutrophil migration into I/R myocardium [29]. Accordingly, an O-desulphated non-anticoagulant high molecular weight heparin has been recently demonstrated to attenuate myocardial I/R injury in pigs and dogs [30, 31]. Here, for the first time, we demonstrated that protective effects against myocardial infarction can be obtained by using K5-N,OSepi, a new bacterial polysaccharide derivative, structurally related to low molecular weight heparins, with a very high degree of N- and O-sulphation and devoid of any anticoagulant activity. This semi-synthetic glycosaminoglycan K5 derivative was obtained through highly selective chemical and enzymatic modifications of the capsular K5 polysaccharide from E. coli. [15]. The presence of O-sulphated glucuronic acid residues, not detectable in extractive heparin, and the conformational change induced by the enzymatic modification of K5 by C5-epimerase enable its strong anti-inflammatory activity, as shown in a rat model of carrageenan-induced pleurisy [18] and in human LPS-stimulated mononuclear cells [16]. Our group has also demonstrated that the K5 derivative K5-N,OSepi protects the brain against I/R injury [17]. In this study, we extend the understanding of the pharmacology of the heparin-like compound, K5-N,OSepi, in a rat model of regional myocardial I/R injury. Our results demonstrate that intravenous administration of K5-N,OSepi 15 min. before the onset of reperfusion caused a significant reduction in the degree of I/R-induced myocardial injury. The highest dose of K5-N,OSepi used in this study (1 mg/kg) reduced myocardial infarct size by approximately 60%, when evaluated at 1 hr reperfusion, without affecting haemodynamic parameters. Interestingly, quantitatively similar reduction in infarct size has been previously reported in different animal models of ischaemic preconditioning, which is recognized as one of the strongest forms of in vivo protection against myocardial I/R injury [32, 33]. In addition, we demonstrated that a substantial reduction in the mean size of ischaemic lesions was still detectable at 24 hrs reperfusion, thus further supporting the clinical relevance of the present study. The persisting protective effects exerted by K5-N,OSepi administration can be, at least in part, due to the high resistance to heparanase digestion, previously documented for similar N-deacetylated/N-sulphated, C5epimerizated K5 polysaccharide derivatives [34]. In contrast, the biologically inactive non-sulphated polysaccharide B4/100, used as negative control molecule, was ineffective in preventing myocardial injury at both 1 hr and 24 hrs reperfusion. Our findings support previous studies showing that, in the ischaemic myocardium, reperfusion accelerates inflammation, which may cause further injury [35]. Here, we confirmed that neutrophil infiltration of previously ischaemic sections of the heart was higher in animals exposed to I/R, when compared with sham-operated animals. Most notably, administration of K5-N,OSepi suppressed the expression of the adhesion molecule ICAM-1, and thus significantly reduced neutrophil migration out of the vessels. In addition, the elevated levels of MPO activity and PGE2 and TNF-α production, used as markers of tissue leucocyte infiltration and activation, were inhibited by K5-N,OSepi treatment. These data are in agreement with other reports showing that heparin and heparin-like derivatives reduce neutrophil adhesion to I/R coronary and vascular endothelium [30, 36]. We can thus speculate that the decreased myocardial neutrophil infiltration may have significantly contributed to the reduced heart injury in rats treated with K5-N,OSepi. As previous studies have shown that prostacyclin (PGI2) plays an important role in reducing neutrophil infiltration during myocardial infarction [37] and defibrotide, an antithrombotic drug with no anticoagulant activity, exert protective effects against myocardial I/R injury by increasing PGI2 endothelial production [38], we may suppose that K5-N,OSepi inhibits local leucocyte activation through an increase in PGI2 levels in the damaged heart tissue. However, further study is required to better elucidate the protective mechanism(s). The marked reduction in the local inflammatory infiltrate caused by K5-N,OSepi treatment may also account for the decrease in calcium overload and tissue markers of oxidative stress, thus supporting the notion that release of ROS from activated neutrophils is responsible at least in part for peroxidation of lipid membranes and free radical-induced DNA damage in the myocardium. An additional contribution to the cardioprotection evoked by the non-anticoagulant heparin-like derivative may rely on its effects on cardiac mast cells. In fact, resident mast cells undergo degranulation during post-ischaemic reoxygenation of the heart, releasing powerful mediators, such as histamine, serotonin and leukotrienes that can increase tissue oedema and induce cardiac arrhythmias [39]. The reduced occurrence of cardiac arrhythmias in the K5-N,OSepi-treated rats is probably related to the decrease in mast cell histamine release, which can also account for the slight, not-significant decrease in HR. In addition, Frangogiannis et al. [40] have shown that in response to I/R, cardiac mast cells degranulate and release TNF-α, which subsequently induces IL-6 expression in infiltrating mononuclear cells. IL-6 in turn up-regulates myocardial ICAM-1 expression, increasing susceptibility to neutrophil-induced adhesion, extravasation and cytotoxicity [41]. Therefore, we may speculate that the inhibitory effects of K5-N,OSepi on mast cell degranulation contribute at least in part to the reduction in leucocyte activation and infiltration and, hence, to myocardial salvage.
Complementary to the attenuation in the inflammatory response, K5-N,OSepi further reduced myocardial apoptosis. After prolonged ischaemia, cardiomyocyte cell death is attributable to necrosis, whereas apoptosis occurs after short coronary occlusion and reperfusion appears to accelerate the timing of apoptosis [42]. Cardiac-specific overexpression of anti-apoptotic regulator Bcl-2 substantially reduces infarct size, cardiac myocyte apoptosis and cardiac dysfunction following I/R [43]. Similarly, mice lacking the multi-domain pro-apoptotic Bax demonstrate reductions in infarct size and dysfunction following I/R [44], while activation of Bid (a pro-apoptotic marker) into truncated forms of tBid has been shown to operate during myocardial I/R [45]. In addition, several pharmacological interventions have been previously reported to exert protection in animal models of myocardial I/R injury, at least in part, by affecting apoptotic cell death [46–48]. Our results indicate that K5-N,OSepi mediated significant anti-apoptotic effects, which are secondary to the prevention by K5-N,OSepi of Bax expression and caspase 3 and Bid activation caused by I/R. In addition, K5-N,OSepi ameliorated the expression of the anti-apoptotic marker Bcl-2 protein, whose levels were drastically reduced by I/R injury. Recently, evidence has been accumulating to show that there is continual cross-talk between local I/R-related inflammatory response and cardiomyocyte apoptosis [49, 50]. For instance, in cardiomyocytes, apoptotic cell death in response to TNF-α involves calcium-mediated calpain activation [51]. TNF-α also activates the extrinsic death pathway resulting in the recruitment and activation of caspase-3 [52]. We might thus speculate that suppression of apoptosis by K5-N,OSepi is a secondary event through its modulation of inflammatory response parameters, mainly TNF-α production. Nevertheless, in the absence of direct experimental data, we cannot rule out the possibility that K5-N,OSepi directly affect specific apoptotic pathways. Further investigation is required to determine the mechanism(s) more precisely.
Taken together, the above-described results indicate that K5-N,OSepi protects against I/R-induced myocardial cell injury and cardiac dysfunction via several different mechanisms, including inhibition of leucocyte infiltration and modulation of the apoptosis evoked by regional myocardial I/R in the rat. The results of this study offer good perspectives for a novel therapeutic approach in the medical treatment of myocardial infarction, based on the use of new heparin derivatives, with low anticoagulant and high anti-inflammatory activities. These heparin-like derivatives may have several advantages over heparin and low-molecular-weight heparins, mainly less risk of haemorrhagic and thrombotic complications, thus, meeting some crucial requirements for clinical use.
Acknowledgments
This work was supported by grants from Regione Piemonte, Ente Cassa di Risparmio of Florence, Cardiovascular Research Institute and the Italian Ministry of Education, University and Research (Progetti di Ricerca di Interesse Nazionale, 2007).
Author contributions
Collino, Pini, Mastroianni, Benetti and Lanzi performed the experiments; Manoni and Chini contributed heparin-like derivatives and provided information on K5-N,OSepi half life; Collino, Bani, Fantozzi and Masini analysed the data and wrote the manuscript; Masini designed and acquired funding for the research.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Steffens S, Montecucco F, Mach F. The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thromb Haemost. 2009;102:240–7. doi: 10.1160/TH08-12-0837. [DOI] [PubMed] [Google Scholar]
- 3.Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–97. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Mannaioni PF, Masini E. The release of histamine by free radicals. Free Radic Biol Med. 1988;5:177–97. doi: 10.1016/0891-5849(88)90080-9. [DOI] [PubMed] [Google Scholar]
- 5.Masini E, Bianchi S, Gambassi F, et al. Ischaemia reperfusion injury and histamine release in isolated and perfused guinea-pig heart: pharmacological interventions. Agents Actions. 1990;30:198–201. doi: 10.1007/BF01969037. [DOI] [PubMed] [Google Scholar]
- 6.Hearse DJ, Tosaki A. Free radicals and calcium: simultaneous interacting triggers as determinants of vulnerability to reperfusion-induced arrhythmias in the rat heart. J Mol Cell Cardiol. 1988;20:213–23. doi: 10.1016/s0022-2828(88)80054-3. [DOI] [PubMed] [Google Scholar]
- 7.Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol Med. 2003;9:177–82. doi: 10.1016/s1471-4914(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 8.Di Napoli P, Taccardi AA, Grilli A, et al. Left ventricular wall stress as a direct correlate of cardiomyocyte apoptosis in patients with severe dilated cardiomyopathy. Am Heart J. 2003;146:1105–11. doi: 10.1016/S0002-8703(03)00445-9. [DOI] [PubMed] [Google Scholar]
- 9.Bialik S, Geenen DL, Sasson IE, et al. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–72. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akasaka Y, Morimoto N, Ishikawa Y, et al. Myocardial apoptosis associated with the expression of proinflammatory cytokines during the course of myocardial infarction. Mod Pathol. 2006;19:588–98. doi: 10.1038/modpathol.3800568. [DOI] [PubMed] [Google Scholar]
- 11.Yan AT, Goodman SG. Low-molecular-weight heparins in ischemic heart disease. Curr Opin Cardiol. 2004;19:309–16. doi: 10.1097/01.hco.0000129665.55707.f6. [DOI] [PubMed] [Google Scholar]
- 12.Friedrichs GS, Kilgore KS, Manley PJ, et al. Effects of heparin and N-acetyl heparin on ischaemia/reperfusion-induced alterations in myocardial function in the rabbit isolated heart. Circ Res. 1994;75:701–10. doi: 10.1161/01.res.75.4.701. [DOI] [PubMed] [Google Scholar]
- 13.Das P, Ziada K, Steinhubl SR, et al. Heparin-induced thrombocytopenia and cardiovascular diseases. Am Heart J. 2006;152:19–26. doi: 10.1016/j.ahj.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Pevni D, Frolkis I, Shapira I, et al. Heparin added to cardioplegic solution inhibits tumour necrosis factor-alpha production and attenuates myocardial ischemic-reperfusion injury. Chest. 2005;128:1805–11. doi: 10.1378/chest.128.3.1805. [DOI] [PubMed] [Google Scholar]
- 15.Rusnati M, Oreste P, Zoppetti G, et al. Biotechnological engineering of heparin/heparan sulphate: a novel area of multi-target drug discovery. Curr Pharm Des. 2005;11:2489–99. doi: 10.2174/1381612054367553. [DOI] [PubMed] [Google Scholar]
- 16.Gori AM, Attanasio M, Gazzini A, et al. Cytokine gene expression and production by human LPS-stimulated mononuclear cells are inhibited by sulfated heparin-like semi-synthetic derivatives. J Thromb Haemost. 2004;2:1657–62. doi: 10.1111/j.1538-7836.2004.00866.x. [DOI] [PubMed] [Google Scholar]
- 17.Collino M, Castiglia S, Manoni M, et al. Effects of a semi-synthetic N-,O-sulfated glycosaminoglycan K5 polysaccharide derivative in a rat model of cerebral ischaemia/reperfusion injury. Thromb Haemost. 2009;102:837–45. doi: 10.1160/TH09-01-0012. [DOI] [PubMed] [Google Scholar]
- 18.Ceccarelli M, Bani D, Cinci L, et al. Anti-inflammatory effects of low molecular weight heparin derivative in a rat model of carrageenan-induced pleurisy. J Cell Mol Med. 2009;13:2704–12. doi: 10.1111/j.1582-4934.2008.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbinati C, Bugatti A, Oreste P, et al. Chemically sulfated Escherichia coli K5 polysaccharide derivatives as extracellular HIV-1 Tat protein antagonists. FEBS Lett. 2004;568:171–7. doi: 10.1016/j.febslet.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 20.Guerrini M, Bisio A, Torri G. Combined quantitative (1)H and (13)C nuclear magnetic resonance spectroscopy for characterization of heparin preparations. Semin Thromb Hemost. 2001;27:473–82. doi: 10.1055/s-2001-17958. [DOI] [PubMed] [Google Scholar]
- 21.Gelvan D, Saltman P, Powell SR. Cardiac reperfusion damage prevented by a nitroxide free radical. Proc Natl Acad Sci USA. 1991;88:4680–4. doi: 10.1073/pnas.88.11.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masini E, Cuzzocrea S, Mazzon E, et al. Protective effects of M40403, a selective superoxide dismutase mimetic, in myocardial ischaemia and reperfusion injury in vivo. Br J Pharmacol. 2002;136:905–17. doi: 10.1038/sj.bjp.0704774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bani D, Masini E, Bello MG, et al. Relaxin protects against myocardial injury caused by ischaemia and reperfusion in rat heart. Am J Pathol. 1998;152:1367–76. [PMC free article] [PubMed] [Google Scholar]
- 24.Aruoma OI, Halliwell B, Laughton MJ, et al. The mechanism of initiation of lipid peroxidation. Evidence against a requirement for an iron(II)-iron(III) complex. Biochem J. 1989;258:617–20. doi: 10.1042/bj2580617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masini E, Bani D, Vannacci A, et al. Reduction of antigen-induced respiratory abnormalities and airway inflammation in sensitized guinea pigs by a superoxide dismutase mimetic. Free Radic Biol Med. 2005;39:520–31. doi: 10.1016/j.freeradbiomed.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem. 1997;272:25719–23. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- 27.Collino M, Thiemermann C, Mastrocola R, et al. Treatment with the glycogen synthase kinase-3beta inhibitor, TDZD-8, affects transient cerebral ischaemia/reperfusion injury in the rat hippocampus. Shock. 2008;30:299–307. doi: 10.1097/SHK.0b013e318164e762. [DOI] [PubMed] [Google Scholar]
- 28.Magee KD, Campbell SG, Moher D, et al. Heparin versus placebo for acute coronary syndromes. Cochrane Database Syst Rev. 2008:CD003462. doi: 10.1002/14651858.CD003462.pub2. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Brown JR, Varki A, et al. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. 2002;110:127–36. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thourani VH, Brar SS, Kennedy TP, et al. Nonanticoagulant heparin inhibits NF-kappaB activation and attenuates myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2000;278:H2084–93. doi: 10.1152/ajpheart.2000.278.6.H2084. [DOI] [PubMed] [Google Scholar]
- 31.Barry WH, Zhang XQ, Halkos ME, et al. Nonanticoagulant heparin reduces myocyte Na+ and Ca2+ loading during simulated ischaemia and decreases reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298:H102–11. doi: 10.1152/ajpheart.00316.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hide EJ, Thiemermann C. Limitation of myocardial infarct size in the rabbit by ischaemic preconditioning is abolished by sodium 5-hydroxydecanoate. Cardiovasc Res. 1996;31:941–6. [PubMed] [Google Scholar]
- 33.Schultz JE, Hsu AK, Gross GJ. Ischemic preconditioning in the intact rat heart is mediated by delta1- but not mu- or kappa-opioid receptors. Circulation. 1998;97:1282–9. doi: 10.1161/01.cir.97.13.1282. [DOI] [PubMed] [Google Scholar]
- 34.Pikas DS, Li JP, Vlodavsky I, et al. Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem. 1998;273:18770–7. doi: 10.1074/jbc.273.30.18770. [DOI] [PubMed] [Google Scholar]
- 35.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 36.Silvestro L, Viano I, Macario M, et al. Effects of heparin and its desulfated derivatives on leukocyte-endothelial adhesion. Semin Thromb Hemost. 1994;20:254–8. doi: 10.1055/s-2007-1001910. [DOI] [PubMed] [Google Scholar]
- 37.Rossoni G, Manfredi B, De Gennaro Colonna V, et al. Nitric oxide and prostacyclin pathways: an integrated mechanism that limits myocardial infarction progression in anaesthetized rats. Pharmacol Res. 2006;53:359–66. doi: 10.1016/j.phrs.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Thiemermann C, Thomas GR, Vane JR. Defibrotide reduces infarct size in a rabbit model of experimental myocardial ischaemia and reperfusion. Br J Pharmacol. 1989;97:401–8. doi: 10.1111/j.1476-5381.1989.tb11967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaggi AS, Singh M, Sharma A, et al. Cardioprotective effects of mast cell modulators in ischaemia-reperfusion-induced injury in rats. Methods Find Exp Clin Pharmacol. 2007;29:593–600. doi: 10.1358/mf.2007.29.9.1161005. [DOI] [PubMed] [Google Scholar]
- 40.Frangogiannis NG, Lindsey ML, Michael LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischaemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 41.Sanceau J, Kaisho T, Hirano T, et al. Triggering of the human interleukin-6 gene by interferon-gamma and tumour necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J Biol Chem. 1995;270:27920–31. doi: 10.1074/jbc.270.46.27920. [DOI] [PubMed] [Google Scholar]
- 42.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Chua CC, Ho YS, et al. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–20. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 44.Hochhauser E, Cheporko Y, Yasovich N, et al. Bax deficiency reduces infarct size and improves long-term function after myocardial infarction. Cell Biochem Biophys. 2007;47:11–20. doi: 10.1385/cbb:47:1:11. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, He H, Zhan S, et al. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischaemia/reperfusion. J Biol Chem. 2001;276:30724–8. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 46.Vecsernyes M, Juhasz B, Der P, et al. The administration of alpha-melanocyte-stimulating hormone protects the ischemic/reperfused myocardium. Eur J Pharmacol. 2003;470:177–83. doi: 10.1016/s0014-2999(03)01780-1. [DOI] [PubMed] [Google Scholar]
- 47.Das S, Lekli I, Das M, et al. Cardioprotection with palm oil tocotrienols: comparison of different isomers. Am J Physiol Heart Circ Physiol. 2008;294:H970–8. doi: 10.1152/ajpheart.01200.2007. [DOI] [PubMed] [Google Scholar]
- 48.Sivarajah A, Collino M, Yasin M, et al. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;31:267–74. doi: 10.1097/SHK.0b013e318180ff89. [DOI] [PubMed] [Google Scholar]
- 49.Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH. Molecular biology of apoptosis in ischaemia and reperfusion. J Invest Surg. 2005;18:335–50. doi: 10.1080/08941930500328862. [DOI] [PubMed] [Google Scholar]
- 50.Yan L, Tang Q, Shen D, et al. SOCS-1 inhibits TNF-alpha-induced cardiomyocyte apoptosis via ERK1/2 pathway activation. Inflammation. 2008;31:180–8. doi: 10.1007/s10753-008-9063-5. [DOI] [PubMed] [Google Scholar]
- 51.Bajaj G, Sharma RK. TNF-alpha-mediated cardiomyocyte apoptosis involves caspase-12 and calpain. Biochem Biophys Res Commun. 2006;345:1558–64. doi: 10.1016/j.bbrc.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 52.Haudek SB, Taffet GE, Schneider MD, et al. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117:2692–701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]