

Abstract
Atherosclerosis is a chronic inflammatory disease occurring within the artery wall and is an underlying cause of cardiovascular complications, including myocardial infarction, stroke and peripheral vascular disease. Its pathogenesis involves many immune cell types with a well accepted role for monocyte/macrophages. Cholesterol-loaded macrophages are a characteristic feature of plaques and are major players in all stages of plaque development. As well as modulating lipid metabolism, macrophages secrete inflammatory cytokines, chemokines and reactive oxygen and nitrogen species that drive pathogenesis. They also produce proteases and tissue factor that contribute to plaque rupture and thrombosis. Macrophages are however heterogeneous cells and when appropriately activated, they phagocytose cytotoxic lipoproteins, clear apoptotic bodies, secrete anti-inflammatory cytokines and synthesize matrix repair proteins that stabilize vulnerable plaques. Pharmacological modulation of macrophage activity therefore represents a potential therapeutic strategy for atherosclerosis. The aim of this review is to provide an overview of the current understanding of the different macrophage subsets and their monocyte precursors, and, the implications of these subsets for atherosclerosis. This will present a foundation for highlighting novel opportunities to exploit the heterogeneity of macrophages as important diagnostic and therapeutic targets for atherosclerosis and its associated diseases.
Keywords: monocyte, macrophage, M1, M2, activation, atherosclerosis, inflammation, immunomodulation, imaging, plaque stability
Introduction
Atherosclerosis is now well recognized as a chronic inflammatory disorder involving many immune cell types [1]. In the 1960s macrophages were the first inflammatory cells to be identified within atherosclerotic plaques [2]. Macrophages are a key feature of all stages of atherogenesis where they have a very significant impact on lesion progression. As part of their innate immune role, macrophages infiltrate developing lesions and respond to and phagocytose oxidized low-density lipoprotein (oxLDL). This results in the secretion of cytokines, chemokines and toxic oxygen and nitrogen radicals that not only direct and amplify the local immune response but also leads to tissue injury. Moreover macrophages cause plaque destabilization, rupture and thrombosis, highlighting their destructive role [3]. However, macrophages are heterogeneous cells and can also develop functions that facilitate tissue repair, remodelling and restoration of normal tissue homeostasis [4, 5]. Importantly, macrophages as professional phagocytes remove senescent and apoptotic cells as well as cytotoxic oxidized lipids from developing lesions. Macrophages also secrete anti-inflammatory mediators to down-regulate inflammatory responses within the lesion; the consequences of removing all macrophages are therefore likely to be significant. Indeed, the functions of macrophages in developing and advanced plaques are dictated by several factors including the subset of monocytes from which they derive, how they integrate signals from their local tissue environment and the exact stage in the disease process in which they infiltrate.
The marked infiltrate of macrophages within atherosclerotic plaques and the contradictory functions macrophages can develop have significant clinical implications. The development of therapies that switch off macrophage cytotoxic and plaque destabilizing functions and exploit their natural reparative properties would prove a powerful new approach to curtail macrophage-mediated injury and enhance plaque stability. Moreover, macrophage-rich areas are more frequently associated with unstable plaques. Detection of macrophage-rich areas using new imaging tools and metabolic labelling could therefore provide non-invasive methods for estimating plaque stability, permitting earlier detection of high-risk patients and more effective assessment of new therapies. The purpose of this review is firstly, to outline the different subsets of monocyte and macrophages and their activation and function; secondly, to discuss the role of macrophage subsets in atherogenesis and the heterogeneity of plaque macrophages and thirdly, to examine the potential of pharmacological modulation and non-invasive imaging of plaque macrophages to provide novel and better therapeutic, diagnostic and more importantly preventative regimes for cardiovascular disease.
Current concepts in monocyte heterogeneity
An early event in atherosclerosis is monocyte recruitment to the activated endothelium and subsequent diapedesis in response to chemoattractive stimuli. These events have been extensively reviewed elsewhere [6–8]. Circulating monocytes originate from a common monoblast precursor in the bone marrow and are rapidly released into the circulation, especially in response to inflammatory stimuli. Upon infiltrating tissue, monocytes differentiate into macrophages, dendritic cells or, in some cases, osteoclasts [9]. The mechanisms controlling monocyte emigration from bone marrow is an area of active investigation but the chemokine receptor CCR2 and its ligand CCL7 are thought to be important. Circulating monocytes are heterogeneous in that they express distinctive chemokine receptors and have different migratory and differentiation properties. At least two blood monocyte populations, based on expression of chemokine and other receptors, adhesion molecules and differences in size and granularity, have been classified in human beings and rodents [10]. Different subsets use different chemokine receptors for plaque entry [reviewed in 11]. In human beings, the majority of monocytes (about 90%) express high levels of CD14, low levels of the IgG receptor CD16 (CD14++CD16−), high levels of CCR2 and low levels of the chemokine receptor CX3CR1 and CCR5 [12–14]. These cells are larger (18.4 μm) [15] and phagocytic, and demonstrate low pro-inflammatory cytokine production and high interleukin (IL)-10 when activated in vitro with lipopolysaccharide (LPS). A recent genomic study concluded that that in patients with coronary heart disease these monocytes express a more anti-inflammatory profile [16]. The remainder are CD14+CD16+ or CD14dimCD16+. These CD16+ monocytes are smaller (13.8 μm), express high levels of CX3CR1 and CCR5 and lower levels of CCR2 [14] and scavenger receptor type-A [17]. Their numbers are reported to be increased in patients with acute inflammation [18], obesity [19], hypercholesterolemia [20] and coronary heart disease [21]. CD16+ monocyte numbers are decreased in the blood after treatment with glucocorticoids [22], again suggesting that their abundance is related to inflammatory stimuli. Interestingly, however, in one study the percentage of CD16+ monocytes was reduced in high-risk coronary artery disease patients whereas the percentage of CD14++CD16− monocytes increased with the number of risk factors [23]. Information on the change in numbers or proportions of the circulating CD14++, CD16− subset in other human pathologies is lacking.
In mice, two equally represented subsets are divided into Ly-6Chigh CCR2+ CX3CR1low and Ly6ClowCCR2−CX3CR1high and these probably represent different stages of a continuous maturation pathway [24]. Ly6Chigh monocytes are short lived in the circulation and rapidly infiltrate inflamed sites (e.g. atherosclerotic plaques) where they differentiate into inflammatory macrophages [25–27]. They are phenotypic equivalents to human CD14++CD16−, CCR2+ CX3CR1low monocytes [28]. Although the Ly-6Chigh monocytes are phenotypic equivalents to human CD14++, CD16−, CCR2highCX3CR1low monocytes there is a discrepancy in the functional properties of the respective subtypes of cells. This is important, leading to caution in directly extrapolating any of the findings from mouse to human beings. Numbers of Ly6Chigh monocytes increase in response to inflammation or hypercholesterolemia [25, 26]. In contrast, the Ly6Clow monocytes persist longer in the circulation and can differentiate into tissue resident macrophages or dendritic cells. Under steady state conditions, Ly6Clow monocytes patrol the endothelium without extravasation. These monocytes play important functions in scavenging oxidized lipids, dead cells and potential pathogens. In response to endothelial injury, Ly6Clow monocytes rapidly extravasate and develop pro-inflammatory properties. Some monocyte subsets therefore heighten disease while others attenuate it. Consequently, studying the kinetics of monocyte recruitment into tissue would represent an effective prognostic tool. A recent study has devised methods using magnetic nano-sensors to profile and quantify peripheral monocyte subsets (e.g. phagocytic activity) and the fluctuations that occur in patients with atherosclerosis [29], highlighting a potential screening method for ‘at risk’ patients.
Monocytes migrating into inflamed sub-endothelial tissue differentiate into macrophages and become activated to exhibit distinct gene expression patterns and functions [30–32]. It is still debated whether a transition from a distinct monocyte subpopulation to a specific macrophage type exists or whether macrophage phenotypes within plaques are influenced by the heterogeneity of circulating monocytes [33]. Studies in inflamed murine kidney have demonstrated, however, that recruitment of a single lineage of Ly6Chigh monocytes differentiate into three populations of kidney macrophages, including a pro-fibrotic Ly6Clow population. This suggests macrophage heterogeneity is complex and not due to a simple transition of specific monocyte subtypes [34].
Pro-atherogenic role of plaque macrophages
Activated macrophages up-regulate their expression of both scavenger receptors and toll-like receptors (TLR) to enhance phagocytosis and remove harmful substances (e.g. dying cells or oxLDL) as part of the normal inflammatory response to sub-intimal pathogenic lipoproteins [35]. Usually, inflammation is self-limiting and homeostasis is restored. If engulfed cholesterol cannot exit the cell then macrophages become lipid-laden foam cells. Foam cells secrete additional extracellular matrix (ECM) components that further endorse lipoprotein retention within the sub-endothelium. CD36 signalling via oxLDL promotes adhesion and trapping of foam cell macrophages [36], provoking an inflammatory response at the local site. Foam cells within the developing fatty streak produce chemokines, cytokines, proteases, growth factors, bioreactive lipids and angiogenic factors that perpetuate the inflammatory process. This also results in migration of smooth muscle cells from the media to the intima of the artery. Subsequent proliferation of smooth muscle cells in response to foam cell secreted mediators contributes to the enlarged fibro-fatty plaque and fibrous cap. Moreover, activated macrophages in developing plaques produce reactive oxygen species (ROS), which cause cell apoptosis and lipoprotein oxidation [37]. OxLDL engulfed by macrophages is digested, processed and peptide antigens presented to T lymphocytes, activating the adaptive immune system and initiating a cytotoxic T helper type 1 (Th1) immune response. The interferon-γ (IFN-γ) produced by Th1, as well as amplifying inflammation, causes induction of pro-coagulant tissue factor expression in macrophages.
More advanced plaques contain large numbers of macrophages and can develop into complex atherosclerotic lesions that are classified as stable or unstable [38]. Stable plaques are characterized by a thick fibrous cap overlying a plaque that does not contain a cholesterol-rich necrotic core. Unstable plaques are lipid-filled necrotic core, have a thin fibrous cap and a high ratio of macrophages to smooth muscle cells [39]. Here, macrophages secrete proteolytic enzymes that degrade matrix components, as well as the protective fibrous cap, leading to plaque destabilization and increased risk of plaque rupture and thrombosis [40]. Indeed, the number of macrophages present within plaques correlates with plaque stability and it is well known that plaques tend to rupture at sites of increased macrophage content [41]. Lesional macrophages also contribute to death of surrounding cells by release of toxic oxygen and nitrogen radicals and via Fas-Fas ligand interactions [42]. Large lipid-laden cells undergoing apoptosis present a difficult target for phagocytosis and failed apoptosis results in necrotic death. This leads to accumulation of insoluble lipids and other cellular components, causing increased pro-inflammatory responses. Pro-inflammatory mediators cause further apoptosis of smooth muscle cells, endothelial cells and leucocytes that account for much of the plaque-disrupting core of vulnerable plaques. Uncleared apoptotic cells shed plasma membrane microparticles that stimulate thrombosis [43]. Advanced plaque macrophages also contain large quantities of tissue factor which are responsible for much of the pro-coagulant and pro-thrombotic activity following plaque rupture [44].
Anti-atherogenic role of plaque macrophages
The highly pathogenic role of macrophages in atherosclerosis suggests their removal from vulnerable, rupture prone plaques would be beneficial. Indeed, animal studies have demonstrated that macrophage depletion in developing plaques provides resistance to atherosclerosis [45, 46]. However, macrophages also have important anti-inflammatory, reparative effects and importantly are essential for the normal inflammatory response protecting the host from infection. Their depletion is therefore not a strong therapeutic option. In the early stages of atherogenesis, macrophages scavenge cytotoxic lipoproteins and senescent and apoptotic cells preventing accumulation and cytotoxicity within developing lesions. Efficient clearance of apoptotic cells is essential for preventing secondary necrosis and also triggers an anti-inflammatory response through induction of transforming growth factor (TGF)-β, IL-10 and other anti-inflammatory cytokines [47] to down-regulate ongoing inflammation. In advanced plaques, appropriately activated macrophages promote tissue repair by promoting ECM synthesis and smooth muscle cell proliferation that enhance plaque stability. Macrophages are also necessary for resolution of injury that occurs within the plaque or at sites of rupture. Here they secrete anti-inflammatory cytokines, matrix repair proteins and angiogenic factors, and clear tissue debris. Data from other tissues including lung [48], skin [49], heart [50] and kidney [51, 52] demonstrate the importance of macrophages for repair processes, and data are emerging that shows a similar reparative role in atherosclerosis. Intraplaque haemorrhage accelerates atherosclerosis and contributes to lesion development and destabilization. Normally, macrophages scavenge haemoglobin–haptoglobin complexes via CD163, and this process provokes the secretion of the anti-inflammatory atheroprotective mediators IL-10 and heme oxygenase-1 [53]. Finally, macrophages secrete u-PA and t-PA; these plasminogen activators activate the serine protease plasmin, which plays a key role in fibrinolysis of blood clots [54]. The role of monocytes/ macrophages in atherosclerosis is therefore critically dependent on the exact stage of plaque development. A similar phenomenon is described in liver injury where functionally distinct subpopulations of macrophages were shown to exist in the same tissue favouring ECM accumulation during ongoing injury but enhancing matrix degradation during recovery [55]. Regulating macrophage function is therefore a fundamental approach for atherosclerosis treatment but each step in atherogenesis creates a new microenvironment which in turn influences apoptotic, cytotoxic and phagocytotic processes differently. Consequently, strategies targeting macrophage functions may need to be tailored to the stage in the disease process.
Macrophage heterogeneity and in vitro classification
The extent of macrophage heterogeneity and cues that induce polarization in atherosclerotic plaques are still being unravelled. Activation studies in vitro have, however, phenotypically and functionally characterized several polarized macrophage subtypes. Classical or M1a macrophage activation requires priming by IFN-γ together with a second activating signal such as a microbial product (e.g. LPS, CpG-DNA) ligating TLR, or a pro-inflammatory cytokine such as TNF-α or IL-1 [5, 56, 57]. M1a macrophages produce copious amounts of reactive oxygen and nitrogen intermediates and release chemokines and pro-inflammatory cytokines including TNF-α and IL-6 that direct endothelial injury. M1a-activated macrophages mediate resistance against intracellular parasites and tumours but also elicit tissue damage. They express high levels of MHC class II and co-stimulatory molecules CD80/86, which increases their capacity as antigen presenting cells, and IL-12 that supports Th1-driven immune responses [51]. They also secrete a variety of tissue-degrading proteases. Functionally, M1a macrophages exhibit enhanced phagocytic capacity and bacterial (and cell) killing mechanisms [5]. Innate or M1b activated macrophages are consequences of pathogen-associated molecular patterns, e.g. LPS, CpG-DNA or flagellin engaging pattern recognition receptors [58], such as TLRs and nucleotide oligomerization domain receptors [59]. They display a phenotype similar to M1a activated cells but do not exhibit enhanced phagocytosis and they secrete low levels of IL-12.
Alternative or M2-activated macrophages, in contrast to M1 macrophages, kill intracellular pathogens poorly and are anti-inflammatory, having a critical role in resolution of injury [5, 57, 60]. The precise properties of M2 macrophages vary depending on the activating conditions and they have been divided into M2a, M2b and M2c subtypes [5]. M2a activation occurs when macrophages are stimulated by IL-4 or IL-13, typically associated with Th2 responses. They provoke increased deposition of ECM, e.g. by increased fibronectin and TGF-β production [57] and consequently have also been referred to as ‘tissue reparative’ or ‘wound healing’ macrophages [33]. M2a macrophages are strongly associated with extracellular parasite infections, allergy, humoural immunity and fibrosis. They show increased expression of C-type lectins including dectin-1 and macrophage mannose receptor, as well as both the IL-1 receptor antagonist (IL-1ra) and the decoy IL-1β type II receptor, which inhibits inflammation elicited by IL-1. Arginase expression is enhanced in M2a activated mouse (not human) macrophages, and in contrast to M1 activated macrophages, the expression of inducible nitric oxide synthase (iNOS) and production of nitric oxide is reduced [5]. M2b macrophages develop after exposure to LPS, CD40 ligand or IL-1β in cells primed by IgG immune complexes that are recognized by Fc gamma receptors [60, 61]. They secrete enhanced IL-10 and decreased IL-12 but produce the pro-inflammatory cytokines TNF-α, IL-6 and IL-1. M2c macrophages similarly produce high levels of IL-10 and thus can down-regulate pro-inflammatory cytokines to limit inflammation. They are induced by IL-10, TGF-β or glucocortocoids [5] and have increased debris scavenging activity and a pro-healing functional program. M2b and M2c have recently been described as ‘regulatory macrophages’ due to their high secretion of IL-10 [33]. Apoptotic cell uptake induces another type of anti-inflammatory macrophage outwith the M1/2 classification [62, 63] although these have been referred to as regulatory cells due to production of IL-10 [33]. These are characterized by transient expression of small amounts of pro-inflammatory cytokines and chemokines, followed by later and sustained TGF-β and PGE2 synthesis and other anti-inflammatory lipid mediators that down-regulate inflammatory responses and dominate resolution of tissue injury [63].
Macrophage heterogeneity exists in tissue in vivo although the phenotype observed may not directly correspond to simplistic phenotypes generated in vitro. In vivo, macrophages are exposed to a complex microenvironment generated from several cell types. The way infiltrating macrophages are activated is dictated by a multitude of signals impinging on their receptors (see Fig. 1). These signals change during the evolution of the underlying disease process and have the capacity to influence the outcome of the disease. Other less-defined macrophage subtypes induced by metabolic factors are liable to be present in atherosclerotic plaques. For example, uptake of oxidized or acetylated LDL, as a model of foam cell formation increases the expression of commonly used M1 markers and transcription factors such as iNOS, metalloproteinase-1 and NF-κB [64]. Fatty acids present in developing plaques activate an inflammatory programme; saturated fatty acids are robustly pro-inflammatory, polyunstaturated fatty acids are weakly inflammatory or neutral while omega-3 unsaturated fatty acids induce more anti-inflammatory functions [65]. Moreover, under appropriate conditions, infiltrating monocytes differentiate into osteoclast-like cells within plaques and promote lesion calcification [9], whereas macrophages exposed to particulate calcium mineral have been reported to undergo osteoclastic differentiation [66].
Fig 1.
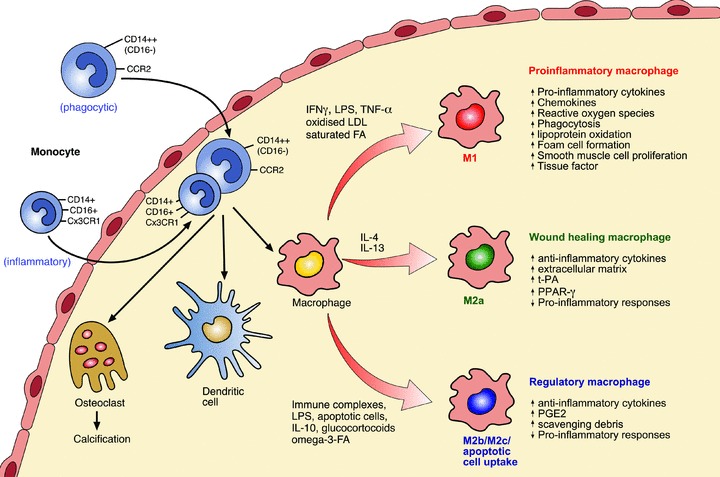
Activation of macrophages in developing and advanced human atherosclerotic plaques. Monocytes (CD14++CD16− or CD16+ subsets) enter the developing atheroma guided by adhesion molecules and chemokines and once infiltrated can differentiate into macrophages, dendritic cells or osteoclasts. Differences in monocyte subsets also exist for mice (see text for details). In response to microenvironmental stimuli, macrophages become activated to develop either atherogenic or atheroprotective functions. The factors controlling entry of the different monocyte subsets into plaque and whether specific monocyte subsets differentiate into distinct functional macrophage subsets requires further investigation. M1 macrophages are microbicidal and involved in host defence. They are pro-inflammatory and cause tissue injury and promote lesion development as well as enhancing plaque vulnerability. M2a macrophages are involved in tissue repair. They are anti-inflammatory and can stabilize vulnerable plaques. M2b (e.g. immune complex/LPS activated) and M2c macrophages, as well as macrophages that take up apoptotic cells in the presence of pro-inflammatory stimuli, are immunoregulatory and anti-inflammatory and stabilize or even regress atherosclerotic plaques, see text for further details. Abbreviations: IFN-γ, interferon-γ; LPS, lipopolysaccharide; LDL, low-density lipoprotein; FA fatty acid; t-PA, tissue plasminogen activator; PPAR-γ, peroxisome proliferator activated receptor-gamma; PGE2, prostaglandin-2.
Macrophage heterogeneity within atherosclerotic plaques
Studies in several rodent models have shown evidence of macrophage heterogeneity within the atherosclerotic plaque [26, 27, 67]. Heterogeneity amongst macrophages infiltrating human atherosclerotic lesions has also been recognized for many years [68] although it has been difficult to distinguish the specific activation phenotypes at the single cell level. This is due to lack of markers that clearly delineate different activation states in plaque tissue although MCP-1 has been used as an M1 marker and macrophage mannose receptor and CD163 as M2 markers. The differences in phenotype of plaque macrophages are reported to be due to the time of residence within the plaque [68], the initial phenotype of infiltrating monocytes, or site of location within the plaque (inflamed/non-inflamed) [69]. Waldo et al. have characterized CD14+ macrophages (CD68+) within active plaques whereas CD14− macrophages were abundant in disease-free regions [69]. They propose this could be due to dissimilar activating environments but different CD14 expressions could also be due to time of residence within plaques given CD14 is lost on maturation of monocytes. Interestingly, macrophages expressing low levels of CD14 express five times more peroxisome proliferator activated receptor-gamma (PPAR-γ), a nuclear hormone receptor, than macrophages expressing high levels of CD14, and PPAR-γ is associated with an M2-anti-inflammatory/wound healing phenotype [70]. The haptoglobin/haemoglobin scavenger receptor CD163 has also been associated with an anti-inflammatory macrophage. In a separate study, scattered macrophages within diffuse intimal lesions showed strong positivity for CD163 whereas foamy plaque macrophages were weakly positive [71]. Distinct populations of CD163+ macrophages have also been identified in haemorrhaged atherosclerotic plaques and importantly these had low levels of human leucocyte antigen-DR and were unlike classical lipid core macrophages [53]. These were thought to suppress the impact of haemorrhage on atherosclerotic progression.
Bouhlel et al. also provide evidence for macrophage heterogeneity in atherosclerosis since both M1 and M2 macrophage markers (MCP-1 and macrophage mannose receptor, respectively) were detectable in human atherosclerotic lesions, and these markers were present in distinct locations [70]. Interestingly, they showed that PPAR-γ expression correlated with the expression of M2 activation markers and PPAR-γ activation skewed human monocytes towards an anti-inflammatory phenotype [70] (see also section ‘Pharmacological modulation of macrophage function’). Taken together, these observations provide evidence for the presence of several macrophage phenotypes within developing and advanced plaques and their complexity in promoting, as well as inhibiting, plaque progression. Phenotyping isolated plaque macrophages through several ‘omics’ approaches will undoubtedly provide a more detailed picture of the complex functional role of individual subtypes in the progression and stabilization of the plaque.
Macrophage heterogeneity in obesity and obesity-associated disorders
Obesity is well known to contribute strongly to the risk of atherosclerosis as well as insulin resistance, type II diabetes and the metabolic syndrome, all of which are associated with atherothrombotic vascular disease [72, 73]. Macrophages are also important players in the development of obesity and are coupled with low grade inflammation in adipose tissue [74]. High numbers of circulating CD16+ monocytes are associated with obesity [19] and macrophages are present in much higher numbers in adipose tissue of obese patients than in that of lean patients [75]. Both M1 and M2 populations of macrophages have been identified in rodent and human adipose tissue with M2-type macrophages as the main population in tissue from lean rodents [76] and human beings [77]. Importantly, Lumberg et al. reported that high fat diet-induced obesity caused a switch in adipose tissue macrophages from an M2 anti-inflammatory state to M1 pro-inflammatory cells [76] favouring inflammation. Further studies by Fujisaka et al. confirmed that insulin resistance is associated with both increased numbers of M1 macrophages and an increased M1/M2 ratio in adipose tissue [78]. On the other hand, weight reduction promotes the occurrence of M2-like macrophages in adipose tissue of obese patients [79, 80]. The nuclear receptor PPAR-γ is required for maturation of M2 macrophages and deletion of the PPAR-γ gene in myeloid cells results in a shift of macrophage differentiation towards M1, predisposing mice to diet-induced obesity, insulin resistance and glucose intolerance [81, 82]. Agonizing PPAR-γ could therefore promote M2 polarization in adipose tissue protecting from insulin resistance [83]. Together, these studies suggest, as is the case for atherosclerosis, M1 macrophages are pathogenic and polarization of macrophages towards an M2-status has potential as a new strategy for treatment of obesity-induced metabolic disorders.
Macrophage modulation by pathogens and tumours
Several pathogens have developed systems to dampen the innate response and survive the hostile environment produced by M1 macrophages. This is achieved by inhibiting pro-inflammatory intracellular pathways and/or enhancing anti-inflammatory responses (M1- to M2-macrophage switch). Moreover, to avoid macrophage-mediated destruction, pathogens can interfere with receptor-mediated recognition, phagocytosis and trafficking of bacteria to degradative lysosomes (reviewed in [84, 85]). Tumours, like pathogens, can skew macrophage functions by reducing their ability to produce IL-12 while enhancing autocrine IL-10 production [86] and by decreasing their antigen presenting ability. Tumours also cause enhanced production of matrix-degrading proteases by macrophages that favour metastasis, and they induce enhanced angiogenic factors that promote vascular growth [87]. The capacity of tumours and pathogens to subvert macrophage function from M1 to M2 to avoid immunosurveillance and promote their own survival provides a clear precedent for altering macrophage activation to favour resolution of plaque-induced tissue injury and enhance plaque stability. A key challenge is to devise methods to therapeutically switch M1 to M2 macrophages within developing or unstable plaques (or adipose tissue of obese patients) as effectively as pathogens and tumours.
Pharmacological modulation of macrophage function
Many available standard therapies for atherosclerotic vascular disease (e.g. angiotensin converting enzyme inhibitors, β-blockers, aspirin, corticosteroids) influence general immune responses but these lack specific macrophage targeting, and are usually only mild modifiers of macrophage activity [88–91]. General immunosuppressive therapies also leave patients vulnerable to infection and cancer and highly effective therapies targeting macrophage activation are thus more desirable. Several common pharmacological agents have already been proposed to modulate macrophage activity for prevention and treatment of inflammatory-related diseases, including atherosclerosis.
PPAR-γ is a key factor in regulating macrophage lipid metabolism and inflammatory responses. It is induced by natural ligands such as prostaglandins and some pharmacologicals including anti-diabetic thiazolidinediones (TZD), which slow progression of atherosclerosis. As discussed previously, PPAR-γ is highly up-regulated in M2 macrophages and PPAR-γ agonists have been shown directly to induce M2-like differentiation of monocytes in vivo and in vitro[70]. Conversely, in mice, selective inactivation of macrophage PPAR-γ impairs M2 activation and exacerbated diet-induced obesity [82], suggesting that PPAR-γ agonists have therapeutic potential. However PPAR-γ activation did not switch the function of M1 macrophages to M2 in atherosclerotic lesions [70]. PPAR-γ activation by TZD also increased transcription of many genes that cause weight gain and increased LDL cholesterol [92] and modifications of this compound are required should its therapeutic potential be developed for atherosclerosis.
Liver X receptors (LXRs) are up-regulated in M2 macrophages and like PPAR-γ, exert important atheroprotective effects by regulating cholesterol metabolism and M1 macrophage-induced inflammatory gene responses [93]. In experimental models macrophage-specific loss of LXRs resulted in a marked increase in lesion size [94] while LXR agonists reduced the size of pre-existing plaques and this reduction was dependent on macrophage LXR activity [95]. The exact mechanisms controlling LXR-induced macrophage responses are unknown but they are thought to up-regulate PPAR-γ, thus a switch from M1 to M2 is likely. However, LXR activation induces lipogenesis and hypertriglyceridemia and new LXR ligands need to be designed without these undesirable side effects [96].
Statins are effective cholesterol-lowering agents that also dampen immune responses through inhibition of macrophage inflammatory activity [97], again possibly via PPARs [98]. In one study, statin treatment slightly increased the number of plaque macrophages yet other markers of inflammation were reduced with the authors concluding that it is not the presence of macrophages but activation and subsequent protease and cytokine release that is attenuated by statin use [98]. Interestingly, the orally available drug FTY720, a mimetic of sphingosine-1-phosphate (a biologically active sphingolipid) has been shown to increase the proportion of M2 macrophages in atherosclerotic lesions and slow lesion progression in mice [99], indicating significant potential for therapy.
Pharmacological agents that selectively target macrophages are not widely available. Small interfering RNA (siRNA) therapeutics offers a potential new class of pharmaceutical drug to target inflammatory genes and signalling pathways that skew macrophage function [100]. Our group, for example, has shown that modulating expression of the suppressor of cytokine signalling proteins by siRNA efficiently switches macrophage function from M1 to M2 [32]. New methods have been devised for delivering siRNA specifically to macrophages [101] and an exciting study by Aouadi et al. [102] has shown that targeting macrophage mitogen-activated protein kinase-4 (using siRNA) via an oral delivery system suppresses systemic inflammation. It will be fascinating to see whether this approach could open up a novel avenue to manipulate macrophage function to treat patients suffering from atherosclerosis and related diseases.
Macrophages as a target to non-invasively image vulnerable plaques
The abundance of macrophages in unstable plaques makes their visualization very relevant for both diagnostic purposes and for the evaluation of therapeutic interventions [103]. This could present an important adjunct to morphological markers for the detection of high-risk plaques using methods such as computed tomography. Several non-invasive imaging techniques have been described for macrophage detection in tissue, including atherosclerotic plaques, and advances in these suggest they could soon provide an excellent new diagnostic option.
A promising approach for imaging vulnerable plaques involves superparamagnetic iron oxide particles (SPIOS) and ultra SPIOS that are rapidly taken up by macrophages via phagocytosis. When clumped within the phagolysosomes, these nanoparticles produce a strong MRI signal. Their use permits the direct visualization of plaque macrophage in models of atherosclerosis and in patients with severe carotid disease [104, 105]. It is thought that uptake only occurs in recently migrated cells and accumulation is indicative of acute ongoing inflammation that is present in rupture-prone or already ruptured plaques, thus providing a good means to assess plaque activity [106]. Inflammatory cytokines within the inflamed lesion enhance phagocytic uptake of USPIOs [107] and after uptake there is the added advantage in that production of TNF-α is decreased and anti-inflammatory IL-10 is enhanced. [108]. New MRI probes for imaging plaque macrophages include macrophage scavenger receptor-targeted immunomicelles loaded with gadolinium [109]. Iodine-containing contrast agents have also labelled lesional macrophages for detection with computed tomography [110].
Nuclear agents, e.g.64Cu-labelled nanoparticles, are efficiently taken up by macrophages and detected by positron emission tomography (PET) [111]. Radioactive fluorodeoxyglucose (18F-FDG) competes with glucose for uptake into activated macrophages and is proportional to macrophage density when imaged by PET [112, 113]. Furthermore, increased 18F-FDG uptake occurs in symptomatic carotid plaques compared to the asymptomatic contra-lateral carotid artery [95]. However, not all atherosclerotic plaques showed high 18F-FDG uptake [114] reflecting the fact that some patients were receiving statin therapy which can alter macrophage metabolic activity and 18F-FDG uptake in human aortic and carotid arteries [115]. Another PET-imaged biomarker successfully used to detect plaque macrophage activity is the peripheral benzodiazepine receptor ligand, a mitochondrial protein that is highly expressed in activated macrophages [116]. New technologies are being developed to selectively image macrophage subsets, e.g. M2, [117, 118] and whether this is clinically translatable for more efficiently monitoring plaque vulnerability will be an exciting area of future research.
Summary
It is now clear that monocytes and macrophages are heterogeneous and that their subsets have either harmful or beneficial functions in atherogenesis. Clinical trials for anti-atherogenic drugs should not only determine the effect of therapy on macrophage numbers but also their activation status and whether macrophages can be skewed to a more reparative phenotype. Elucidating the exact roles of macrophages localizing within lesions and the molecular cues that drive differential activation will undoubtedly aid in the development of novel strategies for early diagnosis, stabilization or even regression of vulnerable atherosclerotic plaques, which in the long term will effectively help reduce the burden of cardiovascular disease on modern societies.
Acknowledgments
This work was supported by the Medical Research Council, (grant no. 74804), NHS Grampian Endowments Research Trust (grant 09/02) and by the Cunningham Trust (grant no. ACC/KWF/CT08/03).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Hansson GK. Atherosclerosis–an immune disease: the Anitschkov Lecture 2007. Atherosclerosis. 2009;1:2–10. doi: 10.1016/j.atherosclerosis.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Gerrity RG, Naito HK. Ultrastructural identification of monocyte-derived foam cells in fatty streak lesions. Artery. 1980;3:208–14. [PubMed] [Google Scholar]
- 3.Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;6:309–15. doi: 10.1038/ncpcardio0195. [DOI] [PubMed] [Google Scholar]
- 4.Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci. 2003;104:27–38. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 5.Martinez FO, Sica A, Mantovani A, et al. Macrophage activation and polarisation. Frontiers in Bioscience. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 6.Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;6:228–32. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koenen RR, Weber C. Therapeutic targeting of chemokine interactions in atherosclerosis. Nat Rev Drug Discov. 2010;2:141–53. doi: 10.1038/nrd3048. [DOI] [PubMed] [Google Scholar]
- 8.Galkina E, Ley K. Leukocyte influx in atherosclerosis. Curr Drug Targets. 2007;8:1239–48. doi: 10.2174/138945007783220650. [DOI] [PubMed] [Google Scholar]
- 9.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- 10.Strauss-Ayali D, Conrad SM, Mosser DM. Monocyte subpopulations and their differentiation patterns during infection. J Leukoc Biol. 2007;2:244–52. doi: 10.1189/jlb.0307191. [DOI] [PubMed] [Google Scholar]
- 11.Gautier EL, Jakubzick C, Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;10:1412–8. doi: 10.1161/ATVBAHA.108.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 13.Ziegler-Heitbrock HW. Definition of human blood monocytes. J Leukoc Biol. 2000;5:603–6. doi: 10.1002/jlb.67.5.603. [DOI] [PubMed] [Google Scholar]
- 14.Weber C, Belge KU, Von Hundelshausen P, et al. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 15.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 16.Schirmer SH, Fledderus JO, Van Der Laan AM, et al. Suppression of inflammatory signaling in monocytes from patients with coronary artery disease. J Mol Cell Cardiol. 2009;46:177–85. doi: 10.1016/j.yjmcc.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Draude G, Von Hundelshausen P, Frankenberger M, et al. Distinct scavenger receptor expression and function in the human CD14(+)/CD16(+) monocyte subset. Am J Physiol. 1999;276:H1144–9. doi: 10.1152/ajpheart.1999.276.4.H1144. [DOI] [PubMed] [Google Scholar]
- 18.Mizuno K, Toma T, Tsukiji H, et al. Selective expansion of CD16highCCR2− subpopulation of circulating monocytes with preferential production of haem oxygenase (HO)-1 in response to acute inflammation. Clin Exp Immunol. 2005;142:461–70. doi: 10.1111/j.1365-2249.2005.02932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogacev KS, Ulrich C, Blömer L, et al. Monocyte heterogeneity in obesity and subclinical atherosclerosis. Eur Heart J. 2010;3:369–76. doi: 10.1093/eurheartj/ehp308. [DOI] [PubMed] [Google Scholar]
- 20.Rothe G, Gabriel H, Kovacs E, et al. Peripheral blood mononuclear phagocyte subpopulations as cellular markers in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1996;12:1437–47. doi: 10.1161/01.atv.16.12.1437. [DOI] [PubMed] [Google Scholar]
- 21.Schlitt A, Heine GH, Blankenberg S, et al. CD14+CD16+ monocytes in coronary artery disease and their relationship to serum TNF-alpha levels. Thromb Haemost. 2004;2:419–24. doi: 10.1160/TH04-02-0095. [DOI] [PubMed] [Google Scholar]
- 22.Fingerle-Rowson G, Angstwurm M, Andreesen R, et al. Selective depletion of CD14+ CD16+ monocytes by glucocorticoid therapy. Clin Exp Immunol. 1998;112:501–6. doi: 10.1046/j.1365-2249.1998.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hristov M, Leyendecker T, Schuhmann C, et al. Circulating monocyte subsets and cardiovascular risk factors in coronary artery disease. Thromb Haemost. 2010 doi: 10.1160/TH10-01-0069. Doi: 10.1371/journal.pone.0005663. [DOI] [PubMed] [Google Scholar]
- 24.Sunderkötter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;7:4410–7. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 25.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;12:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 29.Wildgruber M, Lee H, Chudnovskiy A, et al. Monocyte subset dynamics in human atherosclerosis can be profiled with magnetic nano-sensors. PLoS One. 2009;5:e5663. doi: 10.1371/journal.pone.0005663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson HM, Minto AW, Brown PA, et al. Transforming growth factor-beta isoforms and glomerular injury in nephrotoxic nephritis. Kidney Int. 2000;57:2434–44. doi: 10.1046/j.1523-1755.2000.00102.x. [DOI] [PubMed] [Google Scholar]
- 31.Wilson HM, Barker RN, Erwig LP. Macrophages: promising targets for the treatment of Atherosclerosis. Current Vascular Pharmacology. 2009;2:234–43. doi: 10.2174/157016109787455635. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Stewart KN, Bishop E, et al. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol. 2008;180:6270–8. doi: 10.4049/jimmunol.180.9.6270. [DOI] [PubMed] [Google Scholar]
- 33.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;12:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin SL, Castaño AP, Nowlin BT, et al. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;10:6733–43. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 35.Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 36.Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119:136–45. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugiyama S, Okada Y, Sukhova GK, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–91. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:503–16. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 39.Davies MJ, Richardson PD, Woolf N, et al. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J. 1993;69:377–81. doi: 10.1136/hrt.69.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69:614–24. doi: 10.1016/j.cardiores.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Van Der Wal AC, Becker AE, Van Der Loos CM, et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. doi: 10.1161/01.cir.89.1.36. [DOI] [PubMed] [Google Scholar]
- 42.Martinet W, Kockx MM. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol. 2001;12:535–41. doi: 10.1097/00041433-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Mallat Z, Hugel B, Ohan J, et al. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999;3:348–53. doi: 10.1161/01.cir.99.3.348. [DOI] [PubMed] [Google Scholar]
- 44.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 45.Danenberg HD, Fishbein I, Gao J, et al. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599–605. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- 46.Stoneman V, Braganza D, Figg N, et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–93. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Lorenzo BH, Godoy LC, Novaes E, et al. Macrophage suppression following phagocytosis of apoptotic neutrophils is mediated by the S100A9 calcium-binding protein. Immunobiology. 2009 doi: 10.1016/j.imbio.2009.05.013. DOI: 10.1016/j.imbio.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 48.Teder P, Vandivier RW, Jiang D, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–8. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 49.Nagaoka T, Kaburagi Y, Hamaguchi Y, et al. Delayed wound healing in the absence of intercellular adhesion molecule-1 or L-selectin expression. Am J Pathol. 2000;157:237–47. doi: 10.1016/S0002-9440(10)64534-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;2:147–58. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson HM, Chettibi S, Jobin C, et al. Inhibition of macrophage nuclear factor-kappa B (NF-κB) leads to a dominant anti-inflammatory phenotype that attenuates glomerular inflammation. In vivo. Am J Pathol. 2005;167:27–37. doi: 10.1016/s0002-9440(10)62950-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson HM, Stewart KN, Brown PAJ, et al. Bone marrow derived macrophages genetically modified to produce IL-10 reduce injury in experimental glomerulonephritis. Molecular Therapy. 2002;6:710–17. doi: 10.1006/mthe.2002.0802. [DOI] [PubMed] [Google Scholar]
- 53.Boyle JJ, Harrington HA, Piper E, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. 2009;3:1097–108. doi: 10.2353/ajpath.2009.080431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Booth NA. Fibrinolysis and thrombosis. Baillieres Best Pract Res Clin Haematol. 1999;12:423–33. doi: 10.1053/beha.1999.0034. [DOI] [PubMed] [Google Scholar]
- 55.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;111:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;12:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 57.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 58.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 59.Mukhopadhyay S, Gordon S. The role of scavenger receptors in pathogen recognition and innate immunity. Immunobiology. 2004;209:39–49. doi: 10.1016/j.imbio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 60.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 61.Anderson CF, Mosser DM. Cutting edge: biasing immune responses by directing antigen to macrophage Fc gamma receptors. J Immunol. 2002;168:3697–701. doi: 10.4049/jimmunol.168.8.3697. [DOI] [PubMed] [Google Scholar]
- 62.Savill J, Dransfield I, Gregory C, et al. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 63.Erwig LP Henson PM. Immunological consequences of apoptotic cell phagocytosis. Am J Pathol. 2007;171:2–8. doi: 10.2353/ajpath.2007.070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chase AJ, Bond M, Crook MF, et al. Role of nuclear factor-kappa B activation in metalloproteinase-1, -3, and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler Thromb Vasc Biol. 2002;5:765–71. doi: 10.1161/01.atv.0000015078.09208.92. [DOI] [PubMed] [Google Scholar]
- 65.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 66.Merkel KD, Erdmann JM, McHugh KP, et al. Tumor necrosis factor-alpha mediates orthopedic implant osteolysis. Am J Pathol. 1999;1:203–10. doi: 10.1016/s0002-9440(10)65266-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khallou-Laschet J, Varthaman A, Fornasa G, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;25(5):e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poston RN, Hussain IF. The immunohistochemical heterogeneity of atheroma macrophages: comparison with lymphoid tissues suggests that recently blood-derived macrophages can be distinguished from longer-resident cells. J Histochem Cytochem. 1993;41:1503–12. doi: 10.1177/41.10.7504008. [DOI] [PubMed] [Google Scholar]
- 69.Waldo SW, Li Y, Buono C, Zhao B, et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–26. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 71.Komohara Y, Hirahara J, Horikawa T, et al. AM-3K, an anti-macrophage antibody, recognizes CD163, a molecule associated with an anti-inflammatory macrophage phenotype. J Histochem Cytochem. 2006;7:763–71. doi: 10.1369/jhc.5A6871.2006. [DOI] [PubMed] [Google Scholar]
- 72.Haffner SM, Lehto S, Rönnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 73.Malik S, Wong ND, Franklin SS, et al. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–50. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 74.Bourlier V, Bouloumie A. Role of macrophage tissue infiltration in obesity and insulin resistance. Diabetes Metab. 2009;4:251–60. doi: 10.1016/j.diabet.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 75.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;11:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;1:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeyda M, Farmer D, Todoric J, et al. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes. 2007;9:1420–8. doi: 10.1038/sj.ijo.0803632. [DOI] [PubMed] [Google Scholar]
- 78.Fujisaka S, Usui I, Bukhari A, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;11:2574–82. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clément K, Viguerie N, Poitou C, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;14:1657–69. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 80.Aron-Wisnewsky J, Tordjman J, Poitou C, et al. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;11:4619–23. doi: 10.1210/jc.2009-0925. [DOI] [PubMed] [Google Scholar]
- 81.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chawla A, Barak Y, Nagy L, et al. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;1:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 84.Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol. 2003;5:385–96. doi: 10.1038/nrm1104. [DOI] [PubMed] [Google Scholar]
- 85.Benoit M, Desnues B, Mege J-L. macrophage polarization in bacterial infection. J Immunol. 2008;181:3733–9. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 86.Vicari AP, Chiodoni C, Vaure C, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:541–9. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Allavena P, Sica A, Garlanda C, et al. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 88.Li C, Yang CW, Park CW, et al. Long-term treatment with ramipril attenuates renal osteopontin expression in diabetic rats. Kidney Int. 2003;63:454–63. doi: 10.1046/j.1523-1755.2003.00751.x. [DOI] [PubMed] [Google Scholar]
- 89.Mizuno M, Sada T, Kato M, et al. The effect of angiotensin II receptor blockade on an end-stage renal failure model of type 2 diabetes. J Cardiovasc Pharmacol. 2006;48:135–42. doi: 10.1097/01.fjc.0000245241.79959.d6. [DOI] [PubMed] [Google Scholar]
- 90.Schölkens BA, Landgraf W. ACE inhibition and atherogenesis. Can J Physiol Pharmacol. 2002;80:354–9. doi: 10.1139/y02-038. [DOI] [PubMed] [Google Scholar]
- 91.Guo Y, Wang QZ, Tang BS, et al. Effects of aspirin on atherosclerosis and the cyclooxygenase-2 expression in atherosclerotic rabbits. Chin Med J (Engl) 2006;119:1808–14. [PubMed] [Google Scholar]
- 92.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;4:507–14. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 93.Joseph SB, Castrillo A, Laffitte BA, et al. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–9. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 94.Tangirala RK, Bischoff ED, Joseph SB, et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci. USA. 2002;99:11896–901. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Levin N, Bischoff ED, Daige CL, et al. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol. 2005;25:135–42. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 96.Schultz JR, Tu H, Luk A, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–8. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morimoto K, Janssen WJ, Fessler MB, et al. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 2006;176:7657–65. doi: 10.4049/jimmunol.176.12.7657. [DOI] [PubMed] [Google Scholar]
- 98.Verhoeven BA, Moll FL, Koekkoek JA, et al. Statin treatment is not associated with consistent alterations in inflammatory status of carotid atherosclerotic plaques: a retrospective study in 378 patients undergoing carotid endarterectomy. Stroke. 2006;37:2054–60. doi: 10.1161/01.STR.0000231685.82795.e5. [DOI] [PubMed] [Google Scholar]
- 99.Nofer JR, Bot M, Brodde M, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2007;115:501–8. doi: 10.1161/CIRCULATIONAHA.106.641407. [DOI] [PubMed] [Google Scholar]
- 100.Bumcrot D, Manoharan M, Koteliansky V, et al. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–9. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim SS, Ye C, Kumar P, Chiu I, et al. Targeted Delivery of siRNA to Macrophages for Anti-inflammatory Treatment. Mol Ther. 2010 doi: 10.1038/mt.2010.27. DOI: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aouadi M, Tesz GJ, Nicoloro SM, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;7242:1180–4. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lipinski MJ, Amirbekian V, Frias JC, et al. MRI to detect atherosclerosis with gadolinium-containing immunomicelles targeting the macrophage scavenger receptor. Magn Reson Med. 2006;56:601–10. doi: 10.1002/mrm.20995. [DOI] [PubMed] [Google Scholar]
- 104.Trivedi RA, Mallawarachi C, U-King-Im JM, et al. Identifying inflamed carotid plaques using in vivo USPIO-enhanced MRI imaging to label plaque macrophages. Arterioscler Thromb Vasc Biol. 2006;26:1601–6. doi: 10.1161/01.ATV.0000222920.59760.df. [DOI] [PubMed] [Google Scholar]
- 105.Trivedi RA, U-King-Im JM, Graves MJ, et al. In vivo detection of macrophages in human carotid atheroma: temporal dependence of ultrasmall superparamagnetic particles of iron oxide-enhanced MRI. Stroke. 2004;35:1631–5. doi: 10.1161/01.STR.0000131268.50418.b7. [DOI] [PubMed] [Google Scholar]
- 106.Kooi ME, Cappendijk VC, Cleutjens KB, et al. Accumulation of ultrasmall superparamagnetic particles of iron oxide in human atherosclerotic plaques can be detected by in vivo magnetic resonance imaging. Circulation. 2003;19:2453–8. doi: 10.1161/01.CIR.0000068315.98705.CC. [DOI] [PubMed] [Google Scholar]
- 107.Litovsky S, Madjid M, Zarrabi A, et al. Superparamagnetic iron oxide-based method for quantifying recruitment of monocytes to mouse atherosclerotic lesions in vivo: enhancement by tissue necrosis factor-alpha, interleukin-1beta, and interferon-gamma. Circulation. 2003;107:1545–9. doi: 10.1161/01.cir.0000055323.57885.88. [DOI] [PubMed] [Google Scholar]
- 108.Siglienti I, Bendszus M, Kleinschnitz C, et al. Cytokine profile of iron-laden macrophages: implications for cellular magnetic resonance imaging. J Neuroimmunol. 2006;173:166–73. doi: 10.1016/j.jneuroim.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 109.Mulder WJ, Strijkers GJ, Briley-Saboe KC, et al. Molecular imaging of macrophages in atherosclerotic plaques using bimodal PEG-micelles. Magn Reson Med. 2007;6:1164–70. doi: 10.1002/mrm.21315. [DOI] [PubMed] [Google Scholar]
- 110.Hyafil F, Cornily JC, Rudd JH, et al. Quantification of inflammation within rabbit atherosclerotic plaques using the macrophage-specific CT contrast agent N1177: a comparison with 18F-FDG PET/CT and histology. J Nucl Med. 2009;6:959–65. doi: 10.2967/jnumed.108.060749. [DOI] [PubMed] [Google Scholar]
- 111.Nahrendorf M, Zhang H, Hembrador S, et al. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation. 2008;117:379–87. doi: 10.1161/CIRCULATIONAHA.107.741181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ben-Haim S, Kupzov E, Tamir A, et al. Evaluation of 18F-FDG uptake and arterial wall calcifications using 18F-FDG PET/CT. J Nucl Med. 2004;45:1816–21. [PubMed] [Google Scholar]
- 113.Davies JR, Rudd JH, Fryer TD, et al. Identification of culprit lesions after transient ischemic attack by combined 18F fluorodeoxyglucose positron-emission tomography and high-resolution magnetic resonance imaging. Stroke. 2005;36:2642–7. doi: 10.1161/01.STR.0000190896.67743.b1. [DOI] [PubMed] [Google Scholar]
- 114.Okane K, Ibaraki M, Toyoshima H, et al. 18F-FDG accumulation in atherosclerosis: use of CT and MR co-registration of thoracic and carotid arteries. Eur J Nucl Med Mol Imaging. 2006;33:589–94. doi: 10.1007/s00259-005-0005-2. [DOI] [PubMed] [Google Scholar]
- 115.Tahara N, Kai H, Ishibashi M, et al. Simvastatin attenuates plaque inflammation: evaluation by fluorodeoxyglucose positron emission tomography. J Am Coll Cardiol. 2006;48(1):825–31. doi: 10.1016/j.jacc.2006.03.069. [DOI] [PubMed] [Google Scholar]
- 116.Fujimura Y, Hwang PM, Trout Iii H, et al. Increased peripheral benzodiazepine receptors in arterial plaque of patients with atherosclerosis: an autoradiographic study with [(3)H]PK 11195. Atherosclerosis. 2008;1:108–11. doi: 10.1016/j.atherosclerosis.2008.02.032. [DOI] [PubMed] [Google Scholar]
- 117.Leimgruber A, Berger C, Cortez-Retamozo V, et al. Behavior of endogenous tumor-associated macrophages assessed in vivo using a functionalized nanoparticle. Neoplasia. 2009;5:459–68. doi: 10.1593/neo.09356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weissleder R, Kelly K, Sun EY, et al. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;11:1418–23. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]