

Abstract
Dendritic cells (DC) are known to develop from macrophage dendritic progenitors (MDP) in bone marrow (BM), which give rise to conventional (c)DC and monocytes, both dominant antigen presenting cell (APC) subsets in spleen. This laboratory has however defined a distinct dendritic-like cell subset in spleen (L-DC), which can also be derived in long-term cultures of spleen. In line with the restricted in vitro development of only L-DC in these stromal cultures, we questioned whether self-renewing HSC or progenitors exist in spleen with restricted differentiative capacity for only L-DC. Neonatal spleen and BM were compared for their ability to reconstitute mice and to give rise to L-DC, as well as other splenic APC. Neonatal spleen cells were transplanted into allotype-distinct lethally irradiated hosts along with host-type competitor BM cells, and assayed over 8 to 51 weeks for haematopoietic reconstitution of L-DC and cDC subsets, along with other lymphoid and myeloid cells. In this study, neonatal spleen showed multilineage haematopoietic reconstitution in mouse chimeras, rather than specific or restricted ability to differentiate into L-DC. However, the representation of individual APC subsets was found to be unequal in chimeras partially reconstituted with donor cells, such that more donor-derived progeny were seen for L-DC than for myeloid and cDC subsets. The ability of HSC in spleen to develop into L-DC was indicated by a strong bias in the subset size of these cells over other splenic APC subsets. This type of evidence supports a model whereby spleen represents an important site for haematopoiesis of this distinct DC subset. The conditions under which haematopoiesis of L-DC occurs in spleen, or the progenitors involved, will require further investigation.
Keywords: haematopoiesis, dendritic cells, haematopoietic stem cells, spleen
Introduction
The mouse is a valuable model for haematopoiesis and many studies involve bone marrow (BM) as a niche for maintenance and differentiation of haematopoietic stem cells (HSC). While spleens of adult mice [1] and human beings [2] contain HSC, the relative contribution of spleen versus BM HSC to haematopoiesis has not been fully investigated. A common view is that spleen HSC undergo haematopoiesis only during times of stress or myeloablation, and it has long been assumed that they contribute mainly to erythropoiesis [3]. Haematopoiesis in spleen appears to be redundant, since neonatally splenectomized mice display normal haematopoietic development, presumably from BM [1, 4]. Similarly, asplenic human beings have no major haematopoietic deficiency, and their immunity is adequate except for enhanced susceptibility to infection with encapsulated bacteria like Streptococcus[5]. It was recently determined in splenectomized mice that the spleen is essential for generation of the B-1a lineage of B cells which provides innate immunity to early bacterial and viral infection [6].
In vitro studies of haematopoiesis have generally involved BM rather than spleen as a source of progenitors. For several years now, our laboratory has been studying in vitro haematopoiesis occurring in long-term cultures (LTC) derived from murine spleen that support continuous haematopoiesis of immature dendritic cells (DC) [7–9]. Our recent studies have identified LTC-DC as phenotypically immature CD11bhiCD11cloMHC-II−CD8a− DC which are phenotypically and functionally distinct from known conventional (c)DC and plasmacytoid (p)DC subsets in murine spleen and also monocytes/macrophages [10]. LTC-DC have very high capacity for endocytosis and cross-presentation of antigen for CD8+ T cell activation, but are only weakly capable of activating CD4− T cells [8]. A cell equivalent to LTC-DC has now been identified in vivo in spleen, verifying LTC as a physiologically relevant culture system for production of this novel DC subset [11]. The in vivo subset has been tentatively named L-DC.
Spleen LTC are unique in vitro culture systems that support development of both progenitors and progeny myeloid DC-like cells within the same culture [7, 12]. They can be maintained continuously over many years [13, 14], implicating a progenitor or stem cell with self-renewing properties. We have also derived a splenic endothelial cell line, STX3 from one LTC, and have characterized it as an immature, endothelial-like cell line [15, 16] which supports continuous haematopoiesis from overlaid BM or spleen cells [17]. To date, we have characterized stroma as endothelial cells on the basis of gene and marker expression and ability to form tube-like structures in Matrigel [15, 16, 18]. They also produce high levels of CXCL12, a factor that recruits HSC into BM niches [19, 20].
In line with the restricted in vitro development of only L-DC in spleen stromal cocultures, we questioned whether HSC developing in different sites are perhaps subject to different niche signals, and distinct in terms of their haematopoietic potential. We compared spleen and BM for their ability to reconstitute mice and to give rise to all haematopoietic cell lineages including DC. In particular, we tested whether spleen HSC and BM HSC have equal reconstitution potential for antigen presenting cells, and particularly for the L-DC subset.
Materials and methods
Animals
C57BL/6J and C57BL/6.SJL-PtprcaPep3b/BoyJ (B6.SJL) mice were bred at the John Curtin School of Medical Research (JCSMR: Canberra, Australia) under specific pathogen-free conditions. Neonatal B6.SJL mice were used at age 7–9 days, and adult B6.SJL and C57BL/6J mice used at 4–8 weeks of age. Mice were housed and handled according to the guidelines of the Animal and Experimental Ethics Committee at the Australian National University (ANU: Canberra, Australia).
Preparation of irradiation chimeras
Mouse chimeras were generated using marker distinct haematopoietic cells. Donor haematopoietic cells of interest (104–105) were prepared from B6.SJL mice (CD45.1+) and resuspended in Hanks Balanced Salt Solution (HBSS; 200 ml). Cells were intravenously injected via the tail vein of C57BL/6J recipients (CD45.2+) aged 4–6 weeks using a 1 ml Tuberculin syringe (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with a 26.5 gauge needle (Becton Dickinson). In lethal-irradiation chimeras, mice were pre-conditioned by 9.5 Grays whole body irradiation using a Cobalt60 source (Commonwealth Scientific and Industrial Research Organisation, Black Mountain, ACT, Australia). Mice were rested for 2–3 hours prior to cell transfer. Chimeras were given host (CD45.21) BM (1 × 105 cells) together with donor (CD45.1+) haematopoietic cells to ensure survival. All irradiated mice were maintained on antibiotic water (Neomycin sulphate, 1.1 g/l, Sigma-Aldrich and Polymyxin B sulphate, 106U/L; Sigma-Aldrich, St Louis, MO, USA) for 4 weeks.
Analysis of cell surface marker expression
Antibody staining and flow cytometry were performed to distinguish cell subsets based on marker expression. In order to inhibit non-specific antibody binding via Fc receptors, cells (∼106) were incubated with anti-CD16/32 (FcR block; eBioscience, San Diego, CA, USA) for 15 min. on ice in a final volume of 25 μl FACS buffer [DMEM/1%FCS/0.1%NaN3] in the wells of a flexible 96-well polystyrene microtitre plate (Corning, NY, USA), or in 14 ml Falcon tubes. Cells were then pelleted by centrifugation (300 g, 5 min. at 48°C; Beckman, Palo Alto, CA, USA), and the supernatant flicked off. Specific antibody diluted in FACS buffer (25 μl total) was then absorbed to cells for 20 min. on ice. Biotin- or fluorochrome-conjugated antibodies used for flow cytometry were specific for CD11c (N418; allophycocyanin [APC]), ckit (2B8; phycoerythrin [PE]), CD11b (M1/70; phycoerythrin-Cy7 [PE-Cy7]), CD45.1 (A20; fluorescein isothiocyanate [FITC]) and CD19 (1D3; biotin) (eBioscience). Antibodies specific for CD8 (53–6.7; PE) and MHC-II (25–9-17; biotin) were purchased from BD Biosciences (San Jose, CA, USA). Following absorption of antibody, cells were washed twice by addition of 100 ml FACS buffer, centrifugation, and discard of supernatant. For addition of second-stage streptavidin (SA)-APC-Cy7 (eBioscience), the same process was repeated. For flow cytometric analysis, cells were resuspended at 107 cells/ml in FACS buffer in a 1.2 ml cluster tube (Corning). Propidium iodide (PI: 1 μg/ml, Sigma-Aldrich) was added prior to flow cytometry for discrimination of live and dead cells. Flow cytometry was performed immediately on a BD LSRII flow cytometer (Becton Dickinson). For flow cytometric analysis, between 3 × 104 and 1 × 106 events were collected for each sample. BD FACSDiva Software (Becton Dickinson) was used to acquire data. Data analysis involved post-acquisition gating using FlowJo software (Tree Star, Ashland, OR, USA).
T and B cell depletion of spleen
For depletion of T and B cells, splenocytes were labelled on ice for 15 min. with biotinylated monoclonal antibodies (mAbs) specific for CD19 (clone: eBio1D3), Thy1.2 (30-H12) and TER-119, purchased from eBioscience, and previously titrated for optimum binding. Cells were then washed twice to remove unbound antibody and resuspended at 108 cells/ml in separation buffer [phosphate-buffered saline (PBS)/0.5%BSA/2mM EDTA/0.1%NaN3]. Cells were incubated with anti-biotin MACS microbeads (13 μl beads/108 cells; Miltenyi Biotec, Gladbach, Germany) for 20 min. on ice. After one wash, splenocytes were resuspended in separation buffer (500 μl/108 cells). T cells and B cells were depleted by running cells through a LS column (Miltenyi Biotec) in a SuperMACS II Separation Unit (Miltenyi Biotec), washing thrice with separation buffer and collecting unbound cells as flow-through.
Results
In order to compare the capacity of HSC in neonatal spleen versus adult BM for capacity to produce L-DC, adoptive transfer of neonatal splenocytes was followed by flow cytometric analysis to delineate APC subsets in spleens of chimeras established across 8 to 51 weeks. Reconstitution of other cell lineages including T cells, B cells and monocytes/macrophages was also assessed in chimeras established for >17 weeks to determine long-term (LT) multi-lineage cell reconstitution potential reflective of LT-HSC. The seeding of HSC into BM niches in host mice was also determined by assessing the frequency of host- and donor-type cells expressing the ckit marker.
Investigating the in vivo differentiative capacity of spleen-derived HSC for L-DC
Spleen (SPL) chimeras were prepared by co-injecting equal numbers of whole neonatal B6.SJL splenocytes (CD45.1+), along with unfractionated C57BL/6J host-type (CD45.2+) BM competitor cells into C57BL/6J (CD45.2+) lethally irradiated recipient mice. Control BM chimeras were prepared following transplantation of only whole CD45.1+ BM into CD45.21 mice. For analysis of reconstitution due to HSC with short-term reconstitution capacity, multiple APC subsets were identified in spleens of 8 week chimeras (Fig. 1A), and then assessed for donor-derived (CD45.11) or host-type (CD45.1−) cell contribution (Fig. 1B). The procedure for identification of L-DC shown in Figure 1A involves gating live cells from T and B cell depleted spleen, followed by delineation of FSCloCD11bhiCD11c−MHC-II− myeloid cells (monocytes/macrophages), FSChiCD11bhiCD11cloMHC-II− L-DC, and the CD8− and CD8+ subsets of cDC: namely CD11bloCD11chiMHC-II+CD8− and CD11b−CD11chiMHC-II+CD8+.
Fig 1.
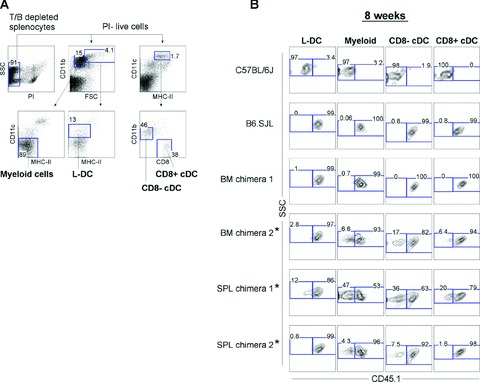
Haematopoietic reconstitution potential of spleen HSC for dendritic cells. Chimeras were prepared by adoptive transfer of marker-distinct neonatal spleen (SPL) cells along with host-type bone marrow (BM) cells into lethally irradiated mice. Whole splenocytes (1 × 105) from 8-day-old B6.SJL mice (CD45.1+) were co-injected into lethally irradiated (9.5Gy) C57BL/6J mice (CD45.2+) along with 1 × 105 host-type (CD45.2+) adult BM cells. Control BM chimeras were prepared by injection of only 1 × 105 dissociated CD45.1+ (B6.SJL) adult BM cells. For ‘short-term’ reconstitution, spleens were collected at 8 weeks, enriched for DC by depletion of T and B cells, and stained with antibodies specific for CD45.1, CD11c, CD11b, CD8 and MHC-II. Prior to flow cytometry, cells were incubated with propidium iodide (PI; 1 mg/ml) for gating of live (PI−) cells. (A) Spleen APC subsets were gated as follows: L-DC (CD11cloCD11bhiMHC-II−FSChi), myeloid cells (CD11c−CD11bhiMHC-II−FSClo), CD8a− cDC (CD11chiCD11b+CD8−MHC-II+) and CD8a+ cDC (CD11chiCD11b−CD8+MHC-II+). (B) Spleen APC subsets were assessed for donor (CD45.1+) cell composition. Numbers in gates indicate per cent positive cells. C57BL/6J and B6.SJL splenocytes were included as negative and positive controls, respectively, in order to set gates to define CD45.1− and CD45.1+ cell populations. Labelled chimeras (*) indicate mice showing only partial donor cell reconstitution.
As expected, very high levels of donor-reconstitution of APC subsets were observed in control BM chimeras (nos. 1 and 2; Fig. 1B). This was complete in the case of BM chimera 1, and partially complete for BM chimera 2, with donor cell contribution highest for L-DC. SPL chimeras (also given host-type BM competitor cells) displayed high levels of reconstitution with donor-derived (CD45.1+) spleen HSC, particularly for SPL chimera 2. However in both SPL chimeras, host-derived (CD45.1−) CD8− cDC and myeloid cells were present in higher proportionate numbers than were L-DC and CD8+ cDC. This same trend was seen for BM chimera 2 that was also only partially reconstituted with donor-type cells. This same trend was therefore common to chimeras given only BM, as well as SPL chimeras given equal numbers of both CD45.1+ spleen cells and host-type CD45.2+ BM cells. Like BM, neonatal spleen must contain progenitors/HSC which can reconstitute the four APC subsets analysed here after 8 weeks, L-DC, myeloid (macrophages/monocytes) cells, CD8+ and CD8− cDC. Such high donor cell reconstitution in two out of two SPL chimeras indicates that HSC in neonatal spleen effectively out-compete BM HSC for short-term reconstitution of all APC subsets following intravenous administration.
Long-term reconstitution of DC subsets by spleen HSC
The superior reconstitution potential of HSC in neonatal spleen was long-lasting, providing CD45.1+ donor-type haematopoietic cells in all SPL chimeras tested (3 of 3) for more than 25 weeks (Fig. 2A). As with short-term reconstitution analyses shown in Figure 1B, all long-term SPL chimeras gave almost complete donor (CD45.1+) cell reconstitution. This demonstrates that donor-type neonatal spleen cells (CD45.1+) out-compete an equal number of host-type BM cells (CD45.1−) for haematopoietic reconstitution, under the adoptive transfer protocol used here. Complete (.99%) reconstitution of spleen APC subsets by donor-type spleen HSC was observed in two out of three SPL chimeras analysed after 25 weeks. Only one chimera displayed partial donor reconstitution of APC subsets in spleen at 51 weeks, with proportions of subsets varying from 67% to 96%.
Fig 2.
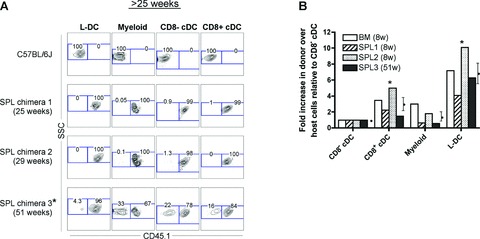
Dendritic cell reconstitution by neonatal spleen HSC. Spleen (SPL) and bone marrow (BM) chimeras were prepared as described in Figure 1. For analysis of long-term (.25 week) reconstitution, spleens were collected, enriched for DC by depletion of T and B cells, and stained with antibodies specific for CD45.1, CD11c, CD11b, CD8 and MHC-II. Prior to flow cytometry, cells were incubated with propidium iodide (PI; 1 mg/ml) for gating of live (PI−) cells. C57BL/6J splenocytes were stained as a CD45.1− control. (A) Individual spleen APC subsets (L-DC, myeloid cells, CD8− cDC and CD8+ cDC) were gated as shown in Figure 1A and assessed for donor-cell (CD45.1+) composition. Numbers in gates indicate per cent positive cells. Labelled chimeras (*) indicate mice showing only partial donor reconstitution with some evidence of host-type cell development. (B) The relative prevalence of donor- to host-type cells was calculated for each spleen APC subset in animals showing partial donor reconstitution as indicated in Figures 1 and 2 (*). The fold-increase of donor- to host-type cells was calculated relative to CD8− cDC assigned a fold-increase of 1.0. Data represent mean 6 SE (n= 4). Cell subsets having significantly higher representation of donor- over host-type cells, compared with CD8− cDC, are indicated (*) (P≤ 0.02; Wilcoxon Rank Sum Test).
Chimeras having only partial donor cell reconstitution were observed in both short- and long-term experiments (Figs 1B and 2A). For each of these chimeras, variability was seen in the proportional donor (CD45.1+) cell reconstitution amongst the four APC subsets studied. Analysis of partially reconstituted chimeras allowed us to address the question of whether spleen HSC have higher propensity to differentiate into L-DC than other myeloid subsets. The relative contribution of donor- and host-type cells for each APC subset was calculated and expressed as a factor relative to the CD8a− cDC subset (taken as 1.0). For SPL chimeras, this calculation will determine whether reconstitution by SPL as opposed to BM HSC was biased to any particular APC subset (Fig. 2B). If all spleen APC subsets were reconstituted equally, then values for cDC, myeloid cells and L-DC would be expected to equal 1.
By comparison with CD8a− cDC, 3.1-fold more donor-type CD8a+ cDC were produced than host-type CD8a+ cDC. For myeloid cells, this increase was 1.5-fold. For L-DC, 6.9-fold more donor- than host-type cells was produced. The increased ratios for L-DC and CD8+ cDC over CD8− cDC were statistically significant (P≤ 0.02; Wilcoxon Rank Sum Test). Thus, the unequal donor-to-host cell reconstitution originally observed in spleen APC subsets (Figs 1B and 2A) could be interpreted as a consistent and significant bias in the production of L-DC and to a lesser extent CD8+ cDC over CD8− cDC and myeloid cells. The higher donor HSC derivation of L-DC was observed in three SPL chimeras and one BM chimera, suggesting that the origin of the HSC from neonatal spleen as opposed to BM was not related to this outcome. It is possible, therefore, that HSC which have more propensity to localize in spleen, e.g. spleen-derived HSC, or HSC which enter spleen directly through blood, e.g. intravenously transferred BM HSC, may give rise to higher numbers of L-DC. The spleen may therefore be a preferred environment for L-DC development from HSC, which enter spleen through blood.
Spleen HSC also give multi-lineage, long-term haematopoietic reconstitution
Further analysis of haematopoietic cell subsets in both SPL and BM chimeras shown in Figures 1 and 2 indicated that neonatal spleen, like BM, contains HSC which are multi-potential and long-term reconstituting. First, for all chimeras studied between 8 and 51 weeks after reconstitution, the four APC compartments were found to be present at near homeostatic levels when compared with control untreated mice (Fig. 3A). Furthermore, in all chimeras showing long-term donor-type (CD45.1+) reconstitution of APC subsets in spleen, donor-type lymphoid T and B cells were also detected in mesenteric lymph nodes and spleen (Fig. 3B), confirming long-term multi-lineage reconstitution by spleen HSC of multiple lymphoid and myeloid cell lineages. Another measure of LT-HSC reconstitution of chimeric mice is the colonization of host lymphoid tissue by donor-type HSC. Both SPL and BM chimeras showed spleen colonization by donor-type HSC spleen cells. Both donor-derived Lin−ckitlo and Lin−ckithi progenitor populations were detected in three out of four SPL chimeras which are shown in Figure 3C.
Fig 3.
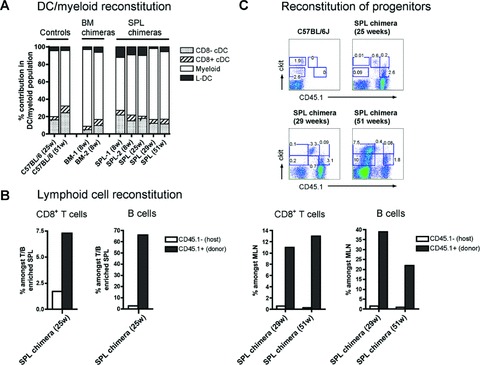
Neonatal spleen HSC give multilineage long-term reconstitution. Mouse chimeras described in Figures 1 and 2 were analysed for donor-derived (CD45.1+) subsets of L-DC, myeloid cells, CD8− cDC, CD8+ cDC, CD8+ T cells, B cells, and ckit+ haematopoietic progenitors in spleen, as well as CD8+ T cells and B cells in mesenteric lymph nodes (MLN). APC subsets were identified by antibody staining and flow cytometry as described in Figures 1 and 2. Lymphoid subsets in spleen were identified amongst the enriched subset discarded after T/B cell depletion to enrich for APC subsets. Cells were then stained for CD45.1, CD19 and CD8. Analysis of cells in MLN involved direct staining with antibodies specific for CD45.1, CD19 and CD8. For detection of haematopoietic progenitors, T/B cell depleted splenocytes were stained with antibodies specific for CD45.1, CD11c, CD11b, ckit and MHC-II. In all analyses, propidium iodide (PI; 1 mg/ml) staining was performed for gating of live (PI−) cells. (A) The prevalence of donor-derived spleen L-DC, myeloid cells, CD8− cDC and CD8+ cDC subsets in SPL and BM chimeras was calculated as a percentage of total (DC plus myeloid cells) in spleen. Chimeras were compared with control C57BL/6J mice for distribution of spleen APC subsets. (B) The presence of lymphoid cell subsets was analysed in spleen and MLN. For spleen, total [CD8+ T cells and CD19+CD8− B cells] are shown as per cent amongst T/B cell enriched spleen cells. For MLN, total [CD8+ T cells and CD19+CD8− B cells] are represented as per cent amongst total MLN. (C) Spleens collected from long-term SPL chimeras were analysed for the presence of Lin−ckit+ progenitors. T/B cell depleted splenocytes (CD19−Thy1.2−) were gated as CD11c−CD11b−MHC-II− (Lin−) cells, prior to analysis of donor (CD45.1+) ckit expression. Numbers in gates indicate per cent positive cells.
Discussion
Murine spleen HSC show long-term reconstitution upon adoptive transfer into irradiated mice. This was reported by Wolber et al.[1], and has been verified here for neonatal spleen, which gives multilineage reconstitution of lethally irradiated host mice for up to 51 weeks post-transplantation. In this report, the reconstitution potential of neonatal spleen progenitors was investigated with particular emphasis on capacity to generate a novel spleen APC subset previously defined in this laboratory, namely L-DC [11]. Our in vitro studies showed sustained and restricted development of L-DC when spleen cells were cocultured over endothelial stroma derived from spleen [17]. This may have been due to spleen containing a specific progenitor for L-DC. However, upon adoptive transfer of neonatal splenocytes into host mice, no evidence was found for a progenitor with restricted developmental capacity for L-DC. Our in vitro culture system based on splenic endothelial cells therefore represents a restricted environment supporting haematopoiesis of only L-DC and not other haematopoietic cells.
In this study, we confirmed that neonatal spleen contains LT-HSC, and that their reconstitution potential for splenic APC subsets was more efficient than for BM-derived LT-HSC in a competitive situation where equal numbers of spleen and BM cells were given intravenously. However, spleen chimeras reconstituted with both donor-type spleen and host-type BM, as well as chimeras reconstituted with only donor-type BM, generated L-DC and all other APC subsets tested. All spleen and BM chimeras studied acquired homeostatic proportions of CD8a− cDC, CD8a+ cDC, L-DC and myeloid cells in spleen. These chimeras also showed donor-type reconstitution of multiple myeloid and lymphoid cell lineages, as well as HSC subsets present in spleen. These results confirmed the presence of LT-HSC in neonatal spleen.
However, neonatal spleen did show specific bias towards L-DC development in spleen over other APC subsets. This finding was confirmed for chimeras in which intravenously administered neonatal spleen or BM was used as a source of HSC. Such a bias could only be detected in chimeras partially reconstituted with donor-type cells. These chimeras all showed variable proportions of donor-derived APC subsets in spleen (Figs 1B and 2A). For spleen chimeras, the L-DC subset consistently gave the highest donor to host cell ratio (Fig. 2B), indicating that donor spleen HSC produced more L-DC relative to CD8a− cDC, CD8a+ cDC, or myeloid subsets. Since HSC from donor BM also gave rise to higher levels of donor-derived L-DC than other APC in one BM chimera, the tissue origin of HSC from BM or spleen appears to be unrelated to L-DC development.
The lineage origin of splenic APC subsets including monocytes and cDC has been the subject of recent investigations. These cells share a distinct BM origin, and derive from a distinct macrophage/dendritic cell progenitor, which can differentiate to give both monocytes and cDC [21]. A more differentiated common dendritic cell progenitor (CDP) has also been identified in BM which gives rise to only cDC [22]. However, both these progenitors are specific to BM and are not found in spleen [23]. Spleen has however been shown to contain pre-cDC [24] and precursors of CD8+ cDC [25] which are short-lived precursors with specific differentiative capacity for cDC. Since spleen does not contain CDP and MDP, the reconstitution of spleen with cDC and monocytes would be dependent on localization of donor-type HSC in BM, and subsequent development of progenitors and precursors, which would subsequently seed spleen [23]. The higher relative frequency of L-DC over cDC and monocytes (myeloid cells) serves as further indirect evidence that L-DC have a different lineage origin to cDC and monocytes. However, the distinct contribution of early spleen progenitors to endogenous APC development is unknown, although there has been some evidence supporting such a hypothesis [7, 26].
While a specific bias in L-DC development was observed from transplanted HSC, the mechanism for this outcome is unclear. Based on a model for development of L-DC from endogenous spleen progenitors, it can be hypothesized that adoptively transferred HSC of either spleen or BM origin, given to mice that have undergone myeloablative irradiation treatment, will engraft into specific splenic niches that support haematopoiesis of L-DC. Simultaneously, and in line with a BM origin for cDC and monocyte progenitors [21–23], HSC will also migrate to BM niches for cDC and monocyte (myeloid cell) development. Such a division of haematopoietic sites could lead to differences in the subset size of output cells. However, the haematopoietic pathway for L-DC development from HSC is unknown and requires identification using a lineage analysis approach to identify a specific progenitor.
Overall, this study provides important evidence for a role of spleen in haematopoiesis of a novel lineage of spleen APC. Further experiments will need to investigate specific progenitors for L-DC in spleen, and to resolve the mechanism behind preferential L-DC development from intravenously transferred stem cells over other DC and APC subsets. The findings reported here also impact on the importance of vascular niches that support haematopoiesis present in both spleen and BM [27], and their function in relation to osteoblastic niches, which exist only in BM [28, 29].
Acknowledgments
This work was supported by ANU institutional funding to HO. JT was supported by an ANU Postgraduate Research Award.
References
- 1.Wolber FM, Leonard E, Michael S, et al. Roles of spleen and liver in development of the murine hematopoietic system. Exp Hematol. 2002;30:1010–9. doi: 10.1016/s0301-472x(02)00881-0. [DOI] [PubMed] [Google Scholar]
- 2.Dor FJ, Ramirez ML, Parmar K, et al. Primitive hematopoietic cell populations reside in the spleen: studies in the pig, baboon, and human. Exp Hematol. 2006;34:1573–82. doi: 10.1016/j.exphem.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Yanai N, Satoh T, Obinata M. Endothelial cells create a hematopoietic inductive microenvironment preferential to erythropoiesis in the mouse spleen. Cell Struct Funct. 1991;16:87–93. doi: 10.1247/csf.16.87. [DOI] [PubMed] [Google Scholar]
- 4.Shatry AM, Jones M, Levy RB. The effect of the spleen on compartmental levels and distribution of donor progenitor cells after syngeneic and allogeneic bone marrow transplants. Stem Cells Dev. 2004;13:51–62. doi: 10.1089/154732804773099254. [DOI] [PubMed] [Google Scholar]
- 5.Brendolan A, Rosado MM, Carsetti R, et al. Development and function of the mammalian spleen. Bioessays. 2007;29:166–77. doi: 10.1002/bies.20528. [DOI] [PubMed] [Google Scholar]
- 6.Wardemann H, Boehm T, Dear N, et al. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J Exp Med. 2002;195:771–80. doi: 10.1084/jem.20011140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Neill HC, Wilson HL, Quah B, et al. Dendritic cell development in long-term spleen stromal cultures. Stem Cells. 2004;22:475–86. doi: 10.1634/stemcells.22-4-475. [DOI] [PubMed] [Google Scholar]
- 8.Quah B, Ni K, O’Neill HC. In vitro hematopoiesis produces a distinct class of immature dendritic cells from spleen progenitors with limited T cell stimulation capacity. Int Immunol. 2004;16:567–77. doi: 10.1093/intimm/dxh060. [DOI] [PubMed] [Google Scholar]
- 9.Wilson HL, Ni K, O’Neill HC. Identification of progenitor cells in long-term spleen stromal cultures that produce immature dendritic cells. Proc Nat Acad USA. 2000;97:4784–9. doi: 10.1073/pnas.080278897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 11.Tan JKH, Quah BJC, Griffiths KL, et al. Identification of a novel antigen cross-presenting cell in spleen: a counterpart to cells produced in long-term culture. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01089.x. Doi: 10.1111/j.1582-4934.2010.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson HL, O’Neill HC. Dynamics of dendritic cell development from precursors maintained in stroma-dependent long-term cultures. Immunol Cell Biol. 2003;81:144–51. doi: 10.1046/j.0818-9641.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 13.Ni K, O’Neill H. Establishment of long-term stromal cultures producing dendritic cells. Br J Haematol. 1997;97:710–25. doi: 10.1046/j.1365-2141.1997.00135.x. [DOI] [PubMed] [Google Scholar]
- 14.Ni K, O’Neill H. Spleen stromal cells support haemopoiesis and in vitro growth of dendritic cells from bone marrow. Br J Haematol. 1999;105:58–67. [PubMed] [Google Scholar]
- 15.Despars G, Ni K, Bouchard A, et al. Molecular definition of an in vitro niche for dendritic cell development. Exp Hematol. 2004;32:1182–93. doi: 10.1016/j.exphem.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 16.Despars G, O’Neill HC. Splenic endothelial cell lines support development of dendritic cells from bone marrow. Stem Cells. 2006;24:1496–504. doi: 10.1634/stemcells.2005-0530. [DOI] [PubMed] [Google Scholar]
- 17.Periasamy P, Tan JKH, Griffiths KL, et al. Splenic stromal niches support hematopoiesis of dendritic-like cells from precursors in bone marrow and spleen. Exp Hematol. 2009;30:1060–71. doi: 10.1016/j.exphem.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Despars G, Periasamy P, Tan J, et al. Gene signature of stromal cells which support dendritic cell development. Stem Cells Dev. 2008;17:917–27. doi: 10.1089/scd.2007.0170. [DOI] [PubMed] [Google Scholar]
- 19.Sugiyama T, Kohara H, Noda M, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 20.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity. 2006;25:862–4. doi: 10.1016/j.immuni.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Fogg DK, Sibon C, Miled C, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311:83–7. doi: 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- 22.Onai N, Obata-Onai A, Schmid MA, et al. Identification of clonogenic common Flt3+M-CSFR1 plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. 2007;8:1207–16. doi: 10.1038/ni1518. [DOI] [PubMed] [Google Scholar]
- 23.Liu K, Victora GD, Schwickert TA, et al. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–7. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naik SH, Metcalf D, Van Nieuwenhuijze A, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol. 2006;7:663–71. doi: 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 25.Bedoui S, Prato S, Mintern J, et al. Characterization of an immediate splenic precursor of CD8+ dendritic cells capable of inducing antiviral T cell responses. J Immunol. 2009;182:4200–7. doi: 10.4049/jimmunol.0802286. [DOI] [PubMed] [Google Scholar]
- 26.Kabashima K, Banks TA, Ansel KM, et al. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–50. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Kiel MJ, Yilmaz OH, Iwashita T, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 28.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 29.Chan CK, Chen CC, Luppen CA, et al. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature. 2009;457:490–4. doi: 10.1038/nature07547. [DOI] [PMC free article] [PubMed] [Google Scholar]