

Abstract
Cells derived from the amniotic foetal membrane of human term placenta have drawn particular attention mainly for their plasticity and immunological properties, which render them interesting for stem-cell research and cell-based therapeutic applications. In particular, we have previously demonstrated that amniotic mesenchymal tissue cells (AMTC) inhibit lymphocyte proliferation in vitro and suppress the generation and maturation of monocyte-derived dendritic cells. Here, we show that AMTC also significantly reduce the proliferation of cancer cell lines of haematopoietic and non-haematopoietic origin, in both cell–cell contact and transwell co-cultures, therefore suggesting the involvement of yet-unknown inhibitory soluble factor(s) in this ‘cell growth restraint’. Importantly, we provide evidence that the anti-proliferative effect of AMTC is associated with induction of cell cycle arrest in G0/G1 phase. Gene expression analyses demonstrate that AMTC can down-regulate cancer cells' mRNA expression of genes associated with cell cycle progression, such as cyclins (cyclin D2, cyclin E1, cyclin H) and cyclin-dependent kinase (CDK4, CDK6 and CDK2), whilst they up-regulate cell cycle negative regulator such as p15 and p21, consistent with a block in G0/G1 phase with no progression to S phase. Taken together, these findings warrant further studies to investigate the applicability of these cells for controlling cancer cell proliferation in vivo.
Keywords: amniotic membrane, amnion-derived cells, placenta, mesenchymal stem/stromal cells, cancer cell lines, cell proliferation
Introduction
In recent years, mesenchymal stromal/stem cells (MSCs) have been extensively studied as a valuable tool for developing novel therapeutic cell-based approaches [1]. MSCs have been isolated from bone marrow (BM) as well as from almost all other sites of the body, including placenta [2]. One of the most intriguing properties of these cells is their immunomodulatory activity, which targets virtually all types of immune cells. Indeed, it has been demonstrated that MSCs are able to suppress T-lymphocyte activation and proliferation in vitro, and modulate B-cell functions and the proliferation and differentiation of specific T-cell subsets [e.g. regulatory T cells, T helper (Th)1 and Th2 cells]. MSCs can also affect the cytotoxic activity of natural killer cells, and also interfere with differentiation, maturation and function of dendritic cells (DCs) [3–5].
The question of whether cell types other than immune cells could be targets of the inhibitory effects of MSCs is attracting ever increasing attention. Of particular interest are the anti-proliferative actions of MSCs on cancer cells, which could have important implications on cancer treatment. In this regard, conflicting results have been obtained to date for both MSCs and cancer cells of different origins. For example, it has been shown that BM-derived MSCs are able to inhibit the in vitro proliferation of cancer cells of both haematopoietic and non-haematopoietic origin [6–9]. Meanwhile, other authors have reported that adipose tissue-derived MSCs do not suppress lymphoblastic leukemic cell line proliferation [10], but may exert different effects (either inhibition, increase or no effect on proliferation), on various cancer cells derived from human or murine sources [11].
Conflicting results have also been obtained regarding the effect of MSCs on cancer cell apoptosis, i.e. promotion of survival from spontaneous or induced apoptosis [12, 13] versus increased apoptosis of human hepatoma cell lines [14, 15] or lymphoma cells [15–17].
Several groups have also shown that MSCs are capable of homing towards primary and metastatic tumour locations within the body [18–20], suggesting that MSCs might be an attractive tool for developing novel cancer treatments. However, although some studies on MSC tumour homing and treatment in vivo have led to demonstrations that MSCs possess anti-tumoural effects (e.g. injection of low number of MSCs completely abolished adenocarcinoma tumour formation) [11], it has also been reported that these cells may enhance growth and development, as well as metastatic potency, of tumours of different origins [3, 12, 13, 21, 22].
Through in vitro studies, we have previously shown that human amniotic membrane-derived MSCs (herein referred to as AMTC, for amniotic mesenchymal tissue cells), strongly inhibit lymphocyte proliferation induced by allo-antigens or via T-cell receptor cross-linking [23, 24], suppress the generation and maturation of monocyte-derived DCs and abolish the production of inflammatory cytokines [25]. In addition, we and others have demonstrated that foetal membrane-derived cells (including AMTC) can migrate and successfully engraft long-term in several organs and tissues [23], and display enormous potential for treating inflammatory and fibrotic diseases after transplantation in vivo [26]. Notably, the potential effects of AMTC on cancer cells have never been investigated before.
In this study, we evaluated the effects of AMTC on the proliferation of different cancer cell lines and provide evidence that these cells block cancer cells in the G0/G1 phase of the cell cycle, but they do not induce apoptotic cell death.
Materials and methods
All biological samples (placenta, BM and skin biopsy) were obtained with informed consent according to the guidelines of the Ethical Committee of the hospital Fondazione Poliambulanza-Istituto Ospedaliero (Brescia, Italy).
Isolation of amniotic mesenchymal tissue cells
Placentas (n = >30) were obtained from healthy women after vaginal delivery or caesarean section. AMTC were isolated according to a well-established protocol, as previously described [24, 27]. Immediately after isolation, AMTC were plated in RPMI complete medium composed of RPMI 1640 medium (Lonza, Basel, Switzerland), supplemented with 10% heat-inactivated foetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA), 2 mM l-glutamine (Lonza), 100 U/ml penicillin and 100 μg/ml streptomycin (both from Euroclone, Whetherby, UK). The cells were used according to the different experimental settings as described below. The phenotype of cells used is described in supplementary section (Table S1 and Fig. S1).
Isolation of human dermal fibroblasts
Human fibroblasts were isolated from a skin biopsy. The biopsy was diced into small fragments, layered onto six-well plates (Corning, NY, USA) and incubated in DMEM complete medium: DMEM (Sigma-Aldrich) supplemented with 20% FBS, 2 mM l-glutamine and 1 mM Na-Pyruvate (Lonza), 100 U/ml penicillin and 100 μg/ml streptomycin, at 37°C in a humidified atmosphere of 5% CO2. When fibroblasts reached confluency (after about 10 days), cells were trypsinized (0.25% trypsin-EDTA solution; Sigma-Aldrich) and from passage 2 expanded in RPMI complete medium to be consistent with AMTC, cancer cell lines and all co-culture experiments. Cells were sub-cultured at a density of 10–15 × 103 cells/cm2 and used for experiments during passages 6 to 20.
Isolation of BM-derived MSCs
The BM was aspirated from the femoral heads of patients undergoing orthopaedic surgery and BM-MSCs were isolated as previously described [27], with some modifications. BM samples were diluted (1:4) in phosphate-buffered saline (PBS) (Sigma-Aldrich) and centrifuged at 900 × g for 15 min. After discarding the fat layer and supernatant, the cells were layered on a Lymphoprep gradient (Axis Shield, Oslo, Norway) and centrifuged at 670 × g for 30 min. Recovered mononuclear cells were plated at a density of 1 × 106 cells/cm2 in DMEM complete medium, and incubated at 37°C, 5% CO2. After 3 days, non-adherent cells were removed and adherent cells were cultured until they reached confluency. Cells were then trypsinized and sub-cultured at a density of 8 × 103 cells/cm2, and were used for experiments during passages 6 to 11.
Cancer cell lines
The following cancer cell lines were obtained from the Centro Substrati Cellulari, Istituto Zooprofilattico of Brescia (Italy): KG1 cells (human acute myelogenous leukaemia cell line), KG1a cells (an undifferentiated variant of the KG-1 cell line) [28], Jurkat cells (human T-cell leukaemia cell line), U937 cells (human monocytic cell line obtained from histiocytic lymphoma), Girardi heart cells (human heart cell line), HeLa cells (human cervical epithelioid carcinoma cell line) and Saos cells (human osteosarcoma cell line). All cells were cultured in RPMI complete medium at 37°C in 5% CO2.
Proliferation assays
Effects of AMTC on the proliferation of cancer cell lines
Cancer cell lines (KG1, KG1a, Jurkat, U937, Girardi heart, HeLa and Saos) were cultured alone or in the presence of different concentrations of AMTC (at a ratio of cancer cells: AMTC of 1:0.25, 1:0.5, 1:1, 1:2 or 1:4), either in direct contact or with physical separation using transwell chambers. For direct contact experiments, 1.25 × 104, 2.5 × 104, 5 × 104, 10 × 104 or 20 × 104 AMTC at passage 0 were plated in flat-bottom 96-well plates (Corning) in RPMI complete medium, and then gamma-irradiated (30 Gy). AMTC were irradiated to block cell proliferation, so that any proliferation observed could be attributed solely to the not-irradiated cell line. After 1 day, 5 × 104 tumour cells were added to each well. For non-contact experiments, co-cultures were established by using a transwell (0.4 μm pore, polycarbonate membrane, Corning) of 24-wells. AMTC (6.25 × 104, 12.5 × 104, 25 × 104, 50 × 104 or 100 × 104 cells in 300 μl of RPMI complete medium) were plated in the upper compartment and cancer cell lines were plated in the lower compartment (25 × 104 cells in 1 ml of RPMI complete medium).
After 3 days, proliferation of cancer cells was assessed by adding [3H]-thymidine (1 μCi/well; Perkin Elmer, Life Sciences, Zaventem, Belgium) for 16–18 hrs. Cells were then harvested with a Filtermate Harvester (Perkin Elmer) and thymidine incorporation was measured by using a microplate scintillation and luminescence counter (Top Count NXT; Perkin Elmer).
Effects of different cells on the proliferation of cancer cell lines
Cancer cells (Jurkat and U937) were cultured alone or in the presence of AMTC, fibroblast dermal cells, BM-MSCs, KG1a, Jurkat or U937 cells (herein referred to as ‘modulators’), in either direct contact or transwell settings, at a ratio of tumour cells: modulators of 1:2. For contact experiments, 10 × 104 modulators were plated in flat-bottom 96-well plates in RPMI complete medium, and then gamma-irradiated (30 Gy for AMTC, fibroblast dermal cells and BM-MSCs; 60 Gy for KG1a, Jurkat and U937 cells). After 1 day, 5 × 104 tumour cells were added to each well. For non-contact experiments, modulators (10 × 104 cells in 80 μl of RPMI complete medium) were plated in the upper compartment (0.4-μm pore, polycarbonate membranes, 96-well plates, Corning) and cancer cells were plated in the lower compartment of transwell chambers (5 × 104 cells in 200 μl of RPMI complete medium). Proliferation of cancer cells was assessed after 1, 2 and 3 days by measuring thymidine incorporation, as described above.
Apoptosis analysis
Jurkat and U937 cells (25 × 104) were cultured alone or in the presence of AMTC in 24-well plates (Corning), in either direct contact or transwell settings, at a tumour cell: AMTC ratios of 1:1 and 1:2. After 5, 24 and 48 hrs, cancer cells were harvested and analysed for cell apoptosis by Annexin-V and propidium iodide (PI) staining, using the FITC Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA, USA), according to the manufacturer's instructions. To avoid the possibility that the analysis of cancer cell apoptosis might be confounded by the presence of AMTC (collected when plated in contact with cancer cells), the cancer cells were stained with an allophycocyanin (APC)-conjugated antibody specific for human CD45 (APC-CD45), (clone 2D1, BD Biosciences) prior to the Annexin V/PI staining, and the analysis was restricted to the CD45-positive cancer cells. Specifically, cells were collected and washed with FACS buffer [0.1% sodium azide (Sigma-Aldrich) and 0.1% bovine serum albumin (BSA), (Promega Corporation, Madison, WI, USA) in PBS]. To block non-specific binding, cells were then incubated with 20 mg/ml polyglobin (Gammagard#x00AE;; Baxter, Deerfield, IL, USA) prepared in PBS with 1% BSA, and then with APC-CD45 for 20 min. at 4°C. After two washes in PBS, cells were incubated with FITC-Annexin V and PI for 15 min at room temperature. Samples were acquired and CD45-positive tumour cells were analysed with a FACS Calibur and the CellQuest Software (BD Biosciences).
Cell cycle analysis
Jurkat and U937 cells were synchronized in the G1/S phase of the cell cycle by aphidicolin blocking [29]. Specifically, U937 and Jurkat cells (6 × 105 cells/ml) were incubated for 24 hrs with 2.5 and 5 μg/ml aphidicolin (Sigma-Aldrich), respectively, in RPMI complete medium containing 1% heat-inactivated FBS instead of 10%, at 37°C and 5% CO2. Synchronized Jurkat and U937 cells (25 × 104) were cultured alone or in the presence of AMTC in 24-well plates, in either direct contact or transwell settings, at a cancer cell: AMTC ratio of 1:2. After 16, 24 and 48 hrs, cancer cells were harvested and analysed for cell cycle with the APC bromodeoxyuridine (BrdU) flow kit (BD Biosciences), according to the manufacturer's instructions. Samples were acquired and analysed with a FACS Calibur and the CellQuest Software (BD Biosciences).
Quantitative (real time) RT-PCR-based gene array assays
Jurkat and U937 cells (25 × 104 cells/ml in RPMI complete medium) that were synchronized in the G1/S phase were cultured in the presence of AMTC in a transwell system, at a cancer cell: AMTC ratio of 1:2. After 24 hrs, cancer cells were harvested to analyse the expression of a panel of genes related to cell cycle by using the Cell Cycle PCR Array kit (SuperArray Biosciences, Qiagen, Frederick, MD, USA). Samples were analysed according to the manufacturer's instructions by using an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA, USA). Total RNA was purified from 1 × 106 cells by using the EZ1 RNA cell Mini Kit protocol (Qiagen), in a BioRobot EZ1 Workstation. The RT2 First Strand Kit was used for cDNA synthesis (Sabiosciences, Qiagen) from 500 ng of RNA. The cDNA template was mixed with 2X SABiosciences RT2 qPCR Master Mix and water, in a final volume of 25 μl. The real-time PCR cycling program was as follows: 10 min. at 95°C, 40 cycles of 15 sec. at 95°C, 1 min. at 60°C. Data were analysed with the Excel-based PCR Array Data Analysis Template provided by the manufacturer.
Statistical analysis
Data are expressed as mean ± S.D. Analysis of variance was used to assess differences between groups. Raw P-values were adjusted by Holm-Bonferroni's procedure for multiple comparison. P-values < 0.05 were considered statistically significant.
Results
Effect of AMTC on the proliferation of cancer cell lines
We first investigated the effects of AMTC on the proliferation of different cancer cell lines of both haematopoietic and non-haematopoietic origin. Specifically, AMTC were co-cultured in direct contact with KG1, KG1a, Jurkat, U937, Girardi heart, HeLa or Saos cells at different ratios. AMTC reduced the proliferative activity of all of the cancer cell lines investigated and this effect was cell-dose-dependent. The proliferation of all of the cancer cell lines tested was significantly inhibited at a cancer cells: AMTC ratio of 1:1. Meanwhile, the proliferation of KG1 and U937 cell lines was also significantly reduced with a ratio of 1:0.25 (Fig. 1, upper panel).
Fig 1.
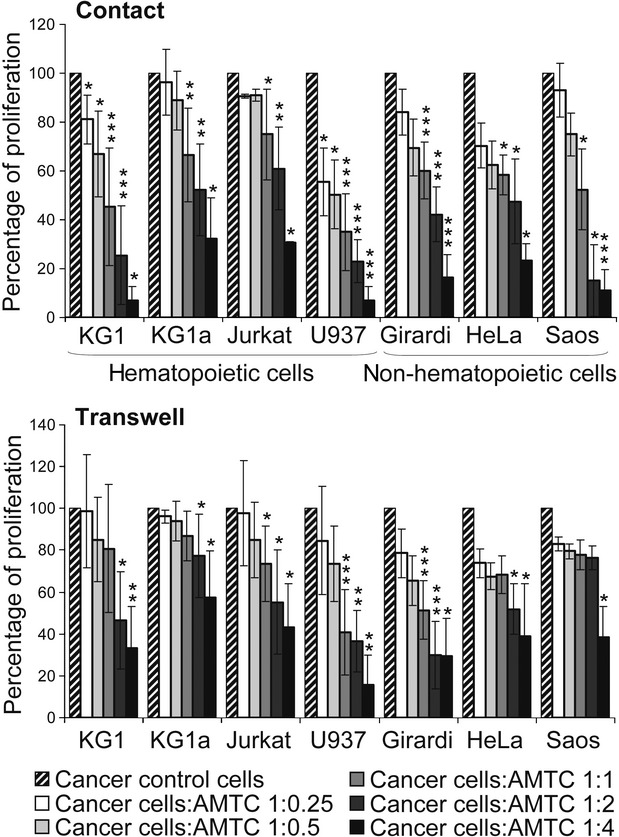
AMTC reduce the proliferation of cancer cells of both haematopoietic and non-haematopoietic origin. Haematopoietic and non-haematopoietic cancer cell lines were cultured alone (control), or with AMTC either in direct contact (upper panel) or with physical separation (transwell setting, lower panel). Different cancer cell: AMTC ratios were used. After 3 days of culture, cancer cell proliferation was assessed by [3H]-thymidine incorporation and reported as a percentage of cell proliferation in comparison with control cancer cell proliferation. Data are expressed as mean ± S.D. of at least four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding control sample.
This anti-proliferative effect was also observed when AMTC and cancer cell co-cultures were physically separated in a transwell system (Fig. 1, lower panel). In the non-contact setting, proliferation was significantly reduced when the ratio of cancer cells: AMTC was 1:1 for Jurkat, U937 and Girardi heart cells and 1:2 for KG1, KG1a, and HeLa cells. AMTC were able to significantly alter the proliferation of Saos cells only at a ratio of 1:4 (Fig. 1, lower panel). Taken together, these data indicate that AMTC-induced reduction of cancer cell proliferation is not strictly dependent on cell-to-cell contact, but may be mediated by soluble factors released by AMTC.
Effect of different cells on the proliferation of cancer cell lines
To further explore the anti-proliferative properties of AMTC, we focused our studies on the effects of AMTC on representative haematopoietic cell lines of lymphocyte origin and monocyte precursor origin, namely, Jurkat and U937 cells.
First, we compared the effects of AMTC on the proliferation of these cells with the effects exerted by other cells, namely, primary stromal cells (BM-MSCs, dermal fibroblast cells) and cancer cell lines (KG1a, as well as Jurkat and U937 themselves) after 1, 2 and 3 days of co-culture. A statistically significant inhibitory effect was detected for AMTC on both Jurkat and U937 cells in both contact and transwell co-culture settings, at all of the different time points analysed (Fig. 2). BM-MSCs significantly reduced the proliferative ability of the two tumour cell lines only in a contact co-culture system, and meanwhile, under this same condition, human dermal fibroblasts only had a significant effect on U937 proliferation (Fig. 2). When co-cultures were performed with combinations of two different cancer cell lines, no inhibitory effects on cancer cell proliferation were observed (Fig. 2).
Fig 2.
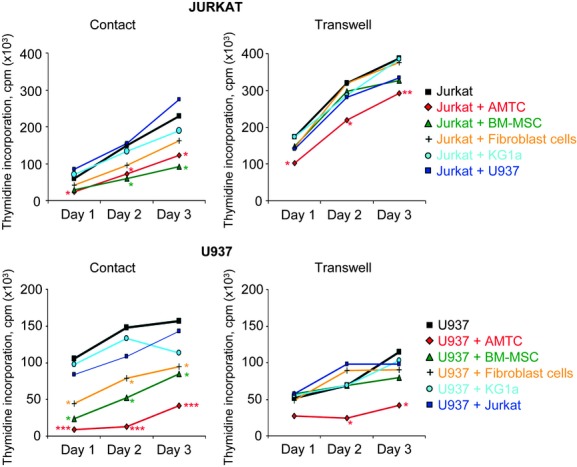
Alteration in cancer cell proliferation by AMTC and other cell lines. Jurkat and U937 cells were cultured alone or in the presence of different γ-irradiated cell types (AMTC or BM-MSC, dermal fibroblast cells, KG1a, U937 and Jurkat). Co-cultures were performed in both contact and transwell settings, at cancer cell: different cell types ratios of 1:2. After 1, 2 and 3 days of culture, Jurkat and U937 cell proliferation was assessed by [3H]-thymidine incorporation. Data are expressed as mean of more than four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding control sample.
Effect of AMTC on cancer cell apoptosis
To assess whether the reduction of cancer cell proliferation induced by AMTC was associated with an increase in cell apoptosis, we analysed the apoptotic rate of Jurkat and U937 cells at different time points (5, 24 and 48 hrs) after they had been co-cultured with AMTC in both contact and transwell settings. The percentages of both early and late apoptotic cells were not significantly increased in comparison with those registered when Jurkat and U937 cells were cultured alone (Table 1).
Table 1.
AMTC-induced inhibition of cancer cell proliferation is not mediated by tumour cell apoptosis
| Cancer cell line, % ± S.D. | Cancer cell line + AMTC contact, % ± S.D. | Cancer cell line + AMTC transwell, % ± S.D. | |||
|---|---|---|---|---|---|
| JURKAT | 5 hrs | Early apoptosis | 20.3 ± 2.88 | 8.03 ± 1.95 | 15.6 ± 4.42 |
| Late apoptosis | 17.1 ± 8.27 | 11.6 ± 8.25 | 16.0 ± 8.58 | ||
| 24 hrs | Early apoptosis | 11.8 ± 5.00 | 6.10 ± 3.38 | 9.69 ± 1.27 | |
| Late apoptosis | 18.1 ± 6.08 | 16.0 ± 10.2 | 26.3 ± 8.21 | ||
| 48 hrs | Early apoptosis | 7.25 ± 1.17 | 2.61 ± 1.41 | 4.69 ± 2.11 | |
| Late apoptosis | 12.9 ± 7.25 | 10.0 ± 7.19 | 21.3 ± 11.7 | ||
| U937 | 5 hrs | Early apoptosis | 2.47 ± 0.75 | 4.89 ± 4.14 | 3.36 ± 1.69 |
| Late apoptosis | 4.10 ± 4.35 | 3.40 ± 3.10 | 4.40 ± 4.71 | ||
| 24 hrs | Early apoptosis | 2.65 ± 1.21 | 5.99 ± 0.56 | 7.01 ± 2.92 | |
| Late apoptosis | 3.13 ± 2.24 | 4.30 ± 1.18 | 4.34 ± 2.88 | ||
| 48 hrs | Early apoptosis | 2.42 ± 0.35 | 3.00 ± 2.33 | 8.1 ± 5.52 | |
| Late apoptosis | 3.72 ± 0.81 | 5.73 ± 4.01 | 11.3 ± 7.30 |
The results are expressed as percentages and represent the mean ± S.D. of at least three independent experiments.
Cell cycle analysis in cancer cells co-cultured with AMTC
To determine whether AMTC could affect cell cycle progression, Jurkat and U937 cells, which had been aphidicolin-synchronized in the G1/S phase, were co-cultured in contact or in transwell setting with AMTC. After 16, 24 and 48 hrs, cell cycle assay by flow cytometry showed that for Jurkat cells cultured with AMTC, the percentage of cells in the G0/G1 phase was higher than that of cells cultured alone (Fig. 3). This was also accompanied by a considerable reduction in the percentage of Jurkat cells in the S phase (Fig. 3 and Table 2). When U937 cells were co-cultured with AMTC, there was a statistically significant accumulation of U937 cells in the G0/G1 phase, and the percentage of cells that entered the S phase was clearly reduced in comparison with control cells cultured without AMTC (Fig. 3 and Table 2).
Fig 3.
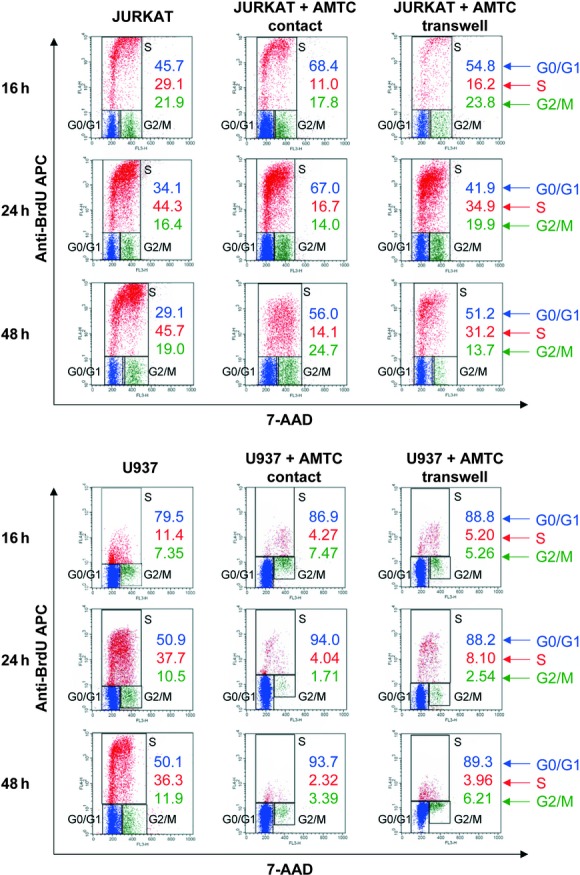
AMTC arrest the cell cycle of cancer cells. Jurkat (upper panel) and U937 (lower panel) cells were cultured alone or in the presence of AMTC, in either contact or transwell settings, at a cancer cell: AMTC ratio of 1:2. At 16, 24 and 48 hrs, cultures were pulsed with BrdU for 1h, and cancer cells were then collected and stained with anti-BrdU APC and 7-AAD. The percentage of cells in the G0/G1, S or G2/M phase of the cell cycle is indicated. The FACS profiles shown are representative of three independent experiments.
Table 2.
AMTC arrest the cell cycle of cancer cells
| Cancer cell line, % ± S.D. | Cancer cell line + AMTC contact, % ± S.D. | Cancer cell line + AMTC transwell, % ± S.D. | |||
|---|---|---|---|---|---|
| JURKAT | 16 hrs | G0/G1 phase | 48.2 ± 6.65 | 61.6 ± 12.0 | 55.27 ± 1.84 |
| S phase | 21.9 ± 7.41 | 18.1 ± 9.76 | 15.7 ± 0.46 | ||
| G2/M phase | 27.8 ± 5.44 | 18.8 ± 2.46 | 25.5 ± 1.51 | ||
| 24 hrs | G0/G1 phase | 44.3 ± 8.99 | 70.2 ± 3.77* | 51.9 ± 10.0 | |
| S phase | 30.3 ± 12.1 | 13.47 ± 2.85 | 24.8 ± 8.91 | ||
| G2/M phase | 21.2 ± 4.18 | 13.3 ± 0.99 | 18.3 ± 1.66 | ||
| 48 hrs | G0/G1 phase | 41.2 ± 10.8 | 62.2 ± 5.69* | 56.7 ± 5.09 | |
| S phase | 34.0 ± 10.2 | 11.86 ± 4.32* | 17.8 ± 11.6 | ||
| G2/M phase | 20.0 ± 1.00 | 21.2 ± 8.36 | 19.5 ± 5.17 | ||
| U937 | 16 hrs | G0/G1 phase | 74.3 ± 5.58 | 86.0 ± 11.41 | 85.6 ± 4.45 |
| S phase | 12.1 ± 0.99 | 2.88 ± 2.67* | 6.18 ± 1.39* | ||
| G2/M phase | 11.3 ± 5.62 | 7.39 ± 0.11 | 7.27 ± 2.84 | ||
| 24 hrs | G0/G1 phase | 61.2 ± 6.52 | 86.5 ± 6.29** | 81.7 ± 8.40*** | |
| S phase | 20.9 ± 11.9 | 3.01 ± 1.95* | 6.58 ± 3.57* | ||
| G2/M phase | 16.3 ± 9.25 | 9.41 ± 7.45 | 10,4 ± 7.27 | ||
| 48 hrs | G0/G1 phase | 60.1 ± 7.56 | 90.4 ± 4.79** | 83.9 ± 6.17*** | |
| S phase | 23.5 ± 12.1 | 4.10 ± 4.40** | 4.01 ± 3.7** | ||
| G2/M phase | 14.4 ± 6.97 | 4.83 ± 1.25 | 10.7 ± 4.13 |
The results are expressed as percentages and represent the mean ± S.D. of at least three independent experiments.
P < 0.05,
P < 0.01,
P < 0.001 versus corresponding control sample.
Cell cycle gene expression in cancer cells co-cultured with AMTC
To obtain further insight into the AMTC-induced cell cycle arrest of Jurkat and U937 cells, we compared the expression of a panel of genes related to cell cycle in cells cultured alone versus cells cultured in transwell with AMTC. We adopted a cell cycle PCR array because of its advantage in concurrently detecting the expression of many genes (84 pathway-focused genes), which are key to cell cycle regulation.
Our preliminary analysis indicates that in the presence of AMTC, the expression of genes involved in the G0/G1 transition, as well as the expression of genes important in the S phase and DNA replication were all down-regulated in comparison with cells cultured without AMTC. Specifically, the down-regulated genes were cyclins (CCND2, CCNE1, CCNH), cyclin-dependent kinases (CDK4, CDK6 and CDK2), components of the minichromosome maintenance complex (MCM2, MCM4, MCM5), and proliferating cell nuclear antigen (PCNA).
Moreover, the expression of Cullin1, which has an important role in ubiquitination and degradation of different proteins, including p21 [30, 31], was also down-regulated. In contrast, the expressions of cyclin-dependent kinase inhibitor 1A (also known as p21), cyclin-dependent kinase inhibitor 2B (p15) and cyclin G2 were all up-regulated.
The pattern of gene up- or down-regulation in the presence of AMTC was comparable between U937 and Jurkat cells (Table 3).
Table 3.
Cell cycle gene expression in Jurkat and U937 cell lines cultured with AMTC
| Jurkat | U937 | ||||||
|---|---|---|---|---|---|---|---|
| Functional gene groupings | mRNA | Mean of fold change | P value | mRNA | Mean of fold change | P value | |
| Positive regulators of cell cycle | G1 phase and G1/S transition | CCNE1 | −2.30 | * | CCNE1 | −2.13 | ** |
| CCNH | −1.28 | * | CCNH | −1.32 | ** | ||
| CCND2 | 1.03 | n.s. | CCND2 | −5.60 | *** | ||
| CDK2 | 1.03 | n.s. | CDK2 | −1.66 | * | ||
| CDK4 | −1.53 | ** | CDK4 | −2.70 | *** | ||
| CDK6 | −2.26 | * | CDK6 | −2.82 | ** | ||
| CUL1 | −1.67 | * | CUL1 | −1.33 | * | ||
| SKP2 | −1.45 | * | SKP2 | −3.08 | * | ||
| RBL1 | −1.46 | * | RBL1 | −2.50 | * | ||
| S phase and DNA replication | MCM2 | −4.92 | * | MCM2 | −2.48 | * | |
| MCM3 | 1.09 | n.s. | MCM3 | −2.42 | ** | ||
| MCM4 | −1.49 | * | MCM4 | −3.40 | * | ||
| MCM5 | −3.71 | * | MCM5 | −2.40 | ** | ||
| PCNA | −1.31 | * | PCNA | −2.16 | ** | ||
| DDX11 | −3.68 | * | DDX11 | −4.32 | * | ||
| G2 phase and G2/M transition | CCNH | −1.28 | * | CCNH | −1.32 | ** | |
| CDK5R1 | −2.35 | * | CDK5R1 | −2.67 | * | ||
| DDX11 | −3.68 | * | DDX11 | −4.32 | * | ||
| Negative regulators of cell cycle | G1 phase and G1/S transition | CCNG2 | 2.00 | * | CCNG2 | 2.08 | * |
| CDKN1A | 1.66 | ** | CDKN1A | 12.2 | ** | ||
| CDKN2B | 2.66 | * | CDKN2B | 3.14 | * | ||
| RB1 | 1.22 | * | RB1 | 2.10 | * | ||
| G2 phase and G2/M transition | CDKN1A | 1.66 | ** | CDKN1A | 12.2 | ** | |
Jurkat and U937 cell lines were cultured alone (control) or in the presence of AMTC (treated group). Data are expressed as mean fold change in mRNA expression in the treated group versus control from at least three separate experiments. n.s.: not significant; CCNE1: Cyclin E1; CCNH: Cyclin H; CCND2: Cyclin D2; CDK: Cyclin-dependent kinase; CUL1: Cullin 1; SKP2: S-phase kinase-associated protein 2 (p45); MCM: Minichromosome maintenance complex component; PCNA: Proliferating cell nuclear antigen; DDX11: DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 11; CDK5R1: Cyclin-dependent kinase 5, regulatory subunit 1 (p35); RBL1: Retinoblastoma-like 1 (p107); CCNG1: Cyclin G1; CCNG2: Cyclin G2; CDKN1A: Cyclin-dependent kinase inhibitor 1A (p21, Cip1); CDKN2B: Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4); RB1: Retinoblastoma 1.
P < 0.05,
P < 0.01,
P < 0.001.
Discussion
In this study, we demonstrate for the first time that cells isolated from the mesenchymal region of the amniotic membrane (AMTC) exert a significant anti-proliferative action on different cancer cell lines, and that AMTC halt these cells in the G0/G1 phase of the cell cycle.
In recent years, placenta-derived cells have drawn particular attention mainly for their plasticity and immunological properties, which render them interesting for stem-cell research and cell-based therapeutic applications [26, 32]. In particular, we have previously demonstrated that AMTC inhibit lymphocyte proliferation in vitro [23, 24] and, more recently, that these cells suppress the generation and maturation of monocyte-derived DCs [25]. The mechanism that underlies inhibition of DC differentiation by AMTC involves arrest of the stimulated monocyte in the G0 phase of the cell cycle and may occur in the absence of cell–cell contact, strongly suggesting the involvement of some soluble factor(s) [25]. In this study, we provide clear evidence that the anti-proliferative activity of AMTC can also target cancer cell lines of both haematopoietic [lymphoid (KG1a, Jurkat) and myeloid (KG1, U937)] and non-haematopietic origin (Girardi heart, Hela, Saos). This anti-proliferative effect was observed not only when the AMTC and cancer cells were cultured in direct contact, but also when they were physically separated (transwell system). This strongly suggests that the inhibitory effects evoked by AMTC may entail the release of yet-unknown soluble factor(s) by these cells, consistent with similar results obtained for cancer cells cultured with MSCs isolated from sources other than placenta [6, 33]. This hypothesis is also supported by co-culture experiments, which we have performed with AMTC fixed with paraformaldehyde, where AMTC were not able to inhibit the proliferation of KG1, KG1a and Jurkat cells (data not shown). Studies aimed at characterizing the soluble anti-proliferative factor(s) secreted by MSC are ongoing in different laboratories, but no clear results have yet been obtained. Cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-6 (IL-6), and α/β interferons (IFNα/β) are reported to be constitutively secreted by MSC [22]. We and others have demonstrated production of IL-6 also in amniotic mesenchymal cells [25, 34]. These factors have been described to exert anti-tumour effects [35–37] and therefore could participate in the mechanisms involved in the control of cancer cell proliferation, even though it remains controversial because the use of antibodies against TNFa, IFNα and TGFβ did not inhibit the MSC-anti-proliferative effect [33, 38]. A recent report suggests that the dickkopf-1 (DKK-1), secreted by MSC obtained from adipose tissue, plays an important role in suppressing K562 proliferation, but the growth rate of K562 was not fully restored to the normal level by RNAi for DKK-1 or by neutralizing antibodies against DKK-1 [33]. It is likely that a complex milieu is necessary for the antitumour mechanism of MSC, and more studies are needed for the characterization of this factor(s).
Consistent with other results reported for the effects of BM-MSCs on cancer cells [6], the ratio of cancer cells: AMTC sufficient to obtain a significant inhibitory effect was lower in contact co-cultures than that required in transwell settings (in contact KG1 and U937, cell lines were significantly inhibited also with a ratio of 1:0.25, while the same cancer cells need at least a ratio of 1:1 in the transwell system; Fig. 1). It is conceivable therefore that cell–cell contact might enhance/stimulate the production of inhibitory soluble factors by AMTC, and/or that cell–cell contact may engage AMTC in other pathways regulating cancer cell proliferation. On the other hand, the observation that the anti-proliferative effect of AMTC is also exerted in a transwell system excludes the possibility that the effect may be due to radiation-induced bystander effects of irradiated AMTC in contact experiments. Moreover, given that the addition of cancer cells (KG1a, Jurkat and U937) as modulators did not result in the inhibition of cancer cell proliferation, another conclusion of this study is that the reduction in proliferation observed with AMTC is due to specific effects of these cells, and not to medium exhaustion of nutrients or molecular and cellular crowding in the well.
Our analyses demonstrated that both BM-MSCs and AMTC possess anti-proliferative effects on cancer cells under contact co-culture conditions; while in transwell culture settings, only AMTC were able to inhibit cancer cell proliferation. Previous studies have indicated that BM-MSCs do not modulate T-cell proliferation [39, 40] or exert anti-proliferative effects on cancer cell lines [20] in a transwell system. Our findings regarding the inhibitory effects of dermal fibroblasts are in accordance with results indicating that human fibroblasts share immunosuppressive properties with MSCs, probably associated with the common mesodermal origin of these cell types [41, 42].
Conflicting data have been reported with regard to the effects of MSCs on cancer cell apoptosis and cell cycle regulation [13]. These differences are likely due to different experimental designs and different origin of MSCs tested. The diversity of the investigational outcomes is also indicative of the complexity of mechanisms by which MSCs interact with and regulate tumour cell growth. In this study, we show that AMTC-induced inhibition of cancer cell proliferation in vitro is not mediated by promotion of cancer cell apoptosis, but rather, by cell cycle arrest. In contrast, it has been reported that the use of supernatant from amniotic epithelial cells enhance Jurkat apoptosis [43], and that placenta (i.e. trophoblast)-derived MSCs increase myeloma cell apoptosis [44], therefore suggesting that, within placental tissues, different cell types may employ different mechanisms to inhibit cancer cell proliferation. Our FACS analyses clearly indicate that in the presence of AMTC, Jurkat and U937 cells accumulated in the G0/G1 phase of cell cycle. This arrest was particularly evident in the U937 cells, for which we observed that the proportion of cells which remain in the G0/G1 phase was always higher than 80%, while the percentage of cells which enter the S phase was very low. The differences in magnitude of the anti-proliferative effect of AMTC on U937 and Jurkat cells could be attributed to the fact that AMTC may employ different regulatory mechanisms on these two cell types and/or, more likely, that these cells respond to differing extent to the same inhibitory cues.
To gain further insight into the mechanisms involved in cell cycle arrest induced by AMTC, we explored the effects of AMTC on the expression of a panel of genes that both positively and negatively regulate the transition between each of the cell cycle phases, DNA replication, checkpoints and arrest of cell cycle. We found that the pattern of genes whose expression appeared modulated after exposition to AMCT was similar in both Jurkat and U937 cells, even though the fold increase/decrease levels obtained were often higher in U937 cells.
Although this preliminary analysis of gene expression was performed at the transcriptional level and requires further exploration in terms of changes in protein expression and phosphorylation, the results obtained are consistent with a block in G0/G1 phase and with no progression to S phase (Table 3). Notably, we observed a down-regulation in the expression of some of the cyclins and CDKs that promote cell cycle progression, such as cyclin D2, cyclin E1, cyclin H, CDK4, CDK6 and CDK2 [45]. Moreover, genes important in S phase and DNA replication were also down-regulated. The down-regulation of these positive regulators of cell cycle progression at G1 phase was accompanied by the up-regulation of negative regulators of cell cycle, specifically, the CDK inhibitor 1A (CDKN1A, or p21) and CDKN2B (p15) [45–47]. These data are consistent with previous studies reporting the ability of MSCs to inhibit cancer cell proliferation through down-regulation of the expression of cyclin D2, cyclin E and CDK4 accompanied by an up-regulation of p21 [6, 15, 33]. These authors also reported the up-regulation of p27. In our setting, p21 and p15 (and not p27) seem to play a major role in blocking of the cell cycle. Interestingly, we observed a down-regulation of Cullin-1 (CUL1), involved in the ubiquitination process and, consequently, in the degradation of different proteins, including p21 [31]. The down-regulation of CUL1 may contribute to the block of cell cycle progression. Furthermore, we also observed that the expression of cyclin G2 was up-regulated. Cyclin G2 is an unconventional cyclin whose expression is not associated with promotion of cell proliferation, but with cell cycle inhibition and arrest [48–50]. Finally, the expression of the retinoblastoma protein (pRB), which is important for proper cell cycle arrest in G0/G1 phase, was also up-regulated [51], while RB-like 1 (p107), whose levels are generally low in quiescent cells and high when cells proliferate [52], was down-regulated in both U937 and Jurkat.
In conclusion, the present study demonstrates that AMTC exert inhibitory effects on the proliferation of different cancer cells in vitro, probably via the release of yet-unknown soluble factors. Although further studies are required to identify these factors and their signalling pathways, here we have demonstrated that the inhibitory effects of AMTC entail the arrest of tumour cells in the G0/G1 phase of the cell cycle, with modulation of the expression of key genes involved in cell cycle regulation, but that AMTC do not induce cancer cell apoptosis. Indications from pre-clinical studies support the beneficial effects of amniotic membrane-derived cells as anti-inflammatory and anti-fibrotic agents for the treatment of different pathologies [2]. The findings reported in the present study increase the body of knowledge regarding the properties of amniotic MSCs and highlight that these cells could exert a profound effect also in the regulation of cancer cell proliferation, fostering studies on their potential anti-cancer activities in animal models and, prospectively, in clinical applications. However, extensive investigations should be conducted into different aspects, both for these cells and for MSC isolated from different sources. Indeed, due to discrepancies reported in studies that directly investigated the effects of MSCs on tumours in animal models [22], further in vitro experiments aimed at isolating better-characterized and more homogenous MSC populations should be carried out. In vivo studies to evaluate and compare the effects of MSC isolated from different sources, passages and cell concentrations, as well as to monitor the safety and lack of side effects of their transplantation are also fundamental.
Acknowledgments
The authors thank the physicians and midwives of the Department of Obstetrics and Gynecology of Fondazione Poliambulanza-Istituto Ospedaliero, Brescia, Italy, and all the mothers who donated placenta. The authors thank Dr. Maddalena Caruso for support in writing the manuscript. The authors also thank Dr. Marco Evangelista for help in editing the manuscript.
This work was supported in part by Fondazione Cariplo (Progetto Nobel 2006), Ministero dell'Istruzione, dell'Università e della Ricerca (FIRB RBNE06JBCW_002) and Fondazione Poliambulanza.
Author contribution
MM, SDM and EV performed the research and collected the data; MM and OP designed the research study, analysed and interpreted the data and wrote the paper; OP provided financial support and gave final approval of the manuscript.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1 Surface marker expression of AMTC.
Table S1 Phenotypic analysis of AMTC at different time points.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Garcia-Gomez I, Elvira G, Zapata AG, et al. Mesenchymal stem cells: biological properties and clinical applications. Expert Opin Biol Ther. 2010;10:1453–68. doi: 10.1517/14712598.2010.519333. [DOI] [PubMed] [Google Scholar]
- 2.Parolini O, Alviano F, Bergwerf I, et al. Toward cell therapy using placenta-derived cells: disease mechanisms, cell biology, preclinical studies, and regulatory aspects at the round table. Stem Cells Dev. 2010;19:143–54. doi: 10.1089/scd.2009.0404. [DOI] [PubMed] [Google Scholar]
- 3.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 4.Pistoia V, Raffaghello L. Potential of mesenchymal stem cells for the therapy of autoimmune diseases. Expert Rev Clin Immunol. 2010;6:211–8. doi: 10.1586/eci.09.86. [DOI] [PubMed] [Google Scholar]
- 5.Bernardo ME, Locatelli F, Fibbe WE. Mesenchymal stromal cells. Ann N Y Acad Sci. 2009;1176:101–17. doi: 10.1111/j.1749-6632.2009.04607.x. [DOI] [PubMed] [Google Scholar]
- 6.Ramasamy R, Lam EW, Soeiro I, et al. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007;21:304–10. doi: 10.1038/sj.leu.2404489. [DOI] [PubMed] [Google Scholar]
- 7.Sarmadi VH, Heng FS, Ramasamy R. The effect of human mesenchymal stem cells on tumour cell proliferation. Med J Malaysia. 2008;63:63–4. [PubMed] [Google Scholar]
- 8.Zhang HM, Zhang LS. Influence of human bone marrow mesenchymal stem cells on proliferation of chronic myeloid leukemia cells. Chin J Cancer. 2009;28:29–32. [PubMed] [Google Scholar]
- 9.Motaln H, Schichor C, Lah TT. Human mesenchymal stem cells and their use in cell-based therapies. Cancer. 2010;116:2519–30. doi: 10.1002/cncr.25056. [DOI] [PubMed] [Google Scholar]
- 10.Mousavi Niri N, Jaberipour M, Razmkhah M, et al. Mesenchymal stem cells do not suppress lymphoblastic leukemic cell line proliferation. Iran J Immunol. 2009;6:186–94. [PubMed] [Google Scholar]
- 11.Djouad F, Bony C, Apparailly F, et al. Earlier onset of syngeneic tumors in the presence of mesenchymal stem cells. Transplantation. 2006;82:1060–6. doi: 10.1097/01.tp.0000236098.13804.0b. [DOI] [PubMed] [Google Scholar]
- 12.Kidd S, Spaeth E, Klopp A, et al. The (in) auspicious role of mesenchymal stromal cells in cancer: be it friend or foe. Cytotherapy. 2008;10:657–67. doi: 10.1080/14653240802486517. [DOI] [PubMed] [Google Scholar]
- 13.Feng B, Chen L. Review of mesenchymal stem cells and tumors: executioner or coconspirator? Cancer Biother Radiopharm. 2009;24:717–21. doi: 10.1089/cbr.2009.0652. [DOI] [PubMed] [Google Scholar]
- 14.Qiao L, Xu Z, Zhao T, et al. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008;18:500–7. doi: 10.1038/cr.2008.40. [DOI] [PubMed] [Google Scholar]
- 15.Lu YR, Yuan Y, Wang XJ, et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther. 2008;7:245–51. doi: 10.4161/cbt.7.2.5296. [DOI] [PubMed] [Google Scholar]
- 16.Xu G, Zhang Y, Zhang L, et al. Bone marrow stromal cells induce apoptosis of lymphoma cells in the presence of IFNgamma and TNF by producing nitric oxide. Biochem Biophys Res Commun. 2008;375:666–70. doi: 10.1016/j.bbrc.2008.08.077. [DOI] [PubMed] [Google Scholar]
- 17.Loebinger MR, Janes SM. Stem cells as vectors for antitumour therapy. Thorax. 2010;65:362–9. doi: 10.1136/thx.2009.128025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung SC, Deng WP, Yang WK, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11:7749–56. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 19.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 20.Khakoo AY, Pati S, Anderson SA, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi's sarcoma. J Exp Med. 2006;203:1235–47. doi: 10.1084/jem.20051921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwyer RM, Khan S, Barry FP, et al. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klopp AH, Gupta A, Spaeth E, et al. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11–9. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailo M, Soncini M, Vertua E, et al. Engraftment potential of human amnion and chorion cells derived from term placenta. Transplantation. 2004;78:1439–48. doi: 10.1097/01.tp.0000144606.84234.49. [DOI] [PubMed] [Google Scholar]
- 24.Magatti M, De Munari S, Vertua E, et al. Human amnion mesenchyme harbors cells with allogeneic T-cell suppression and stimulation capabilities. Stem Cells. 2008;26:182–92. doi: 10.1634/stemcells.2007-0491. [DOI] [PubMed] [Google Scholar]
- 25.Magatti M, De Munari S, Vertua E, et al. Amniotic mesenchymal tissue cells inhibit dendritic cell differentiation of peripheral blood and amnion resident monocytes. Cell Transplant. 2009;18:899–914. doi: 10.3727/096368909X471314. [DOI] [PubMed] [Google Scholar]
- 26.Parolini O, Caruso M. Review: Preclinical studies on placenta-derived cells and amniotic membrane: an update. Placenta. 2011;32:S186–95. doi: 10.1016/j.placenta.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 27.Soncini M, Vertua E, Gibelli L, et al. Isolation and characterization of mesenchymal cells from human foetal membranes. J Tissue Eng Regen Med. 2007;1:296–305. doi: 10.1002/term.40. [DOI] [PubMed] [Google Scholar]
- 28.Koeffler HP, Billing R, Lusis AJ, et al. An undifferentiated variant derived from the human acute myelogenous leukemia cell line (KG-1) Blood. 1980;56:265–73. [PubMed] [Google Scholar]
- 29.Jost JP, Oakeley EJ, Zhu B, et al. 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation. Nucleic Acids Res. 2001;29:4452–61. doi: 10.1093/nar/29.21.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci USA. 1998;95:11324–9. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W, Shang F, Liu Q, et al. Differential regulation of components of the ubiquitin-proteasome pathway during lens cell differentiation. Invest Ophthalmol Vis Sci. 2004;45:1194–201. doi: 10.1167/iovs.03-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parolini O, Alviano F, Bagnara GP, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells. 2008;26:300–11. doi: 10.1634/stemcells.2007-0594. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Y, Sun Z, Han Q, et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23:925–33. doi: 10.1038/leu.2008.384. [DOI] [PubMed] [Google Scholar]
- 34.Kronsteiner B, Wolbank S, Peterbauer A, et al. Human mesenchymal stem cells from adipose tissue and amnion influence T-cells depending on stimulation method and presence of other immune cells. Stem Cells Dev. 2011;20:2115–26. doi: 10.1089/scd.2011.0031. [DOI] [PubMed] [Google Scholar]
- 35.Eisenthal A, Kashtan H, Rabau M, et al. Antitumor effects of recombinant interleukin-6 expressed in eukaryotic cells. Cancer Immunol Immunother. 1993;36:101–7. doi: 10.1007/BF01754409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kojiro S, Yano H, Ogasawara S, et al. Antiproliferative effects of 5-fluorouracil and interferon-alpha in combination on a hepatocellular carcinoma cell line in vitro and in vivo. J Gastroenterol Hepatol. 2006;21:129–37. doi: 10.1111/j.1440-1746.2005.04154.x. [DOI] [PubMed] [Google Scholar]
- 37.Driessens G, Nuttin L, Gras A, et al. Development of a successful antitumor therapeutic model combining in vivo dendritic cell vaccination with tumor irradiation and intratumoral GM-CSF delivery. Cancer Immunol Immunother. 2011;60:273–81. doi: 10.1007/s00262-010-0941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maestroni GJ, Hertens E, Galli P. Factor(s) from nonmacrophage bone marrow stromal cells inhibit Lewis lung carcinoma and B16 melanoma growth in mice. Cell Mol Life Sci. 1999;55:663–7. doi: 10.1007/s000180050322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Potian JA, Aviv H, Ponzio NM, et al. Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol. 2003;171:3426–34. doi: 10.4049/jimmunol.171.7.3426. [DOI] [PubMed] [Google Scholar]
- 40.Krampera M, Glennie S, Dyson J, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–9. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 41.Haniffa MA, Wang XN, Holtick U, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol. 2007;179:1595–604. doi: 10.4049/jimmunol.179.3.1595. [DOI] [PubMed] [Google Scholar]
- 42.Haniffa MA, Collin MP, Buckley CD, et al. Mesenchymal stem cells: the fibroblasts' new clothes? Haematologica. 2009;94:258–63. doi: 10.3324/haematol.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Niederkorn JY, Neelam S, et al. Immunosuppressive factors secreted by human amniotic epithelial cells. Invest Ophthalmol Vis Sci. 2005;46:900–7. doi: 10.1167/iovs.04-0495. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Ling W, Pennisi A, et al. Human placenta-derived adherent cells prevent bone loss, stimulate bone formation, and suppress growth of multiple myeloma in bone. Stem Cells. 2011;29:263–73. doi: 10.1002/stem.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 46.Hengst L, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol. 1998;227:25–41. doi: 10.1007/978-3-642-71941-7_2. [DOI] [PubMed] [Google Scholar]
- 47.Harper JW, Adami GR, Wei N, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 48.Horne MC, Donaldson KL, Goolsby GL, et al. Cyclin G2 is up-regulated during growth inhibition and B cell antigen receptor-mediated cell cycle arrest. J Biol Chem. 1997;272:12650–61. doi: 10.1074/jbc.272.19.12650. [DOI] [PubMed] [Google Scholar]
- 49.Bennin DA, Don AS, Brake T, et al. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B' subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J Biol Chem. 2002;277:27449–67. doi: 10.1074/jbc.M111693200. [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Gac L, Marques M, Garcia Z, et al. Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol Cell Biol. 2004;24:2181–9. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodrich DW, Lee WH. Abrogation by c-myc of G1 phase arrest induced by RB protein but not by p53. Nature. 1992;360:177–9. doi: 10.1038/360177a0. [DOI] [PubMed] [Google Scholar]
- 52.Wirt SE, Sage J. p107 in the public eye: an Rb understudy and more. Cell Div. 2010;5:9. doi: 10.1186/1747-1028-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.