

Abstract
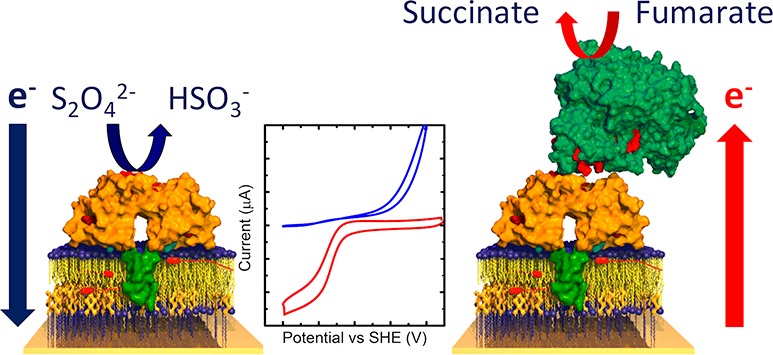
Protein–protein interactions are well-known to regulate enzyme activity in cell signaling and metabolism. Here, we show that protein–protein interactions regulate the activity of a respiratory-chain enzyme, CymA, by changing the direction or bias of catalysis. CymA, a member of the widespread NapC/NirT superfamily, is a menaquinol-7 (MQ-7) dehydrogenase that donates electrons to several distinct terminal reductases in the versatile respiratory network of Shewanella oneidensis. We report the incorporation of CymA within solid-supported membranes that mimic the inner membrane architecture of S. oneidensis. Quartz-crystal microbalance with dissipation (QCM-D) resolved the formation of a stable complex between CymA and one of its native redox partners, flavocytochrome c3 (Fcc3) fumarate reductase. Cyclic voltammetry revealed that CymA alone could only reduce MQ-7, while the CymA-Fcc3 complex catalyzed the reaction required to support anaerobic respiration, the oxidation of MQ-7. We propose that MQ-7 oxidation in CymA is limited by electron transfer to the hemes and that complex formation with Fcc3 facilitates the electron-transfer rate along the heme redox chain. These results reveal a yet unexplored mechanism by which bacteria can regulate multibranched respiratory networks through protein–protein interactions.
Introduction
Protein–protein complexes are fundamental to life where they are key to processes ranging from central metabolism to cell signaling. Transient protein–protein interactions generally underpin the electron-transfer (ET) pathways of respiration.1 One of the many well-characterized examples of a transient ET complex is that between cytochrome c and cytochrome c oxidase.2−5 The interaction between these partner proteins is weak and dynamic. This ensures the frequent exchange of partner proteins as required to support electron flux in cases where the sole function of one of the proteins is to shuttle electrons between redox partners.1 While it is generally assumed that such transient protein–protein interactions are specific, for Paracoccus denitrificans it has recently been shown that seven proteins in a respiratory network interact in a seemingly ill-defined manner.6 This results in an intricate electron-transfer network that may be better suited to successful colonization of habitats with changing resources.
Many quinol dehydrogenases are multisubunit enzymes. Typical examples are the quinol nitrate oxidoreductase, NarGHI, and the quinol nitrite oxidoreductase, NrfHA.7,8 Quinol oxidation occurs in a trans-membrane subunit that is permanently bound to one or more periplasmic subunits that contain the site for catalytic reduction of the water-soluble substrate. A chain of redox centers extends between the catalytic sites to support electron exchange between them. However, during anaerobic respiration in Shewanella, a single enzyme, CymA, oxidizes the membrane-bound menaquinol-7 pool and donates the electrons to a variety of terminal reductases, which can, for instance, reduce periplasmic nitrate, nitrite, fumarate, or extracellular ferric oxides and dimethyl sulfoxide (DMSO).9,10 The promiscuity with which CymA passes electrons from MQ-7 to a wide variety of proteins poses an interesting question: Does CymA form long-lived ET complexes, analogous to the NrfHA complex and quinol dehydrogenases in other species, or transient complexes of the type commonly observed when a redox protein functions as a shuttle to transfer electrons between two enzymes? This question is particularly pertinent as CymA donates electrons to proteins that function solely in ET, such as ScyA, and to enzymes, including tetraheme flavocytochrome c3 (Fcc3) fumarate reductase (Figure 1A), iron-reducing small tetraheme cytochrome (STC), and a nitrite reductase (NrfA).9
Figure 1.
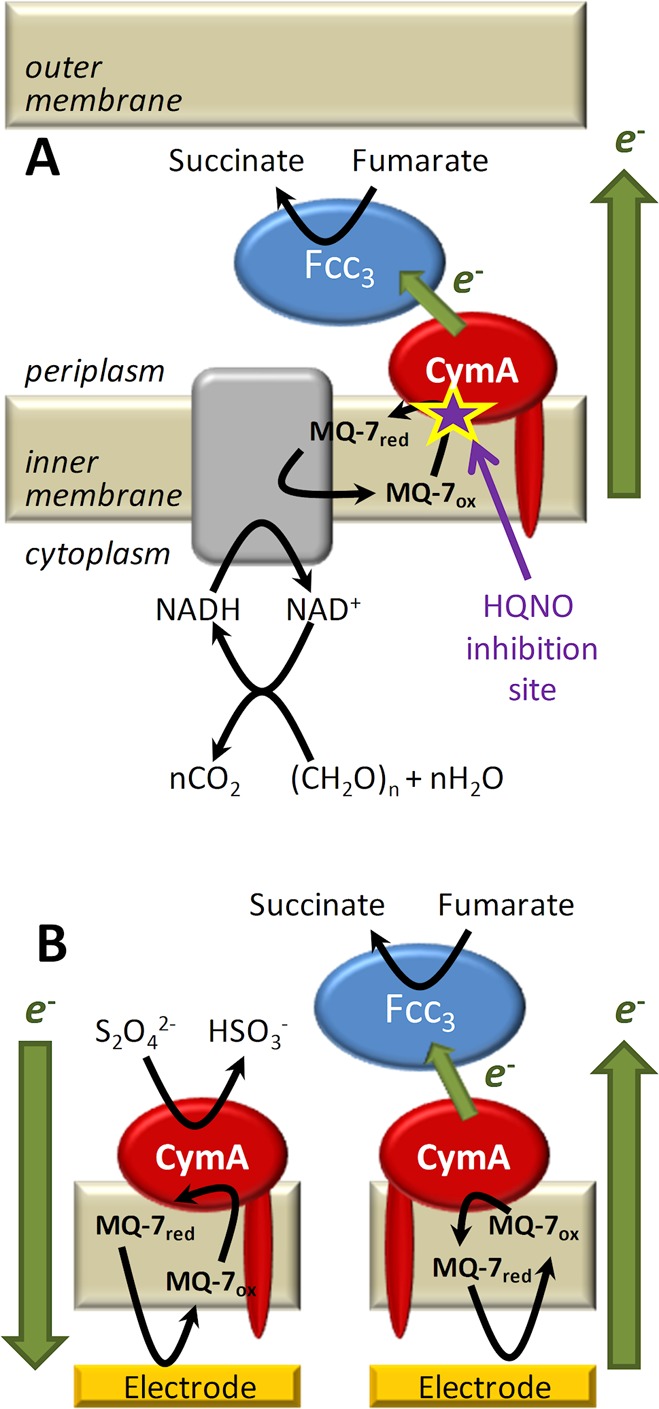
(A) Schematic of the inner membrane respiratory chain of Shewanella grown anaerobically with fumarate as the terminal electron acceptor. Electrons generated during catabolism are donated to the MQ-7 pool, which is reoxidized by CymA. Based on primary sequence analysis,46 homology to NrfH for which a crystal structure is available,38 and biochemical analysis of both CymA and other NapC/NirT superfamily members, CymA is known to contain a single N-terminal transmembrane α-helix with a single globular “head” domain facing the periplasm. CymA transfers the electrons to the periplasmic enzyme flavocytochrome c3 (Fcc3), which reduces fumarate to succinate. The site of action of the competitive inhibitor, 2-n-heptyl-4-hydroxyquinoline N-oxide (HQNO), is indicated in magenta. (B) Schematic of CymA containing inner-membrane architecture on an electrode surface in the presence of Fcc3 and a chemical reducing agent dithionite (S2O42–). In both panels, chemical reactions and ET steps are shown with black and green arrows, respectively.
Previously, we have studied the interaction between CymA and its native quinol substrate by adsorbing CymA on gold electrodes and exposing the protein film to liposomes containing MQ-7. Cyclic voltammetry revealed that CymA in isolation catalyzed the reduction of MQ-7 but failed to perform the reaction that underpins anaerobic respiration, the oxidation of MQ-7.11 Here, we report the extension of these studies in which the quinone oxidoreductase activity of CymA is determined in the absence and presence of its ET partner, Fcc3 fumarate reductase.12 Our approach was to construct a CymA containing inner-membrane architecture on planar surfaces (Figure 1B). The interaction between CymA and Fcc3 was characterized with a quartz-crystal microbalance with dissipation (QCM-D) and the quinol dehydrogenase activity of CymA monitored by electrochemistry. Formation of a long-lived complex between CymA and Fcc3 was found to retune the catalytic bias of CymA toward oxidation of MQ-7 (schematically shown in Figure 1B). This ability of protein–protein interactions to modulate the catalytic bias of redox enzymes reveals a new mechanism by which the magnitude and direction of electron flux through respiratory ET networks can be regulated.
Methodology
Treatments of Specialist Chemicals
EO3-cholesteryl was made as previously described.13 All solvents were HPLC grade (Fisher) and used as received. Menaquinone-7 (MQ-7, Wako) stock solutions were prepared at concentrations of 1 mg/mL in chloroform. 2-n-Heptyl-4-hydroxyquinoline N-oxide (HQNO) was purchased from Alexis Biochemicals, and a stock solution of 50 mM was prepared in DMSO. Mixtures of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Avanti Polar Lipids) and cardiolipin (CL; Avanti Polar Lipids) at a 90/10 ratio were dissolved in chloroform, aliquoted to 5 mg total lipid, and dried under an N2 stream before storage under an N2 atmosphere at −20 °C.
Expression and Purification of CymA, CymAsol, and Fcc3
MR-1 cultures were grown microaerobically, and Fcc3 and membranous CymA were isolated as described elsewhere.14 A CymA truncation mutant (CymAsol), in which the N-terminal transmembrane helix was removed and replaced with a his-tagged maltose-binding protein (MBP) and a TEV cleavage motif between the CymAsol and the MBP, was expressed and purified as described in the Supporting Information of ref (15). The plasmid used to express the MBP-CymA construct is described by Londer et al.16 The MPB was cleaved from CymA by incubating with TEV protease, purified as described previously,17 at 1:2 (w/w) ratio for 16 h at 4 °C. CymAsol was then purified by passing the protein mixture though a Ni-sepharose (Fast flow, GE Healthcare) column, to remove His6-tagged MPB, His6-tagged TEV protease, and any remaining uncleaved CymAsol-MBP from the cleaved CymAsol fraction. Full-length recombinant CymA, CymAsol-MBP fusion protein, and Fcc3 were confirmed by mass spectrometry, N-terminal sequencing, and/or immunoblotting. SDS-PAGE analysis identified approximately 90% pure hemoprotein with a band at 24 kDa (CymA), 64 kDa (CymAsol-MBP fusion), or 62 kDa (Fcc3). Electronic absorption spectroscopy characterized CymA and CymAsol as typical c-type cytochromes (A407 nm/A275 nm = 4) consistent with four inserted hemes per protein monomer. Protein concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Sigma) with bovine serum albumin as the standard.
Vesicle Preparation
MQ-7 was introduced into aliquots of a mixture of 90%/10% (w/w) POPC and CL using a 50:50 chloroform/methanol mixture before being dried under a stream of nitrogen for at least 1 h. The resulting film was resuspended by vortexing in buffer (20 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 30 mM Na2SO4, pH 7.4) to a concentration of 5 mg/mL. A homogeneous vesicle solution was prepared by extrusion through a 200 nm nucleopore track-etched polycarbonate membrane (Whatman) using a mini-extruder (Avanti) at 20 °C.
Reconstitution of CymA
CymA was reconstituted by the method based on that of Carter et al.18 On the day of reconstitution, vesicles were prepared as described above except a lipid concentration of 20 mg/mL was used. Lipid vesicles, octylglucoside (OG), and CymA were mixed by inversion to final concentrations of 16.4 mg/mL lipid, 45 mM OG, and 0.16 or 0.32 mg/mL CymA (i.e., 1 or 2% (w/w) CymA to lipid). QCM-D experiments were performed with 2% (w/w) CymA, while the electrochemistry was done with 1% (w/w). After 15 min incubation on ice, the lipid/protein mixture was rapidly diluted 200-fold with buffer, precooled to 4 °C, and centrifuged at 142 000g for 1 h to pellet the proteoliposomes. The proteoliposomes were resuspended in the same volume of fresh cold buffer and centrifuged again at 142 000g for 1 h. Proteoliposomes were resuspended in 1 mL of buffer and extruded through a 200 nm track-etched polycarbonate membrane using a mini-extruder (Avanti).
Quartz Crystal Microbalance with Dissipation (QCM-D)
QCM-D experiments were conducted using a Q-Sense E4 (Q-Sense AB). Experiments were performed using silicon-oxide sensor crystals at 21 °C, with the flow rate held at 70 μL/min. Experiments were conducted outside and inside an N2-filled glovebox (MBraun LabMaster; <1 ppm O2), and no difference in behavior was observed. All solutions were purged with N2 and stored in the glovebox at least 24 h before use. Silicon-oxide QCM-D crystals were cleaned by bath sonicating them with Milli-Q water (30 min), 0.4% SDS detergent (20 min), and again Milli-Q water (20 min). After that, crystals were treated for 20 min with UV/ozone (UV/ozone cleaning system, low pressure quartz-mercury vapor lamp emitting 254 and 185 nm UV; UVOCS) followed by a 30 min bath sonication with Milli-Q water. All bilayers were formed using 0.5 mg/mL lipid vesicles in water containing 10 mM CaCl2. After being rinsed with water, the formed SSMs were rinsed with 20 mM MOPS, 30 mM Na2SO4, pH 7.4 (buffer) containing 1 mM EDTA and then buffer alone to remove excess vesicles and calcium ions. All protein-binding experiments were performed in buffer. On graphs, changes in the dissipation (ΔD) and frequency (Δf) of the seventh overtone are presented, while third, fifth, ninth, eleventh, and thirteenth overtones were also recorded. ΔD and Δf values are given as the shift as compared to values obtained in buffer. Δf was used to calculate adsorbed weight under the assumptions of the Sauerbrey equation (i.e., 17.7 ng cm–1 Hz–1 for the equipment and crystals used) and then into protein coverages by taking into account that approximately 25% of the adsorbed weight is due to water entrapped within the protein matrix.
Electrode Preparation and Modification
Routinely, electrochemical experiments were carried out with ultraflat template stripped gold (TSG) working electrodes, prepared as described previously.19 150 nm of 99.99% gold (Goodfellows) was evaporated on silicon wafers using an Edwards Auto 306 evaporator at <2 × 10–6 mbar. 1.2 cm2 glass slides were glued to the gold layer with Epo-Tek 377 and cured for 2 h at 120 °C. Before use, the glass slides were detached from the silicon wafers to expose the TSG surface. The formation of the self-assembled monolayers (SAMs) containing the cholesterol “tether” and the formation of the SSM onto the electrode were performed as described previously.20 SAMs were formed by incubating a freshly exposed TSG slide in 0.11 mM EO3-cholesteryl and 0.89 mM 6-mercaptohexanol (6MH) in propanol for 16 h. After incubation, the excess thiol was gently washed away with isopropanol and methanol, and the electrodes were then dried in a stream of N2. This procedure results in a 60%/40% EO3-cholesteryl/6-mercaptohexanol area ratio on the surface as confirmed by impedance spectroscopy before each experiment. To form solid-supported lipid membranes (SSMs), vesicles or proteoliposomes were added to the SAM surface at a final concentration of 0.5 mg/mL in the presence of 10 mM CaCl2 and incubated for 1–2 h until a capacitance drop to less than 1.2 μF/cm2 was observed. The surface was then rinsed three times with water, then buffer containing 1 mM EDTA to remove any traces of calcium ions in the cell. Finally, the SSM-modified electrode was rinsed three times with buffer and used in the electrochemistry experiments. Care was taken to keep the electrodes immersed in an aqueous environment at all times during rinsing.
Cyclic Voltammetry (CV) and Electrochemical Impedance Spectroscopy (EIS)
A bespoke glass spectro-electrochemical cell was assembled using a standard three-electrode setup: the ultraflat template-stripped gold (TSG) working electrode was embedded in a PTFE holder with a rubber O-ring seal, (A = 0.2 cm2); a platinum wire counter electrode and a saturated silver/silver chloride electrode (Ag/AgCl) completed the circuit in the buffer volume. All potentials are quoted versus standard hydrogen electrode (SHE) using 0.199 V vs SHE for the Ag/AgCl reference electrode. Routinely, experiments were conducted in 20 mM MOPS, 30 mM Na2SO4, pH 7.4 (buffer). The cell was used in a steel mesh Faraday cage to minimize electrical noise, and all experiments were conducted inside an N2-filled glovebox (MBraun LabMaster) where the O2 levels were <1 ppm. All solutions were purged with N2 and stored in the glovebox at least 24 h before use. Electrochemical measurements were recorded at 21 °C using an Autolab electrochemical analyzer with a PGSTAT30 potentiostat, SCANGEN and ADC750 modules, and FRA2 frequency analyzer (Ecochemie). Analogue cyclic voltammetry experiments were routinely carried out by holding the potential at 0.5 or 0.3 V for 5 s before cycling at a scan rate of 1 or 10 mV/s in the potential window from 0.5 or 0.3 to −0.4 V. After addition of CymAsol, Fcc3, or HQNO to the buffer, an incubation time of 30 min was allowed. After incubating the SSM with CymAsol or Fcc3, protein that was not bound to the SSM was washed out by rinsing the electrochemical cell five times with buffer. Where HQNO was introduced in DMSO, DMSO concentration remained below 5% of the total cell volume.
Results
CymA-Fcc3 Complex Formation
QCM-D was used to monitor the association of Fcc3 with CymA-containing inner membrane architectures assembled on silicon-oxide surfaces (Figure 2A). The QCM-D technique gives simultaneous and real-time information about changes in adsorbed mass, through the resonant frequency (Δf), and the “rigidity” or viscoelastic properties of the adsorbed layer through the dissipation (ΔD). Solid-supported bilayer membranes (SSMs) were formed on the silicon-oxide surfaces from vesicles comprised of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cardiolipin (CL) that contained 2% (w/w) CymA to lipid. SSM formation was confirmed by a drop in frequency of ∼26–27 Hz and a small change in dissipation (Figure 2A; for clarity, the formation of the bilayer itself is not shown and the trace starts after the bilayer is formed). The frequency shift is very similar to values reported for the formation of phosphatidylcholine-only SSMs under similar conditions.21 This is expected because the low CymA content of the SSM-forming vesicles corresponds to ∼0.3 pmol of CymA/cm2, and this would only cause a frequency shift of approximately 0.5 Hz.
Figure 2.

The assembly and catalytic activities of CymA(sol) containing SSMs. (A) QCM-D results of a SiO2 surface in buffer, plotting (black line, left axis) frequency and (red line, right axis) dissipation against time. For clarity, only the results after formation of the SSM are shown. Changes in the solution composition flowing over the SSM are indicated: (Fcc3) 5 μM Fcc3/1 mM fumarate; (wash 1) 1 mM fumarate; (wash 2) buffer only. SSM was formed with CymA proteoliposomes (90:10 POPC:cardiolipin; 2% (w/w) CymA; 1% (w/w) MQ-7). The plot shown is representative of triplicate experiments. (B) CVs (10 mV/s) of a SSM on a gold electrode modified with cholesterol tethers. The SSM was formed with CymA proteoliposomes (90:10 POPC:cardiolipin; 1% (w/w) CymA; 1% (w/w) MQ-7). CVs are shown (a) before and after addition of (b) 5 μM Fcc3, (c) 1 mM fumarate (after rinsing unbound Fcc3 from solution), and (d) 1 mM fumarate/10 μM HQNO. (C) Relative current measured from CVs as in (B, trace c) at −0.25 V vs SHE of the reductive scan as a function of MQ-7 concentration in the SSM. MQ-7 content is given in weight percentages relative to the lipid weight in the proteoliposomes. The line represents a fit to the Michaelis–Menten equation. (D) QCM-D results of a SiO2 surface in buffer, plotting (black line, left axis) frequency and (red line, right axis) dissipation against time. For clarity reasons, only the traces after the formation of the SSM are shown. Changes in the solution composition flowing over the SSM are indicated: (CymAsol) 0.1 μM CymAsol; (wash 1), 1 mM fumarate; (Fcc3) 5 μM Fcc3/1 mM fumarate; (wash 2) 1 mM fumarate. SSM was formed with liposomes (90:10 POPC:cardiolipin; 1% (w/w) MQ-7). The plot shown is representative of triplicate experiments. (E) CVs (1 mV/s) of a SSM on a gold electrode modified with cholesterol tethers. The SSM was formed with liposomes (90:10 POPC:cardiolipin; 2% (w/w) MQ-7). CVs are shown (a) before and after addition of (b) 0.1 μM CymAsol, (c) 5 μM Fcc3, (d) 1 mM fumarate (after rinsing unbound Fcc3 from solution), and (e) 1 mM fumarate/10 μM HQNO. (F) Relative current measured from CVs as in (E, trace d) at −0.25 V vs SHE of the reductive scan as a function of MQ-7 concentration in the SSM. MQ-7 content is given in weight percentages relative to the lipid weight in the liposomes. The line represents a fit to the Michaelis–Menten equation.
Exposure of CymA-containing SSMs to Fcc3 results in a further decrease in frequency to ∼ –55 Hz, and the dissipation rises by approximately 2 units (Figure 2A). This indicates that Fcc3 binds to SSMs as a “layer” exhibiting some viscosity, such that it is not fully elastic, which suggests that some of the Fcc3 is loosely bound. This was confirmed when subsequently the SSM assemblies were washed with buffer solutions lacking Fcc3 (Figure 2A). The dissipation returned to its previous value, and the frequency settled at a value of ∼ –35 Hz indicating that some, but not all, Fcc3 had detached from the membrane. The shift in frequency (∼9 Hz) due to remaining tightly bound Fcc3 is equivalent to the adsorption of approximately 0.16 μg Fcc3/cm2 and so approximately 2 pmol Fcc3/cm2.
Control experiments with SSMs that did not contain CymA revealed that no Fcc3 remained bound to the membrane after washing (Figure S1 in the Supporting Information). Thus, CymA is required for tight association of Fcc3 to the SSMs. Trans-membrane proteins are known to introduce defects into SSMs,22 and although the QCM-D data indicate that the CymA-containing SSMs are essentially homogeneous, it cannot be excluded that a small number of defects cause some a-specific adsorption of Fcc3. Thus, in terms of the molecular architecture of the SSM and its associated proteins, the QCM-D data do not allow for unambiguous interpretation of the ratio of bound Fcc3 to CymA. Furthermore, Fcc3 is reported to be a monomer in solution, and it is unlikely that CymA forms complexes with Fcc3 oligomers of the high order that is implied by the frequency shifts (approximately six Fcc3 per CymA). A more reasonable interpretation is that CymA forms complexes with Fcc3 with a stoichiometry close to 1:1 (or 1:2 as observed for NrfHA), while additional Fcc3 molecules are bound to defects in the SSM induced by the presence of CymA.
Defects in the SSM that are capable of binding Fcc3 may arise because the location of CymA in the SSMs used for these experiments cannot be controlled. Previous electrophoresis of CymA-containing SSMs equivalent to those studied here has shown that approximately one-half the CymA is immobile.23 One-half the population of CymA may present its head domain toward the silicon oxide surface leading to interactions that immobilize the protein, while the mobile population of CymA presents its head domain to the solution. To eliminate the possibility that this issue influences the QCM-D results, a series of experiments were performed with a truncated form of CymA that lacks the N-terminal transmembrane helix. This protein, termed CymAsol, is water-soluble. As a consequence, CymAsol could be introduced to SSMs after their formation such that the protein can only associate with the solvent-exposed face of the SSM. Tight binding of CymAsol to SSMs was observed within minutes from solutions containing approximately 0.1 μM CymAsol (Figure 2D). After washing the CymAsol-containing SSM assembly to remove loosely bound protein, the frequency was ∼21 Hz lower than that displayed by the SSM alone. This shift in frequency corresponds to the adsorption of 12 pmol CymAsol/cm2. On exposing the CymAsol-containing SSMs to Fcc3, followed by washing to remove loosely bound material, the frequency dropped by a further ∼18 Hz. This behavior is consistent with Fcc3 binding tightly to CymAsol-containing SSMs and confirms the data obtained with the full-length CymA.
Assuming that all CymAsol remains bound to the SSM upon Fcc3 adsorption, the drop in frequency on Fcc3 binding corresponds to 4 pmol Fcc3/cm2. From this, the ratio of Fcc3 bound per CymAsol is 1:3. Even allowing for the possibility that Fcc3 partly replaces CymAsol on the SSM surface, the Fcc3 to CymAsol ratio is still significantly lower than that for Fcc3 bound to full-length CymA. These observations most likely reflect steric constraints on the binding of Fcc3 to CymAsol that are not present for CymA-containing SSMs. In fact, assuming that CymAsol has dimensions comparable to those of the NrfH head domain, the QCM-D data correspond to the formation of a close-packed monolayer of CymAsol covered by another close-packed monolayer of Fcc3.
In summary, the QCM-D is consistent with Fcc3 binding strongly to SSMs containing either CymA or CymAsol, but not to SSMs without CymA(sol), such that an inner membrane architecture may be present on the solid surfaces as shown schematically in Figure 1B. If physiologically relevant CymA(sol)-Fcc3 complexes are formed, both enzymes should be catalytically active and should support intermolecular ET to couple MQ-7 oxidation to fumarate reduction. To assess whether this was the case, SSMs containing CymA(sol)-Fcc3 complexes and MQ-7 were assembled on gold electrode surfaces as described below and studied by electrochemistry.
Catalytic Activity of CymA-Fcc3 and CymA
SSMs were formed from CymA proteoliposomes by self-assembly on ultrasmooth gold electrodes that had been modified with submonolayers of cholesterol tethers. Formation of planar SSMs was confirmed by electrochemical impedance spectroscopy, which showed a drop in capacitance to below 1.2 μF/cm2 after addition of the proteoliposomes (Figure S2 in the Supporting Information).24 Cyclic voltammograms (CVs) of the SSMs containing CymA and MQ-7 showed little change after exposure to Fcc3 (Figure 2B). However, addition of fumarate had a large effect on the appearance of the CV that was converted to a waveshape typical for catalytic reduction reactions.
The catalytic CV was unaltered whether Fcc3 remained present in solution or not. This confirmed that the catalytic response had its origin in Fcc3 bound irreversibly to the CymA-containing SSM. To exclude the possibility that Fcc3 received electrons directly from the electrode, or MQ-7, an inhibitor of CymA’s quinol-dehydrogenase activity was added, HQNO. The addition of HQNO inhibited catalytic fumarate reduction (Figure 2B), demonstrating that the electrons for fumarate reduction are indeed supplied to Fcc3 from MQ-7 oxidation by CymA as illustrated in Figure 1. In agreement with this conclusion, when the MQ-7 concentration within the lipid membrane was altered, the catalytic current increased in a manner consistent with Michaelis–Menten behavior (Figure 2C). The maximum catalytic current (∼0.25 μA or ∼1.25 μA/cm2 at −250 mV) is achieved at MQ-7 contents between 0.75% and 2% (w/w). The turnover frequency for MQ-7 oxidation can be calculated if the current can be related to the number of catalytically active CymA-Fcc3 complexes on the surface. As described above, the precise number of such complexes in these experiments is not defined. An upper limit for the concentration of complex is 0.15 pmol/cm2 based on the SSMs being formed from vesicles containing 1% (w/w) CymA. From this, we calculate a minimum turnover number for MQ-7 oxidation by the CymA-Fcc3 complex of 40 s–1.
The experiments described above reveal that the CymA-Fcc3 complex is able to oxidize MQ-7 in SSMs. This is in stark contrast to our previous studies of CymA in contact with MQ-7 containing liposomes where only MQ-7 reduction was observed.11 To establish whether Fcc3 is responsible for the change in the catalytic bias of CymA, the activity of CymA in the membrane-modified electrodes was measured in the absence of Fcc3 using chemical reductants and oxidants (Figure 3). Addition of ferricyanide had almost no effect on the voltammogram (Figure 3A). Electronic absorbance spectroscopy demonstrates that solutions of reduced CymA are rapidly oxidized by ferricyanide (not shown). As a consequence, the absence of a reductive signal in the CV confirms that CymA without Fcc3 is unable to oxidize MQ-7. The reduction potential of ferricyanide (+420 mV) is higher than that of fumarate (−30 mV), indicating that MQ-7 oxidation by the CymA-Fcc3 complex is not driven solely by thermodynamics. Instead, formation of the long-lived CymA-Fcc3 complex shifts the catalytic bias of CymA toward MQ-7 oxidation. When, instead of ferricyanide, dithionite is added to the SSM, a catalytic oxidation signal appears that is inhibited by addition of HQNO. This confirms our previous findings, that CymA is only able to reduce MQ-7 in the absence of Fcc3.
Figure 3.
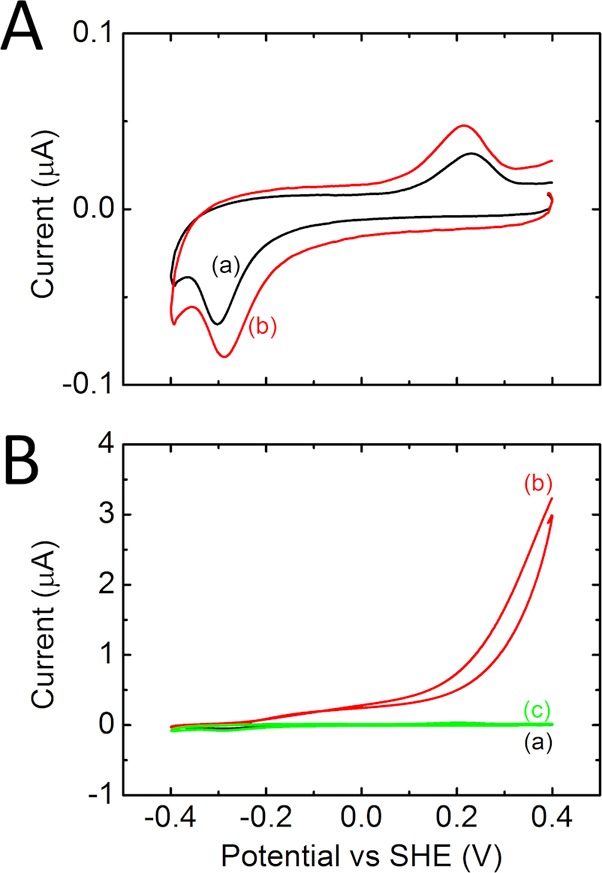
CVs (10 mV/s) of a SSM on a gold electrode modified with cholesterol tethers. The SSM was formed with CymA proteoliposomes (90:10 POPC:cardiolipin; 1% (w/w) CymA; 1% (w/w) MQ-7). (A) CVs (a) before and (b) after addition of 1 mM potassium ferricyanide. (B) CVs (a) before and (b,c) after addition of (b) 1 mM sodium dithionite and (c) 1 mM sodium dithionite/10 μM HQNO.
Very similar results were obtained when experiments were repeated with CymAsol (in these experiments, CymAsol is adsorbed after SSM formation; Figure 2E and F and Figure S3 in the Supporting Information). This demonstrates that structural details of the interaction between the lipid membrane and CymA are not important determinants of the enzyme’s catalytic bias. It is notable that the catalytic currents displayed by CymAsol are 4–5-fold lower than those for the SSMs containing full-length CymA. This is despite the fact that the surface coverages of CymAsol and Fcc3 are higher than those for the CymA assemblies. This lower activity could reflect very slow quinol oxidation by CymAsol and/or adsorption of CymAsol in a range of orientations, many of which preclude efficient interaction with MQ-7 and/or ET to Fcc3. The latter possibility is consistent with the absence of a significant change in the CV from the SSMs after immobilization of CymAsol. A turnover frequency of ∼0.1 s–1 for MQ-7 oxidation can be calculated on the basis of a CymAsol coverage of 12 pmol/cm2. However, on the basis of arguments discussed above, this activity represents a lower limit.
Discussion
S. oneidensis MR-1 has become an important model organism for bioreactor, bioengineering, and bioremediation studies.25−29 Its ability to transfer electrons from the oxidation of organic compounds or hydrogen to extracellular minerals and electrodes has inspired many to investigate applications in microbial-fuel cell technology. Underpinning these capabilities is a conduit for electron transfer that extends from the quinol pool and CymA at the inner membrane to extracellular minerals and electrodes.30 However, CymA also distributes electrons from quinol oxidation to the terminal reductases for fumarate, nitrate, nitrite, and extracellular DMSO.9,10 CymA thus plays a significant role in the highly branched network that supports anaerobic respiration in S. oneidensis MR-1. Our finding that complex formation between CymA and one of its partner proteins is required for CymA to act as a quinol dehydrogenase significantly advances our understanding of the factors that regulate the distribution of electrons across the respiratory network of S. oneidensis MR-1.
That CymA does not oxidize MQ-7 in the absence of a partner protein will occlude ET to nonphysiological redox partners and prevent side-reactions such as the production of radical oxygen species. Because complex formation between CymA and each of its partner proteins is required to trigger MQ-7 oxidation, respiratory electron flux should be dependent to some extent on the lifetime of the complexes formed by CymA. The CymA-STC ET complex is believed to be transient because it has not been possible to cross-link these proteins.31 However, for CymA-Fcc3, evidence has been presented to support transient as well as long-lived ET complexes.32−34 A recent NMR study by Fonseca et al. shows that detergent-solubilized CymA forms a transient low-affinity complex with Fcc3 with a Kd of 0.4 mM.32 Schwalb et al. describe a solution assay in which CymAsol is continuously oxidized by a much smaller number of Fcc3 enzymes in the presence of fumarate.33 In contrast, Ross and colleagues provided evidence for static CymA-Fcc3 and CymA-MtrCAB complexes in whole cells of S. oneidensis MR-1.34 The results presented here also suggest static complexes are formed. Given the critical role of protein–protein complex formation in activating the MQ-7-dehydrogenase activity of CymA, it is now important to fully understand those factors that determine the lifetime of the complexes formed by CymA under cellular conditions.
The electrochemical data indicate that the MQ-7 oxidation activity of the CymA-Fcc3 complex is at least 40 s–1. If we assume that CymA oxidizes MQ-7 at similar rates in complex with its other redox partners, we anticipate that the electron flux through CymA will be sufficient to support anaerobic respiration of S. oneidensis MR-1, including respiration on inorganic minerals and electrodes where a flux of 1 electron s–1 per MtrC is reported.35−37 Previously, the rate of fumarate reduction by Fcc3 and CymA was measured as ∼0.01 s–1 in a spectrophotometric assay that employed the water-soluble quinol analogue menadiol.14 The turnover frequencies resolved here for CymA with the natural substrate, MQ-7, are orders of magnitude greater. Similar behavior has been observed in studies with NrfHA where rates of menaquinol oxidation coupled to nitrite reduction are of the order of 390 s–1 but drop to 17 s–1 when assayed with the water-soluble analogue 2,3-dimethyl-1,4-naphthoquinone.38,39 Together these studies illustrate the care that should be taken when interpreting results obtained with water-soluble analogues.
That the formation of a protein–protein complex can influence the catalytic activity or “bias” of redox enzymes, or their subunits, adds an intriguing dimension to the mechanisms by which organisms regulate their ET networks. The group of Léger has recently suggested that in a multicentered redox enzyme, NiFe-hydrogenase, the forward (H2-formation) and reverse (H2-oxidation) reactions are limited by different rate-limiting steps and that this phenomena results in a tuning of catalytic bias, which can be altered by a series of mutations that do not alter the reduction potential of the active site.40 For NiFe-hydrogenase, it is proposed that the rate of H2 oxidation is defined by the rate of ET along a redox chain consisting of three Fe–S clusters.41 The reduction potentials of the hemes in CymA lie between −110 and −265 mV,11,14 which is lower than that of MQ-7 (−70 mV). This is also the case for other members of the NapC/NirT superfamily.42−44 We have previously shown that menadione, which has a reduction potential equal to MQ-7, is unable to reduce the hemes in CymA.14 The results presented here extend this observation to the native substrate MQ-7 and demonstrate that MQ-7 oxidation in CymA is limited by ET to the heme groups. Fcc3 binding to CymA is unlikely to change the properties of the buried MQ-7 active site, but will influence the properties of the surface-exposed hemes that are presented to the periplasm. Consequently, we propose that MQ-7 oxidation by CymA, and possibly all members of the NapC/NirT superfamily, is regulated by an increase in the reduction potential of these hemes upon complex formation. We note that not all heme reduction potentials need to be higher than that of MQ-7 as “rollercoaster” profiles, in which redox sites of alternating high and low potentials are commonly observed to make up a redox chain. An extreme example of the latter is the iron–sulfur redox chain in complex I.45
In conclusion, we find that inner membrane architectures can be constructed on planar surfaces and that this provides a powerful approach to resolve protein complex formation and quinone oxidoreductase activity of membrane enzymes. Our finding that protein–protein interactions modulate the catalytic bias of CymA reveals a new mechanism by which the magnitude and direction of electron flux through respiratory ET networks can be regulated. MQ-7 oxidation is regulated by controlling the rate of ET along the heme redox chain, which is rate limiting for CymA in isolation.
Acknowledgments
This research was supported by the Biotechnology and Biological Sciences Research Council (BB/G009228), the U.S. Department of Energy, Office of Biological and Environmental Research (BER) through the Subsurface Biogeochemical Research (SBR) Program, and the Research Corporation for the Advancement of Science. The manuscript represents a contribution from the Pacific Northwest National Laboratory (PNNL) SBR SFA. PNNL is operated for the Department of Energy by Battelle. We thank Evan T. Judd of Boston University for help with shipping protein samples, Jim Fredrickson and John Zachara of PNNL for critical reading of the manuscript, and Frank Collart and colleagues of Argonne National Laboratories for the plasmid for the expression of CymAsol-MBP. We also thank Prof. Baldwin and Dr. Postis (University of Leeds) for their kind gift of TEV protease.
Supporting Information Available
Figures S1, S2, and S3. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Supplementary Material
References
- Bashir Q.; Scanu S.; Ubbink M. FEBS J. 2011, 278, 1391. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Kamiya M.; Imai M.; Shinzawa-Itoh K.; Uchida T.; Kawano K.; Yoshikawa S.; Ishimori K. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov A. N.; van Nuland N. A. J. PLoS Comput. Biol. 2012, 8, e1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. F.; Zhen Y. J.; Sadoski R.; Grinnell S.; Geren L.; Ferguson-Miller S.; Durham B.; Millett F. J. Biol. Chem. 1999, 274, 38042. [DOI] [PubMed] [Google Scholar]
- Zhen Y.; Hoganson C. W.; Babcock G. T.; Ferguson-Miller S. J. Biol. Chem. 1999, 274, 38032. [DOI] [PubMed] [Google Scholar]
- Meschi F.; Wiertz F.; Klauss L.; Blok A.; Ludwig B.; Merli A.; Heering H. A.; Rossi G. L.; Ubbink M. J. Am. Chem. Soc. 2011, 133, 16861. [DOI] [PubMed] [Google Scholar]
- Bertero M. G.; Rothery R. A.; Palak M.; Hou C.; Lim D.; Blasco F.; Weiner J. H.; Strynadka N. C. J. Nat. Struct. Biol. 2003, 10, 681. [DOI] [PubMed] [Google Scholar]
- Rodrigues M. L.; Oliveira T. F.; Pereira I. A. C.; Archer M. EMBO J. 2006, 25, 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marritt S. J.; McMillan D. G. G.; Shi L.; Fredrickson J. K.; Zachara J. M.; Richardson D. J.; Jeuken L. J. C.; Butt J. N. Biochem. Soc. Trans. 2012, 40, 1217. [DOI] [PubMed] [Google Scholar]
- Simon J.; Klotz M. G. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 114. [DOI] [PubMed] [Google Scholar]
- McMillan D. G. G.; Marritt S. J.; Butt J. N.; Jeuken L. J. C. J. Biol. Chem. 2012, 287, 14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb C.; Chapman S. K.; Reid G. A. Biochem. Soc. Trans. 2002, 30, 658. [DOI] [PubMed] [Google Scholar]
- Boden N.; Bushby R. J.; Clarkson S.; Evans S. D.; Knowles P. F.; Marsh A. Tetrahedron 1997, 53, 10939. [Google Scholar]
- Marritt S. J.; Lowe T. G.; Bye J.; McMillan D. G. G.; Shi L.; Fredrickson J.; Zachara J.; Richardson D. J.; Cheesman M. R.; Jeuken L. J. C.; Butt J. N. Biochem. J. 2012, 444, 465. [DOI] [PubMed] [Google Scholar]
- Firer-Sherwood M.; Pulcu G. S.; Elliott S. J. J. Biol. Inorg. Chem. 2008, 13, 849. [DOI] [PubMed] [Google Scholar]
- Londer Y. Y.; Giuliani S. E.; Peppler T.; Collart F. R. Protein Expression Purif. 2008, 62, 128. [DOI] [PubMed] [Google Scholar]
- van den Berg S.; Lofdahl P. A.; Hard T.; Berglund H. J. Biotechnol. 2006, 121, 291. [DOI] [PubMed] [Google Scholar]
- Carter K.; Gennis R. B. J. Biol. Chem. 1985, 260, 10986. [PubMed] [Google Scholar]
- Stamou D.; Gourdon D.; Liley M.; Burnham N. A.; Kulik A.; Vogel H.; Duschl C. Langmuir 1997, 13, 2425. [Google Scholar]
- Jeuken L. J. C.; Daskalakis N. N.; Han X.; Sheikh K.; Erbe A.; Bushby R. J.; Evans S. D. Sens. Actuators, B 2007, 124, 501. [Google Scholar]
- Reimhult E.; Höök F.; Kasemo B. Langmuir 2003, 19, 1681. [Google Scholar]
- Granéli A.; Rydström J.; Kasemo B.; Höök F. Langmuir 2003, 19, 842. [Google Scholar]
- Cheetham M. R.; Bramble J. P.; McMillan D. G. G.; Krzeminski L.; Han X.; Johnson B. R. G.; Bushby R. J.; Olmsted P. D.; Jeuken L. J. C.; Marritt S. J.; Butt J. N.; Evans S. D. J. Am. Chem. Soc. 2011, 133, 6521. [DOI] [PubMed] [Google Scholar]
- Jeuken L. J. C.; Bushby R. J.; Evans S. D. Electrochem. Commun. 2007, 9, 610. [Google Scholar]
- Biffinger J. C.; Pietron J.; Ray R.; Little B.; Ringeisen B. R. Biosens. Bioelectron. 2007, 22, 1672. [DOI] [PubMed] [Google Scholar]
- Fredrickson J. K.; Romine M. F.; Beliaev A. S.; Auchtung J. M.; Driscoll M. E.; Gardner T. S.; Nealson K. H.; Osterman A. L.; Pinchuk G.; Reed J. L.; Rodionov D. A.; Rodrigues J. L. M.; Saffarini D. A.; Serres M. H.; Spormann A. M.; Zhulin I. B.; Tiedje J. M. Nat. Rev. Microbiol. 2008, 6, 592. [DOI] [PubMed] [Google Scholar]
- Jensen H. M.; Albers A. E.; Malley K. R.; Londer Y. Y.; Cohen B. E.; Helms B. A.; Weigele P.; Groves J. T.; Ajo-Franklin C. M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J.; Park H. S.; Hyun M. S.; Chang I. S.; Kim M.; Kim B. H. Enzyme Microb. Technol. 2002, 30, 145. [Google Scholar]
- McLean J. S.; Wanger G.; Gorby Y. A.; Wainstein M.; McQuaid J.; Ishii S. I.; Bretschger O.; Beyenal H.; Nealson K. H. Environ. Sci. Technol. 2010, 44, 2721. [DOI] [PubMed] [Google Scholar]
- Richardson D. J.; Butt J. N.; Fredrickson J. K.; Zachara J. M.; Shi L.; Edwards M. J.; White G.; Baiden N.; Gates A. J.; Marritt S. J.; Clarke T. A. Mol. Microbiol. 2012, 85, 201. [DOI] [PubMed] [Google Scholar]
- Ross D. E.; Ruebush S. S.; Brantley S. L.; Hartshorne R. S.; Clarke T. A.; Richardson D. J.; Tien M. Appl. Environ. Microbiol. 2007, 73, 5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca B. M.; Paquete C. M.; Neto S. E.; Pacheco I.; Soares C. M.; Louro R. O. Biochem. J. 2013, 449, 101. [DOI] [PubMed] [Google Scholar]
- Schwalb C.; Chapman S. K.; Reid G. A. Biochemistry 2003, 42, 9491. [DOI] [PubMed] [Google Scholar]
- Ross D. E.; Flynn J. M.; Baron D. B.; Gralnick J. A.; Bond D. R. PLoS One 2011, 6, e16649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon C. L.; Dohnalkova A. C.; Nachimuthu P.; Kennedy D. W.; Saffarini D. A.; Arey B. W.; Shi L.; Wang Z.; Moore D.; Mclean J. S.; Moyles D.; Marshall M. J.; Zachara J. M.; Fredrickson J. K.; Beliaev A. S. Geobiology 2010, 8, 56. [DOI] [PubMed] [Google Scholar]
- Ross D. E.; Brantley S. L.; Tien M. Appl. Environ. Microbiol. 2009, 75, 5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron D.; LaBelle E.; Coursolle D.; Gralnick J. A.; Bond D. R. J. Biol. Chem. 2009, 284, 28865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues M. L.; Scott K. A.; Sansom M. S. P.; Pereira I. A. C.; Archer M. J. Mol. Biol. 2008, 381, 341. [DOI] [PubMed] [Google Scholar]
- Simon J.; Gross R.; Einsle O.; Kroneck P. M. H.; Kroger A.; Klimmek O. Mol. Microbiol. 2000, 35, 686. [DOI] [PubMed] [Google Scholar]
- Abou Hamdan A.; Dementin S.; Liebgott P. P.; Gutierrez-Sanz O.; Richaud P.; De Lacey A. L.; Roussett M.; Bertrand P.; Cournac L.; Leger C. J. Am. Chem. Soc. 2012, 134, 8368. [DOI] [PubMed] [Google Scholar]
- Dementin S.; Belle V.; Bertrand P.; Guigliarelli B.; Adryanczyk-Perrier G.; De Lacey A. L.; Fernandez V. M.; Rousset M.; Leger C. J. Am. Chem. Soc. 2006, 128, 5209. [DOI] [PubMed] [Google Scholar]
- Todorovic S.; Rodrigues M. L.; Matos D.; Pereira I. A. C. J. Phys. Chem. B 2012, 116, 5637. [DOI] [PubMed] [Google Scholar]
- Roldan M. D.; Sears H. J.; Cheesman M. R.; Ferguson S. J.; Thomson A. J.; Berks B. C.; Richardson D. J. J. Biol. Chem. 1998, 273, 28785. [DOI] [PubMed] [Google Scholar]
- Gon S.; Giudici-Orticoni M. T.; Mejean V.; Iobbi-Nivol C. J. Biol. Chem. 2001, 276, 11545. [DOI] [PubMed] [Google Scholar]
- Hirst J. Biochem. J. 2010, 425, 327. [DOI] [PubMed] [Google Scholar]
- Myers C. R.; Myers J. M. J. Bacteriol. 1997, 179, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.