

Abstract
End-stage hypertensive heart disease is an increasing cause of cardiac mortality. Therefore, the current study focused on the cardiac remodelling from hypertrophy to fibrosis in old-aged spontaneously hypertensive rats (SHRs), and explored the therapeutic effects of Rosuvastatin and its possible mechanism(s) of action. Spontaneously hypertensive rats at age 52 weeks were randomly divided into three groups, the first two to receive Rosuvastatin at a dose of 20 mg/kg/day and 40 mg/kg/day, respectively, and the third to receive placebo, which was to be compared with Wistar-Kyoto as controls. After 2-month treatment, SBP, heart to body weight ratio (HW/BW%) and echocardiographic features were evaluated, followed by haematoxylin and eosin and Masson trichrome staining in conjunction with qPCR of foetal gene expressions. Transferase-mediated dUTP nick-end labelling assay and immunofluorescent labelling for active caspase-3 were used to detect the apoptotic cardiomyocytes. Signaling pathways involved were examined by using western blot. Old-aged SHR developed end-stage hypertensive heart disease characterized by significant enhancement of HW/BW%, LVAWd and LVPWd, and decreased LVEF and LVFS, accompanied by cardiomyocytes enlargement and fibrosis along with activation of foetal gene programme. Cardiac apoptosis increased significantly during the transition process. Rosuvastatin reduced hypertrophy significantly via AT1 Receptor-PKCβ2/α-ERK-c-fos pathway; protected myocardium against apoptosis via Akt-FOXO1, Bcl-2 family and survivin pathways and consequently suppressed the caspase-3 activity. The present study revealed that old-aged SHRs developed cardiac remodelling from hypertrophy to fibrosis via cardiac apoptosis during the end stage of hypertensive heart disease. These pathological changes might be the consequence of activation of AT1 Receptor-PKCβ2/α-ERK-c-fos and AKT-FOXO1/Bcl-2/survivin/Caspase3 signaling. Rosuvastatin effectively attenuated the structural changes by reversing the signaling transductions involved.
Keywords: old-aged spontaneously hypertensive rats, cardiac remodelling, hypertrophy, fibrosis, apoptosis, Rosuvastatin, 3-hydroxy-3-methylglutaryl-coenzyme-A (HMG-CoA)-reductase inhibitors
Introduction
Arterial hypertension, a major health problem for its high frequency and concomitant risks of the cardiovascular system, has been identified as the leading risk factor for cardiac mortality, ranked third as a cause of disability-adjusted life years [1]. Cardiac remodelling including ventricular hypertrophy and the subsequent congestive heart failure are the common outcome of human essential hypertension [2]. The consequent cardiac malfunction leads to poor clinical prognosis and ultimately cardiovascular-related death. During the pathophysiological progress, cardiac apoptosis has been reported as one of the reasons for reduced myocardial mass [3]. However, the exact role of apoptosis in cardiac remodelling during the end-stage hypertensive heart disease has remained unknown.
The HMG-CoA-reductase inhibitors (statins) are the most commonly prescribed agents for hyperlipidemia. In addition to their lipid-lowering action, statins exert important effects on the cardiovascular system [4], as indicated by the heart function restoration in the hypertensive patients with or without coronary diseases observed by clinical trials [5]. These effects are independent of lipid-lowering effect [6–8]. However, the underlying mechanisms of these beneficial effects are not completely clear. Furthermore, JUPITER trial suggested that Rosuvastatin could effectively decrease C-reactive protein level, thereby reducing cardiovascular event rates [9], which has provoked furthermore studies on the potential effects of Rosuvastatin.
The present study was divided into three parts: Firstly, we observed the transition of myocardium from hypertrophy to fibrosis in old-aged spontaneously hypertensive rats (SHRs); next, we sought to determine the possible mechanisms and underlying signaling pathways involved in the cardiac remodelling during the end stage of hypertension in SHRs; finally, we examined our hypothesis that Rosuvastatin could reverse the impairments in the end-stage hypertensive heart disease by investigating the candidate signaling pathways.
Materials and methods
Animals
Twenty-one adult male SHRs at age 52 weeks, weighing approximately 300 g and seven Wistar-Kyoto (WKY) rats were obtained from and bred on standard chow at Animal Administration Center of Fudan University. SHRs were randomly divided into three groups: SHR control group (SHR, n = 7); low-dose Rosuvastatin group (SHR + LD, 20 mg/kg/day, n = 7); and high-dose Rosuvastatin group (SHR + HD, 40 mg/kg/day, n = 7), with an additional group of WKY as control (WKY, n = 7). Rosuvastatin was administered daily through an intra-gastric tube for 8 weeks, while the two control groups were treated with saline. All experimental protocols were approved by the Animal Care and Use Committee of Fudan University and in compliance with Guidelines for the Care and Use of Laboratory Animals published by the National Academy Press (NIH Publication No.85-23, revised 1996).
Measurement of systolic blood pressure and heart to body weight ratio
Systolic blood pressures (SBP) were measured at the beginning, and then at two 4-week intervals when all the animals were conscious at the maintained temperatures of 30°C [10]. Before the measurement, the animals were trained to adapt themselves to the restraining cages and tail-cuff apparatus for the standard non-invasive tail-cuff. The animals scarified by decapitation, the heart to body weight ratio (HW/BW%) was calculated.
Evaluation of cardiac structure and heart function
Trans-thoracic echocardiographic analysis was performed using an animal specific instrument (VisualSonics#x00AE; Vevo770#x00AE;; VisualSonicsInc.,Toronto, Canada), as previously described [11]. The rats were anesthetized with isoflurane. M-mode images of the left ventricle were recorded when the heart rate was near 400 bpm. All measurements were averaged for five consecutive cardiac cycles and repeated three times. Left ventricular anterior and posterior wall thickness of diastolic phase (LVAWd and LVPWd), ejection fraction and fractional shortening (LVEF and LVFS) were measured to evaluate the hypertrophy and function of the hearts.
Haematoxylin and eosin staining
The extent of cardiac myocyte hypertrophy and myocyte cross-sectional area was determined on the haematoxylin-eosin stained sections, as described by Frustaci [12].
Masson trichrome staining
The extent of cardiac fibrosis was determined by Masson's trichrome staining. The sections were stained for collagen fibres using the Masson's trichrome method (Sigma-Aldrich, St. Louis, USA), with the images observed under the Leica DM-RE microscope and analysed via LeicaQwin software (Leica Imaging Systems, Cambridge, UK).
dUTP nick-end labelling assay
To determine cardiac apoptosis, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) assay was performed with a commercial kit (FragEL™ DNA Fragmentation Detection Kit; Calbiochem, Merck, Darmstadt, German) according to the manufacturer's instructions. The deparaffinized sections were pretreated with protease K (20 μg/ml) for 20 min. at room temperature. The endogenous peroxidase activity inactivated, the slides were incubated with equilibration buffer for 30 min., followed by incubation with terminal deoxynucleotidyl transferase in a moist chamber for 90 min. at 37°C. The reaction was visualized using streptavidin-biotin-peroxidase complex and diaminobenzidine. The normal nuclei with a relatively insignificant number of DNA-3-OH ends were not stained. The sections of the negative controls were incubated without enzyme or nucleotide. From each of the four groups, 3–4 slides were chosen for TUNEL assays, the average number of TUNEL-positive cells examined in six randomly chosen high-power fields (×400) and normalized to that of WKY.
Immunofluorescent labelling for active caspase-3
Immunofluorescent labelling was performed on the frozen sections. The samples were fixed in acetone for 30 min., washed in PBS, and then incubated in PBS containing 3% BSA for 30 min. Afterwards, incubation with anti-active caspase3 antibody (rabbit polyclonal, 1:100; ABCAM, Cambridge, Massachusetts, USA) was performed at 4°C overnight. After rinsing in PBS thrice, the primary antibody was detected with secondary goat anti-rabbit antibodies conjugated with Alexa Fluor#x00AE;488 (1:300; ABCAM), before the nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, 2 μg/ml). The samples were examined under a fluorescence microscope (Olympus, Tokyo, Japan) to count the cells. The sections derived from 3–4 slides of each group were analysed, and the percentage of active caspase 3-positive cells was calculated as follows: 200× (the number of positive cells counted/total number of nuclei counted and then normalized to that of WKY).
Real-time quantitative reverse transcriptional PCR
RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), its concentrations determined with a NanoDrop instrument (NanoDrop Technologies, Wilmington, DE, USA). SYBR RT-PCR kit (Takara, Dalian, China) was employed for real-time quantitative PCR analyses. The PCR reaction was monitored by ABI PRISM 7900 System (Applied Biosystems, Carlsbad, CA, USA). The primers for skeletal isoform of α-actin (SAA), β-isoform of myosin heavy chain (β-MHC), atrial natriuretic peptide (ANP) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed by Takara [13]:
For SAA gene, forward, 5′-AGCAGATGTGGATCACCAAG -3′; reverse, 5′- CTGCAACCACAGCACGATTG-3′.
For ANP gene, forward, 5′- ATGGGCTCCTTCTCCATCAC -3′; reverse, 5′- TCTTCGGTACCGGAAGCT -3′.
For β-MHC gene, forward, 5′-GTGCCAAGGGCCTGAATGAG-3′; reverse, 5′-GCAAAGGCTCCAGGTCTGA-3′.
For GAPDH gene, forward, 5′-GTGCAGTGCCAGCCTCGTC-3′, reverse, 5′-GGCAGCACCAGTGGATGCAG-3′.
The relative expression levels of the genes were normalized to that of GAPDH using 2−ΔΔCt cycle threshold method.
Western blot
The ventricle tissues, rapidly removed from rats, were stored at −80°C. The expression of phosphorylated (phos-) protein kinase C (PKC) β1, phos-PKCβ2, phos-PKCα, survivin (1:1000, Santa Cruz, CA, USA), AT1 Receptor, phos-extracellular signal-regulated kinase (ERK), phos-Jun N-terminal kinase (JNK), c-fos, Akt, phos-Akt, forkhead box O (FOXO1), phos-FOXO1, B cell leukaemia-x long (Bcl-xl), B cell leukaemia-2 (Bcl-2), Bcl-2-associated X protein (Bax) (1:1000; Cell Signal Technology, Danvers, MA, USA), active caspase-3 (1:1000; ABCAM), were measured by western blot and normalized to the protein level of GAPDH or β-actin. From the frozen ventricle tissues were extracted proteins, their concentrations determined with a BCA protein assay. Totally, 40 mg of protein was loaded and resolved on an 8–10% sodium-dodecyl sulphate–polyacrylamide gel before being transferred electrically onto Immobilon#x00AE;-P Transfer membranes. The blots were blocked with Tris-Buffered Saline containing 5% BSA and 0.05% Tween-20 for 2 hrs at room temperature, and then incubated, respectively. Primary antibodies against these factors were applied to the blots with gentle rocking overnight at 4°C. The filters blots were washed thrice with TBST buffer (0.05% Tween 20 in TBS, pH 7.4) for 10 min. each, and were incubated with horseradish peroxidase (HRP)-conjugated rabbit or goat secondary antibodies (1:5000 dilution; Kang Chen Biotechnology, GuangZhou, China) for 1 hr at room temperature, followed by three times of 10-min washing. The antigen-antibody complexes were detected using an ECL Western-blotting Detection Reagents (catalogue RPN2106; GE Healthcare, Fairfield, CT, USA) with LAS-3000 detect system. Quantification of immunoreactivity was performed through a densitometric analysis using ImagePro 5.0 (Media Cybernetics, Inc., Silver Spring, MD, USA).
Statistical analysis
The statistical analyses were performed using spss software package, version 16.0 (SPSS Inc. Chicago, IL, USA), the results expressed as mean ± SED. The differences in functional parameters between the groups were investigated via one-way anova followed by Fisher's LSD test for pairs of means. A value of P < 0.05 was accepted as statistically significant.
Results
Animal characteristics
Heart to body weight ratio was used as a parameter reflecting the size of the entire heart. In the SHR controls, the average HW/BW% was found to be higher than that of WKY by 98.9%, 0.358 ± 0.037% versus 0.180 ± 0.022% (P < 0.05). Rosuvastatin at a low- and high-dose resulted in reductions by 21.8% and 24.6%, respectively, compared with untreated SHR, 0.280 ± 0.030% and 0.270 ± 0.008% versus 0.358 ± 0.037% (P < 0.05). However, there were no significant differences between SHR + LD and SHR+ HD (P > 0.05) (Fig. 1). To exclude the interference of after-load on LVEF and LVFS, SBP were measured; consequently, no significant changes were observed before and after the administration of Rosuvastatin (P > 0.05) (Table 1).
Fig 1.
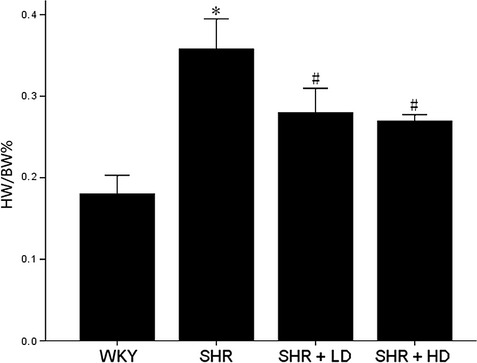
The heart to body weight ratio (HW/BW%) of WKY controls, SHR controls, SHR + LD and SHR + HD; Hypertrophy significant in SHR controls compared with that of WKY ones (P < 0.05); significantly reduced hypertrophy in SHR + LD and SHR + HD as indicated by the HW/BW% (P < 0.05); no significant difference between SHR + LD and SHR + HD (P > 0.05). Values, mean ± SED; n = 5; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Table 1.
Changes of systolic blood pressure (SBP) during the study
| SBP (mmHg) | |||
|---|---|---|---|
| Group | Base line | At 4 weeks | At 8 weeks |
| WKY | 125.60 ± 6.462 | 126.20 ± 6.232 | 117.20 ± 2.059 |
| SHR | 210.00 ± 4.604* | 206.20 ± 5.083* | 201.80 ± 2.223* |
| SHR + LD | 205.60 ± 4.069* | 203.20 ± 2.437* | 200.00 ± 8.343* |
| SHR + LD | 204.00 ± 6.626* | 193.60 ± 4.915* | 197.60 ± 2.249* |
Data were presented as mean ± SED, n = 5.
P < 0.01 versus WKY group.
SHR, spontaneously hypertensive rat; WKY, Wistar-Kyoto.
Changes in ventricular wall thickness and heart function
Echocardiographic measurements were conducted in vitro to prove whether or not myocardium of SHR underwent hypertrophy and malfunction. The results indicated an increase in LVAWd and LVPWd in SHR control by 56.6% and 29.7%, respectively, when compared with WKY, 3.43 ± 0.28 mm versus 2.19 ± 0.41 mm; 2.75 ± 0.52 mm versus 2.12 ± 0.01 mm (P < 0.05). In SHR + LD, significant reductions developed in LVAWd and LVPWd by 21.3% and 8.7%, respectively, 2.63 ± 0.32 mm versus 3.43 ± 0.28 mm; 2.51 ± 0.35 mm versus 2.75 ± 0.52 mm (P < 0.05), while in SHR + HD, by 31.8% and 36.7% in LVAWd and LVPWd, 2.34 ± 0.08 mm versus 3.43 ± 0.28 mm; 1.74 ± 0.14 mm versus 2.75 ± 0.52 mm (P < 0.05) (Fig. 2).
Fig 2.
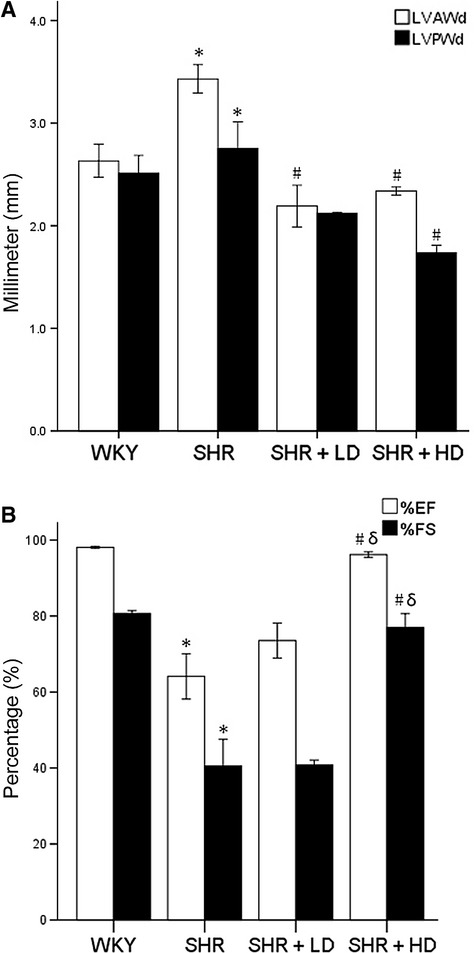
Echocardiographic data of LVAWd, LVPWd, LVEF and LVFS of WKY controls, SHR controls, SHR + LD and SHR + HD; (A) Hypertrophy significant in SHR controls compared with WKY controls as indicated by both LVAWd and LVPWd (P < 0.05); LVAWd and LVPWd significantly reduced in SHR + HD (P < 0.05); no significant difference in LVAWd and LVPWd between SHR + LD and SHR + HD (P > 0.05); (B) heart function decreased significantly in SHR controls compared with WKY ones as indicated by LVEF and LVFS (P < 0.05), but reversed in SHR + HD (P < 0.05). Values, mean ± SED; n = 4; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
It was found that LVEF and LVFS were lower in SHR controls than in WKY by 34.6% and 49.8%, respectively, 64.13 ± 11.9% versus 98.09 ± 0.4%, 40.52 ± 14.0% versus 80.70 ± 1.5% (P < 0.05). In SHR + HD, LVEF and LVFS increased by 50% and 90%, respectively, when compared with SHR controls, 96.16 ± 1.4% versus 64.13 ± 11.9%, 76.97 ± 7.3% versus 40.52 ± 14.0% (P < 0.05).
Cardiac myocyte enlargement
There was an increase in cross-sectional area of SHR controls by 95.2% compared with that of WKY, 320.32 ± 14.28 μm2 versus 164.10 ± 28.13 μm2 (P < 0.05) (Fig. 3). These changes were attenuated by 22.9% and 32.3% following a low- and high-dose treatment, 246.85 ± 25.57 μm2 and 216.75 ± 50.91 μm2 versus 320.32 ± 14.28 μm2 (P < 0.05); the latter suppressed the augmentation more powerfully than the former, but without any significance (P > 0.05).
Fig 3.
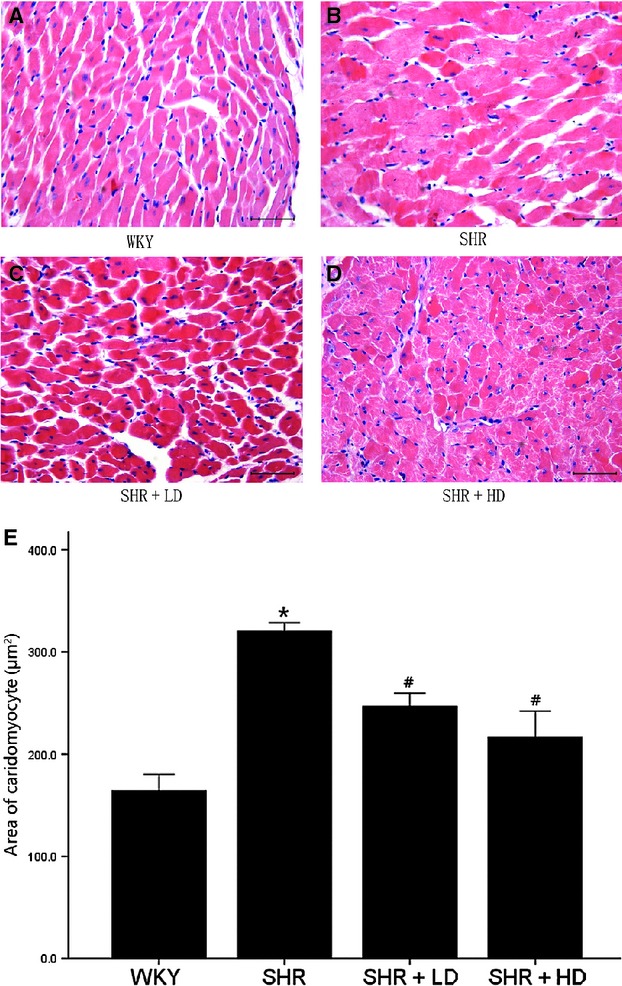
Histopathological analysis of left ventricle of WKY controls, SHR controls, SHR + LD and SHR + HD; Paraffin-embedded hearts sectioned and subjected to haematoxylin and eosin staining. (A) WKY controls exhibiting myocytes with normal size; (B) SHR controls, significantly enlarged myocytes compared with WKY ones (P < 0.05); (C) SHR + LD, significantly reduced myocyte size compared with SHR controls (P < 0.05); (D) SHR + HD, reduced myocyte size even more significantly compared with SHR controls (P < 0.05); Images ×400 power, scale bars = 50 μm; (E). quantitative analysis of the cell size (μm2) of cardiac myocytes in the four groups. Values, mean ± SED; n = 3–4; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Myocardium fibrosis
Masson Trichrome staining clearly showed increased interstitial collagen and fibrin accumulation in SHR controls compared with WKY, 5.08 ± 1.88% versus 1.22 ± 0.51% (P < 0.05). The extracellular matrix changes were significantly reversed in SHR + LD and SHR + HD by 49.8% and 62.6%, respectively, but without significant differences between them, 2.55 ± 0.25% and 1.90 ± 0.47% versus 5.08 ± 1.88% (P < 0.05) (Fig. 4).
Fig 4.
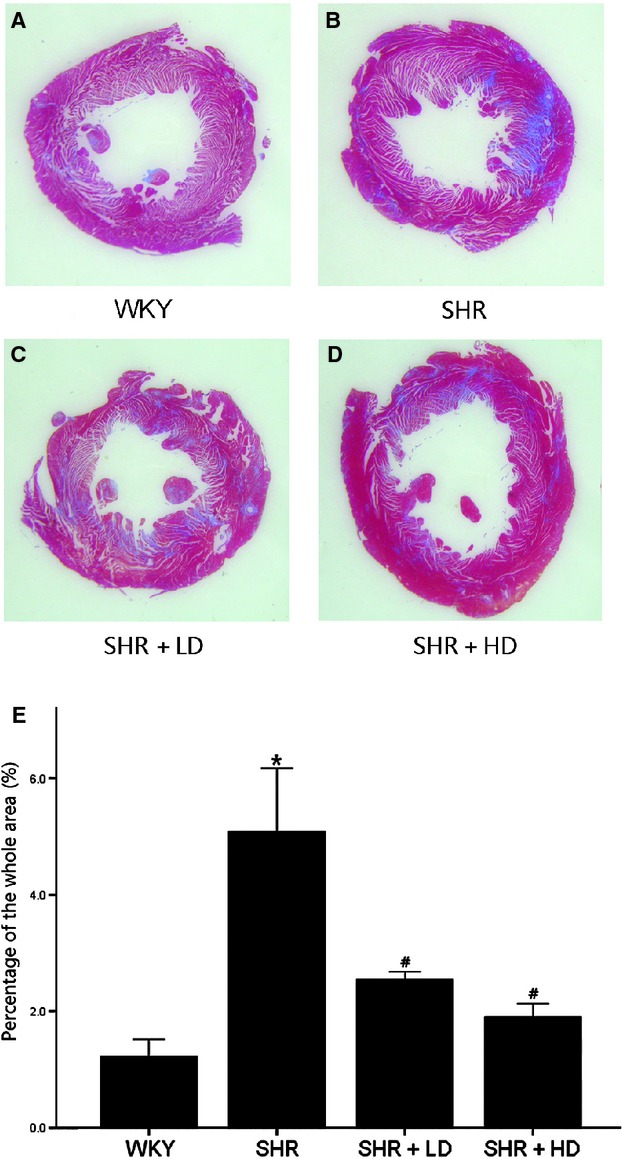
Masson's trichrome stain of hearts of WKY controls, SHR controls, SHR + LD and SHR + HD, Blue = fibrous collagen, red = myocytes. (A) the comparative fibrosis of WKY controls; (B) SHR controls showing an increasing amount of collagen deposition compared with WKY controls (P < 0.05); (C & D). SHR + LD and SHR + HD, less collagen accumulation compared with SHR controls (P < 0.05); representative histological sections; (E) quantitative analysis of fibrosis in the four groups by the ratio of the fibrosis area to the whole. Values, mean ± SED; n = 3–4; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Cardiac myocyte apoptosis detected by TUNEL assay
TUNEL assay showed some DNA strand breaks with dark brown nuclei in cardiac myocytes of all SHR groups, whereas in WKY controls, few TUNEL positive myocytes were observed, 5.12-folds versus WKY (P < 0.05). Apoptotic cells were not uniformly seen across sections but were randomly scattered. SHR + LD demonstrated a reduction in TUNEL-positive cell count by 43.7%, and SHR + HD, a reduced ratio by 46.7%, with no significant difference in SHR + LD and SHR + HD (P > 0.05) (Fig. 5).
Fig 5.
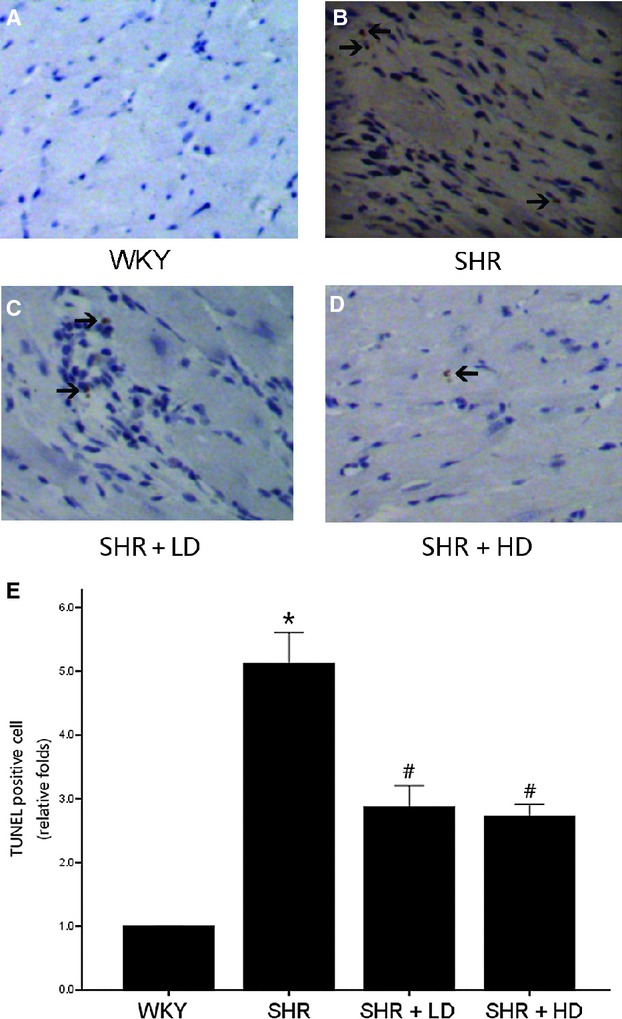
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining from rat heart tissue sections of WKY controls, SHR controls, SHR + LD and SHR + HD; arrow indicating a TUNEL-positive cardiomyocyte in brown colour. (A) no TUNEL-positive cardiomyocyte in the representative TUNEL staining of WKY controls; (B) multiple TUNEL-positive cardiomyocyte in the representative TUNEL staining of SHR controls compared with WKY controls (P < 0.05); (C & D) less TUNEL-positive cardiomyocyte in SHR + LD and SHR + LD (P < .05); (E) quantitative analysis of TUNEL-positive cardiac myocytes in the four groups by the ratio of TUNEL-positive cell number to the total and normalized to the WKY controls. Values, mean ± SED; n = 4; * P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Cardiac myocyte apoptosis detected by immunofluorescent labelling for active caspase-3
To further determine the apoptosis status, we stained tissue sections for active caspase-3 by immunofluorecent labelling, finding higher caspase-3 immunoreactivity and increased number of caspase-3 positive cells in SHR group than in WKY controls, 5.18-folds versus WKY (P < 0.05); significantly reduced caspase-3 immunoreactivity and caspase-3 positive cell count in SHR + LD and SHR + HD (P < 0.05); and caspase-3 staining predominantly in the cytoplasm at the examining point within the myocardial cells (Fig. 6).
Fig 6.
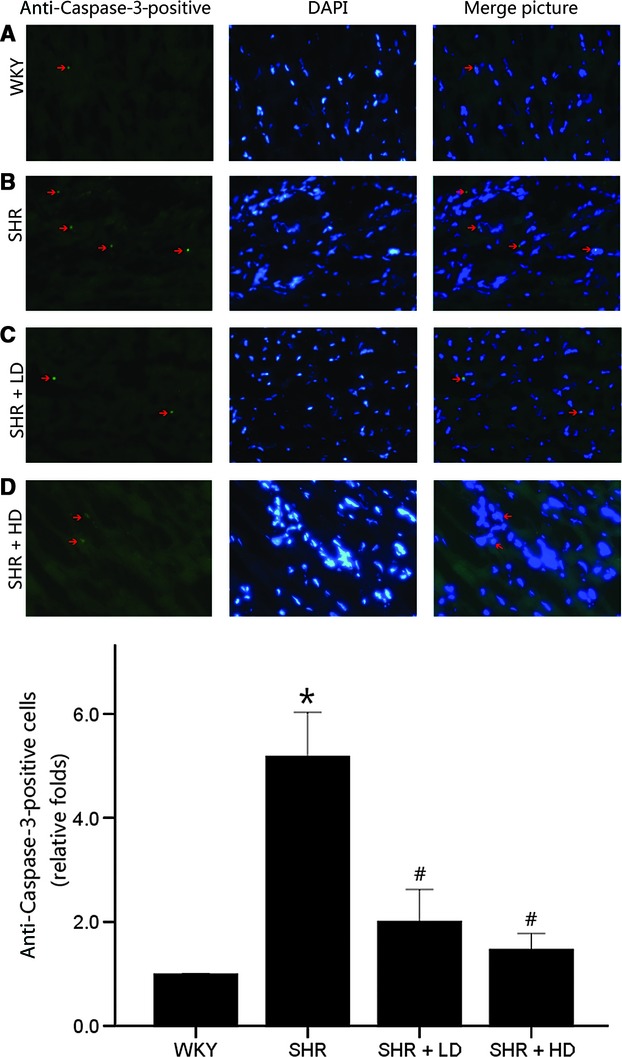
Immunofluorescent labelling for active caspase-3 on rat heart tissue sections of WKY controls, SHR controls, SHR + LD and SHR + HD; arrow indicating an active caspase-3-positive cardiomyocyte in green colour. (A) few active caspase-3-positive cardiomyocytes in the representative immunofluorescent staining of WKY controls; (B) multiple active caspase-3-positive cardiomyocytes in the representative immunofluorescent staining of SHR controls compared with WKY controls (P < 0.05); (C & D) less active caspase-3-positive cardiomyocytes in SHR + LD and SHR + HD (P < 0.05); (E) quantitative analysis of active caspase-3-positive cardiomyocytes in the four groups by the ratio of active caspase-3-positive number to the total and normalized to the WKY controls. Values, mean ± SED; n = 3–4; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Altered expressions of foetal cardiac genes
In addition to the morphological alterations, expressions of foetal cardiac genes associated with cardiac hypertrophy were measured using qPCR and examined by relative levels of the threshold cycle [Δthreshold cycle (ΔCt), relative to WKY controls, arbitrarily assigned a value of 1]. ANP and SAA mRNA significantly increased in SHR controls compared with their WKY controls, 6.35 ± 1.85 and 7.53 ± 2.79 folds versus WKY (P < 0.05), whereas β-MHC expression declined in SHR controls compared with WKY ones, 0.15 ± 0.09 versus WKY (P > 0.05). Furthermore, ANP mRNA significantly decreased in SHR + LD and SHR + HD by 74.8% and by 59.8%, respectively, and SAA mRNA decreased significantly in SHR + LD and SHR + HD by 42.1% and 61.5%, respectively, but with no significant difference between these two groups (Fig. 7).
Fig 7.
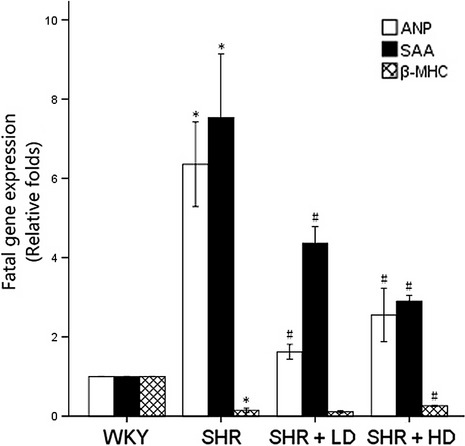
Expressions of cardiac-specific foetal genes of WKY controls, SHR controls, SHR + LD and SHR + LD; real-time RT-qPCR analysis of ANP, SAA and β-MHC; SHR controls showing significantly higher ANP and SAA expressions than WKY ones (P < 0.05); SHR + LDand SHR + LD, significantly reduced mRNA level (P < 0.05); SHR controls, reduced β-MHC (P < 0.05); SHR + LD, significantly elevated mRNA level of β-MHC (P < 0.05). Values, mean ± SED; n = 3; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls.
Expressions of signaling proteins
Mitogen-activated protein kinase (MAPK) signaling was detected by using western blot analysis and examined by the relative levels of integrated optical density (IOD) (ΔIOD, relative to WKY controls, arbitrarily assigned a value of 1) (Fig. 8). AT1 Receptor, a specific receptor for angiotensin II (Ang II), decreased significantly in SHR + LD and SHR + HD by 57.0% and 63.2% respectively. This indicated that Rosuvastatin might exert a therapeutic effect by interfering with the local rennin-angiotensin system (RAS) (P < 0.05). PKCβ2 and PKCα signaling was significantly inhibited in SHR + LD and SHR + HD, but with no impact on PKCβ1 activation, as indicated by the down-regulation of phos-PKCβ2 by 61.6% and 72.6%, respectively (P < 0.05), and the down-regulation of phos-PKCα by 47.7% and 41.3%, respectively (P < 0.05), and with consistent phos-PKCβ1 expression when compared with that in SHR controls (P > 0.05) (Fig. 8A).
Fig 8.
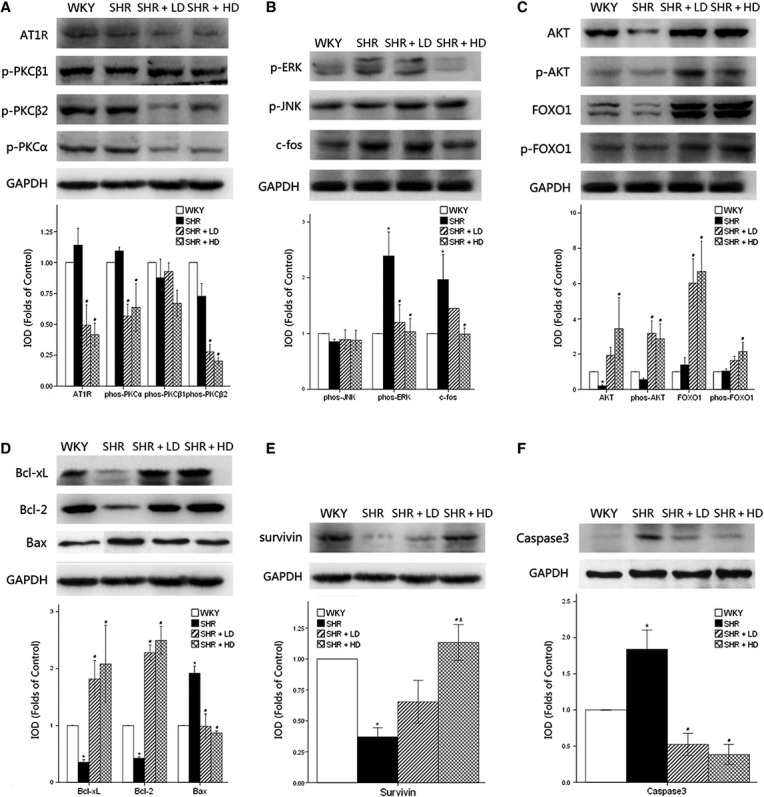
Western blot analysis of hypertrophy-associated signaling pathways and anti-apoptotic signaling pathways in myocardium of WKY controls, SHR controls, SHR + LD and SHR + HD; the relative level of integrated optical density (ΔIOD) measured. (A) the expressions of AT1 receptor, phos-PKCα and phos-PKCβ2 significantly down-regulated in SHR + LD and SHR + HD (P < 0.05); no significant changes in expression of phos-PKCβ1 (P > 0.05); (B) the expressions of phos-ERK and c-fos significantly up-regulated in SHR controls compared with WKY controls (P < 0.05), the former reversed in SHR + LD and SHR + LD, the latter reversed in SHR + LD (P < 0.05); the expression of phos-JNK remained consistent (P > 0.05); (C) Akt, phos-Akt, FOXO1 and phos-FOXO1 up-regulated in SHR + LD (P < 0.05); (D) the expressions of Bcl-xl and Bcl-2 significantly suppressed, and the expression of Bax significantly augmented in SHR controls compared with WKY controls (P < 0.05), and reversed in SHR + LD and SHR + LD (P < 0.05); (E) survivin down-regulated significantly in SHR controls and reversed in SHR + HD (P < 0.05), with significant difference between SHR + LD and SHR + HD (P < 0.05); (F) Caspase-3 expression augmented in SHR (P < 0.05) and suppressed in SHR + LD and SHR + HD (P < 0.05). Values, mean ± SED; n = 3; *P < 0.05 versus WKY controls; #P < 0.05 versus SHR controls; δ P < 0.05 versus SHR + LD.
Phos-extracellular signal-regulated kinase and phos-Jun N-terminal kinase are the two parallel downstream effectors of AT1 Receptor-PKC cascade. Their phosphorylation is crucial for their biological activity and the translocation from cytoplasm into nucleus. ERK was significantly activated in SHR controls compared with WKY ones, 2.39 ± 0.75 folds versus WKY (P < 0.05), and the phosphorylation of ERK was reduced in SHR + LD and SHR + HD by 49.8% and 56.9% respectively (P < 0.05). However, no significant difference in phos-JNK was observed among WKY controls and three SHR groups (P > 0.05), indicating AT1 Receptor-PKCβ2/α might propagate its signal to the downstream signaling pathway via ERK rather than JNK. C-fos, modulated by ERK activation, was up-regulated by nearly two-folds in SHR controls, but significantly suppressed in SHR + HD by 49.7% (P < 0.05) (Fig. 8B).
Akt expression was down-regulated in SHR controls, but significantly up-regulated in SHR + HD, 0.20 ± 0.07 versus 3.43 ± 3.05 (P < 0.05). In SHR + LD and SHR + HD, the phosphorylation of Akt underwent similar alterations, 0.56 ± 0.12 versus 3.18 ± 1.22 and 2.87 ± 1.46 (P < 0.05); FOXO1, a downstream signaling factor of Akt pathway, was subjected to a higher level of expression, 1.39 ± 0.72 versus 6.02 ± 2.38 and 6.69 ± 2.97 (P < 0.05); and more importantly, the Akt-mediated phosphorylation of FOXO1, which translocated into the nucleus and suppressed the apoptosis of cardiac cells, was augmented significantly in SHR + HD, 1.04 ± 0.22 versus 2.15 ± 0.90 (P < 0.05) (Fig. 8C).
Western blot analysis showed low expressions of Bcl-2 and Bcl-xl in SHR controls compared with WKY ones. In SHR + LD and SHR + HD, however, the expressions increased significantly, 0.42 ± 0.04 versus 2.28 ± 0.23 and 2.49 ± 0.42 (P < 0.05) and 0.35 ± 0.08 versus 1.82 ± 0.57 and 2.08 ± 1.18 respectively (P < 0.05). Bax and the ratio of Bax to Bcl-2 were significantly up-regulated in SHR controls compared with WKY ones, and reversed to normal ranges in SHR + LD and SHR + HD, 1.92 ± 0.22 versus 0.99 ± 0.37 and 0.87 ± 0.06 (P < 0.05). These data suggested that mitochondria-associated apoptotic signaling was involved (Fig. 7D).
In the present study, survivin decreased significantly in SHR controls, whereas Rosuvastatin exerted does-dependent up-regulatory effects on its expression (P < 0.05) (Fig. 8E).
Akt pathway, Bcl-2 family and survivin were down-regulated in SHR controls, but up-regulated in SHR + LD and SHR + HD, which resulted in changes in their caspase-3 expression, with an increase in the former and a decrease in the latter, 1.84 ± 0.46 versus 0.52 ± 0.27 and 0.38 ± 0.24 (P < 0.05) (Fig. 8).
Discussion
Cardiac remodelling including ventricular hypertrophy and the subsequent congestive heart failure are the common outcome of essential hypertension. Cardiac apoptosis has been reported as one of the reasons for reduced myocardial mass. However, the exact role of apoptosis in cardiac remodelling from hypertrophy to fibrosis during the end-stage hypertensive heart disease remained unknown. The present study provided functional, structural and molecular evidence for the cardiac remodelling, from hypertrophy to fibrosis and ultimately heart failure, during the end-stage hypertensive heart disease in SHRs. Cardiac hypertrophy and fibrosis were significantly reversed in SHR + LD and SHR + HD, as indicated by echocardiography, haematoxylin and eosin staining, Masson Trichrome staining and the altered expressions of ANP and SAA. The consistent SBP among the three SHR groups indicated that Rosuvastatin acted on the myocardium without changing it significantly. Its anti-hypertrophic effects were accompanied by the decreased expression of AT1 receptor on cardiac myocytes, and the suppression of PKCβ2 and PKCα activation, ERK phosphorylation and c-fos expression. These findings indicated that Rosuvastatin may favourably modify the development of cardiac remodelling by affecting the outside-in signaling of the local rennin-angiotensin system on the cardiac cells. Furthermore, we revealed the crucial role of cardiac apoptosis in cardiac remodelling during the end-stage hypertensive heart disease by TUNEL staining and active caspase-3 immunofluorescent labelling, and found the anti-apoptotic effects of Rosuvastatin, which involved the attenuation of the three anti-apoptotic pathways: AKT-FOXO1, Bcl Family and survivin signaling, which contributed together to the final down-regulation of caspase-3. These findings indicated that Rosuvastatin might prevent the transition from cardiac hypertrophy to heart failure via its anti-apoptotic effect.
High blood pressure stimulates both fibroblasts and cardiomyocytes to express cytokines such as angiotensin II and results in hypertrophy and fibrosis [14]. Reduction in SBP could attenuate these pathological changes and improve the prognosis of patients with cardiac malfunction, which has been supported by numerous randomized studies, such as ACCOMPLISH, ADVANCE, HYVET and PRoFESS [15]. Statins have been reported to reverse cardiac remodelling; however, there have been controversies on their anti-hypertensive effects. Loch and Saka reported that statin treatment failed to alter blood pressure in Dahl salt-sensitive rats [16, 17]. However, Susic et al. proved that statin reduced total peripheral vascular resistance in both SHR and WKY/L-NAME rats [18] and resulted in a reduction in blood pressure. In the current study, no significant changes in SBP were observed in the three SHR groups. It could be that 52-week-old SHRs had developed severe perivascular fibrosis, arterial stiffness and endothelial compromise. In this case, Rosuvastatin, possessing weak anti-hypertensive effect, could not alter significantly the peripheral resistance and the resultant SBP. Prandin MG also pointed out that the confounding factors, such as age and baseline SBP, might influence the blood pressure of each individual [19]. Thus, differences in animal models, statins and experimental protocols might account for these divergent findings. In the current study, nevertheless, the consistent SBP demonstrated that Rosuvastatin was capable of attenuating cardiac hypertrophy without changing SBP levels. In addition, different loading conditions would interfere with LVEF and LVFS. The consistent SBP indicated the LV function was significantly reversed by Rosuvastatin without interference of the after-load difference.
In the present study, SHR + LD and SHR + HD developed a reduction of cardiac remodelling to a certain degree. The correlation observed between the doses of Rosuvastatin and the levels of regression further supported the therapeutic effect of Rosuvastatin, even without statistical significance in some cases, providing more evidence for Rosuvastatin as a modulator of the pathological changes in myocardium. However, previous studies concerning the effects of Rosuvastatin on cardiac remodelling have been controversial. Luo et al. reported that statins could reduce cardiac mass and ventricular collagen concentration in rats [20] and exerted its antioxidant effects by Rac1 inhibition [21]. Conversely, Frohlich et al. noted no effect of Rosuvastatin on cardiovascular mass and collagen deposition in 20-week-old SHR with Rosuvastatin treatment [18]. Furthermore, Chang et al. proposed that Rosuvastatin could have limited therapeutic value when used to prevent progression from LV hypertrophy (LVH) to heart failure in Dahl-Iwai salt-sensitive hypertensive hearts [14]. It is noteworthy that differences in animal models and experimental protocols might lead to different conclusions. In Chang's study, Dahl-Iwai salt-sensitive rats used as a model of hypertensive cardiomyopathy, with salt-induced hypertension [22], might respond to Rosuvastatin by means other than AT1 receptor. Comparatively, SHR, in which RAS activity was characteristically altered [23], might respond to Rosuvastatin via AT1 receptor and its downstream signaling pathways, as indicated in our study. This might explain the different results and further supported the possible mechanisms of Rosuvastatin via AT1 receptor. It can also be that the severity of cardiac remodelling and different degrees of pathological changes in SHRs may be responsible for these divergent findings. These outcome suggest that the time of statins administration might be crucial to the management of hypertensive heart disease.
Clinically, the therapeutic effects of statins on cardiac remodelling have also been controversial. Many nonrandomized studies have suggested that the use of statins is associated with preserved LV function, reduced adverse cardiovascular events and better outcome in patients with LV dysfunction [6, 24, 25]. A recent study demonstrated the veterans who were treated with statin at any time after the diagnosis of heart failure were much less likely to suffer all-cause mortality with mean follow-up of 2.66 years [26]. Conversely, in John Kjekshus' trial, even Rosuvastatin could reduce the total number of hospitalizations for heart failure, but produced no effect on heart failure mortality [27]. The UNIVERSE trials also failed to demonstrate an therapeutic effect of statin on LV remodelling in patients with chronic heart failure [28]. One of the reasons might be that those enrolled had been well treated for heart failure with a standard therapy other than Rosuvastatin, which reduced significantly the population with potentially fatal heart failure enrolled in the trial. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, the commonly prescribed anti-heart failure medications, which could also affect the local RAS, might interfere with the observation of the therapeutic effects of Rosuvastatin. It can also be that the patients' follow-up was not long enough to see a beneficial effect of treatment. In addition, the patients in these trials, with heart failure of ischaemic or non-ischaemic aetiology, were not differentially enrolled, and those with hypertrophic cardiomyopathy and decompensated heart failure were excluded, Thus, different inclusion and exclusion criteria can cause different pathophysiological and etiological changes in heart failure, confusing the consistence of the results in the cohort, and study durations and different methods might be responsible for these divergent conclusions.
The tissue renin-angiotensin system has been demonstrated by several studies as an independent risk factor for cardiac remodelling and congestive heart failure during the development of essential hypertension. Fabris and Kossmehl suggested that angiotensin II stimulation of AT1 receptors in the heart in vivo can be associated with an increased rate of apoptosis, suggesting RAS activation might lead to cardiac apoptosis [29, 30]. Nickening et al. reported that statins exert protective effects on hypertensive patients, such as effectively retrieving heart function, the mechanisms of which might involve RAS suppression [31]. Therefore, we postulated that Rosuvastatin could exert a beneficial effect on cardiac remodelling by interfering with apoptosis via tissue AT1 receptor expressed on the myocardial cells of hypertensive patients. We observed lower percentages of TUNEL-positive and active caspase 3-positive cells in myocardium, accompanied by an elevated expression of Akt-FOXO1, Bcl-2 family and survivin signaling upon treatment. We also found that AT1 receptors on myocaridac cells were suppressed. Therefore, we provided convincible evidence proving that Rosuvastatin can reverse cardiac apoptosis during cardiac remodelling, and slower the transition from hypertrophy to heart failure [32] via local RAS depression and apoptotic signaling regulation.
Our study is not devoid of limitations. The doses of Rosuvastatin were much higher in SHRs than those in clinical patients. However, the physiological relevance of the findings cannot be belittled even though higher doses are generally administered in animal studies for better therapeutic effects in a short period. As SBP was not changed by treatment in the current study, it seems unlikely that changes in SBP are central to the attenuation of cardiac remodelling in SHR by Rosuvastatin, but clinically, SBP lowering is taken into consideration at first. In addition, Rosuvastatin was exclusively applied to the animal model; therefore, its potential therapeutic effect on the end-stage hypertensive heart disease may not apply to the patients who have undergone standard treatments.
Conclusively, statins have many pleiotropic effects beyond lipid lowering, including decreasing oxygen radicals production [21], inhibition of inflammatory responses [33], stimulating stem cells [34, 35] and even attenuation of myocardial fibrosis via microRNAs [24, 36–41]. It can be concluded from the present study that anti-apoptosis via AT1 receptors and the anti-apoptotic signaling pathways, and the subsequent prevention of the transition from cardiac hypertrophy to fibrosis, and ultimately to heart failure we discovered can be one of the pleiotropic effects, which can make statins of potential benefit to patients with LVH and even with heart failure of both iscahemic and nonischaemic aetiologies of which hypertension is the main risk factor [21].
Acknowledgments
This work was supported by the Outstanding Youth Grant from National Natural Science Foundation of China (30725036). The authors are thankful to Dr. Xinxing Xie from the Department of Cardiology of Qianfoshan Hospital, Shandong Province, for his animal dissections, and to Dr. Bingyu Li from Shanghai Institute of Cardiovascular Diseases of Zhongshan Hospital, Fudan University, for her kind help in immunofluorescence and TUNEL study. We are also grateful to Dr. Jianguo Jia and Jian Wu from Shanghai Institute of Cardiovascular Diseases of Zhongshan Hospital, Fudan University, for their technical assistance in echocardiographic evaluation.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Reference
- 1.Kearney PM, Whelton M, Reynolds K, et al. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–23. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 2.Heagerty AM, Heerkens EH, Izzard AS. Small artery structure and function in hypertension. J Cell Mol Med. 2010;14:1037–43. doi: 10.1111/j.1582-4934.2010.01080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu HR, Tao L, Gao E, et al. Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc Res. 2004;62:135–44. doi: 10.1016/j.cardiores.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 4.Marzoll A, Melchior-Becker A, Cipollone F, et al. Small leucine-rich proteoglycans in atherosclerotic lesions: novel targets of chronic statin treatment? J Cell Mol Med. 2009;15:232–43. doi: 10.1111/j.1582-4934.2009.00986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horwich TB, MacLellan WR, Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol. 2004;43:642–8. doi: 10.1016/j.jacc.2003.07.049. [DOI] [PubMed] [Google Scholar]
- 6.Sicard P, Zeller M, Dentan G, et al. Beneficial effects of statin therapy on survival in hypertensive patients with acute myocardial infarction: data from the RICO survey. Am J Hypertens. 2007;20:1133–9. doi: 10.1016/j.amjhyper.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Domanski M, Coady S, Fleg J, et al. Effect of statin therapy on survival in patients with nonischemic dilated cardiomyopathy (from the Beta Blocker Evaluation of Survival Trial [BEST]) Am J Cardiol. 2007;99:1448–50. doi: 10.1016/j.amjcard.2006.12.080. [DOI] [PubMed] [Google Scholar]
- 8.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Danielson E, Fonseca FAH, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–82. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 10.Zhao LL, Chen HJ, Chen JZ, et al. Losartan reduced connexin43 expression in left ventricular myocardium of spontaneously hypertensive rats. J Zhejiang Univ Sci B. 2008;9:448–54. doi: 10.1631/jzus.B0820050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–8. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 12.Frustaci A, Kajstura J, Chimenti C, et al. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123–32. doi: 10.1161/01.res.87.12.1123. [DOI] [PubMed] [Google Scholar]
- 13.Connelly KA, Kelly DJ, Zhang Y, et al. Functional, structural and molecular aspects of diastolic heart failure in the diabetic (mRen-2)27 rat. Cardiovasc Res. 2007;76:280–91. doi: 10.1016/j.cardiores.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 14.Chang SA, Kim YJ, Lee HW, et al. Effect of rosuvastatin on cardiac remodelling, function, and progression to heart failure in hypertensive heart with established left ventricular hypertrophy. Hypertension. 2009;54:591–7. doi: 10.1161/HYPERTENSIONAHA.109.131243. [DOI] [PubMed] [Google Scholar]
- 15.Mourad J, Bertrand M. Impact of antihypertensive treatment on mortality: a late-break analysis of recent clinical trials in hypertension. J Hypertens. 2010;28:395–6. [Google Scholar]
- 16.Loch D, Levick S, Hoey A, et al. Rosuvastatin attenuates hypertension-induced cardiovascular remodelling without affecting blood pressure in DOCA-salt hypertensive rats. J Cardiovasc Pharmacol. 2006;47:396–404. doi: 10.1097/01.fjc.0000210072.48991.f6. [DOI] [PubMed] [Google Scholar]
- 17.Saka M, Obata K, Ichihara S, et al. Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J Cardiovasc Pharmacol. 2006;47:770–9. doi: 10.1097/01.fjc.0000211791.22411.0d. [DOI] [PubMed] [Google Scholar]
- 18.Susic D, Varagic J, Ahn J, et al. Beneficial pleiotropic vascular effects of rosuvastatin in two hypertensive models. J Am Coll Cardiol. 2003;42:1091–7. doi: 10.1016/s0735-1097(03)00926-4. [DOI] [PubMed] [Google Scholar]
- 19.Prandin MG, Cicero AF, Dormi A, et al. Prospective evaluation of the effect of statins on blood pressure control in hypertensive patients in clinical practice. Nutr Metab Cardiovasc Dis. 2010;20:512–8. doi: 10.1016/j.numecd.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Luo JD, Zhang WW, Zhang GP, et al. Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clin Exp Pharmacol Physiol. 1999;26:903–8. doi: 10.1046/j.1440-1681.1999.03165.x. [DOI] [PubMed] [Google Scholar]
- 21.Takemoto M, Node K, Nakagami H, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–37. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomohiro A, Kimura S, He H, et al. Regional blood flow in Dahl-Iwai salt-sensitive rats and the effects of dietary L-arginine supplementation. Am J Physiol. 1997;272:1013–9. doi: 10.1152/ajpregu.1997.272.4.R1013. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki J, Matsubara H, Urakami M, et al. Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circ Res. 1993;73:439–47. doi: 10.1161/01.res.73.3.439. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Tsitsiou E, Herrick SE, et al. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–21. doi: 10.1111/j.1742-4658.2010.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sola S, Mir MQ, Lerakis S, et al. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J Am Coll Cardiol. 2006;47:332–7. doi: 10.1016/j.jacc.2005.06.088. [DOI] [PubMed] [Google Scholar]
- 26.Thambidorai SK, Deshmukh AR, Walters RW, et al. Impact of statin use on heart failure mortality. Int J Cardiol. 2011;147:438–43. doi: 10.1016/j.ijcard.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 27.Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357:2248–61. doi: 10.1056/NEJMoa0706201. [DOI] [PubMed] [Google Scholar]
- 28.Krum H. Presented at the annual meeting of the American College of Cardiology. Atlanta, Georgia: 2006. UNIVERSE: rosuvastatin has no effect on ventricular remodelling. March 13. [Google Scholar]
- 29.Fabris B, Candido R, Bortoletto M, et al. Stimulation of cardiac apoptosis in ovariectomized hypertensive rats: potential role of the renin-angiotensin system. J Hypertens. 2011;29:273–81. doi: 10.1097/HJH.0b013e328340d0d3. [DOI] [PubMed] [Google Scholar]
- 30.Kossmehl P, Kurth E, Faramarzi S, et al. Mechanisms of apoptosis after ischemia and reperfusion: role of the renin-angiotensin system. Apoptosis. 2006;11:347–58. doi: 10.1007/s10495-006-4350-9. [DOI] [PubMed] [Google Scholar]
- 31.Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis. Circulation. 2002;105:530–6. doi: 10.1161/hc0402.102619. [DOI] [PubMed] [Google Scholar]
- 32.Reeve JLV, Duffy AM, O'Brien T, et al. Don't lose heart – therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J Cell Mol Med. 2005;9:609–22. doi: 10.1111/j.1582-4934.2005.tb00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109:III39–43. doi: 10.1161/01.CIR.0000131517.20177.5a. [DOI] [PubMed] [Google Scholar]
- 34.Revenco D, Morgan JP. Metabolic modulation and cellular therapy of cardiac dysfunction and failure. J Cell Mol Med. 2009;13:811–25. doi: 10.1111/j.1582-4934.2009.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tateishi K, Takehara N, Matsubara H, et al. Stemming heart failure with cardiac- or reprogrammed-stem cells. J Cell Mol Med. 2008;12:2217–32. doi: 10.1111/j.1582-4934.2008.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cismasiu VB, Radu E, Popescu LM. miR-193 expression differentiates telocytes from other stromal cells. J Cell Mol Med. 2011;15:1071–4. doi: 10.1111/j.1582-4934.2011.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–72. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- 38.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419–29. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 39.Bauersachs J. Regulation of myocardial fibrosis by microRNAs. J Cardiovasc Pharmacol. 2010;56:454–9. doi: 10.1097/FJC.0b013e3181ee81df. [DOI] [PubMed] [Google Scholar]
- 40.Ohtani K, Dimmeler S. Control of cardiovascular differentiation by microRNAs. Basic Res Cardiol. 2011;106:5–11. doi: 10.1007/s00395-010-0139-7. [DOI] [PubMed] [Google Scholar]
- 41.Mishra PK, Tyagi N, Kumar M, et al. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778–89. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]