

Abstract
The present study identified a novel mechanism of induction of apoptosis in glioblastoma cells by scriptaid – a histone deacetylase (HDAC) inhibitor. Scriptaid reduced glioma cell viability by increasing Jun N-terminal kinase (JNK) activation. Although scriptaid induced activation of both p38MAPK and JNK, it was the inhibition of JNK that attenuated scriptaid-induced apoptosis significantly. Scriptaid also increased the expression of (i) p21 and p27 involved in cell-cycle regulation and (ii) γH2AX associated with DNA damage response in a JNK-dependent manner. Treatment with scriptaid increased Ras activity in glioma cells, and transfection of cells with constitutively active RasV12 further sensitized glioma cells to scriptaid-induced apoptosis. Scriptaid also inhibited telomerase activity independent of JNK. Taken together, our findings indicate that scriptaid (i) induces apoptosis and reduces glioma cell proliferation by elevating JNK activation and (ii) also decreases telomerase activity in a JNK-independent manner.
Keywords: Ras, JNK, telomerase, scriptaid, glioblastoma, HDAC inhibitors
Introduction
The lack of effective therapy for glioblastoma multiforme (GBM), which remains one of the most challenging solid cancers to treat because of its highly proliferative and invasive nature, has necessitated identification of new treatment strategies. Histone deacetylase (HDAC) inhibitors with potent chemopreventive and chemotherapeutic activities inhibit tumour development through inhibition of cell proliferation, induction of cell-cycle arrest and apoptosis of cancer cells [1]. HDAC inhibition has been reported to promote growth arrest in glioma cells [2–4]. Scriptaid, a recently identified HDAC inhibitor [5], arrests growth of endometrial and ovarian cancer cells [6]. As HDAC inhibitors are emerging as a promising anti-neoplastic strategy, we assessed the effect of scriptaid on glioma cell apoptosis.
Activation of c-Jun-NH2-terminal kinase/stress-activated protein kinase (JNK) plays a functional role in HDAC inhibitor-mediated apoptosis [7]. Importantly, JNK activation potently sensitizes glioma cells to apoptosis [8] by triggering apoptotic cascades [9]. JNK is activated by MAP kinase kinases (MAPKK), and the GTP-binding protein, Ras, plays a crucial role in MAP kinase activation [10]. We have recently demonstrated that increased Ras activation can play an active role in glioma cell apoptosis [11], and the pro-apoptotic effects of Ras are mediated by MAPK [12]. As HDAC inhibitors are known to induce cell death selectively in cells that harbour activated kRasV12 [13], we evaluated the ability of scriptaid to affect Ras/JNK activity in glioma cells. HDAC inhibitors are also known to suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells [14] and JNK is a key regulator of telomerase activity [15]. Moreover, telomerase inhibition is a promising approach for the targeting GBMs [16] and HDAC inhibitor has been shown to inhibit telomerase activity and hTERT expression in brain cancer cells [17]. Therefore, the ability of scriptaid to effect telomerase activity was also investigated.
Here, we show that scriptaid-induced JNK activation plays a crucial role in triggering glioma cell death. Although scriptaid affected the expression of molecules involved in cell-cycle regulation and DNA damage repair response in a JNK-dependent manner, it decreased telomerase activity independent of JNK activation. Our results indicate that scriptaid not only (i) induces glioma cell death and inhibit their proliferation but also (ii) decreases telomerase activity.
Materials and methods
Antibodies, inhibitors and plasmids
Antibodies to Ras, JNK and phospho-p38MAPK were purchased from Cell Signaling Technologies (Beverly, MA, USA). Antibodies to p21 and p27 were purchased from BD Biosciences (San Diego, CA, USA) and Abcam (Cambridge, UK), respectively. Antibodies to c-Myc, p38MAPK and phospho-JNK were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibody to β-actin was purchased from Sigma (St. Louis, MO, USA). Antibodies to H2AX, γH2AX, acetylated H3 and H4 were purchased from Upstate Biotechnology (Temecula, CA, USA). Inhibitors for p38MAPK (SB203580) and JNK (SP600125) were purchased from Calbiochem (La Jolla, Ca, USA) and Sigma, respectively. Constitutive active Ras (RasV12) and dominant negative (RasN17) were purchased from Clontech (Mountainview, CA, USA). Scriptaid was purchased from Tocris Cookson, UK.
Cell culture and treatment
Glioblastoma cell lines LN229 and T98G obtained from American Type Culture Collection (Manassas, VA, USA) were cultured in DMEM, supplemented with 10% FBS. On attaining semi-confluence, cells were switched to serum-free media, and after 6 hrs, cells were treated with different concentrations of scriptaid (in dimethyl sulphoxide, DMSO) for 24 hrs. DMSO-treated cells were used as controls. To investigate the role of Ras in scriptaid-induced glioma cell death, cells were transfected with dominant negative (RasN17) or constitutive active (RasV12) Ras constructs using Lipofectamine 2000 (Gibco-Invitrogen, Rockville, MD, USA), as described previously [11]. The transfection efficiency was greater than 80% as determined by transfection with GFP-expressing construct. Twenty-four hours after transfection, cells were either left untreated or treated with scriptaid for 24 hrs. To determine the role of MAPK in scriptaid-induced apoptosis, cells were pretreated with 20 μM p38MAPK (SB203580) and JNK (SP600125) inhibitors, respectively, for 1 hr. Following pretreatment, cells were co-treated with inhibitors and scriptaid for 24 hrs. Treated cells were then processed for MTS, Ras, HDAC, telomerase activity and Western blot analysis. All reagents were purchased from Sigma unless otherwise stated.
Determination of cell viability
Cell viability was assessed using the [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] (MTS) according the manufacturer’s instruction (Promega, Madison, WI, USA), as described previously [11]. Following treatment of cells (4 × 103) with scriptaid in 96-well plate, 20 μl of MTS solution was added. After 4 hrs of incubation the absorbance reflecting reduction of MTS by viable cells was determined at 490 nm. Values were expressed as a percentage relative to those obtained in controls.
Assay of caspase-3 activity
The Colorimetric Assay Kit for caspases-3 (Sigma) was used to determine the enzymatic activity of caspase-3 in glioma cells treated with scriptaid, as described previously [18]. The assay is based on spectrophotometric detection of the chromophore p-nitroaniline (pNA) after cleavage from the substrate peptides conjugated to pNA. The substrate peptide used for the colorimetric assay of caspase-3 is Ac-DEVD. Following treatment, cells were lysed with the lysis buffer provided, and 100 μg of the protein lysates were incubated with the colorimetric substrate with assay buffer provided at 37°C for 2 hrs in a 96-well microplate. The pNA released by cleavage of the peptide was quantified spectrophotometrically at 405 nm in a microtitre plate reader.
Western blot analysis
Proteins from whole cell lysates and nuclear extracts were isolated as described previously [19]. Thirty microgram of protein isolated from control and scriptaid-treated cells was electrophoresed on 6–12% polyacrylamide gel and Western blotting was performed as described [19], using the following antibodies: Ras, p38MAPK, pp38MAPK, JNK, pJNK, p21, p27, cMyc, γH2AX, H2AX, acetylated H3 and H4. For histone extraction, cells were lysed in H-buffer (50 mM Tris HCl, 150 mM NACl, 2 mM EDTA, 1% Triton-100) and incubated at 4°C for 15 min before centrifugation at 2000 rpm for 10 min. After a wash step in H-buffer the pellet was dissolved in H-buffer containing 0.2 M HCl and incubated on ice for 30 min, before a second centrifugation at 10,000 rpm for 10 min. The supernatant was directly used for immunoblotting. Secondary antibodies were purchased from Vector Laboratories Inc (Burlingame, CA, USA). After addition of chemiluminescence reagent (Amersham, Buckinghamshire, UK), blots were exposed to Chemigenius Bioimaging System (Syngene, Cambridge, UK) for developing and images were captured using Genesnap software (Syngene). The blots were stripped and reprobed with anti-β-actin to determine equivalent loading as described [19].
Comet assay
Comet assay was performed as described previously [20]. Briefly, untreated and scriptaid-treated cells were mixed with 120 ml of low-melting-temperature agarose (0.5% in PBS) and added to microscope slides that had been covered with a bottom layer of 1.5% agarose. Slides were processed using alkali denaturation and electrophoresis (0.86 V/cm) for 20 min each. Images of 50 randomly selected cells stained with ethidium bromide were analysed.
HDAC activity assay
The HDAC activity assay was performed using the HDAC activity kit according to the manufacturer’s instructions (Upstate Biotechnology, Temecula, CA, USA). Briefly, HDAC assay buffer was added to nuclear extract obtained from control and scriptaid-treated glioma cells followed by addition of HDAC assay substrate and incubation at 37°C for 1 hr. Activator solution was then added and samples were incubated for an additional 20 min at room temperature and absorbance was read at 405 nm.
Flow cytometric analysis of DNA content
To determine the effect of scriptaid on glioma cell-cycle progression, FACS analysis of DNA content was performed as described [18]. Control and scriptaid-treated cells were fixed in 70% ethanol and stored at −20°C. The fixed cells were then washed in PBS, resuspended in propidium iodide solution (BD Biosciences) for 20 min at room temperature, and flow cytometric analysis of 106 cells was carried out using Cell Quest program on FACS Calibur (BD Biosciences). The percentages of cells in the G1, S, and G2/M phases of the cell cycle were analysed with the Mod Fit LT program as described [18].
Telomerase activity
Telomerase activity of cells treated with scriptaid in the presence and absence of JNK inhibitor was determined using the TeloTAGGG Telomerase PCR ELISA Plus kit (Roche Diagnostics, Germany). Cells (2 × 105) were lysed according to the manufacturers’ instruction and equal amount of protein (0.5 μg) from cell extracts was added to 25 μl of reaction mixture and 5 μl of internal standard, and the final volume was made 50 μl by adding nuclease-free water. Primer elongation was done for 30 min and amplification reaction was performed for 30 cycles. The amplification product (2.5 μl) was mixed with 10 μl of denaturation reagent and 100 μl of hybridization buffer, transferred to streptavidin-coated microplate, and the reaction was incubated for 2 hrs. The hybridization buffer was then removed, 100 μl TMB substrate solution was added per well and the reaction mixture was incubated for 30 min following which 100 μl of stop reagent was added in each well, and absorbance of samples was measured at 450 nm with reference wavelength at 690 nm using ELISA reader.
Measurement of Ras activity
The Ras activity was performed using a commercially available Ras activation assay kit purchased from Upstate Biotechnology (Temecula, CA, USA), as described previously [11]. Briefly, cells (2 × 106) treated with scriptaid for different time intervals were lysed in Mg2+ lysis buffer. Lysates (500 μg) were incubated for 1 hr at 4°C with beads coated with a fusion protein (GST-Raf1-RBD) consisting of GST fused to the Ras-binding domain of Raf-1. Beads were washed three times with cold Mg2+ containing lysis buffer, and bound protein was eluted by boiling for 5 min with 10× sample buffer and analysed by immunoblotting for Ras.
Results
Scriptaid induces apoptosis in glioma cells
To investigate whether scriptaid affects glioma cell viability, LN229 and T98G glioma cells were treated with increasing concentrations of scriptaid for 24 hrs and cell viability was determined using MTS assay. Although treatment with 5 μM of scriptaid for 24 hrs reduced glioma cell viability to approximately 85%, an approximately 30% decrease in viability was observed in LN229 and T98G, respectively, upon treatment with 10 μM scriptaid, as compared to the untreated control (Fig. 1A). A further decrease in cell viability by approximately 50% was observed in both the cell lines upon treatment with 20 μM scriptaid (Fig. 1A).
Fig 1.
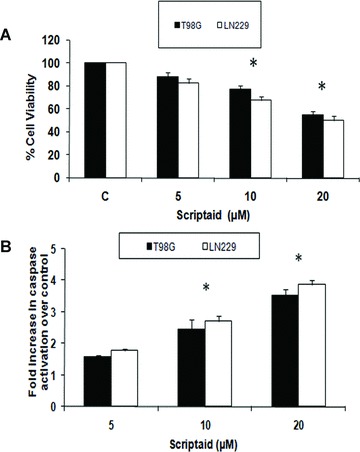
Scriptaid induces apoptosis in glioma cells. (A) Scriptaid decreases viability of glioma cells in a dose-dependent manner. LN229 and T98G cells (5 × 103) were treated with 5–20 μM scriptaid for 24 hrs, and cells were subjected to MTS assay. The graph represents decrease in glioma cell proliferation upon treatment with increasing concentration of scriptaid. (B) Fold increase in caspase-3 activity in LN229 and T98G cells treated with different concentrations of scriptaid for 24 hrs, as determined by the caspase-3 activity assay. Values in (A) and (B) represent the means ± SEM from three independent experiments. *Significant decrease/increase from control (P≤ 0.05).
We next determined the levels of active caspase-3 in cells treated with scriptaid. Although the expression of active caspase-3 in glioma cells was unaffected in cells treated with 5 μM of scriptaid (Fig. 1B), a significant 2.3- and 2.7-fold increase in caspase-3 activity was observed in T98G and LN229 cells, respectively, upon exposure to 10 μM scriptaid as compared to the control. A further increase in caspase-3 activity by approximately 3.4- and 3.8-fold was observed upon treatment of T98G and LN229, respectively, with 20 μM scriptaid (Fig. 1B). Because the activation of caspase-3-like proteases is crucial in apoptotic cell death [21], these results suggest that scriptaid induces apoptosis in glioma cells.
Scriptaid decreases HDAC activity and increases acetylation of H3 and H4 histone in glioma cells
Because scriptaid is a known HDAC inhibitor, we investigated its ability to regulate the HDAC activity in glioma cells. A significant –40, 50 and 60% decrease in HDAC activity was observed in T98G cells upon treatment with 5, 10 and 20 μM scriptaid (Fig. 2A). A similar trend was observed in LN229 cells (Fig. 2A). As increased histone acetylation is associated with diminished HDAC activity, we determined the acetylation status of H3 and H4 in scriptaid-treated glioma cells. Scriptaid potently induced hyperacetylation of both histones H3 and H4 in glioma cells (Fig. 2B). The decrease in HDAC activity was concurrent with increased H3 and H4 acetylation.
Fig 2.
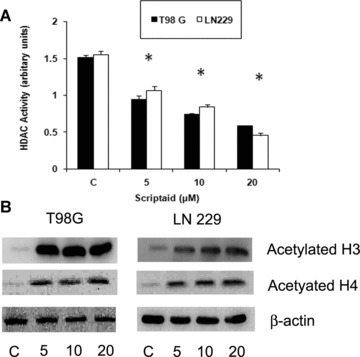
Scriptaid decreases HDAC activity and increases histone acetylation of glioma cells. (A) Scriptaid treatment decreases HDAC activity in glioma cells. HDAC activity of scriptaid-treated LN229 and T98G cells was performed using HDAC activity assay kit according to manufacturer's instruction. A decrease in HDAC activity in LN229 and T98G cells upon exposure to increasing dose of scriptaid was observed. Values represent the means ± SEM from three independent experiments. *Significant decrease from control (P≤ 0.05). (B) Scriptaid induces H3 and H4 hyperacetylation in glioma cells. Western blot analysis was performed on nuclear extracts obtained from LN229 and T98G cells treated with scriptaid. An increase in H3 and H4 acetylation was observed upon exposure of glioma cells to increasing concentration of scriptaid. A representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading.
Scriptaid alters the expression of molecules associated with cell-cycle regulation
HDAC inhibitor-mediated activation of p21WAF1 plays a major role in arresting tumour cell proliferation [22]. As scriptaid significantly inhibited the proliferation of glioma cells, we determined the expression of molecules associated with cell-cycle progression in these cells. Treatment with scriptaid increased the expression of p21 and p27 in a dose-dependent manner (Fig. 3A). The HDAC inhibitor SAHA increases expression of p21 and decreases c-Myc levels in pancreatic cancer cells [23]. As oncoprotein c-Myc suppresses p21WAF1 transcription [24] and because p21 expression was elevated in scriptaid-treated glioma cells, we determined cMyc expression in these cells. Increase in p21 levels was concomitant with decrease in cMyc expression (Fig. 3A).
Fig 3.
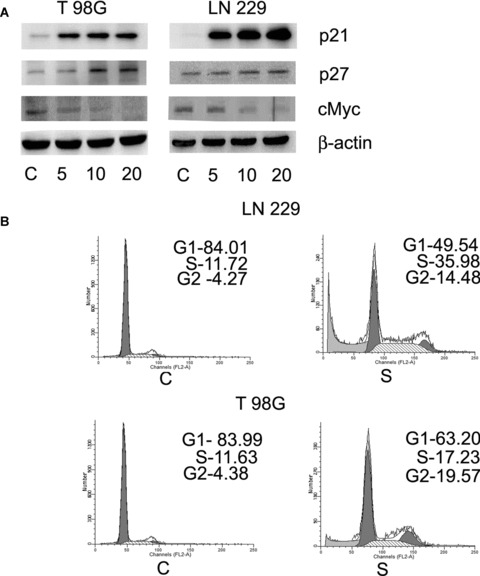
Scriptaid induces glioma cell-cycle arrest. (A) Scriptaid increases the expression of p21, p27 and decreased cMyc level in glioma cells in a dosedependent manner. LN229 and T98G cells were treated with different concentration of scriptaid for 24 hrs and Western blot analysis was performed to detect the expression of cell-cycle regulatory proteins p21, p27 and cMyc. A representative blot from three independent experiments with identical results is shown. Blots were reprobed for β-actin to establish equivalent loading. (B) Treatment of glioma cells with scriptaid induces G2 arrest in the cell cycle. FACS analysis was performed on cells treated with scriptaid and the percentages of cells in the various cell-cycle phases were plotted. FACS analysis profiles of scriptaidtreated cells are shown. Insets indicate percentage cells in G1, S and G2/M phases of the cell cycle in T98G cells. C and S denote control and scriptaid, respectively.
As treatment of glioma cells with scriptaid altered the expression of molecules associated with cell-cycle regulation, we performed FACS analysis to determine the cell-cycle profile of scriptaid-treated cells. An approximately 3- and 4-fold increase of cells at the G2 phase was observed in scriptaid-treated LN229 and T98G cells, respectively (Fig. 3B). Scriptaid also increased the cells arrested in S phase from 11.72 and 11.66% in control to 35.98 and 17.23% in LN229 and T98G cells treated with scriptaid, respectively (Fig. 3B). This increased accumulation indicated that scriptaid causes cell-cycle arrest at S and G2 phases in glioma cells.
Scriptaid induces increased DNA damage signalling response
cMyc induces DNA damage and chromosomal instability by preventing repair of replication-stress-induced DNA lesions [25]. Moreover, HDAC inhibitors have been reported to induce phosphorylation of H2AX in leukaemia cells [26]. Because a decrease in cMyc expression was observed in scriptaid-treated glioma cells, we determined the expression of γ-H2AX in these cells. The expression of γ-H2AX was elevated following scriptaid treatment (Fig. 4A), in a dose-dependent manner. The levels of total H2AX were unaffected by scriptaid treatment (Fig. 4A).
Fig 4.
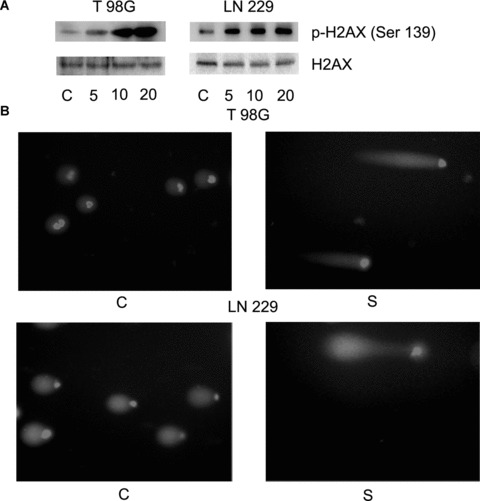
Scriptaid induces DNA damage in glioma cells. (A) Scriptaid affects the ability of glioma cells to repair DNA damage. Ability of scriptaid-treated glioma cells to participate in DNA damage response was detected by determining the expression of γ-H2AX (Ser139) and H2AX by Western blot analysis. A representative blot from three independent experiments with identical results is shown. Blots were reprobed for β-actin to establish equivalent loading. (B) Treatment of glioma cells with scriptaid increases DNA damage as determined by comet assay. The figure is a representative of comet assay performed on control and scriptaid-treated T98G and LN229 cells.
The ability to repair DNA damage is essential to cell survival because maintained DNA breaks induce apoptosis [27]. As expression of DNA damage repair protein γ-H2AX was elevated in scriptaid-treated cells, comet assay was performed to determine the effect of scriptaid on glioma cells. Treatment with scriptaid resulted in increase in comet tail length as compared to untreated control, suggesting an induction of DNA damage by scriptaid (Fig. 4B).
Scriptaid increases Ras expression and activity in glioma cells
Ras induces DNA damage signalling response [28] and cells that senesce in response to oncogenic Ras accumulate DNA damage foci [29]. Disruption of cooperation of Ras and cMyc promotes growth arrest of neuroblastoma cells [30]. Moreover, oncogenic Ras promotes HDAC inhibitor butyrate-induced apoptosis [31]. Because scriptaid-triggered apoptosis was concomitant with decrease in cMyc and increase in γ-H2AX, we investigated the contribution of Ras in scriptaid-induced apoptosis. Western blot analysis demonstrated an increase in Ras expression in LN229 and T98G cells upon increasing exposure to scriptaid (Fig. 5A).
Fig 5.
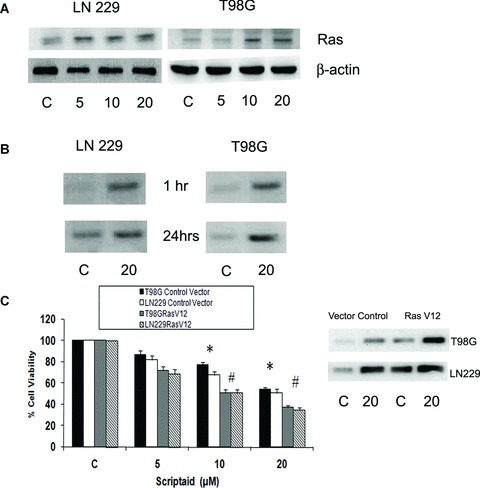
Scriptaid increases the expression and activity of Ras in glioma cells. (A) Scriptaid increases Ras expression in glioma cells in a dose-dependent manner. LN229 and T98G cells were treated with different concentration of scriptaid, and Ras level was determined by Western blot analysis. A representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading. (B) Effects of scriptaid on the levels of GTP-bound Ras. The levels of Ras-GTP in protein extracts of control and scriptaidtreated LN229 and T98G cells were determined by the ability of Ras-GTP to bind to a specific protein domain of Raf in the form of a GST-fusion protein. An increase in Ras activity was observed in glioma cells treated with increasing concentration of scriptaid both at 1 and 24 hrs post-treatment, as compared to the control. The figure is representative from three independent experiments with identical results. (C) Scriptaidinduced cell death is dramatically increased in the presence of constitutive Ras. Glioma cells transfected with constitutive active Ras (RasV12) were treated with scriptaid for 24 hrs and cell viability was determined by MTS assay. Ras activity in cells transfected with RasV12 or control vector in the presence or absence of scriptaid. The graph represents the percentage of viable cells as determined by MTS assay, observed when RasV12-transfected or untransfected glioma cells were treated in the presence and absence of scriptaid for 24 hrs. Values represent the means ± SEM from three independent experiments. *Significant decrease from control (P≤ 0.05); #significant change from scriptaid-treated cells (P≤ 0.05).
As Ras levels were elevated in scriptaid-treated cells, we next investigated the effect of scriptaid on Ras activity. Lysates from cells grown in the presence or absence of scriptaid for different intervals of time were subjected to a Ras-GTP pull-down assay [32]. The levels of activated Ras were dramatically elevated in glioma cells at 1 hr after scriptaid treatment (Fig. 5B). Importantly, this heightened level of activated Ras was sustained even at 24 hrs post-treatment with scriptaid (Fig. 5B). Thus, scriptaid-induced apoptosis of glioma cells was concurrent with increased Ras activity.
Pro-apoptotic effects of scriptaid are mediated by increased Ras activation
Oncogenic Ras induces apoptosis [33] and inducible expression of kRasV12 sensitizes cells to HDAC inhibitor-induced apoptosis [13]. We therefore determined whether elevated Ras was critical in scriptaid-induced cell death. Glioma cells were transfected with the expression plasmid coding for constitutively active Ras (RasV12) in the absence or the presence of scriptaid. The decrease in cell viability observed when glioma cells were treated with scriptaid was further decreased in cells expressing the constitutive active Ras (RasV12). T98G cell viability was reduced from 87, 77 and 54% in mock-transfected cells to 72, 51 and 37% in RasV12-transfected cells, upon treatment with 5, 10 and 20 μM concentration of scriptaid, respectively (Fig. 5C). Similar trend was observed in LN229 cells (Fig. 5C). However, glioma cells transfected with dominant negative RasN17 were only marginally protected from scriptaid-induced apoptosis as compared to untransfected cells (data not shown). This suggests that RasN17 cannot completely override scriptaid-induced increase in Ras activity. To demonstrate that transfection with constitutive RasV12 elevates Ras activity, Ras GTPase activity was determined in glioma cells transfected with RasV12 or control vector in the presence or absence of 20 μM scriptaid. Although transfection with constitutive RasV12 resulted in a increase in Ras activity as compared to the mock-transfected control, the effect was more pronounced in scriptaid-treated cells (Fig. 5C).
Pro-apoptotic effects of Ras are mediated by the JNK pathway
The pro-apoptotic effects of Ras are mediated by the MAPK pathway [12]. Because HDAC inhibitors induce apoptosis in leukaemia cells through JNK activation [7], we investigated the status of p38MAPK and JNK activation in scriptaid-treated glioma cells. An elevation in JNK and the p38MAPK phosphorylation was observed in glioma cells upon increasing exposure to scriptaid (Fig. 6A), as compared to the untreated control. Treatment with scriptaid had no effect on the total p38MAPK and JNK levels in glioma cells (Fig. 6A).
Fig 6.
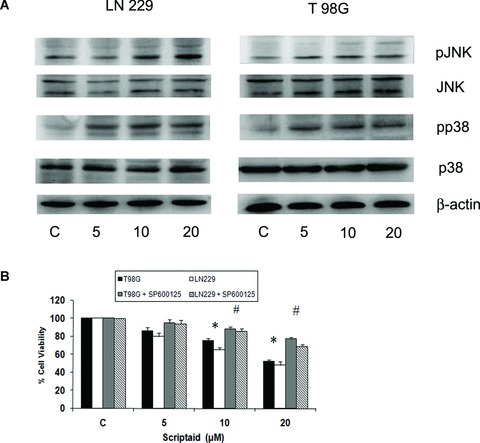
The pro-apoptotic effect of scriptaid is mediated by JNK. (A) Scriptaid increases p38MAPK and JNK phosphorylation in glioma cells. Western blot analysis was performed to detect the expression of p38MAPK, phosphop38MAPK, JNK and pJNK in glioma cells treated with increasing concentration of scriptaid. Representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading. (B) Scriptaid-induced cell death is significantly attenuated in the presence of JNK inhibitor. The graph represents the percentage of viable cells as determined by MTS assay, observed when glioma cells were treated with scriptaid in the presence or absence of JNK inhibitor. Cells were treated with 20 μM scriptaid alone or in combination with 20 μM SP600125 for 24 hrs. Values represent the means ± SEM from three independent experiments. * Significant change from control; #significant change from scriptaid-treated cells (P≤ 0.05).
As p38MAPK and JNK activity was elevated in scriptaid-treated cells, the viability of scriptaid-treated glioma cells in the presence and absence of SB203580 and SP600125-respective inhibitors of p38MAPK and JNK was determined. Treatment of glioma cells with scriptaid in the presence of SB203580 had no effect on scriptaid-induced glioma cell death (data not shown). Although inhibition of p38MAPK had no effect in reversing scriptaid-induced cell death, JNK inhibition significantly decreased scriptaid-induced apoptosis (Fig. 6B). However, inhibition of JNK activity was unable to completely reverse the anti-proliferative effect of scriptaid. Although treatment of T98G cells with 5, 10 and 20 μM scriptaid decreased cell viability by 86, 75 and 52%, respectively, the viability increased to 94, 86 and 69% in cells co-treated with increasing concentration of scriptaid in the presence of JNK inhibitor (Fig. 6B). This suggested the involvement of pathways other than that mediated by increased JNK activation, which triggers the anti-proliferative effect of scriptaid. As we have recently reported that ERK activation plays an active role in mediating Miltefosine-induced apoptosis of glioma cells [11], we determined the status of ERK phosphorylation in scriptaid-treated cells. Treatment with scriptaid had no effect on ERK phosphorylation, which remained comparable to control levels (data not shown).
Scriptaid-induced changes in cell-cycle regulatory and DNA damage response repair proteins are JNK dependent
Inhibition of tumour cell-cycle arrest through JNK activation has been reported [34]. Because JNK activation is involved in scriptaid-induced glioma cell death, we investigated whether JNK pathway modulate cell-cycle regulatory proteins by determining p21 and p27 expression in glioma cells treated with scriptaid in the presence and absence of JNK inhibitors. The increased p21 and p27 expression observed upon scriptaid treatment was reversed in the presence of JNK inhibitor (Fig. 6). Ras induces DNA damage signalling response [28] and JNK activation radiosensitizes colon carcinoma cells through enhanced DNA damage [35]. Because increased Ras and JNK activation was observed in scriptaid-treated cells, we determined the expression of γ-H2AX in cells treated with scriptaid in the presence and absence of JNK inhibitors. The elevated levels of γH2AX observed in scriptaid-treated cells were abrogated to control levels in the presence of JNK inhibitor (Fig. 7). The increase in pJNK levels observed in scriptaid-treated cells was decreased to control levels in the presence of JNK inhibitor. These results indicate the involvement of JNK in scriptaid-induced DSB response in glioma cells.
Fig 7.
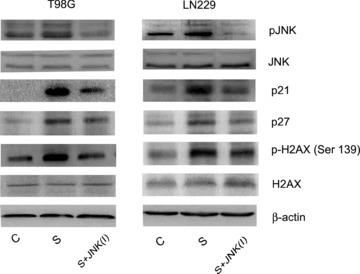
Scriptaid-mediated DNA damage response and alteration in cellcycle regulatory molecules in glioma cells are JNK dependent. Glioma cells were treated with 20 μM scriptaid in the presence or absence of 20 μM JNK inhibitor SP600125, and Western blot analysis was performed. A decrease in pJNK, p21, p27 and H2AX expression respectively was observed in cells treated with scriptaid in the presence of JNK inhibitor, as compared to those treated with Scripatid alone. Representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading.
Scriptaid-mediated suppression of telomerase activity in glioma cells is JNK independent
Telomerase is composed primarily of the catalytic subunit (hTERT) and the RNA template (hTERC), and hallmarks of telomere dysfunction include chromosome end fusion and telomere shortening, which can lead to cell-cycle arrest. Telomerase inhibition is also known to cause apoptosis in human cancers [36]. HDAC inhibitors have been reported to suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells [14]. Besides, JNK is a key regulator of telomerase activity [15]. Because the pro-apoptotic effects of scriptaid are partially mediated by JNK, we investigated the ability of scriptaid to effect telomerase activity in the presence and absence of JNK inhibitor. An approximate 50% reduction in telomerase activity was observed in both LN229 and T98G cells, upon treatment with 20 μM scriptaid (Fig. 8). However, scriptaid-mediated decrease in telomerase activity was not JNK mediated as telomerase activity observed in scriptaid-treated cells was unaffected in the presence of JNK inhibitor (Fig. 8). As inhibition of telomerase activity has been suggested to cause cell death in glioma [16], the ability of scriptaid to abrogate telomerase activity may subsequently result in reduced glioma cell proliferation and cell death.
Fig 8.
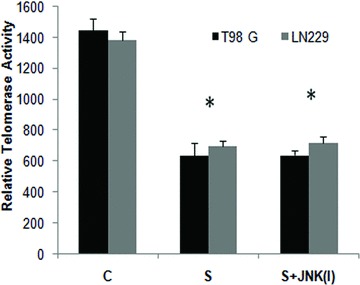
Scriptaid decreases telomerase activity in glioma cells in a JNKindependent manner. Treatment with scriptaid decreases telomerase activity of glioma cells. Glioma cells were treated with 20 μM scriptaid in the presence or absence of 20 μM JNK inhibitor SP600125, and TeloTAGGG Telomerase PCR ELISA was performed. The decrease in telomerase activity observed in scriptaid-treated cells was unaffected by JNK inhibitor. Values represent the means ± SEM from three independent experiments. *Significant decrease from control (P≤ 0.05).
Discussion
HDACs play an important role in the epigenetic regulation of gene expression, and aberrant epigenetic changes are a hallmark of cancer. As a consequence, the efficacy of HDAC inhibitors as promising candidates for cancer therapy has been extensively evaluated for a wide range of malignancies. Although HDAC inhibition is known to promote growth arrest in glioma cells [2–4], no studies have evaluated the effect of scriptaid on glioma cell proliferation. We therefore evaluated the therapeutic potential of HDAC inhibitor scriptaid in the treatment of glioblastoma and investigated its mechanisms of action.
Although the high levels of active Ras has been a target for Ras inhibitor-mediated glioma therapy [37], we have shown that Miltefosine induces apoptosis in glioma cells by increasing Ras/ERK activity [11]. Here, we report that scriptaid-induced glioma cell apoptosis is Ras dependent as overexpression of constitutive Ras enhances scriptaid-induced apoptosis. Although scriptaid elevated both p38MAPK and JNK phosphorylation, it was the inhibition of JNK activation that prevented scriptaid-induced glioma cell apoptosis to a considerable extent. The ability of enhanced JNK activity to amplify the intensity of scriptaid-mediated apoptosis is in line with previous findings that JNK activation enhances apoptosis of transformed cells [38].
HDAC inhibitors have been reported to increase γH2AX expression in leukaemic cells [26], and H2AX is a target of JNK signalling pathway required for apoptotic DNA fragmentation [39]. H2AX is also involved in cellular apoptosis as JNK/H2AX pathway cooperates with the caspase-3 pathway to induce apoptosis [40]. It is possible that scriptaid-mediated JNK-dependent increase in H2AX expression co-operates with increased caspase-3 to induce apoptosis.
HDAC inhibitor suppresses telomerase activity in prostate cancer cells [14] and JNK is a key regulator of telomerase activity [15]. Moreover, dysfunctional telomeres activates DNA damage checkpoint [41] and cellular response to telomere dysfunction is governed by proteins such as γ-H2AX [42]. We also report for the first time that scriptaid inhibits telomerase activity in JNK-independent manner. The ability of scriptaid to inhibit telomerase activity may lead to telomere dysfunction and might contribute to apoptosis. The telomerase activity inhibition, coupled with its ability to induce apoptosis and regulate DNA damage response by scriptaid, warrants further investigation of its role as a promising therapeutic strategy in GBM.
Acknowledgments
This work was supported by a research grant from the Department of Biotechnology (DBT, BT/PR/7047/Med/14/934/2006), Government of India, to ES. VS, NK and DD are recipients of a research fellowship from Council of Scientific and Industrial Research, Government of India. The authors thank Mr. Uttam Kumar Saini for technical assistance.
References
- 1.Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 2.Gensert JM, Baranova OV, Weinstein DE, et al. CD81, a cell cycle regulator, is a novel target for histone deacetylase inhibition in glioma cells. Neurobiol Dis. 2007;26:671–80. doi: 10.1016/j.nbd.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Carroll RS. Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J Neurooncol. 2007;83:267–75. doi: 10.1007/s11060-007-9337-z. [DOI] [PubMed] [Google Scholar]
- 4.Yin D, Ong JM, Hu J, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res. 2007;13:1045–52. doi: 10.1158/1078-0432.CCR-06-1261. [DOI] [PubMed] [Google Scholar]
- 5.Su GH, Sohn TA, Ryu B, et al. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res. 2000;60:3137–42. [PubMed] [Google Scholar]
- 6.Takai N, Ueda T, Nishida M, et al. A novel histone deacetylase inhibitor, scriptaid, induces growth inhibition, cell cycle arrest and apoptosis in human endometrial cancer and ovarian cancer cells. Int J Mol Med. 2006;17:323–9. [PubMed] [Google Scholar]
- 7.Dai Y, Rahmani M, Dent P, et al. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25:5429–44. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia S, Li Y, Rosen EM, et al. Ribotoxic stress sensitizes glioblastoma cells to death receptor induced apoptosis: requirements for c-Jun NH2-terminal kinase and Bim. Mol Cancer Res. 2007;5:783–92. doi: 10.1158/1541-7786.MCR-06-0433. [DOI] [PubMed] [Google Scholar]
- 9.Jin HO, Park IC, An S, et al. Up-regulation of Bak and Bim via JNK downstream pathway in the response to nitric oxide in human glioblastoma cells. J Cell Physiol. 2006;206:477–86. doi: 10.1002/jcp.20488. [DOI] [PubMed] [Google Scholar]
- 10.Minden A, Lin A, McMahon M, et al. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–23. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 11.Tewari R, Sharma V, Koul N, et al. Involvement of Miltefosine mediated ERK activation in glioma cell apoptosis through Fas regulation. J Neurochem. 2008;107:616–27. doi: 10.1111/j.1471-4159.2008.05625.x. [DOI] [PubMed] [Google Scholar]
- 12.Nesterov A, Nikrad M, Johnson T, et al. Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2004;64:3922–7. doi: 10.1158/0008-5472.CAN-03-2219. [DOI] [PubMed] [Google Scholar]
- 13.Klampfer L, Huang J, Shirasawa S, et al. Histone deacetylase inhibitors induce cell death selectively in cells that harbor activated kRasV12: the role of signal transducers and activators of transcription 1 and p21. Cancer Res. 2007;67:8477–85. doi: 10.1158/0008-5472.CAN-07-0210. [DOI] [PubMed] [Google Scholar]
- 14.Suenaga M, Soda H, Oka M, et al. Histone deacetylase inhibitors suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells. Int J Cancer. 2002;97:621–5. doi: 10.1002/ijc.10082. [DOI] [PubMed] [Google Scholar]
- 15.Alfonso-De Matte MY, Yang H, Evans MS, et al. Telomerase is regulated by c-Jun NH2-terminal kinase in ovarian surface epithelial cells. Cancer Res. 2002;62:4575–8. [PubMed] [Google Scholar]
- 16.Mukai S, Kondo Y, Koga S, et al. 2–5A antisense telomerase RNA therapy for intracranial malignant gliomas. Cancer Res. 2000;60:4461–7. [PubMed] [Google Scholar]
- 17.Khaw AK, Silasudjana M, Banerjee B, et al. Inhibition of telomerase activity and human telomerase reverse transcriptase gene expression by histone deacetylase inhibitor in human brain cancer cells. Mutat Res. 2007;625:134–44. doi: 10.1016/j.mrfmmm.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Sharma V, Tewari R, Sk UH, et al. Ebselen sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through two distinct pathways involving NF-kappaB downregulation and Fas-mediated formation of death inducing signaling complex. Int J Cancer. 2008;123:2204–12. doi: 10.1002/ijc.23771. [DOI] [PubMed] [Google Scholar]
- 19.Sharma V, Joseph C, Ghosh S, et al. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol Cancer Ther. 2007;6:2544–53. doi: 10.1158/1535-7163.MCT-06-0788. [DOI] [PubMed] [Google Scholar]
- 20.Rothfuss A, Schutz P, Bochum S, et al. Induced micronucleus frequencies in peripheral lymphocytes as a screening test for carriers of a BRCA1 mutation in breast cancer families. Cancer Res. 2000;60:390–94. [PubMed] [Google Scholar]
- 21.Kumar S, Lavin MF. The ICE family of cysteine proteases as effectors of cell death. Cell Death Differ. 1996;3:255–67. [PubMed] [Google Scholar]
- 22.Gui CY, Ngo L, Xu WS, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumagai T, Wakimoto N, Yin D, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121:656–65. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 24.Gartel AL, Ye X, Goufman E, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA. 2001;98:4510–15. doi: 10.1073/pnas.081074898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menssen A, Epanchintsev A, Lodygin D, et al. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: Identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle. 2007;6:339–52. doi: 10.4161/cc.6.3.3808. [DOI] [PubMed] [Google Scholar]
- 26.Scuto A, Kirschbaum M, Kowolik C, et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008;111:5093–100. doi: 10.1182/blood-2007-10-117762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 28.Weidhaas JB, Eisenmann DM, Holub JM, et al. A conserved RAS/mitogen-activated protein kinase pathway regulates DNA damage-induced cell death postirradiation in Radelegans. Cancer Res. 2006;66:10434–8. doi: 10.1158/0008-5472.CAN-06-2182. [DOI] [PubMed] [Google Scholar]
- 29.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–8. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaari S, Jacob-Hirsch J, Amariglio N, et al. Disruption of cooperation between Ras and MycN in human neuroblastoma cells promotes growth arrest. Clin Cancer Res. 2005;11:4321–30. doi: 10.1158/1078-0432.CCR-04-2071. [DOI] [PubMed] [Google Scholar]
- 31.Klampfer L, Huang J, Sasazuki T, et al. Oncogenic Ras promotes butyrate-induced apoptosis through inhibition of gelsolin expression. J Biol Chem. 2004;279:36680–8. doi: 10.1074/jbc.M405197200. [DOI] [PubMed] [Google Scholar]
- 32.De Rooij J, Bos JL. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene. 1997;14:623–5. doi: 10.1038/sj.onc.1201005. [DOI] [PubMed] [Google Scholar]
- 33.Chen CY, Liou J, Forman LW, et al. Differential regulation of discrete apoptotic pathways by Ras. J Biol Chem. 1998;273:16700–9. doi: 10.1074/jbc.273.27.16700. [DOI] [PubMed] [Google Scholar]
- 34.Shishodia S, Sethi G, Ahn KS, et al. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem Pharmacol. 2007;74:118–30. doi: 10.1016/j.bcp.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou H, Adachi M, Imai K, et al. 2-Methoxyestradiol, an endogenous mammalian metabolite, radiosensitizes colon carcinoma cells through c-Jun NH2-terminal kinase activation. Clin Cancer Res. 2006;12:6532–9. doi: 10.1158/1078-0432.CCR-06-0678. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Mar V, Zhou W, et al. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999;13:2388–99. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldberg L, Kloog Y. A Ras inhibitor tilts the balance between Rac and Rho and blocks phosphatidylinositol 3-kinase-dependent glioblastoma cell migration. Cancer Res. 2006;66:11709–17. doi: 10.1158/0008-5472.CAN-06-1878. [DOI] [PubMed] [Google Scholar]
- 38.Adhikari S, Bhatia M. H(2)S-induced pancreatic acinar cell apoptosis is mediated via JNK and p38 MAP Kinase. J Cell Mol Med. 2008;12:1374–83. doi: 10.1111/j.1582-4934.2008.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sluss HK, Davis RJ. H2AX is a target of the JNK signaling pathway that is required for apoptotic DNA fragmentation. Mol Cell. 2006;23:152–3. doi: 10.1016/j.molcel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 42.Takai H, Smogorzewska A, De Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]