

Abstract
Alzheimer’s disease (AD) is one of the major causes of dementia. The pathogenesis of the disease is not entirely understood, but the amyloid β peptide (Aβ) and the formation of senile plaques seem to play pivotal roles. Oligomerization of the Aβ is thought to trigger a cascade of events, including oxidative stress, glutamate excitotoxicity and inflammation. The kynurenine (KYN) pathway is the major route for the metabolism of the essential amino acid tryptophan. Some of the metabolites of this pathway, such as 3-hydroxykynurenine and quinolinic acid, are known to have neurotoxic properties, whereas others, such as kynurenic acid, are putative neuroprotectants. Among other routes, the KYN pathway has been shown to be involved in AD pathogenesis, and connections to other known mechanisms have also been demonstrated. Oxidative stress, glutamate excitotoxicity and the neuroinflammation involved in AD pathogenesis have been revealed to be connected to the KYN pathway. Intervention at these key steps may serve as the aim of potential therapy.
Keywords: Alzheimer, kynurenine, oxidative stress, glutamate excitotoxicity, neuroinflammation
Alzheimer’s disease
Alzheimer’s disease (AD) is one of the most common causes of dementias. A recent report forecast that the prevalence of AD was set to rise to 35.6 million people globally by 2010 [1, 2], with the imposition of an enormous financial burden. The key feature of the disease is the progressive deficit in several cognitive domains [3–7], paralleled by regionally specific brain atrophy [8–11].
The first breakthrough towards an understanding of the pathomechanism of AD was the identification of amyloid β-peptide (Aβ) in the meningeal vessels of AD patients and later in the senile plaques [12–14]. Aβ is the product of the degradation of the amyloid precursor protein (APP), the gene of which is located on chromosome 21 [15–18]. The APP is cleaved by β- and γ-secretases. Mutations of the presenilin 1 and 2 (the subcomponents of γ-secretase), [19] and the APP [20–24] result in the accumulation of the amyloidogenic form of Aβ and the clinical picture of AD, but the genetically determined form of the disease is relatively rare. However, the oligomerization of Aβ seems to be the pivotal step in the pathogenesis of AD but the role of it was also questioned recently [25]. An intimate interaction between the oligomerization of Aβ and several other pathomechanistic mechanisms leads to the hyperphosphorylation of τ-proteins, the formation of neurofibrillary tangles, synaptic degeneration, oxidative stress, microglial and astrocytic activation, activation of the apoptotic cascade, cell death and transmitter deficiency (Figs 1 and 2). The aim of therapeutic approaches is to modify one or other of these individual steps, generally by anti-amyloid, neuroprotective or neurorestorative means.
Fig 1.
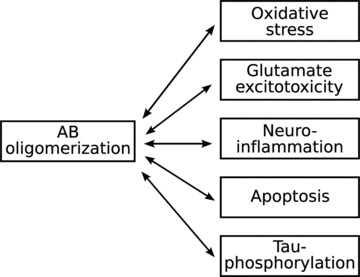
Schematic outline of the pathomechanism of AD.
Fig 2.
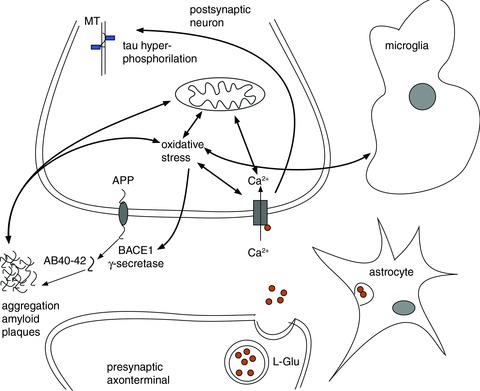
Interactions of the major routes of the AD pathomechanism. The three main cellular components – the neuron, astrocyte and the microglia – are depicted in the figure. The central mechanism in the pathomechanism of AD is the aggregation of Aβ, which in turn activates several parallel but interacting pathomechanistic pathways: oxidative stress, neuroinflammation, τ-hyperphosphorylation, glutamate excitotoxicity.
The kynurenine pathway
Neuroactive kynurenines
The kynurenine (KYN) pathway is the major route for the metabolism of the essential amino acid tryptophan (TRP) [26], the final product of which is nicotinamide adenosine dinucleotide (NAD) (Fig. 3). The first stable metabolite of the pathway is KYN, which is transformed either by KYN aminotransferase (KAT) to kynurenic acid (KYNA) or by KYN hydroxylase to 3-hydroxykynurenine (3-OH-KYN), which is further metabolized to quinolinic acid (QUINA), the precursor of NAD (Fig. 3). These metabolites are usually referred to as neuroactive KYNs [27, 28]. KYNA is an antagonist of the strychnine-insensitive glycine-binding site of the N-methyl-D-aspartate (NMDA) receptor [29, 30], a weak antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) and kainite receptors [31] and also an inhibitor of the α7 nicotinic receptor [32], which is involved in the pre-synaptic regulation of glutamate (L-Glu) release. Conversely, the neuroinhibitory effect of KYNA is concentration dependent: in nanomolar concentrations, it facilitates field excitatory postsynaptic potentials (EPSPs) [33]. QUINA is neurotoxic [34], and has been shown to be a direct activator of NMDA receptors [35], to modulate the release or reuptake inhibition of L-Glu [36] and to be involved in lipid peroxidation [37, 38] and the production of reactive oxygen species (ROS) [38, 39]. 3-OH-KYN also leads to cell death involving apoptotic features by generating ROS [39–42].
Fig 3.
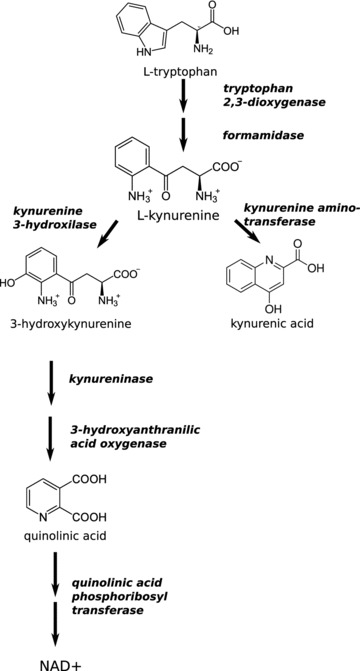
The KYN pathway of the tryptophan metabolism. International Classification Number of the depicted enzymes: tryptophan 2,3-dioxygenase: EC 1.13.11.11; formamidase: EC 3.5.1.9; kynurenine-3-hydroxylase: EC 1.14.13.9; kynurenine aminotransferase: EC 2.6.1.7; kynureninase: EC 3.7.1.3; 3-hydroxyanthranilic acid oxidase: EC 1.13.11.6; quinolinic phosphoribosyltransferase: EC 2.4.2.19.
Enzymes of the kynurenine pathway
The rate-limiting step of the KYN pathway is TRP–KYN transformation, which is catalysed by indoleamine 2,3-dioxygenase (IDO) (Fig. 3.). IDO is known to be expressed by activated astrocytes, microglia and infiltrating macrophages [43], but neuronal expression has also been demonstrated [44].
The key enzyme in the production of putative neuroprotective KYNA is a transaminase. Four isoforms of KAT have been identified in the mammalian brain [45], which contribute differently to KYNA production in the different species [46]. The substrate profile, pH optimum and localization are different for the four isoforms. The pH optimum of KAT I and KAT III is relatively high, at around 9.5 to 10.0, whereas KAT II operates best at physiological pH and has a relative substrate specificity for KYN. KAT II is therefore the major biosynthetic enzyme of KYNA production in the brain. However, recent results indicated that the higher pH optimum of KAT I may well be due to methodological issues [47, 48]. Immunohistochemical studies indicated that KAT I and II are localized preferentially in the astrocytes [49, 50], whereas KAT IV (mitochondrial aspartate aminotransferase) is also present in neurons [51].
Importantly, downstream enzymes of the KYN pathway, such as 3-hydroxyanthranilate oxygenase, which leads to QUINA production, are expressed in the microglia, macrophages and astrocytes, but not in the neurons [52–54]. KYN hydroxylase seems to be an exception as it is not expressed in the astrocytes [55].
Relations of kynurenines to the pathomechanism of AD
Altered activation of the kynurenine pathway in AD
Alterations in the KYN pathway has been identified in several neurological and more specifically neurodegenerative diseases [56, 57], such as Huntington chorea [58], Parkinson’s disease [59–62], multiple sclerosis [63, 64], focal dystonia [65] and migraine [66–69]. An increasing body of evidence indicates that the KYN pathway is involved in the pathogenesis of AD [70, 71]. Baran found slight decreases in the KYN and 3-OH-KYN levels in patients with pathologically confirmed AD [71]. A markedly increased content of KYNA was found selectively in the caudate nucleus and the putamen, which was correlated with increased KAT I activity. The level of aspartate aminotransferase in the cerebrospinal fluid (CSF) was found to be elevated in AD patients [72]. The mitochondrial form of the enzyme was identified as KAT IV [46]. The serum and red blood cell KYNA levels were decreased in AD patients, but there was no alteration in the KAT I or II activity [73]. Furthermore, the serum KYN/TRP ratio was found to be increased in AD patients, indicating and enhanced activity of IDO, the first key enzyme of the pathway [70]. Interestingly the TRP/KYN ratio also proved to be correlated with the cognitive performance of the patients [70]. Another study demonstrated lower KYNA concentration in the lumbar CSF in AD patients [74]. No alteration in QUINA was found either in the CSF [74] or in the examined cortical, subcortical or cerebellar structures [75]. Aβ1–42 induced the expression of IDO and a significant increase in QUINA in human macrophages and microglia [76], but no similar effect of Aβ1–40 was found [77]. A human AD brain preparation involving a subset of senile plaques displayed IDO and QUINA immunoreactivity, and these plaques were characterized by high microglia and reactive astrocytic contents [44].
Connection of oxidative stress and kynurenines
The central nervous system (CNS) is prone to oxidative stress-caused damage as it is rich in polysulphated fatty acids, has a high metabolic oxidative activity, has a high content of transition metals and also exhibits relatively little antioxidant mechanism. Several lines of evidence indicate that oxidative stress has a key role in the pathogenesis of AD and especially in the initiation of pathological processes in sporadic AD [78–80]. In vitro studies have shown that Aβ in aqueous solution fragments and generates free radicals [81]. Post-mortem and animal model studies have confirmed the oxidative stress hypothesis by revealing signs of oxidative damage: changes in antioxidants (Cu/Zn superoxide dismutase [SOD] and glutathione reductase) [82], lipid peroxidation [83], free carbonyls [81] and peroxynitration [84]. A direct connection between Aβ and free radicals was proved by McLellan et al., who demonstrated the co-localization of free radical-induced fluorescent staining with dense core plaques, but not with diffuse plaques in an in vivo transgenic mouse model and in ex vivo human AD tissue [85].
A close connection between APP/Aβ and the mitochondria had already been established. The APP and Aβ were found to be associated with the mithochondrial membrane [86, 87] and to bind to the mitochondrial matrix protein [88]. Aβ1–42 inhibits cytochrome oxidase activity in a Cu-dependent manner [89]. Devi et al. found that the APP accumulates in the protein import channels of the mitochondria of AD patients and inhibits entry of the nuclearly encoded cytochrome c oxidase subunits in association with a decreased cytochrome activity and increased H2O2 production [90]. Similarly, Sirk et al. showed that Aβ25–35 in a sublethal dose can inhibit the import of nuclearly encoded proteins to the mitochondria and that a sustained period of inhibited protein import leads to a reduced mitochondrial membrane potential and an increased level of ROS production [91]. Furthermore, Aβ promotes permeability transition pores in mitochondria [92], this effect seeming to be dependent on cyclophilin D as cyclophilin-deficient mitochondria are resistant to Aβ and Ca2+-induced mitochondrial swelling and permeability transition [93].
In contrast, BACE an aspartyl protease with β-secretase activity [94, 95] can be induced by oxidative stress [96], which in turn leads to a proportional elevation of the carboxyl-terminal fragments of APP. This draws attention to the possible initiating role of oxidative stress in the pathogenesis of sporadic AD.
QUINA is known to cause an increased level of lipid peroxidation [37, 97], an effect that seems to be NMDA receptor dependent: MK-801, an NMDA receptor antagonist, can completely abolish QUINA-induced lipid peroxidation [97]. Another study raised the possibility that the lipid peroxidation effect of QUINA depends on iron and is likely to involve iron chelation by QUINA [98]. QUINA not only induces oxidative stress through the production of ROS, but also appears to influence the antioxidative mechanisms. The concentrations of reduced (GSH) and oxidized (GSSG) glutathione were decreased and increased, respectively, whereas the level of glutathione peroxidase remained stable, indicating a non-enzymatic conversion of GSH to GSSG [99]. The same study also showed that the cytosolic Cu/Zn SOD activity decreased, whereas the mitochondrial Mn SOD was unchanged after intrastriatal QUINA treatment [99], signifying the immediate cytoplasmatic effects of QUINA. Although ROS production seems to be a general feature of QUINA treatment, the lipid peroxidation effect is regionally specific in rat synaptosomes: the striatum and hippocampus displayed increased production of peroxidized lipids after QUINA treatment [38]. Furthermore, lipid peroxidation and oxidative stress could be antagonized by Nω-nitro-L-arginine, a selective antagonist of nitrogen monoxide synthase [100]. Nω-nitro-L-arginine was further shown to diminish KYNA synthesis by reducing the activities of KAT I and II [101, 102].
The importance of ROS production in QUINA toxicity was also demonstrated by the finding that free radical scavengers are able to attenuate the functional structural and behavioural effect of QUINA toxicity [103, 104].
Glutamatergic excitotoxicity
The key feature of glutamatergic neurotransmission is the rapid and efficient removal of L-Glu from the synaptic cleft with high-affinity transporters to prevent receptor over stimulation. L-Glu is taken up by the astrocytes, converted to L-glutamine, transported to the neurons and then recycled to L-Glu and finally packed into synaptic vesicles for reuse. Pathological accumulation of L-Glu leads to prolonged, tonic activation, sustained local depolarization and the influx of cations that trigger the further release of L-Glu. This vicious circle triggers intracellular events [105], primarily swelling of the neurons because of the increased cation concentration and consequent water influx, and secondly a delayed Ca2+-dependent neuronal degeneration [106]. Neuronal degeneration is mediated by calpain I, which brings about cytoskeletal breakdown [107]. Phospholipases break down the cell membranes and generate arachnoidal acid [108], the metabolism of which generates free oxygen radicals and initiates apoptosis [109]. It has been shown that the NMDA receptor is closely linked to protein phosphatase 2A (PP2A), and stimulation of the NMDA receptor leads to the dissociation of PP2A and a reduction of the phosphatase activity [110]. This NMDA receptor-mediated mechanism may be involved in τ-hyperphosphorylation, a key step in the formation of neurofibrillary tangles [111].
As mentioned above, QUINA is the direct activator of NMDA receptors [35] and the neurotoxicity of the compound in sub-physiological concentrations is blocked by the NMDA receptor blockers MK-801 and memantine [112]. The neurotoxicity of QUINA was related in this experiment to the depletion of NAD+, the activation of poly(ADP-ribose) polymerase, extracellular lactate dehydrogenase release and the induction of inducible and neuronal nitric oxide synthase [112]. The other possible mechanism by which QUINA induces AD pathology is PPA2-mediated τ-phosphorylation, which can be abrogated by memantine [111]. Interestingly, this effect of memantine seems to be unrelated to the glycine or L-Glu binding site of the NMDA receptor as PP2A inhibition-induced hyperphosphorylation could not be prevented by the NMDA antagonist 5,7-dichlorokynurenic acid or by D(-)-2-amino-5-phosphopentanoic acid [113].
QUINA was shown to increase the basal L-Glu release in an NMDA receptor-mediated manner [36, 114].
QUINA not only modulates the release of L-Glu but also inhibits the uptake of L-Glu to the astrocytes, which is considered to be one of the major processes in maintaining the L-Glu concentration below toxic levels [36]. A recent experiment demonstrating that KYN pre-treatment, which presumably leads to the production of KYNA in the astrocytes, is able to prevent the neurotoxic effect of L-Glu is indicative of the potential beneficial effect of KYN in neurodegenerative diseases [115].
Inflammation
An increased amount of reactive microglia is commonly found in the brain of AD patients [116, 117]. Most of them are around the Aβ-containing compact plaques [118–120]. Both immunohistochemical and in vivo imaging studies have revealed microgliosis-related signal changes in AD [121]. Furthermore, Edison et al. found that PET detected microglia activation, but not the amyloid burden correlated with the cognitive performance of the patients [122]. A role of the microglia has been proposed in the degradation of Aβ[123], but microglial activation also leads to activation of the complement system and the release of cytokines, chemokines and acute phase proteins (for reviews see [124–126]), which might also play a role in AD pathogenesis.
The KYN pathway is known to be involved in inflammatory processes with various mechanisms. Inflammation due to focal poliovirus is accompanied by the up-regulation of IDO, the rate-limiting step in the KYN pathway that results in increased levels of QUINA, KYN and KYNA [127, 128]. It has also been demonstrated that the sources of QUINA are the macrophages and to a lesser degree microglia. A human foetal brain culture consisting of neurons and astrocytes transformed TRP to KYN when stimulated by γ-interferon, but QUINA was formed only when macrophages were added to the culture [127, 129]. The abilities of macrophages and microglia to produce QUINA differ [130]; this is related to the lower expressions of three key enzymes of the KYN pathway in the microglia: IDO, kynureninase and KYN hydroxylase [131]. Aβ is known to induce phenotypic activation of the microglia and also to modulate the acute and chronic expression of pro-inflammatory genes [132, 133] that may produce potentially toxic products. Interestingly, besides many other pro-inflammatory genes, the expressions of the enzymes of the KYN pathway are also significantly altered by Aβ[124, 132]. Importantly, only Aβ1–42, but not Aβ1–40 or Aβ25–35 activated THP-1 cells (a human monocytic cell line) [134]. Administration of γ-interferon after Aβ1–42 pre-treatment, but not interleukin-1b, tumour necrosis factor-α or interleukin-6, induced the expression of IDO [134]. Microarray analysis of the gene expression profile of the Aβ stimulated microglia indicated an average increase of more than 40-fold (278-fold by real-time PCR) in IDO production at 24 hrs, which remained significantly elevated at 96 hrs [132]. Similarly, the expression of kynureninase was elevated (3.6-fold), but not that of KAT II. These data show that Aβ stimulation of the microglia shifts the KYN pathway in the direction of the production of neurotoxic QUINA relative to the putative neuroprotectant KYNA. In a recent study by Guillemin et al., IDO and QUINA were overproduced in human AD hippocampus preparations [44]. Immunoreactivity of IDO and QUINA was detected in the microglia and astrocytes and also in the neurons. The intracytoplasmatic vesicular neuronal QUINA immunoreactivity is thought to be a result of the uptake rather than the de novo neuronal synthesis of QUINA as it was earlier shown that the neurons produce IDO, but not QUINA [135]. Further, the astrocytes lack KYN hydroxylase and consequently the uptake of QUINA might be part of the neuroprotective mechanism [43]. Additionally, QUINA induces astrogliosis and the production of chemokines such as interleukin 1β, MCP-1 (CCL2), RANTES (CCL5) and interleukine-8 (CXCL8) [136–139].
A future therapeutic approach: modulating the kynurenine pathway
The foregoing data indicate the significant involvement of the KYN pathway in the pathogenesis of AD. The key seems to be the shift in the TRP metabolism in the direction of neurotoxic agents and the relative reduction of neuroprotectant products. This shift has profound, but surely not independent effects on different pathomechanistic pathways in AD: oxidative stress, L-Glu neurotransmission and inflammation. Re-establishment of the physiological metabolite ratios, or even a shift of the TRP metabolism in the neuroprotectant direction may serve as a potential therapeutic approach [27, 140]. Synthetic KYNs such as KYNA are of limited therapeutic use as they penetrate the blood–brain barrier only poorly [141], an exception being 4-Cl-KYN that readily enters the brain and is transformed to 7-Cl-KYN by KAT [142]. The systemic administration of 4-Cl-KYN increased the level of 7-Cl-KYN in the hippocampus and reduced the kainite-induced seizure activity [142]. Similarly, 4-Cl-KYN reduced the neurotoxic effect of QUINA in the rat hippocampus and striatum [143, 144]. In contrast, the synthetic KYN derivative, NMDA antagonist 5,7-dichlorokynurenic acid did not attenuate PP2A inhibition-induced τ-hyperphosporylation [113]. A substantial effort is being made to develop new KYNA derivatives that cross the blood–brain barrier [145]. We recently demonstrated that a novel KYN analogue, 2-(2-N,N-dimethylaminoethylamine-1-carbonyl)-1H-quinolin-4-one hydrochloride, exhibits features similar to those of KYNA [146]. In the micromolar range, its administration decreased the amplitude of the field EPSPs in the CA1 region of the hippocampus. Preclinical and subsequent clinical investigations of the compound are needed to evaluate its usefulness in neurodegenerative diseases such as AD.
Another possibility via which to increase the level of neuroprotectant KYNA is to modulate the activities of the individual enzymes of the KYN pathway. Nicotinylalanine, an agent that inhibits kynureninase and KYN hydroxylase activity, administered together with KYN and probenicid (an inhibitor of organic acid transport), increased the brain KYNA level and inhibited QUINA-induced neurotoxicity [147, 148]. Another such enzyme is KYN hydroxylase, loss of function mutation of which in yeast reduces mutant huntingtin fragment toxicity [149]. Ro 61–8048, a high-affinity inhibitor of KYN hydroxylase significantly reduced the mutant huntingtin-induced production of 3-OH-KYN, but not that of QUINA production, and did not ameliorated ROS production [149]. In a recent study Amori et al. selectively inhibited KAT or KYN hydroxylase and reported the reduction of 3-OH-KYN – QUINA and KYNA production, respectively [150]. Interestingly pre-treatment with intrastriatal QUINA UPF 648 not only decreased the levels of 3-OH-KYN and QUINA, but also moderately elevated KYNA production [150].
Concluding remarks
There is appreciable evidence that the neurodegeneration in AD is mediated, at least partly, by neurotoxic products of the KYN pathway. Possible therapeutic approaches could be to reduce the expression of these neurotoxic agents or to increase the production of putative neuroprotectant KYNA or make use of its analogues. However, the specific involvement of the KYN pathway in AD, it also has to be emphasized that neurodegenerative diseases share several common features. Among other common mechanisms the shift in the KYN pathway seems to be general over different neurodegenerative diseases [27, 56, 58, 62–65] and such, neuroprotective therapies influencing the KYN pathway may be beneficial in several neurological pathologies.
Further research is needed to elucidate the exact role of the KYN pathway in the pathomechanism of these neurodegenerative processes in an effort to promote the development of novel therapeutic agents.
Acknowledgments
Funding of the studies reported in the paper: Teller Ede (NAP-BIO-06-BAYBIOSZ), ETT 026–04, TÁMOP-4.2.2.-08/1/2008–2002, OTKA K75628 and cNEUPRO (LSHM-CT-2007–037950). We thank Dr. David Durham for English editing.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Dartigues JF. Alzheimer’s disease: a global challenge for the 21st century. Lancet Neurol. 2009;8:1082–3. doi: 10.1016/S1474-4422(09)70298-4. [DOI] [PubMed] [Google Scholar]
- 2.Ferri CP, Sousa R, Albanese E. World Alzheimer report 2009. In: Prince M, Jackson J, et al., editors. Alzheimer Disease International. London: 2009. pp. 1–93. [Google Scholar]
- 3.Keri S, Kalman J, Kelemen O, et al. Are Alzheimer’s disease patients able to learn visual prototypes. Neuropsychologia. 2001;39:1218–23. doi: 10.1016/s0028-3932(01)00046-x. [DOI] [PubMed] [Google Scholar]
- 4.Antal A, Keri S, Kincses T, et al. Corticostriatal circuitry mediates fast-track visual categorization. Brain Res Cogn Brain Res. 2002;13:53–9. doi: 10.1016/s0926-6410(01)00089-1. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann I, Nemeth D, Dye CD, et al. Temporal parameters of spontaneous speech in Alzheimer’s disease. Int J Speech Lang Pathol. 2010;12:29–34. doi: 10.3109/17549500903137256. [DOI] [PubMed] [Google Scholar]
- 6.Waltz JA, Knowlton BJ, Holyoak KJ, et al. Relational integration and executive function in Alzheimer’s disease. Neuropsychology. 2004;18:296–305. doi: 10.1037/0894-4105.18.2.296. [DOI] [PubMed] [Google Scholar]
- 7.Salmon DP, Butters N, Chan AS. The deterioration of semantic memory in Alzheimer’s disease. Can J Exp Psychol. 1999;53:108–17. doi: 10.1037/h0087303. [DOI] [PubMed] [Google Scholar]
- 8.Filippini N, Rao A, Wetten S, et al. Anatomically-distinct genetic associations of APOE epsilon4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage. 2009;44:724–8. doi: 10.1016/j.neuroimage.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Zarei M, Patenaude B, Damoiseaux J, et al. Combining shape and connectivity analysis: an MRI study of thalamic degeneration in Alzheimer’s disease. Neuroimage. 2010;49:1–8. doi: 10.1016/j.neuroimage.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Zarei M, Damoiseaux JS, Morgese C, et al. Regional white matter integrity differentiates between vascular dementia and Alzheimer disease. Stroke. 2009;40:773–9. doi: 10.1161/STROKEAHA.108.530832. [DOI] [PubMed] [Google Scholar]
- 11.Smith SM, Rao A, De Stefano N, et al. Longitudinal and cross-sectional analysis of atrophy in Alzheimer’s disease: cross-validation of BSI, SIENA and SIENAX. Neuroimage. 2007;36:1200–6. doi: 10.1016/j.neuroimage.2007.04.035. [DOI] [PubMed] [Google Scholar]
- 12.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 13.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 14.Masters CL, Simms G, Weinman NA, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 16.Goldgaber D, Lerman MI, McBride OW, et al. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–80. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 17.Tanzi RE, Gusella JF, Watkins PC, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–4. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 18.Robakis NK, Ramakrishna N, Wolfe G, et al. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84:4190–4. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–70. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 20.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 21.Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–7. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 22.Hardy J. Framing beta-amyloid. Nat Genet. 1992;1:233–4. doi: 10.1038/ng0792-233. [DOI] [PubMed] [Google Scholar]
- 23.Hendriks L, Van Duijn CM, Cras P, et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat Genet. 1992;1:218–21. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 24.Haass C, Hung AY, Selkoe DJ, et al. Mutations associated with a locus for familial Alzheimer’s disease result in alternative processing of amyloid beta-protein precursor. J Biol Chem. 1994;269:17741–8. [PubMed] [Google Scholar]
- 25.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110:1129–34. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 26.Vamos E, Pardutz A, Klivenyi P, et al. The role of kynurenines in disorders of the central nervous system: possibilities for neuroprotection. J Neurol Sci. 2009;283:21–7. doi: 10.1016/j.jns.2009.02.326. [DOI] [PubMed] [Google Scholar]
- 27.Zadori D, Klivenyi P, Vamos E, et al. Kynurenines in chronic neurodegenerative disorders: future therapeutic strategies. J Neural Transm. 2009;116:1403–9. doi: 10.1007/s00702-009-0263-4. [DOI] [PubMed] [Google Scholar]
- 28.Sas K, Robotka H, Toldi J, et al. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257:221–39. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 29.Kessler M, Terramani T, Lynch G, et al. A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52:1319–28. doi: 10.1111/j.1471-4159.1989.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 30.Birch PJ, Grossman CJ, Hayes AG. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur J Pharmacol. 1988;154:85–7. doi: 10.1016/0014-2999(88)90367-6. [DOI] [PubMed] [Google Scholar]
- 31.Birch PJ, Grossman CJ, Hayes AG. Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur J Pharmacol. 1988;151:313–5. doi: 10.1016/0014-2999(88)90814-x. [DOI] [PubMed] [Google Scholar]
- 32.Hilmas C, Pereira EF, Alkondon M, et al. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–73. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozsa E, Robotka H, Vecsei L, et al. The Janus-face kynurenic acid. J Neural Transm. 2008;115:1087–91. doi: 10.1007/s00702-008-0052-5. [DOI] [PubMed] [Google Scholar]
- 34.Schwarcz R, Whetsell WO, Jr, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–8. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- 35.Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72:411–2. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- 36.Tavares RG, Tasca CI, Santos CE, et al. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int. 2002;40:621–7. doi: 10.1016/s0197-0186(01)00133-4. [DOI] [PubMed] [Google Scholar]
- 37.Rios C, Santamaria A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem Res. 1991;16:1139–43. doi: 10.1007/BF00966592. [DOI] [PubMed] [Google Scholar]
- 38.Santamaria A, Galvan-Arzate S, Lisy V, et al. Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport. 2001;12:871–4. doi: 10.1097/00001756-200103260-00049. [DOI] [PubMed] [Google Scholar]
- 39.Eastman CL, Guilarte TR. Cytotoxicity of 3-hydroxykynurenine in a neuronal hybrid cell line. Brain Res. 1989;495:225–31. doi: 10.1016/0006-8993(89)90216-3. [DOI] [PubMed] [Google Scholar]
- 40.Nakagami Y, Saito H, Katsuki H. 3-Hydroxykynurenine toxicity on the rat striatum in vivo. Jpn J Pharmacol. 1996;71:183–6. doi: 10.1254/jjp.71.183. [DOI] [PubMed] [Google Scholar]
- 41.Eastman CL, Guilarte TR. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem Res. 1990;15:1101–7. doi: 10.1007/BF01101711. [DOI] [PubMed] [Google Scholar]
- 42.Okuda S, Nishiyama N, Saito H, et al. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J Neurochem. 1998;70:299–307. doi: 10.1046/j.1471-4159.1998.70010299.x. [DOI] [PubMed] [Google Scholar]
- 43.Guillemin GJ, Kerr SJ, Smythe GA, et al. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001;78:842–53. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- 44.Guillemin GJ, Brew BJ, Noonan CE, et al. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol Appl Neurobiol. 2005;31:395–404. doi: 10.1111/j.1365-2990.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- 45.Han Q, Cai T, Tagle DA, et al. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell Mol Life Sci. 2010;67:353–68. doi: 10.1007/s00018-009-0166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guidetti P, Amori L, Sapko MT, et al. Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem. 2007;102:103–11. doi: 10.1111/j.1471-4159.2007.04556.x. [DOI] [PubMed] [Google Scholar]
- 47.Han Q, Li J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur J Biochem. 2004;271:4804–14. doi: 10.1111/j.1432-1033.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- 48.Han Q, Robinson H, Cai T, et al. Structural insight into the inhibition of human kynurenine aminotransferase I/glutamine transaminase K. J Med Chem. 2009;52:2786–93. doi: 10.1021/jm9000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guidetti P, Hoffman GE, Melendez-Ferro M, et al. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia. 2007;55:78–92. doi: 10.1002/glia.20432. [DOI] [PubMed] [Google Scholar]
- 50.Roberts RC, Du F, McCarthy KE, et al. Immunocytochemical localization of kynurenine aminotransferase in the rat striatum: a light and electron microscopic study. J Comp Neurol. 1992;326:82–90. doi: 10.1002/cne.903260107. [DOI] [PubMed] [Google Scholar]
- 51.Cechetto JD, Sadacharan SK, Berk PD, et al. Immunogold localization of mitochondrial aspartate aminotransferase in mitochondria and on the cell surface in normal rat tissues. Histol Histopathol. 2002;17:353–64. doi: 10.14670/HH-17.353. [DOI] [PubMed] [Google Scholar]
- 52.Lehrmann E, Molinari A, Speciale C, et al. Immunohistochemical visualization of newly formed quinolinate in the normal and excitotoxically lesioned rat striatum. Exp Brain Res. 2001;141:389–97. doi: 10.1007/s002210100887. [DOI] [PubMed] [Google Scholar]
- 53.Kohler C, Eriksson LG, Flood PR, et al. Quinolinic acid metabolism in the rat brain. Immunohistochemical identification of 3-hydroxyanthranilic acid oxygenase and quinolinic acid phosphoribosyltransferase in the hippocampal region. J Neurosci. 1988;8:975–87. doi: 10.1523/JNEUROSCI.08-03-00975.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts RC, McCarthy KE, Du F, et al. 3-Hydroxyanthranilic acid oxygenase-containing astrocytic processes surround glutamate-containing axon terminals in the rat striatum. J Neurosci. 1995;15:1150–61. doi: 10.1523/JNEUROSCI.15-02-01150.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guillemin GJ, Kerr SJ, Smythe GA, et al. Kynurenine pathway metabolism in human astrocytes. Adv Exp Med Biol. 1999;467:125–31. doi: 10.1007/978-1-4615-4709-9_18. [DOI] [PubMed] [Google Scholar]
- 56.Nemeth H, Toldi J, Vecsei L. Role of kynurenines in the central and peripheral nervous systems. Curr Neurovasc Res. 2005;2:249–60. doi: 10.2174/1567202054368326. [DOI] [PubMed] [Google Scholar]
- 57.Vecsei L, Schwab F. Kynurenine and its metabolites in nervous system diseases. Orv Hetil. 1992;133:1803–7. [PubMed] [Google Scholar]
- 58.Vecsei L, Beal MF. Huntington’s disease, behavioral disturbances, and kynurenines: preclinical findings and therapeutic perspectives. Biol Psychiatry. 1996;39:1061–3. doi: 10.1016/0006-3223(95)00377-0. [DOI] [PubMed] [Google Scholar]
- 59.Nemeth H, Toldi J, Vecsei L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl. 2006:285–304. doi: 10.1007/978-3-211-45295-0_45. [DOI] [PubMed] [Google Scholar]
- 60.Hartai Z, Klivenyi P, Janaky T, et al. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J Neurol Sci. 2005;239:31–5. doi: 10.1016/j.jns.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Rakoczi K, Klivenyi P, Vecsei L. Neuroprotection in Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical findings. Ideggyogy Sz. 2009;62:25–34. [PubMed] [Google Scholar]
- 62.Kincses ZT, Vecsei L. Pharmacological therapy in Parkinson’s disease: focus on neuroprotection. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00150.x. Doi: 10.1111/j.1755-5949.2010.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartai Z, Klivenyi P, Janaky T, et al. Kynurenine metabolism in multiple sclerosis. Acta Neurol Scand. 2005;112:93–6. doi: 10.1111/j.1600-0404.2005.00442.x. [DOI] [PubMed] [Google Scholar]
- 64.Rajda C, Bergquist J, Vecsei L. Kynurenines, redox disturbances and neurodegeneration in multiple sclerosis. J Neural Transm Suppl. 2007;72:323–9. doi: 10.1007/978-3-211-73574-9_40. [DOI] [PubMed] [Google Scholar]
- 65.Hartai Z, Klivenyi P, Janaky T, et al. Peripheral kynurenine metabolism in focal dystonia. Med Chem. 2007;3:285–8. doi: 10.2174/157340607780620707. [DOI] [PubMed] [Google Scholar]
- 66.Knyihar-Csillik E, Chadaide Z, Okuno E, et al. Kynurenine aminotransferase in the supratentorial dura mater of the rat: effect of stimulation of the trigeminal ganglion. Exp Neurol. 2004;186:242–7. doi: 10.1016/j.expneurol.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Knyihar-Csillik E, Toldi J, Mihaly A, et al. Kynurenine in combination with probenecid mitigates the stimulation-induced increase of c-fos immunoreactivity of the rat caudal trigeminal nucleus in an experimental migraine model. J Neural Transm. 2007;114:417–21. doi: 10.1007/s00702-006-0545-z. [DOI] [PubMed] [Google Scholar]
- 68.Vamos E, Pardutz A, Varga H, et al. l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology. 2009;57:425–9. doi: 10.1016/j.neuropharm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 69.Knyihar-Csillik E, Toldi J, Krisztin-Peva B, et al. Prevention of electrical stimulation-induced increase of c-fos immunoreaction in the caudal trigeminal nucleus by kynurenine combined with probenecid. Neurosci Lett. 2007;418:122–6. doi: 10.1016/j.neulet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 70.Widner B, Leblhuber F, Walli J, et al. Tryptophan degradation and immune activation in Alzheimer’s disease. J Neural Transm. 2000;107:343–53. doi: 10.1007/s007020050029. [DOI] [PubMed] [Google Scholar]
- 71.Baran H, Jellinger K, Deecke L. Kynurenine metabolism in Alzheimer’s disease. J Neural Transm. 1999;106:165–81. doi: 10.1007/s007020050149. [DOI] [PubMed] [Google Scholar]
- 72.Tapiola T, Lehtovirta M, Pirttila T, et al. Increased aspartate aminotransferase activity in cerebrospinal fluid and Alzheimer’s disease. Lancet. 1998;352:287. doi: 10.1016/S0140-6736(05)60260-7. [DOI] [PubMed] [Google Scholar]
- 73.Hartai Z, Juhasz A, Rimanoczy A, et al. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem Int. 2007;50:308–13. doi: 10.1016/j.neuint.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 74.Heyes MP, Saito K, Crowley JS, et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain. 1992;115:1249–73. doi: 10.1093/brain/115.5.1249. [DOI] [PubMed] [Google Scholar]
- 75.Sofic E, Halket J, Przyborowska A, et al. Brain quinolinic acid in Alzheimer’s dementia. Eur Arch Psychiatry Neurol Sci. 1989;239:177–9. doi: 10.1007/BF01739651. [DOI] [PubMed] [Google Scholar]
- 76.Guillemin GJ, Williams KR, Smith DG, et al. Quinolinic acid in the pathogenesis of Alzheimer’s disease. Adv Exp Med Biol. 2003;527:167–76. doi: 10.1007/978-1-4615-0135-0_19. [DOI] [PubMed] [Google Scholar]
- 77.Guillemin GJ, Smythe GA, Veas LA, et al. A beta 1–42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport. 2003;14:2311–5. doi: 10.1097/00001756-200312190-00005. [DOI] [PubMed] [Google Scholar]
- 78.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer’s disease. J Neurochem. 2006;96:1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 80.Zhu X, Lee HG, Casadesus G, et al. Oxidative imbalance in Alzheimer’s disease. Mol Neurobiol. 2005;31:205–17. doi: 10.1385/MN:31:1-3:205. [DOI] [PubMed] [Google Scholar]
- 81.Hensley K, Carney JM, Mattson MP, et al. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–4. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leutner S, Czech C, Schindowski K, et al. Reduced antioxidant enzyme activity in brains of mice transgenic for human presenilin-1 with single or multiple mutations. Neurosci Lett. 2000;292:87–90. doi: 10.1016/s0304-3940(00)01449-x. [DOI] [PubMed] [Google Scholar]
- 83.Sayre LM, Zelasko DA, Harris PL, et al. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–7. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 84.Good PF, Werner P, Hsu A, et al. Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol. 1996;149:21–8. [PMC free article] [PubMed] [Google Scholar]
- 85.McLellan ME, Kajdasz ST, Hyman BT, et al. In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J Neurosci. 2003;23:2212–7. doi: 10.1523/JNEUROSCI.23-06-02212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamaguchi H, Yamazaki T, Ishiguro K, et al. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 87.Manczak M, Anekonda TS, Henson E, et al. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 88.Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 89.Crouch PJ, Barnham KJ, Duce JA, et al. Copper-dependent inhibition of cytochrome c oxidase by Abeta(1–42) requires reduced methionine at residue 35 of the Abeta peptide. J Neurochem. 2006;99:226–36. doi: 10.1111/j.1471-4159.2006.04050.x. [DOI] [PubMed] [Google Scholar]
- 90.Devi L, Prabhu BM, Galati DF, et al. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–68. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sirk D, Zhu Z, Wadia JS, et al. Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J Neurochem. 2007;103:1989–2003. doi: 10.1111/j.1471-4159.2007.04907.x. [DOI] [PubMed] [Google Scholar]
- 92.Moreira PI, Santos MS, Moreno A, et al. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci Rep. 2001;21:789–800. doi: 10.1023/a:1015536808304. [DOI] [PubMed] [Google Scholar]
- 93.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hussain I, Powell D, Howlett DR, et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–27. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 95.Hussain I, Powell DJ, Howlett DR, et al. ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol Cell Neurosci. 2000;16:609–19. doi: 10.1006/mcne.2000.0884. [DOI] [PubMed] [Google Scholar]
- 96.Tamagno E, Bardini P, Obbili A, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–88. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 97.Santamaria A, Rios C. MK-801, an N-methyl-D-aspartate receptor antagonist, blocks quinolinic acid-induced lipid peroxidation in rat corpus striatum. Neurosci Lett. 1993;159:51–4. doi: 10.1016/0304-3940(93)90796-n. [DOI] [PubMed] [Google Scholar]
- 98.Stipek S, Stastny F, Platenik J, et al. The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem Int. 1997;30:233–7. [PubMed] [Google Scholar]
- 99.Rodriguez-Martinez E, Camacho A, Maldonado PD, et al. Effect of quinolinic acid on endogenous antioxidants in rat corpus striatum. Brain Res. 2000;858:436–9. doi: 10.1016/s0006-8993(99)02474-9. [DOI] [PubMed] [Google Scholar]
- 100.Santamaria D, Espinoza-Gonzalez V, Rios C, et al. Nomega-nitro-L-arginine, a nitric oxide synthase inhibitor, antagonizes quinolinic acid-induced neurotoxicity and oxidative stress in rat striatal slices. Neurochem Res. 1999;24:843–8. doi: 10.1023/a:1020949812581. [DOI] [PubMed] [Google Scholar]
- 101.Luchowski P, Kocki T, Urbanska EM. N(G)-nitro-L-arginine and its methyl ester inhibit brain synthesis of kynurenic acid possibly via nitric oxide-independent mechanism. Pol J Pharmacol. 2001;53:597–604. [PubMed] [Google Scholar]
- 102.Rozsa E, Robotka H, Nagy D, et al. The pentylenetetrazole-induced activity in the hippocampus can be inhibited by the conversion of L-kynurenine to kynurenic acid: an in vitro study. Brain Res Bull. 2008;76:474–9. doi: 10.1016/j.brainresbull.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 103.Nakai M, Qin ZH, Wang Y, et al. Free radical scavenger OPC-14117 attenuates quinolinic acid-induced NF-kappaB activation and apoptosis in rat striatum. Brain Res Mol Brain Res. 1999;64:59–68. doi: 10.1016/s0169-328x(98)00310-6. [DOI] [PubMed] [Google Scholar]
- 104.Nakao N, Brundin P. Effects of alpha-phenyl-tert-butyl nitrone on neuronal survival and motor function following intrastriatal injections of quinolinate or 3-nitropropionic acid. Neuroscience. 1997;76:749–61. doi: 10.1016/s0306-4522(96)00223-0. [DOI] [PubMed] [Google Scholar]
- 105.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–76. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 106.Koh JY, Choi DW. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991;548:318–21. doi: 10.1016/0006-8993(91)91140-v. [DOI] [PubMed] [Google Scholar]
- 107.Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–87. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 108.Lazarewicz JW, Wroblewski JT, Costa E. N-methyl-D-aspartate-sensitive glutamate receptors induce calcium-mediated arachidonic acid release in primary cultures of cerebellar granule cells. J Neurochem. 1990;55:1875–81. doi: 10.1111/j.1471-4159.1990.tb05771.x. [DOI] [PubMed] [Google Scholar]
- 109.Chen KC, Chang LS. Arachidonic acid-induced apoptosis of human neuroblastoma SK-N-SH cells is mediated through mitochondrial alteration elicited by ROS and Ca(2+)-evoked activation of p38alpha MAPK and JNK1. Toxicology. 2009;262:199–206. doi: 10.1016/j.tox.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 110.Chan SF, Sucher NJ. An NMDA receptor signaling complex with protein phosphatase 2A. J Neurosci. 2001;21:7985–92. doi: 10.1523/JNEUROSCI.21-20-07985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rahman A, Ting K, Cullen KM, et al. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS One. 2009;4:e6344. doi: 10.1371/journal.pone.0006344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Braidy N, Grant R, Adams S, et al. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res. 2009;16:77–86. doi: 10.1007/s12640-009-9051-z. [DOI] [PubMed] [Google Scholar]
- 113.Li L, Sengupta A, Haque N, et al. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–9. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 114.Tavares RG, Schmidt AP, Abud J, et al. In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem Res. 2005;30:439–44. doi: 10.1007/s11064-005-2678-0. [DOI] [PubMed] [Google Scholar]
- 115.Kumar A, Babu GN. In vivo neuroprotective effects of peripheral kynurenine on acute neurotoxicity induced by glutamate in rat cerebral cortex. Neurochem Res. 2010;35:636–44. doi: 10.1007/s11064-009-0114-6. [DOI] [PubMed] [Google Scholar]
- 116.Styren SD, Civin WH, Rogers J. Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer’s disease brain. Exp Neurol. 1990;110:93–104. doi: 10.1016/0014-4886(90)90054-v. [DOI] [PubMed] [Google Scholar]
- 117.McGeer PL, Itagaki S, Tago H, et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 118.Mackenzie IR, Hao C, Munoz DG. Role of microglia in senile plaque formation. Neurobiol Aging. 1995;16:797–804. doi: 10.1016/0197-4580(95)00092-s. [DOI] [PubMed] [Google Scholar]
- 119.Mizoguchi A, Arakawa M, Masutani M, et al. Localization of smg p25A/rab3A p25, a small GTP-binding protein, at the active zone of the rat neuromuscular junction. Biochem Biophys Res Commun. 1992;186:1345–52. doi: 10.1016/s0006-291x(05)81554-2. [DOI] [PubMed] [Google Scholar]
- 120.Wisniewski HM, Wegiel J, Wang KC, et al. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84:117–27. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 121.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–7. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 122.Edison P, Archer HA, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–9. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 123.D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–83. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 124.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–32. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 126.Combs CK. Inflammation and microglia actions in Alzheimer’s disease. J Neuroimmune Pharmacol. 2009;4:380–8. doi: 10.1007/s11481-009-9165-3. [DOI] [PubMed] [Google Scholar]
- 127.Heyes MP, Saito K, Major EO, et al. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from L-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain. 1993;116:1425–50. doi: 10.1093/brain/116.6.1425. [DOI] [PubMed] [Google Scholar]
- 128.Naritsin DB, Saito K, Markey SP, et al. Metabolism of L-tryptophan to kynurenate and quinolinate in the central nervous system: effects of 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. J Neurochem. 1995;65:2217–26. doi: 10.1046/j.1471-4159.1995.65052217.x. [DOI] [PubMed] [Google Scholar]
- 129.Heyes MP, Achim CL, Wiley CA, et al. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–7. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Espey MG, Chernyshev ON, Reinhard JF, Jr, et al. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8:431–4. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- 131.Guillemin GJ, Smith DG, Smythe GA, et al. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol. 2003;527:105–12. doi: 10.1007/978-1-4615-0135-0_12. [DOI] [PubMed] [Google Scholar]
- 132.Walker DG, Link J, Lue LF, et al. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 133.Gan L, Ye S, Chu A, et al. Identification of cathepsin B as a mediator of neuronal death induced by Abeta-activated microglial cells using a functional genomics approach. J Biol Chem. 2004;279:5565–72. doi: 10.1074/jbc.M306183200. [DOI] [PubMed] [Google Scholar]
- 134.Yamada A, Akimoto H, Kagawa S, et al. Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1–42: implications for the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;110:791–800. doi: 10.1111/j.1471-4159.2009.06175.x. [DOI] [PubMed] [Google Scholar]
- 135.Guillemin GJ, Smythe G, Takikawa O, et al. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005;49:15–23. doi: 10.1002/glia.20090. [DOI] [PubMed] [Google Scholar]
- 136.Ting KK, Brew BJ, Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer’s disease. J Neuroinflammation. 2009;6:36–49. doi: 10.1186/1742-2094-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dihne M, Block F, Korr H, et al. Time course of glial proliferation and glial apoptosis following excitotoxic CNS injury. Brain Res. 2001;902:178–89. doi: 10.1016/s0006-8993(01)02378-2. [DOI] [PubMed] [Google Scholar]
- 138.Hanbury R, Charles V, Chen EY, et al. Excitotoxic and metabolic damage to the rodent striatum: role of the P75 neurotrophin receptor and glial progenitors. J Comp Neurol. 2002;444:291–305. doi: 10.1002/cne.10104. [DOI] [PubMed] [Google Scholar]
- 139.Guillemin GJ, Croitoru-Lamoury J, Dormont D, et al. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia. 2003;41:371–81. doi: 10.1002/glia.10175. [DOI] [PubMed] [Google Scholar]
- 140.Schwarcz R. The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol. 2004;4:12–7. doi: 10.1016/j.coph.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 141.Fukui S, Schwarcz R, Rapoport SI, et al. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–17. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
- 142.Wu HQ, Lee SC, Scharfman HE, et al. L-4-chlorokynurenine attenuates kainate-induced seizures and lesions in the rat. Exp Neurol. 2002;177:222–32. doi: 10.1006/exnr.2002.7971. [DOI] [PubMed] [Google Scholar]
- 143.Guidetti P, Wu HQ, Schwarcz R. In situ produced 7-chlorokynurenate provides protection against quinolinate- and malonate-induced neurotoxicity in the rat striatum. Exp Neurol. 2000;163:123–30. doi: 10.1006/exnr.1999.7284. [DOI] [PubMed] [Google Scholar]
- 144.Wu HQ, Salituro FG, Schwarcz R. Enzyme-catalyzed production of the neuroprotective NMDA receptor antagonist 7-chlorokynurenic acid in the rat brain in vivo. Eur J Pharmacol. 1997;319:13–20. doi: 10.1016/s0014-2999(96)00829-1. [DOI] [PubMed] [Google Scholar]
- 145.Fulop F, Szatmari I, Vamos E, et al. Syntheses, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr Med Chem. 2009;16:4828–42. doi: 10.2174/092986709789909602. [DOI] [PubMed] [Google Scholar]
- 146.Marosi M, Nagy D, Farkas T, et al. A novel kynurenic acid analogue: a comparison with kynurenic acid. An in vitro electrophysiological study. J Neural Transm. 2010;117:183–8. doi: 10.1007/s00702-009-0346-2. [DOI] [PubMed] [Google Scholar]
- 147.Miranda AF, Boegman RJ, Beninger RJ, et al. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience. 1997;78:967–75. doi: 10.1016/s0306-4522(96)00655-0. [DOI] [PubMed] [Google Scholar]
- 148.Harris CA, Miranda AF, Tanguay JJ, et al. Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol. 1998;124:391–9. doi: 10.1038/sj.bjp.0701834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Giorgini F, Guidetti P, Nguyen Q, et al. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–31. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Amori L, Guidetti P, Pellicciari R, et al. On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo. J Neurochem. 2009;109:316–25. doi: 10.1111/j.1471-4159.2009.05893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]