

Abstract
CCM3, a product of the cerebral cavernous malformation 3 or programmed cell death 10 gene (CCM3/PDCD10), is broadly expressed throughout development in both vertebrates and invertebrates. Increasing evidence indicates a crucial role of CCM3 in vascular development and in regulation of angiogenesis and apoptosis. Furthermore, loss of CCM3 causes inherited (familial) cerebral cavernous malformation (CCM), a common brain vascular anomaly involving aberrant angiogenesis. This study focused on signalling pathways underlying the angiogenic functions of CCM3. Silencing CCM3 by siRNA stimulated endothelial proliferation, migration and sprouting accompanied by significant downregulation of the core components of Notch signalling including DLL4, Notch4, HEY2 and HES1 and by activation of VEGF and Erk pathways. Treatment with recombinant DLL4 (rhDLL4) restored DLL4 expression and reversed CCM3-silence-mediated impairment of Notch signalling and reduced the ratio of VEGF-R2 to VEGF-R1 expression. Importantly, restoration of DLL4-Notch signalling entirely rescued the hyper-angiogenic phenotype induced by CCM3 silence. A concomitant loss of CCM3 and the core components of DLL4-Notch signalling were also demonstrated in CCM3-deficient endothelial cells derived from human CCM lesions (CCMEC) and in a CCM3 germline mutation carrier. This study defined DLL4 as a key downstream target of CCM3 in endothelial cells. CCM3/DLL4-Notch pathway serves as an important signalling for endothelial angiogenesis and is potentially implicated in the pathomechanism of human CCMs.
Keywords: CCM3/PDCD10, DLL4-Notch signalling, angiogenesis, endothelium, cerebral cavernous malformation
Introduction
Cerebral cavernous malformation (CCM), classified as sporadic and familial (inherited) forms, is one of the most common cerebral vascular anomalies involving aberrant angiogenesis. Loss-of-function mutations in one of the three CCM genes, namely CCM1/KRIT1, CCM2/MGC4607 and CCM3/PDCD10, predispose to CCM and are associated with up to 90% of familial CCM [1]. Of these three genes, CCM3 is the most recently discovered and this gene is highly conserved in both vertebrates and invertebrates [2]. CCM3 is also referred as PDCD10 (programmed cell death 10) owing to the up-regulation of its mRNA expression by apoptotic stimuli in vitro. It has been known that CCM3 is broadly expressed, including in neuronal and endothelial cells [3, 4]. Despite its neuronal expression pattern and the discovery of vascular pathology after targeted deletion of Ccm3 in murine neuroglia [5], intensive studies have been carried out for identifying endothelial functions of these CCM genes. Indeed, it has recently been recognized that human CCM3 mutation carriers display an earlier symptom onset already before 15 years of age and a higher risk for cerebral haemorrhage during childhood [6, 7]. This genotype–phenotype association has raised high interest for researchers to address the detailed functions and signalling pathways underlying CCM3. We have recently shown that silencing CCM3 stimulated angiogenesis with the most prominent effect on endothelial sprouting and tube branching [8]. In contrast, overexpression of CCM3 inhibited endothelial proliferation, migration and tube formation [9]. Endothelia-specific deletion of Ccm3 in mice led to lethal embryos associated with angiogenesis defects and with disruption of vascular integrity [10, 11]. Cardiac and cranial vascular defects were also found in Ccm3-deficient zebrafish [12, 13]. Furthermore, endothelial loss of heterozygosity for Ccm3 in postnatal mice resulted in vascular lesions that resemble typical human CCM [10]. These data indicate that CCM3-deficiency impairs vascular development/maturation, activates angiogenesis and causes CCM-like lesion.
The presence of CCM3 in the protein complex of CCM1-CCM2 has been shown, suggesting common or related pathways for these three proteins [13, 14]. Additional distinct signalling pathways underlying the angiogenic functions of CCM3 have also been demonstrated [10–12, 15, 16]. CCM3 binds the GCK-III family of sterile 20-like serine/threonine kinases STK24, STK25 and MST4 [13, 15, 17–19]. Of note, this signalling context is essential for vascular development in zebrafish [13, 16] and for cell survival after oxidative stress [20]. CCM3 has also been shown to bind paxillin [21] and membrane protein VEGF-R2 [11] and to interact with heart of glass homolog1 (HEG1) [22], thereby, respectively, regulating cell adhesion, angiogenesis and vascular integrity.
Notch signalling is an evolutionarily conserved pathway that is indispensable for cell-fate determination and vascular development [23]. In mammals, four Notch receptors (Notch1-4) and five ligands (DLL1, DLL3-4 and Jagged1-2) have been identified [24]. DLL4 is the most recently identified Notch ligand and also the sole Notch ligand expressed predominantly by the vascular endothelium. In vitro studies have shown that DLL4 is readily able to signal through each of the four human Notch receptors [24]. Regulation of endothelial sprouting is one of the most predominant roles of DLL4-Notch signalling. Activation of DLL4-Notch signalling inhibits excessive tip-cell formation and sprouting in cultured cells, in animal embryos and during tumour angiogenesis [25]. Conversely, loss of DLL4 expression causes dramatic increase in sprouting and branching as a result of excessive tip-cell formation and endothelial proliferation [26]. Therefore, DLL4-Notch is defined as a negative pathway regulating angiogenesis. Indeed, DLL4-Notch pathway has also been implicated in controlling post-angiogenic blood vessel remodelling and to modulate vasoconstriction and blood flow [27]. Interestingly, down-regulation of DLL1 and Notch4 was detected in Ccm1-deficient mouse embryos [1, 28]. Overexpression of CCM1 inhibited sprouting angiogenesis by activating Notch signalling [29].
We have recently studied the angiogenic properties of CCM1-3 genes in different types of endothelial cells and noted that CCM3 silence induced the most potent angiogenic phenotype in comparison with that induced by silencing CCM1 or CCM2 [8, 30]. The distinct and massive angiogenic phenotype caused by CCM3 silence is in accordance with the clinical observation that CCM3 patients showed the most aggressive presentations according to genotype–phenotype analysis [8, 31]. It is noteworthy that the hyper-angiogenic phenotype, including increase in proliferation, migration and particularly massive sprouting and tube brunching, mediated by CCM3 silence was entirely mimetic that induced by inhibition of Dll4-Notch signalling [26, 31]. We have therefore suggested in this study that CCM3 deficiency stimulated endothelial angiogenesis through impairing DLL4-Notch signalling. To address this hypothesis, we studied whether and which core components as well as the potential downstream pathways underlying DLL4-Notch signalling were affected by CCM3 silencing. Furthermore, we investigated whether modulation of endothelial DLL4-Notch signalling was able to rescue the angiogenic phenotype and reverse the altered cellular signalling caused by CCM3 silencing. Finally, the pathological relevance of CCM3 deficiency and DLL4-Notch signalling was further strengthened by the studies using CCM-derived endothelial cells (CCMEC) as well as human CCM specimens.
Materials and methods
Cell culture and CCM gene silencing
Human umbilical vein endothelial cells (HUVECs, Promocell GmbH, Heidelberg, Germany) and human brain microvessel endothelial cells (HBMECs, Provitro GmbH) were cultured according to the manufacture's protocols. Human CCM lesion-derived endothelial cells (CCMECs) were prepared from the surgical specimens of sporadic CCM and cultured as described previously [8, 31]. The diagnosis of sporadic CCM was based on the specific characteristics of magnetic resonance imaging (MRI), multiplicity of the lesion, familial history and genetic background. CCM3 silence was achieved by transfection of the endothelial cells with specific siRNA targeting CCM3 genes (siCCM3; Applied Biosystems/Ambion, Darmstadt, Germany) using established protocols [8, 30]. The Negative control siRNA (Neg. C, Applied Biosystems/Ambion) is comprised of a 19 bp scrambled sequence with 3′dT overhands. The sequence has no significant homology to any known gene sequence from human. The efficiency of CCM3 silencing was confirmed by real-time reverse transcription polymerase chain reaction (RT2-PCR) in all experiments when siRNA transfection was concerned.
Human brain specimens
The diagnosis of CCMs was based on specific characteristics of magnetic resonance imaging, histopathological criteria and genetic background. Operative specimens from six sporadic CCMs including three female and three male with mean age of 42 ± 12 years were used as control. The familial CCM case was a 9-year-old boy and had been shown to carry a frameshift mutation in exon seven of the CCM3 gene (c.350_351insT, p.D118RfsX2) [14, 32]. All participants enrolled in the study provided informed consent. The experimental protocol was approved by the local ethics committee.
Real-time RT-PCR (RT2-PCR)
The RNA extraction, cDNA synthesis, preparation of PCR reaction mixture and the PCR settings were carried out as described previously [30]. Primer sequences and annealing temperature for PCR were shown in Table 1. GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) was stably detected in cell and tissues samples under the conditions of this study and was thus selected as reference gene. Relative mRNA expression of the target gene (fold of change) for each sample was quantified using the cycle threshold (Ct) approach, normalized to the reference gene.
Table 1.
Primer sequences and annealing temperatures for real-time RT-PCR
| Primer | Sequence | T* (°C) | |
|---|---|---|---|
| CCM1 | For. | TGA AGG AAG CAA TTA ACA AAC CA | 60 |
| Rev. | GAG AGA CGC ATT CCT TCC AT | ||
| CCM2 | For. | CCC TGT CGG AGA GTG CAG | 59 |
| Rev. | AGC AGA CAG CAA AGC TCC TC | ||
| CCM3 | For. | TGG CAG CTG ATG ATG TAG AAG | 58 |
| Rev. | TCG TGC CTT TTC GTT TAG GT | ||
| DLL4 | For. | GCG GGG TAC CTT CTC GCTCAT CATC | 60 |
| Rev. | GCC TCC CCA GCC CTC ATC ACA AGT A | ||
| Jagged1 | For. | TCG CTG TAT CTG TCC ACC TG | 60 |
| Rev. | AGT CAC TGG CAC GGT TGT AG | ||
| Notch1 | For. | CAG GCA ATC CGA GGA CTA TG | 60 |
| Rev. | CAG GCG TGT TGT TCT CAC AG | ||
| Notch4 | For. | TCC TGG GGC CCG GGC TGA AGA AAA G | 58 |
| Rev. | ACG CCG GAT GAG CTG GAG GAC GAG A | ||
| HEY2 | For. | GTA CCA TCC AGC AGT GCA TC | 60 |
| Rev. | AGA GAA TTC AGT CAG GGC ATT T | ||
| HES1 | For. | AGT GAA GCA CCT CCG GAA C | 60 |
| Rev. | CGT TCA TGC ACT CGC TGA | ||
| VEGF | For. | GAA GTG GTG AAG TTC ATG GAT GT | 60 |
| Rev. | TGG AAG ATG TCC ACC AGG GTC | ||
| VEGF-R1 | For. | AGC TCC GGC TTT CAG GAA GAT A | 58 |
| Rev. | GAC AGG AAC TCC ATG CCT CTG | ||
| VEGF-R2 | For. | CTC TTG GCC GTG GTG CCT TTG | 58 |
| Rev. | GTG TGT TGC TCC TTC TTT CAA C | ||
| GAPDH | For. | AGC CAC ATC GCT CAG ACA | 58 |
| Rev. | GCC CAA TAC GAC CAA ATC C | ||
T: Annealing temperature; for, forward; rev, reverse.
Western blotting
Total protein extraction and the electrophoresis were performed as described previously [33]. The blots were incubated at 4°C overnight with the following first antibodies: DLL4, p-Erk1/2, p-Akt and GAPDH (each 1:1000 dilution, Cell Signaling, Frankfurt am Main, Germany); Notch4 [1:200, specifically detect the cleaved domain (active form) of the protein, Santa-Cruz Technology, Heidelberg, Germany], VEGF (1:1000; Abcam, Cambridge, UK), Hey1 (1:200, Abcam), CCM3 (1:400; Atlas Antibodies, Stockholm, Sweden) and actin (1:1000; Sigma-Aldrich, Seelze, Germany). After incubation with the corresponding HRP-conjugated secondary antibody, the signal was produced by enhanced chemiluminescence detection reagents. To semi-quantify the blot, integrate optical density (IOD) of the individual blot was measured using Image J software. The IOD ratio of the target protein to the housekeeping protein GAPDH was calculated and the relative expression of the target protein was normalized to the percentage of the control.
Immunofluorescence
Cells were fixed with methanol at −20°C for 20 min. followed by immunofluorescent staining as previously described [31]. For paraffin-embedded brain sections, immunofluorescent staining of CCM3, DLL4 and vWF was simultaneously performed on the adjacent sections. After deparaffinization, the non-specific binding was blocked by incubation of sections with the blocking buffer [31]. The slices were then incubated with respective rabbit anti-human CCM3 (1:65, Atlas Antibodies) or rabbit anti-human DLL4 (1:100, AbD Serotec, Düsseldorf, Germany) or rabbit anti-human vWF (1:200, Dako, Hamburg, Germany) at 4°C overnight. Negative control sections were incubated with non-immune rabbit IgG at the concentrations identical to the primary antibodies. The immunoreactivity of CCM3 was detected using biotinylated goat anti-rabbit IgG followed by incubation with fluorescein avidin D. The signal for the staining of DLL4 and vWF was induced by the incubation of the slices with a texas-red conjugated anti-rabbit IgG. Counterstaining was performed with Hoechst-33258. The sections were analysed by using a fluorescence microscope (Olympus BX51, Hamburg, Germany). Images were acquired using an Olympus DP 70 camera driven by Olympus software.
Proliferation and migration assay
The cell proliferation was detected by WST-1 assay as described previously [31]. For migration assay, cells were seeded onto culture dishes pre-coated with 0.2% gelatin. When the cells reached 95–100% confluence, a thin stripe of cells was scratched by a sterile 1000-μl pipette tip. Six fields per culture dish (4 dishes/group) were randomly taken for photograph and the number of the migrated cells in each field was blindly counted.
Endothelial sprouting assay in 3D gel
The sprouting assay was performed according to Nakatsu et al. [34] with modifications. Cells were mixed with Cytodex 3 beads in a ratio of 400 cells per bead followed by the incubation overnight. After washing, the beads were resuspended in a medium supplemented with 2.5 mg/ml of fibrinogen and 0.15 units/ml of aprotinin. The mixture was quickly added to the well of a 96-well plate containing 0.625 U/ml of thrombin to form clot. The clots were overlaid with 100 μl medium containing 10 ng/ml of VEGF (R&D Systems, Wiesbaden-Nordenstadt, Germany) and 0.15 units/ml of aprotinin. Sprouting was monitored and photographed under a microscope. Length of sprouts was measured in 20 randomly chosen beads of each group using Leica Application Suite software.
Recombinant human DLL4 (rhDLL4) treatment
Cell culture plate was pre-coated with 0.2% gelatin containing different concentrations of rhDLL4 (R&D System) as indicated in individual experiments or the same volume of vehicle (0.1% BSA).
Statistics
Data were presented as mean and standard deviation (means ± SD). Statistical analysis was performed with the WinSTAT program. Differences between multiple groups were analysed using anova followed by the Scheffé test. P value less than 0.05 was considered statistically significant.
Results
Characterization of the angiogenic phenotype after silencing CCM3
To ensure a sufficient time window of CCM3 silence for endothelial behaviour study and for further signalling study, we examined the time course of CCM3 expression after transfection. RT2-PCR revealed a 70%, 90%, 80% and 60% of reduction in CCM3 expression at 24 hrs (P < 0.01), 48 hrs (P < 0.001), 72 hrs (P < 0.001) and 96 hrs (P < 0.01), respectively, after siCCM3 transfection (Fig. 1A). Under this established silencing condition, we then characterized the angiogenic phenotype of endothelial cells. Silencing CCM3 significantly stimulated all tested angiogenic behaviours including endothelial proliferation (Fig. 1B), migration (Fig. 1C) and sprouting (Fig. 1D).
Fig. 1.
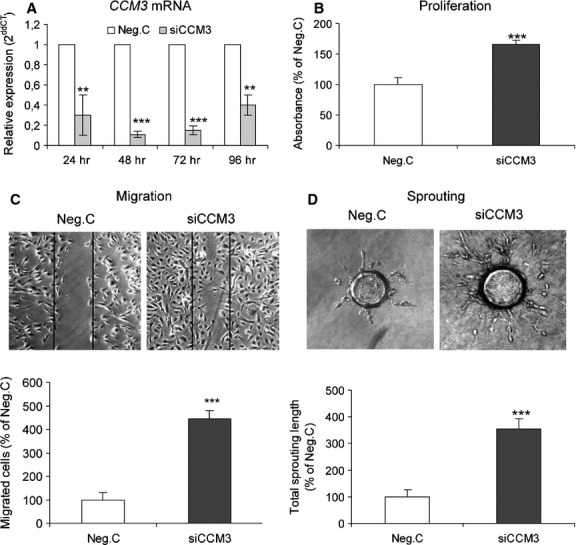
Silencing CCM3-activated angiogenesis. (A) The time course of CCM3 silence. HUVECs were transfected with 70 nM of specific CCM3 siRNA (siCCM3) or with a control siRNA (Neg. C) for different time periods as indicated. The expression of CCM3 was detected by RT2-PCR. Based on this time course, angiogenic assays (B–D) were accordingly performed 72 hrs after the transfection. (B) Silencing CCM3-stimulated endothelial proliferation. HUVEC received 70 nM of siCCM3 or Neg. C. Cell proliferation was detected by using the WST-1 proliferating reagent. (C) Silencing CCM3 promoted cell migration. The cell migration was recorded 10 hrs after scratching and the number of migrated cells was counted randomly in six fields per culture dish. The data were presented as means of four dishes per group. (D) Silencing CCM3 resulted in massive angiogenic sprouting in 3D fibrin gel. The total length of sprouts of 20 beads randomly selected from each group was blindly quantified by using Leica Application Suite software. All data were representative of at least three independent experiments. **P < 0.01 and ***P < 0.001, compared with Neg. C.
Inactivation of DLL4-Notch signalling in CCM3-silenced endothelial cells and in a surgical specimen of familial CCM harbouring a CCM3 germline mutation
To control the efficiency of silencing CCM3 and to rule out unspecific effect of siCCM3 transfection to CCM1 and CCM2, we examined the expression of CCM1, CCM2 and CCM3 in parallel in siCCM3 transfected cells. As shown in Figure 2A, the expression of CCM3, but not of CCM1 and CCM2, was significantly and specifically silenced by the siCCM3 transfection. Thereafter, the core components of DLL4-Notch signalling were examined in the same RNA extracts as used for CCM1-3 detection. Interestingly, transfection of siCCM3 caused a five- and threefold decrease in DLL4 and Notch4 mRNA expression, respectively, but no influence on Notch1 expression. Down-regulation of DLL4-Notch signalling was further proven by detection of 60% and 40% decrease in the levels of target genes HEY2 and HES1, respectively, in CCM3-silenced HUVEC. Western blot confirmed a more than 10- and 3-fold reduction in the protein levels DLL4 and Notch4 (cleaved/active form) when CCM3 was down-regulated to 20% of the control (Fig. 2B). Immunofluorescent staining confirmed a reduced DLL4 immunoreactivity in CCM3-silenced cells (Fig. 2C).
Fig. 2.
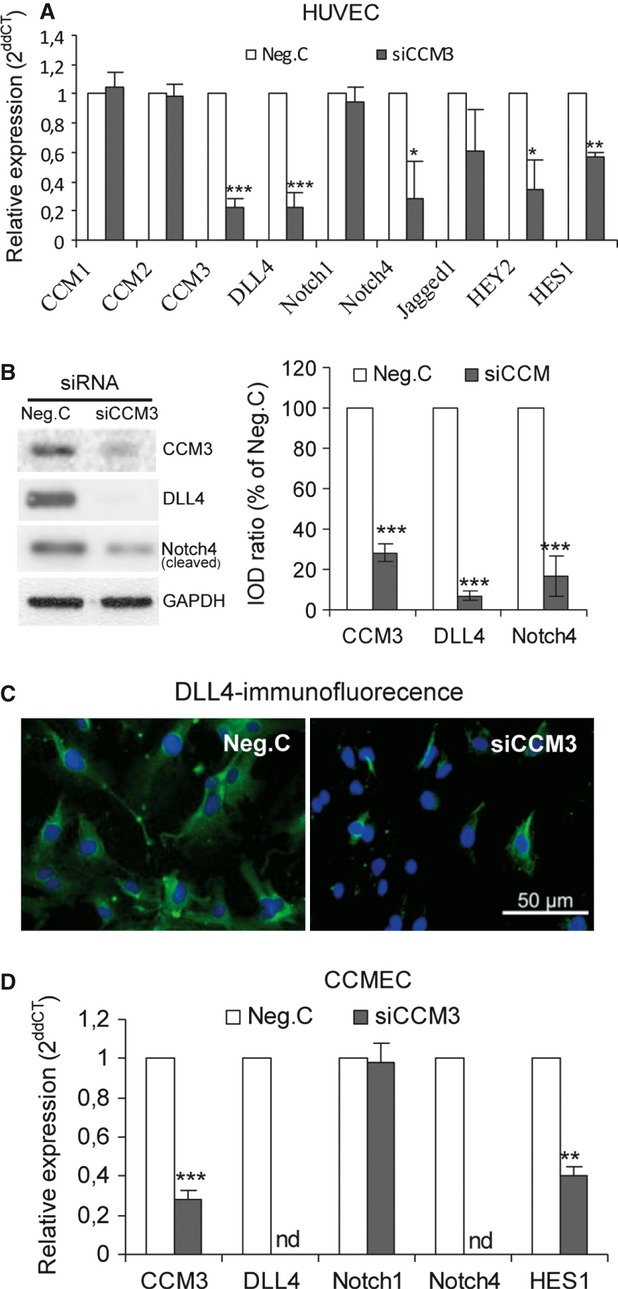
Silencing CCM3 in cultured endothelial cells down-regulated the core components of DLL4-Notch signalling. Cells were transfected with 70 nM of siCCM3 or Neg. C. Total RNA or protein was, respectively, extracted 72 hrs after the transfection. (A) The mRNA expression of core components of Notch signalling after silencing CCM3 in HUVEC. siCCM3 transfection efficiently silenced CCM3 without influencing the expression of CCM1 and CCM2. Under this experiment conditions, silencing CCM3 down-regulated the expression of DLL4, Notch4 and HEY2 and HES1. (B) The down-regulation of DLL4 and Notch4 in CCM3-silenced HUVEC was confirmed by Western blot. To semi-quantify the blot, integrate optical density (IOD) of the individual blot was measured using ImageJ software. The IOD ratio of the target protein to the housekeeping protein actin was calculated and the relative expression of the target protein was normalized to the percentage of the control (Neg. C). (C) Immunofluorescent staining of DLL4. HUVEC was transfected with 70 nM of siCCM3 or Neg. C for 72 hrs. Immunostaining of DLL4 (green) was performed followed by Hoechst 33258 counterstaining (blue). Scale bar: 50 μm. (D) A significant down-regulation of DLL4, Notch4 and HES1 was detected in CCM3-silenced endothelial cells derived from the lesion of human sporadic CCM (CCMEC). The expression of DLL4 and Notch4 was reduced to a non-detectable level (nd) in CCM3-silenced CCMEC, whereas the mRNA level of Notch1 was not altered. All data were representative of at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, compared with Neg. C.
To find out the pathological relevance between CCM3 deficiency and down-regulation of DLL4-Notch signalling, the expression of core components of the DLL4-Notch pathway was further studied in the endothelial cells derived from human CCM lesion (CCMEC) and in surgical specimens of CCMs. As described previously, CCMEC was derived from sporadic CCMs and has been well-established in our laboratory[8, 31]. Silencing CCM genes in CCMEC has been used as a useful in vitro model for analysing CCM gene function and the underlying signalling pathways [8, 31]. As shown in Figure 2D, silencing CCM3 dramatically down-regulated the expression of DLL4 and Notch4 to an undetectable level, and concomitantly accompanied by 60% of reduction in the target gene HES1. It was noted that Notch1 gene expression was not significantly altered by silencing CCM3 in either CCMEC (Fig. 2D) or HUVEC (Fig. 2A).
Next, we examined DLL4-Notch signalling in human CCMs. Because of the extremely rare availability, only one human surgical specimen of a CCM3 germline mutation carrier could be obtained during this study. To draw a more convinced comparison between familial and sporadic CCMs, we used six surgical specimens from sporadic CCM as control. RT2-PCR detected around 5-, 38- and 35-fold decrease in CCM3, DLL4 and Notch4 expression, respectively, in the CCM3-deficient cavernous lesion (Mu-CCM3) in comparison with the human sporadic CCMs (Sp-CCM). A dramatic down-regulation of the target gene HEY2 and HES1 was also observed in Mu-CCM3, indicating an impaired DLL4-Notch signalling in familial CCM with germline mutation in CCM3. Interestingly, the level of Notch1 was similarly detected in Mu-CCM3 and in Sp-CCMs (Fig. 3A). These data drawn from CCM patients were entirely consistent with the results obtained from CCM3-silenced HUVEC (Fig. 2A) and CCMEC (Fig. 2D).
Fig. 3.
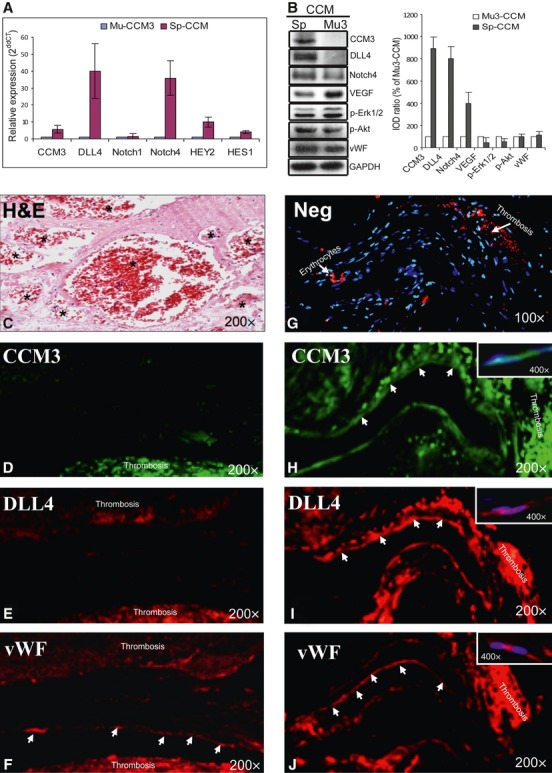
Concomitant down-regulation of CCM3 expression and inactivation of DLL4-Notch signalling in human cavernous tissue derived from a CCM3 mutation carrier in comparison with the controls (sporadic CCMs). (A) Down-regulation of mRNA levels of CCM3 and the core components of DLL4-Notch signalling in a familial CCM harbouring CCM3 mutation. Total RNA was extracted from operative specimens of six sporadic CCMs (Sp-CCM) and a familial CCM3 mutation carrier (Mu-CCM3). The expression of the genes was quantified by RT2-PCR. The relative gene expression was calculated when the gene level in familial CCM was normalized as 1. (B) Down-regulation of the protein levels of CCM3, DLL4 and cleaved Notch4 accompanied with an up-regulation of VEGF and p-Erk1/2 in human CCM3-mutated tissue. The total protein was extracted from the operative specimens of six sporadic CCMs (Sp-CCM) and a familial CCM harbouring CCM3 mutation (Mu-CCM3). Semi-quantification of the blot was performed by measuring integrate optical density (IOD) of each band followed by calculation of the ratio of IOD from the interested protein to that from the housekeeping protein GAPDH. The data were presented as the percentage of the Mu-CCM3. (C) Pathological morphology of CCM revealed by haematoxylin and eosin staining on the section from a sporadic CCM. The lesion consists of immature capillary-like vessels without normal interposed brain parenchyma. These vessels are dilated and often thrombosed (asterisks). (D–J) Immunofluorescent staining of CCM3, DLL4 and vWF on the sections of human CCM lesions. The immunofluorescent staining was simultaneously performed on the adjacent paraffin sections prepared from a CCM harbouring CCM3 mutation (Mu3-CCM; D–F) and from a sporadic CCM (Sp-CCM; G–J). In Mu3-CCM, vWF staining revealed the endothelial layer (arrows) in the thrombosed cavern (F), whereas the immunoreactivity of CCM3 (D) and DLL4 (E) was absent in these endothelial cells. In Sp-CCM, CCM3 (H), DLL4 (I) and vWF (J) were concomitantly detected in the endothelial layer (arrows) of the adjacent sections. Negative control omitting the primary antibody (Neg; G) did not show specific staining, and revealed only unspecific signal (red) in thrombosis and in erythrocytes (arrows) which appeared nuclear staining (blue) negative. The original magnification was 200× in main graphs and 400× in white box.
Western blotting confirmed a significantly lower CCM3 protein level concomitantly with a marked down-regulation of DLL4 and Notch4 in the lesion of the CCM3 mutation carrier (Mu-CCM3). Semi-quantification of the blot using housekeeping protein GAPDH as the reference protein demonstrated nine-, eight- and fourfold higher levels of CCM3, DLL4 and Notch4, respectively, in the sporadic CCM lesions (Sp-CCM) in comparison with that in Mu3. Conversely, the protein levels of VEGF and p-Erk1/2, but not of p-Akt, were clearly elevated in the tissue derived from the CCM3-mutation carrier (Fig. 3B).
It has been known that both sporadic and familial CCMs display undistinguishable histopathological morphology. As revealed by haematoxylin and eosin staining in the section from a sporadic CCM, the lesion typically consists of enriched immature capillary-like vessels often fulfilled with thrombosis (asterisks) without normal interposed brain parenchyma (Fig. 3C). In negative control of immunofluorescent staining omitting the primary antibody (Neg; Fig. 3G), unspecific signal was present in the thrombosis and erythrocytes (arrows), but there was no specific immunoreactivity in the endothelial layer of the covens. To define the association of CCM3 and DLL4 expression in the endothelial layer of CCM covens, immunofluorescent staining of CCM3, DLL4 and vWF was simultaneously performed on adjacent sections from the CCM3-mutation carrier (Mu-CCM3; Fig. 3D–F) and from sporadic CCM (Sp-CCM; Fig. 3H–J). Staining of vWF clearly marked the endothelial layer (arrows) in both Mu-CCM3 (Fig. 3F) and Sp-CCM (Fig. 3J). Immunoreactivity of CCM3 (Fig. 3D) and DLL4 (Fig. 3E) was specifically absent in the endothelial layers of the cavernous lesion on the adjacent sections of Mu-CCM3, but was detected in the endothelial layers (marked by arrows and showed at higher magnification in white box) of Sp-CCM (Fig. 3H–J), indicating a concomitant expression pattern of these two proteins in the endothelial cells of the cavernous lesion.
Recombinant human DLL4 (rhDLL4) recaptured DLL4-Notch signalling and rescued the hyper-angiogenic phenotype of CCM3-silenced endothelial cells
Before testing whether restoration of DLL4 expression by rhDLL4 was able to rescue the hyper-angiogenic phenotype of CCM3-silenced ECs, we first determined the role of rhDLL4 in activation of DLL4-Notch signalling. As shown in Figure 4A, treatment of rhDLL4 (1 μg/ml) to non-transfected cells resulted in a threefold increase in DLL4 mRNA expression and a significantly coincident up-regulation of HEY2 and HES1 mRNA levels, demonstrating an activation of Notch signalling by rhDLL4. Moreover, pre-treatment of rhDLL4 (1 μg/ml) recaptured siCCM3-induced inhibition of DLL4-Notch signalling as demonstrated by a complete recovery of the expression of DLL4, HEY2 and HES1 (Fig. 4A). Of note, rhDLL4 influenced the CCM3 expression neither in non-transfected nor in transfected endothelial cells.
Fig. 4.
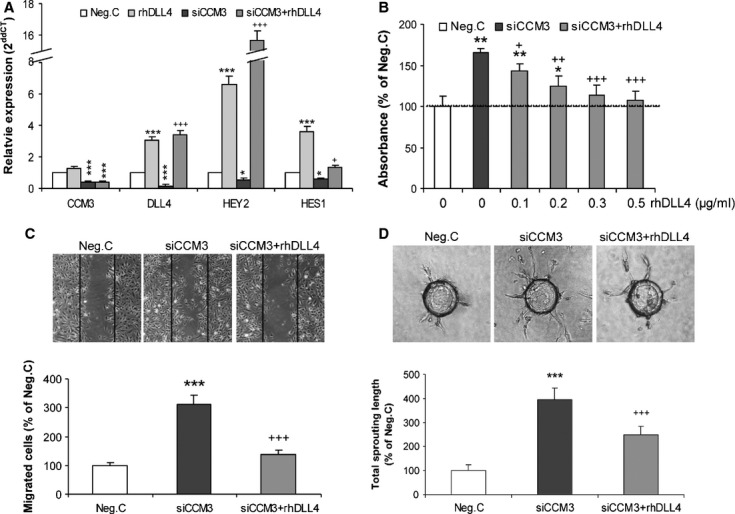
Restoration of DLL4 by treatment with recombinant human DLL4 (rhDLL4) rescued the hyper-angiogenic phenotype of CCM3-silenced ECs. (A) Treatment with rhDLL4 led to up-regulation of mRNA levels of DLL4, HEY2 and HES1 and recaptured CCM3-silence-mediated inactivation of Notch signalling. HUVEC received the pre-treatment of 1 μg/ml of rhDLL4 or vehicle (0.1% BSA) for 24 hrs followed by transfection with either siCCM3 (70 nM) or Neg. C. Total RNA was extracted 72 hrs after the transfection for detecting CCM3 expression and for detecting Notch signalling core components by RT2-PCR in parallel. (B) rhDLL4 abolished the proliferation effect mediated by CCM3 silence. After 48 hrs of transfection of siCCM3 or Neg. C, HUVECs were reseeded and incubated for 24 hrs on the culture wells pre-coated with the different concentration of rhDLL4 as indicated. The proliferation assay was performed by using WST-1 reagent. (C) rhDLL4 inhibited the migration activity of CCM3-silenced endothelial cells. The transfected cells were incubated in culture well pre-coated with 1 μg/ml rhDLL4 or vehicle. Migrated cells were counted in six random fields in each group at 10 hrs after scratching. (D) rhDLL4 suppressed the massive angiogenic sprouting caused by CCM3 silence. The beads formed from the transfected cells were seeded and cultured in 3D fibrin gel immobilized with 1 μg/ml of rhDLL4 or vehicle for 72 hrs. The length of sprouts was measured in 20 randomly selected beads in each group. All data presented in A–D were reproduced in at least three independent experiments. *P < 0.05, ** P < 0.001 and *** P < 0.001, compared with Neg. C; +P < 0.05, ++P < 0.01 and +++P < 0.001, compared with siCCM3 alone.
Under the established rhDLL4 treatment conditions, we were able to further show that pre-treatment of rhDLL4 inhibited activation of proliferation in a dose-dependent manner mediated by siCCM3 silencing. At higher concentrations (0.3 and 0.5 μg/ml), rhDLL4 completely reversed increased proliferation induced by siCCM3 (Fig. 4B). In migration assays, siCCM3 resulted in a threefold higher number of migrated cells in comparison with the control Neg.C (P < 0.001), which was completely abolished by rhDLL4 (P < 0.001; Fig. 4C). Silencing CCM3 largely stimulated sprouting as demonstrated by a fourfold increase in the total length of sprouts (P < 0.001). This excessive sprouting was also significantly suppressed by rhDLL4 (P < 0.001; Fig. 4D).
RhDLL4 reversed the expression of VEGF-R1, -R2 and p-Erk affected by silencing CCM3
To study the downstream pathway(s) influenced by CCM3-silence-mediated inactivation of DLL4-Notch signalling, we examined the expression of VEGF, VEGF-R1 and VEGF-R2 in different types of cultured endothelial cells. In CCMEC, CCM3 siRNA transfection resulted in an efficient down-regulation of CCM3 level to 30% and 20% of the control at 48 hrs (P < 0.001) and 72 hrs (P < 0.001) respectively. Under these silence conditions, the level of VEGF elevated by 76% (P < 0.01) and 55% (P < 0.01), and more interestingly, the expression of VEGF-R2 increased by 82% and 187% of control at 48 hrs (P < 0.01) and 72 hrs (P < 0.001), respectively, after the transfection. In contrast, the VEGF-R1 mRNA level was moderately down-regulated at 72 hrs after siCCM3 transfection (P < 0.05; Fig. 5A). A similar tendency of change in the expression of VEGF, and VEGF-R1 and VEGF-R2 was observed in CCM3-silenced HUVEC (Fig. 5C), whereas CCM3 silencing in HBMEC resulted in only minor elevation of VEGF-R2 level (Fig. 5B).
Fig. 5.
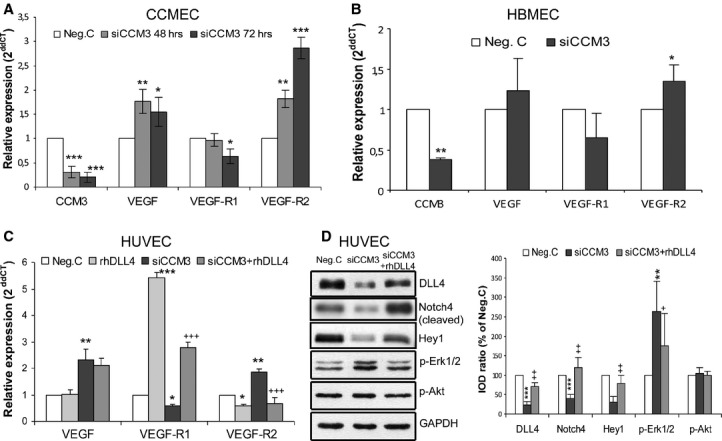
Silencing CCM3 affected multiple signalling components in cultured endothelial cells, which was reversed by rhDLL4. (A) The expression of VEGF, VEGF-R1 and VEGF-R2 after siCCM3 transfection in CCMEC. CCMECs were transfected with either 70 nM of specific CCM3 siRNA (siCCM3) or control siRNA (Neg. C). Total RNA was extracted at 48 and 72 hrs after the transfection for RT2-PCR. CCM3 was detected for controlling an efficient silence induced by siRNA transfection. (B) The expression of VEGF, VEGF-R1 and VEGF-R2 after siCCM3 transfection in HBMEC. HBMEC received the transfection with either 70 nM of siCCM3 or Neg. C. Total RNA was extracted 72 hrs after the transfection for RT2-PCR. CCM3 was detected for controlling an efficient silence induced by siRNA transfection. (C) The treatment of rhDLL4 reversed the down-regulation of VEGF-R1 and the up-regulation of VEGF-R2, but did not influenced the expression of VEGF, in CCM3-silenced HUVEC. Cells were transfected with either 70 nM of specific CCM3 siRNA (siCCM3) or control siRNA (Neg. C) in the presence or the absence of rhDLL4 (1 μg/ml). Total RNA was extracted 72 hrs after the transfection for RT2-PCR. (D) The treatment with rhDLL4 restored DLL4 protein expression, elevated the levels of cleaved Notch4 and target protein Hey1, and reversed increase in p-Erk1/2 expression mediated by silencing CCM3. Total protein was extracted 72 hrs after the transfection in the presence or the absence of rhDLL4 (1 μg/ml). The data presented in A–D were representative of at least three independent experiments. *P < 0.05, **P < 0.001 and ***P < 0.001, compared with Neg. C; +P < 0.05, ++P < 0.01 and +++P < 0.001, compared with siCCM3 alone.
Next, we attempted to test whether rhDLL4 can reverse these effects induced by CCM3 silence. Treatment of rhDLL4 under basal condition caused a dramatic up-regulation of VEGF-R1, and meanwhile a down-regulation of VEGF-R2. Indeed, pre-treatment of rhDLL4 to siCCM3-transfected cells completely reversed the expression of VEGF-R1 and VEGF-R2, but influenced VEGF expression neither in basal control nor after siCCM3 transfection (Fig. 5C).
Western blot revealed that treatment with rhDLL4 not only significantly reversed siCCM3-mediated down-regulation of DLL4 but also the protein levels of the Notch4 (active form) and Hey1 (Fig. 5D), indicating that the inhibition of Notch signalling was resulted from the decreased expression of DLL4 after silencing CCM3. We also examined the activation status of Erk1/2 and Akt in CCM3-silenced ECs. Western blot analyses showed an increased level of p-Erk1/2, but not of p-Akt in CCM3-silenced HUVEC. Importantly, the activation of Erk1/2 was reduced after restoration of DLL4 expression by the treatment of rhDLL4 (Fig. 5D).
Discussion
DLL4-Notch signalling plays a pivotal role in regulating vascularisation, angiogenesis as well as post-angiogenic vessel remodelling and vessel maturation [24, 27]. Compelling evidence indicates that blockade of DLL4-Notch signalling leads to a constitutive angiogenesis [35]. Therefore, DLL4-Notch is defined as a negative pathway regulating angiogenesis. Interestingly, the hyper-angiogenetic phenotype caused by silencing CCM3 (Fig. 1) completely mimetic that induced by inhibition of DLL4 [31]. This study thus focused on study of DLL4-realted-Notch signalling both in cultured endothelial cells and in human CCM tissues. We provided the evidence that loss of CCM3, a gene frequently mutated in familial CCM, significantly down-regulated DLL4 expression and impaired DLL4-Notch signalling thereby activating endothelial angiogenesis. Importantly, a dramatic down-regulation of core components of DLL4-Notch signalling was not only demonstrated in CCM3-silenced endothelial cells including HUVEC (Fig. 2A–C) and to a greater extent CCMEC (Fig. 2D) but also in a human lesion known to harbour CCM3 germline mutation (Mu-CCM3; Fig. 3A and B). The absence of CCM3 immunoreactivity was accompanied by loss of DLL4 expression in the endothelial layer of the cavernous lesion in Mu-CCM3. This suggested a pathological relevance of these two proteins and their potential role in deregulated endothelial function in CCMs (Fig. 3D–F). This proposal was supported by the findings that CCM3-silence-induced hyper-angiogenic phenotype was fully rescued by restoration of DLL4 expression via treatment with rhDLL4 (Fig. 4A–D), and meanwhile, rhDLL4 treatment entirely reversed CCM3-silence-induced down-regulation of core-components DLL4-Notch signalling including DLL4, cleaved (active) Notch4 and target Hey1 (Fig. 5D). Thus, loss of DLL4 seems to be a key downstream event, among others, underlying the pathomechanism of aberrant angiogenesis resulted from CCM3 deficiency.
Besides well-established role of DLL4 in pro-angiogenesis, increasing evidence indicate a pivotal role of DLL4-Notch signalling in post-angiogenic regulation of vessel remodelling and maturation, a process critical for determining the patterning and density of blood vessels in mature tissues. It has been shown that constitutive or conditional genetic deletion of DLL4 or pharmacologic inhibition of Dll4/Notch signalling inhibited the normal developmental pruning of capillaries and up-regulated the expression of vasodilators but suppressed the expression of vasoconstrictor [27]. Of note, all these phenotypes induced by inhibition of Dll4-Notch signalling are very similar to the typical pathology features of human CCM [36]. It is tempting to speculate that CCM3-deficiency-induced down-regulation of DLL4 and the subsequent inactivation of the DLL4-Notch pathway may largely contribute to the severe alteration of the angioarchitecture, which could in part account for the earlier and more severe clinical onset observed in CCM3 mutation carriers [6, 7]. To our knowledge, this is the first report defining DLL4-Notch as a signalling cascade targeted by CCM3 deficiency both in endothelial cells as well as in human-CCM3-deficient tissue. The results on human operative specimen highlight the pathological relevance of the findings obtained from in vitro studies. Meanwhile, we are also aware of the importance to carry out experimental studies on more CCM3 mutation carriers. However, these studies are limited by the fact that such tissue is extremely rare.
Whitehead et al. [28] previously reported that loss of Ccm1 leads to vasculature defects in embryo associated with loss of Dll4 and Notch4; and they further demonstrated the down-regulation of Dll4 and Notch4 in vessels of human CCM lesion associated with loss of function mutations in CCM1. The implication of Notch signalling in the angiogenic function of CCM1 was highlighted by a later study by Wüstehube et al. [29]. In light of these previous and the present studies, it is most likely that loss of either CCM1 or CCM3 commonly inhibited DLL4-Notch signalling, which indirectly supported the previous hypothesis that CCM proteins are present in a complex [13, 14]. On the other hand, increasing evidence indicates that individual CCM proteins may target distinct signalling pathways as well [10, 12], suggesting that CCM may be a common tissue manifestation of distinct mechanistic pathways.
Interestingly, CCM3 has been implicated in oxidative stress. CCM3 was up-regulated by different oxidative stimulations, which protected cells from oxidative stress, whereas silencing CCM3 caused cells death after reactive oxygen species (ROS) exposure [20]. An enhanced ROS levels was also observed in CCM1-deficient cells associated with an increase in cell susceptibility to DNA damage [37]. On the other hand, perturbing Notch signalling has been related to the accumulation of ROS [38]. Based on these, it might be interest to elucidate in the future whether loss of either CCM3 or CCM1 induces oxidative stress in endothelial cells through impairment of Notch signalling.
VEGF is a key angiogenesis regulator and VEGF-mediated angiogenesis is primarily linked to VEGF-receptor 2 (VEGF-R2), whereas VEGF-receptor 1 (VEGF-R1) acts as a functional antagonist of VEGF-R2 negatively regulating the angiogenic effects of VEGF [1, 39, 40]. As VEGF-R1 shows a significantly higher affinity to VEGF in comparison with VEGF-R2, a slight change in VEGF-R1 expression is predictive of a marked influence on VEGF-mediated angiogenesis. Regulating the expression of VEGF receptors by Notch signalling has been suggested as an important mechanism to control vascular patterning [25] and to attenuate sprouting activity [1, 39]. Interestingly, CCM3 has been shown to stabilize VEGF-R2 in a mouse model [11]. However, this effect was not observed in another independent mouse model [10] and in a zebrafish model [12]. This disparity may be because of the difference in knockout strategy and animal species.
Accumulating evidence indicates a crucial role of DLL4 in regulating VEGF-R1 and -R2. Deletion of DLL4 reduced the expression of VEGF-R1 but increased expression of VEGF-R2, thereby stimulating endothelial proliferation, migration and sprouting [41, 42]. In contrast, up-regulation of DLL4 expression suppressed angiogenesis because of regressing VEGF-R2 expression but increasing VEGF–R1 level [31, 43, 44]. These data suggest that the level of DLL4 determines endothelial angiogenic activity through regulating the balance of VEGF-R1/VEGF-R2. In consistence with these findings, we showed that rhDLL4 treatment caused a transcriptional down-regulation of VEGF-R2, and in a much more extent, an up-regulation of VEGF-R1 in HUVEC. Furthermore, this study revealed a tendency of increase in the ratio of the mRNA level of VEGF-R2 to VEGF-R1 in different types of CCM3-silenced endothelial cells and in CCM3-deficient human cavernous tissue. Together with the elevated level of VEGF found in CCM3-silenced endothelial cells, it is feasible to propose that CCM3 deficiency may activate VEGF signalling. It is noteworthy that rhDLL4 treatment completely reversed the down-regulation of VEGF-R1 and up-regulation of VEGF-R2 induced by CCM3 silencing. These findings highlights that CCM3 deficiency in endothelial cells alters the balance of the VEGF-R1 and VEGF-R2 expression owing to down-regulation of DLL4 expression, which may potentially disrupt the homeostasis of VEGF signalling thereby leading to hyper-angiogenesis and vassal hyper-permeability.
Erk1/2 is a widely expressed protein kinase that responds to multiple extracellular stimuli. It has been shown that VEGF regulate variety of cell functions including angiogenesis [45, 46], cell permeability [47] and cell viability [48] through modulating Erk activity. Here, we observed an elevated level of p-Erk1/2, concomitantly with the inactivation of DLL4-Notch signalling and with the up-regulation of VEGF in human-CCM3-deficient cavernous tissue. Restoration of DLL4 by rhDLL4 treatment not only rescued siCCM3-induced inhibition of DLL4-Notch signalling but also blocked siCCM3-induced Erk1/2 activation, suggesting that Erk1/2 lies downstream of Notch signalling and is likely a target of the VEGF pathway.
In summary, this study identified an endothelial signalling axle CCM3/DLL4-Notch/VEGF/Erk1/2. As illustrated in Figure 6, loss of CCM3 impaired DLL4-Notch signalling, disrupted the homeostasis of VEGF pathway and activated Erk1/2, which was crucially involved in the regulation of endothelial proliferation, migration and sprouting. In addition to angiogenesis, DLL4-Notch and VEGF signallings are well-defined pathways for regulation of vessel maturation and vessel permeability respectively. We thus suppose that dysregulation of these pathways may be potentially associated with the enriched immature angioarchitectures and with recurrent haemorrhage typically seen in the lesion of human CCMs. Treatment with rhDLL4 restored DLL4-Notch signalling, inhibited VEGF and Erk1,2 pathways and eventually rescued the hyper-angiogenic phenotype induced by CCM3 silencing. These findings highlight DLL4 as a key downstream modulator in CCM3 endothelial signalling and as a potential target for the medication of CCMs.
Fig. 6.
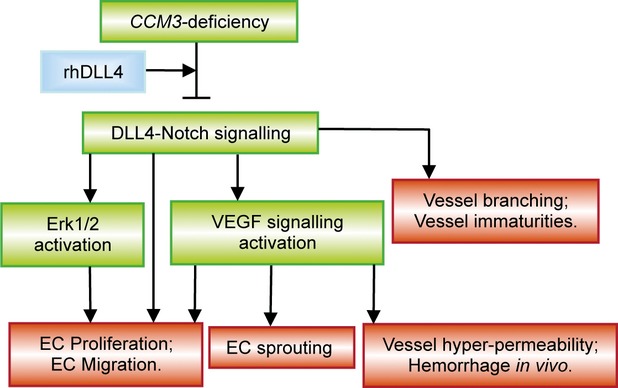
Schematic illustration of the signalling pathways affected by CCM3 deficiency and possible implication in the pathology of CCM. Loss of CCM3 impaired DLL4-Notch signalling, disrupted the homeostasis of VEGF pathway and activated Erk1/2, which leads to stimulation of endothelial proliferation, migration and sprouting. Treatment with rhDLL4 restored DLL4-Notch signalling, inhibited VEGF and Erk1,2 pathways, and eventually rescued the hyper-angiogenic phenotype induced by CCM3 silencing, suggesting DLL4 as a key downstream modulator in CCM3 endothelial signalling. In addition to angiogenesis, DLL4-Notch signalling and VEGF signalling are well-defined pathways for regulation of vessel branching/maturation and vessel permeability respectively. We thus suppose that dysregulation of these pathways may be associated with the impairment of these post-angiogenic functions thereby potentially contributing to the enriched immature angioarchitectures with recurrent haemorrhage typically seen in the lesion of human CCMs.
Acknowledgments
The authors thank Ms. Rita Haase and Ms. Anika Marko for technical assistance and thank Mr. Mike Sucker for graphic assistance. C.Y. received a scholarship from the Medical Faculty, University of Duisburg-Essen. This study was financially supported by University Hospital of Essen and by BayGene (U.F.).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Riant F, Bergametti F, Ayrignac X, et al. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J. 2010;277:1070–5. doi: 10.1111/j.1742-4658.2009.07535.x. [DOI] [PubMed] [Google Scholar]
- 2.Bergametti F, Denier C, Labauge P, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanriover G, Boylan AJ, Diluna ML, et al. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery. 2008;62:930–8. doi: 10.1227/01.neu.0000318179.02912.ca. discussion 8. [DOI] [PubMed] [Google Scholar]
- 4.Petit N, Blecon A, Denier C, et al. Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr Patterns. 2006;6:495–503. doi: 10.1016/j.modgep.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Louvi A, Chen L, Two AM, et al. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci USA. 2011;108:3737–42. doi: 10.1073/pnas.1012617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denier C, Labauge P, Bergametti F, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550–6. doi: 10.1002/ana.20947. [DOI] [PubMed] [Google Scholar]
- 7.Gault J, Sain S, Hu LJ, et al. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59:1278–84. doi: 10.1227/01.NEU.0000249188.38409.03. discussion 84-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Wu Q, Xu JF, et al. Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurg Focus. 2010;29:E1. doi: 10.3171/2010.5.FOCUS1090. Doi: 10.3171/2010.5.FOCUS1090. [DOI] [PubMed] [Google Scholar]
- 9.Schleider E, Stahl S, Wüstehube J, et al. Evidence for anti-angiogenic and pro-survival functions of the cerebral cavernous malformation protein 3. Neurogenetics. 2010;12:83–6. doi: 10.1007/s10048-010-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan AC, Drakos SG, Ruiz OE, et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J Clin Invest. 2011;121:1871–81. doi: 10.1172/JCI44393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Y, Zhang H, Yu L, et al. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010;3:ra26. doi: 10.1126/scisignal.2000722. Doi: 10.1126/scisignal.2000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoruk B, Gillers BS, Chi NC, et al. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev Biol. 2011;362:121–31. doi: 10.1016/j.ydbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Voss K, Stahl S, Schleider E, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249–56. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- 14.Stahl S, Gaetzner S, Voss K, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat. 2008;29:709–17. doi: 10.1002/humu.20712. [DOI] [PubMed] [Google Scholar]
- 15.Fidalgo M, Fraile M, Pires A, et al. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci. 2010;123:1274–84. doi: 10.1242/jcs.061341. [DOI] [PubMed] [Google Scholar]
- 16.Zheng X, Xu C, Di Lorenzo A, et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J Clin Invest. 2010;120:2795–804. doi: 10.1172/JCI39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, Zhao H, Shan J, et al. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18:1965–78. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ceccarelli DF, Laister RC, Mulligan VK, et al. CCM3/PDCD10 heterodimerizes with germinal center kinase III (GCKIII) proteins using a mechanism analogous to CCM3 homodimerization. J Biol Chem. 2011;286:25056–64. doi: 10.1074/jbc.M110.213777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilder TL, Malone MH, Bencharit S, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007;6:4343–55. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- 20.Fidalgo M, Guerrero A, Fraile M, et al. The adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins Ezrin/Radixin/Moesin by Mammalian Ste20-4 to protect cells from oxidative stress. J Biol Chem. 2012;287:11556–65. doi: 10.1074/jbc.M111.320259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Zhang R, Zhang H, et al. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J Biol Chem. 2010;285:24099–107. doi: 10.1074/jbc.M110.128470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleaveland B, Zheng X, Liu JJ, et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15:169–76. doi: 10.1038/nm.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 24.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–18. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 25.Kume T. Novel insights into the differential functions of Notch ligands in vascular formation. J Angiogenes Res. 2009;1:8–16. doi: 10.1186/2040-2384-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hellstrom M, Phng LK, Hofmann JJ, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 27.Lobov IB, Cheung E, Wudali R, et al. The Dll4/Notch pathway controls postangiogenic blood vessel remodeling and regression by modulating vasoconstriction and blood flow. Blood. 2011;117:6728–37. doi: 10.1182/blood-2010-08-302067. [DOI] [PubMed] [Google Scholar]
- 28.Whitehead KJ, Plummer NW, Adams JA, et al. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–48. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 29.Wüstehube J, Bartol A, Liebler SS, et al. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci USA. 2010;107:12640–5. doi: 10.1073/pnas.1000132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Y, Wu Q, Fass M, et al. In vitro characterization of the angiogenic phenotype and genotype of the endothelia derived from sporadic cerebral cavernous malformations (CCMs) Neurosurgery. 2011;69:722–32. doi: 10.1227/NEU.0b013e318219569f. [DOI] [PubMed] [Google Scholar]
- 31.Harrington LS, Sainson RC, Williams CK, et al. Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc Res. 2008;75:144–54. doi: 10.1016/j.mvr.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Pagenstecher A, Stahl S, Sure U, et al. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911–8. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Wloch A, Wu Q, et al. Involvement of PTEN promoter methylation in cerebral cavernous malformations. Stroke. 2009;40:820–6. doi: 10.1161/STROKEAHA.108.526376. [DOI] [PubMed] [Google Scholar]
- 34.Nakatsu MN, Sainson RC, Aoto JN, et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res. 2003;66:102–12. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 35.Ridgway J, Zhang G, Wu Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–7. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 36.Moriarity JL, Wetzel M, Clatterbuck RE, et al. The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery. 1999;44:1166–71. discussion 72-3. [PubMed] [Google Scholar]
- 37.Goitre L, Balzac F, Degani S, et al. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE. 2010;5:e11786. doi: 10.1371/journal.pone.0011786. Doi: 10.1371/journal.pone.0011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bheeshmachar G, Purushotaman D, Sade H, et al. Evidence for a role for notch signaling in the cytokine-dependent survival of activated T cells. J Immunol. 2006;177:5041–50. doi: 10.4049/jimmunol.177.8.5041. [DOI] [PubMed] [Google Scholar]
- 39.Hellstrom M, Phng LK, Gerhardt H. VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adh Migr. 2007;1:133–6. doi: 10.4161/cam.1.3.4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jinnin M, Medici D, Park L, et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236–46. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suchting S, Freitas C, le Noble F, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–30. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lobov IB, Renard RA, Papadopoulos N, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA. 2007;104:3219–24. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams CK, Li JL, Murga M, et al. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107:931–9. doi: 10.1182/blood-2005-03-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hainaud P, Contreres JO, Villemain A, et al. The role of the vascular endothelial growth factor-Delta-like 4 ligand/Notch4-ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res. 2006;66:8501–10. doi: 10.1158/0008-5472.CAN-05-4226. [DOI] [PubMed] [Google Scholar]
- 45.Hashimoto T, Zhang XM, Chen BY, et al. VEGF activates divergent intracellular signaling components to regulate retinal progenitor cell proliferation and neuronal differentiation. Development. 2006;133:2201–10. doi: 10.1242/dev.02385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giuliani N, Lunghi P, Morandi F, et al. Downmodulation of ERK protein kinase activity inhibits VEGF secretion by human myeloma cells and myeloma-induced angiogenesis. Leukemia. 2004;18:628–35. doi: 10.1038/sj.leu.2403269. [DOI] [PubMed] [Google Scholar]
- 47.Breslin JW, Pappas PJ, Cerveira JJ, et al. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–100. doi: 10.1152/ajpheart.00330.2002. Doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 48.Narasimhan P, Liu J, Song YS, et al. VEGF Stimulates the ERK 1/2 signaling pathway and apoptosis in cerebral endothelial cells after ischemic conditions. Stroke. 2009;40:1467–73. doi: 10.1161/STROKEAHA.108.534644. [DOI] [PMC free article] [PubMed] [Google Scholar]