

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder which is characterized by an increasing impairment in normal memory and cognitive processes that significantly diminishes a person's daily functioning. Despite decades of research and advances in our understanding of disease aetiology and pathogenesis, there are still no effective disease-modifying drugs available for the treatment of AD. However, numerous compounds are currently undergoing pre-clinical and clinical evaluations. These candidate pharma-cotherapeutics are aimed at various aspects of the disease, such as the microtubule-associated τ-protein, the amyloid-β (Aβ) peptide and metal ion dyshomeostasis – all of which are involved in the development and progression of AD. We will review the way these pharmacological strategies target the biochemical and clinical features of the disease and the investigational drugs for each category.
Keywords: Alzheimer's disease, amyloid-β, metals, therapeutics
Current pharmacotherapies for the treatment of AD
Alzheimer's disease (AD) is the most prevalent cause of dementia in the elderly population, affecting approximately 35–40 million patients worldwide [1], and is the third leading cause of death in developed countries [2]. As such, AD represents a major socio-economic problem, which requires better diagnostic tools, management and effective therapies in order to ease the burden of this disease. While there are advances being made in all these areas, particularly with the identification of new biomarkers and the development of novel brain imaging compounds for the early detection of disease, it is clear that an effective treatment for AD is as elusive as ever. To date, the only Food and Drugs Administration (FDA)-approved drugs for the treatment of AD patients are the acetylcholinesterase inhibitors (AChEIs) tacrine, donepezil, galantamine and rivastigmine, and the non-competitive N-methyl-D-aspartate (NMDA)-receptor antagonist memantine. The AChEIs exert their affect by preventing the enzymatic degradation of the neurotransmitter acetylcholine (AChE), resulting in increased AChE concentrations in the synaptic cleft and enhanced cholinergic transmission [3]. Memantine, however, protects neurons against NMDA receptor activation-mediated glutamate excitotoxicity [4–6] and also inhibits τ-hyperphosphorylation and aggregation [7]. A new approach, using combination therapy of donepezil and memantine, has been reported to have significant beneficial effects on cognitive function, activities of daily living and behaviour [8]. Meanwhile, potent and more selective AChEIs (Huperzine A, Neuro-Hitech Inc., New York, NY, USA) and NMDA-receptor antagonists (Dimebon, Medivation Inc., San Francisco, CA, USA) are being assessed.
However, irrespective of the form of therapy utilized, the current approaches for the treatment of AD provide only temporary symptomatic relief and do not inhibit and/or reverse the underlying disease mechanisms. This stresses the urgent need for disease-modifying drugs for AD – small, easily administrated, well-tolerated, bioavailable compounds that cross the blood-brain barrier (BBB) and have little or no adverse effects and/or contraindications. There are currently more than 50 compounds in various stages of clinical investigation for the treatment of AD (www.alzforum.org) including: statins [9–12], peroxisome pro-liferator-activated receptor-γ agonists [13–16], non-steroidal anti-inflammatory drugs [17–19], neurotrophic molecules and even metabolic or nutritional drinks (Ketasyn™, Accera, Broomfield, CO, USA; Souvenaid™, Danone Research-Centre for Specialized Nutrition, respectively, Palaiseau, France). In addition, there are many more candidate molecules that are at the pre-clinical stage of development and are likely to proceed into clinical trials. Most of these pharmacological agents have been designed and/or developed based upon a notion that has been dominating the AD field for the past two decades – the ‘amyloid cascade hypothesis’. This theory claims that the metabolism of the amyloid-β (Aβ) peptide (both generation and clearance) is the main initiator of AD, which together with the downstream formation of the τ-protein aggregates, leads to neuronal and synaptic dysfunction and loss, microglial activation and neuronal death [20, 21]. Thus, most of the pharma-cological agents being developed target one or both of the principal cerebral proteins implicated in the pathogenesis of AD: τ and Aβ. In this review, we will provide a broad overview of the therapeutic approaches currently being developed for the treatment of AD.
AD pharmacotherapies targeting τ
Neurofibrillary tangles (NFTs), which are found in AD and other forms of dementia, consist of insoluble, intra-neuronal inclusions [22, 23] comprised paired helical filaments that are formed from hyperphosphorylated τ[24, 25]. Hyperphosphorylation of the microtubule-associated τ-protein is likely to result from an imbalance in kinase and phosphatases activities, and leads to destabilization of microtubules [26], loss of neuronal cytoskeletal architecture and/or plasticity [27], impaired neuronal transport, dystrophy and ultimately neuronal cell death [28, 29]. Based on these findings, small molecules that interfere with the formation of τ-aggregates, selectively inhibit τ-kinases and/or activate τ-phosphatases are being pursued as therapeutic targets (see Fig. 1).
1.
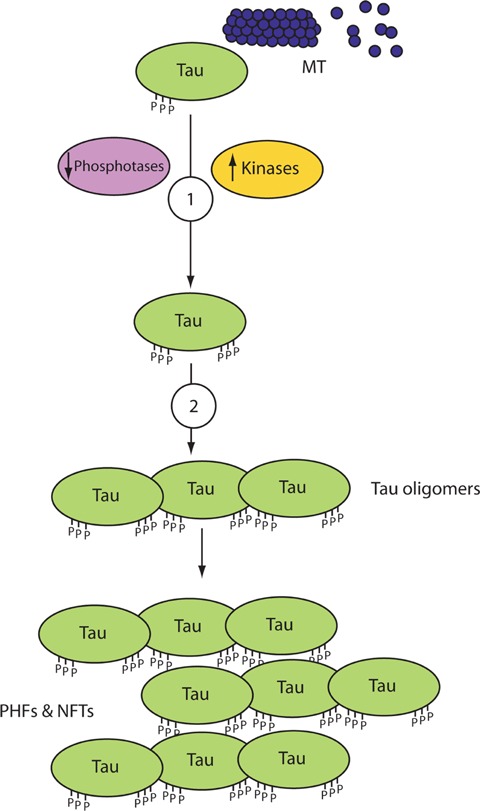
Pharmacotherapeutic strategies for the treatment of Alzheimer's disease targeting τ. Schematic representation of the anti-τ targets for potential pharmacotherapies: (i) Modulators of τ-kinases or phosphatases, (ii) τ-aggregation inhibitors (TAIs). Abbreviations: MT (micro-tubule); NFTs (neurofibrillary tangles); PHFs (paired helical filaments).
Modulators of τ kinases or phosphatases
The biological function of the microtubule-associated τ-protein [30] is regulated by several kinases and phosphatases [31–33]. An imbalance in activity between kinases and phosphatases results in the abnormal phosphorylation of 38 or more serine and/or threonine amino acids on τ in the AD brain [34–37]. Phosphorylation of a tyrosine residue at position 18 (Tyr18) on τ by the tyrosine kinase fyn has also been reported [38]. Decreased mRNA levels [39] and activity of the main τ-protein phosphatases (PP)1 and PP2A, as well as other τ-phosphatases such as PP2B and PP5, have been observed in AD [40–42]. This can lead to a direct reduction in τ-dephosphorylation or indirect hyperphosphorylation by the inability of these phosphatases to inhibit τ-hyperphosphorylation by different kinases [43], therefore τ-phosphatases have been proposed as therapeutic targets [44]. Major kinases, whose protein levels and activities are reported to be up-regulated in AD and other tauopathies [45–48], involved in the phosphorylation of τ include glycogen synthase kinase (GSK)-3, cyclin-dependent protein kinase-5, casein kinase-1, protein kinase A (cyclic adenosine monophosphate (cAMP)-dependent protein kinase), protein kinase C, calcium and calmodulin-dependent protein kinase-II, microtubule-affinity regulation kinase and mitogen-activated protein kinase family members [49–53]. These proteins have also been suggested as therapeutic targets for AD. Recent reports have highlighted the importance of GSK-3β in the developments of both τ and Aβ pathologies in AD and concluded that this kinase is a vital drug target for the treatment of AD and other neurodegenerative diseases [54–57]. Several animal studies, for example, have demonstrated that the inhibition of GSK-3β activity by lithium [58] results in decreased levels of both Aβ (in PDAPP mice) and τ-phospho-rylation, τ-aggregation and NFT formation (in JNPL3 mutant τ-mice) [59–61]. Other GSK-3β inhibitors are being developed, such as AR-A014418 [61], as well as other kinase inhibitors [62–67]; however, this approach is hindered due to the ubiquitous expression of these kinases, their pleiotropic activities in countless cellular functions and the low selectivity of inhibitors for specific kinases, isoforms of a particular kinase, cellular compartment and/or pathological, rather than physiological, activity of the kinase [68–70].
τ aggregation inhibitors (TAIs)
Screening for TAIs started in the early 1990s with reports on the ability of phenothiazines [71], anthraquinones [72] and low molecular weight N-phenylamine derivatives [73] to prevent τ-aggregation and associated toxicity in cell lines [74]. The most clinically advanced TAI is AL-108 or NAP (Allon Therapeutics Inc., Vancouver, BC, Canada), which is an intra-nasal formulation of an 8 amino-acid peptide (NAPVSIPQ) derived from the biological activity-dependent neuroprotective protein secreted by the brain in response to various insults [75]. Studies in transgenic mice suggest that AL-108 interacts with microtubules, reduces τ-hyper-phosphorylation and increases soluble τ levels leading to an improvement in cognition [76, 77]. Data from a recently completed phase IIa trial evaluating AL-108 in 144 patients with amnestic mild cognitive impairment demonstrated that it is safe and well tolerated, and the high dose (15 mg twice a day) resulted in a significant and lasting improvement in short term and working memory (but not in tests that involved executive functions). AL-108 is now being tested as a treatment for other neurodegenerative diseases, mental disorders and ocular disease. An intravenous (IV) formulation of NAP, known as AL-208, is also under clinical investigation for mild cognitive impairment associated with coronary artery bypass graft surgery as well as other indications [78].
A recently announced TAI is Rember™ (TauRx Therapeutics Ltd., Singapore), which has methylthioninium chloride (MTC; also known as the histochemical dye methylene blue) as its active constituent. It is proposed that this compound is not only able to prevent the oligomerization and self-aggregation of τ, but also dissolve pre-formed τ-oligomers and paired helical filaments into truncated τ-fragments, which can then be naturally degraded and eliminated (http://www.taurx.com/). At the 11th International Conference on Alzheimer's Disease (ICAD, Chicago, 2008), pre-clinical data (O1–06-04, P2–383, P2–428) and results of a recently completed 24-week, multi-centred, randomized, double-blind, dose-ranging (30, 60 or 100 mg, three times per day), placebo-controlled phase IIb trial followed by a 60-week, blinded, active treatment extension study were presented (O3–04-07, P4–347, P4–384). Patients with moderate AD who received MTC at 60 mg three times/day showed a significant improvement in the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) scores, compared to placebo control, at the end of the 24-week-long trial. This result was further verified after 50 weeks of treatment and again at the conclusion of the trial (84 weeks in total). Another measure of the drug's efficacy that was utilized was single photon emission computed tomography (SPECT) analysis at week 24 compared to baseline, which revealed that the regional cerebral blood flow decline seen in the hippocampus and entorhinal cortex of individuals treated with placebo, was not observed in individuals treated with MTC (60 mg three times/day). Despite these seemingly encouraging results, great reservations have been expressed, mainly due to unusual trial design and an unconventional method of analysis. However, TauRx Therapeutics Ltd. has announced that it intends to take Rember™ into a phase III clinical trial, and that it is already testing a second generation TAI molecule, LMT-X, in τ-transgenic animal models.
AD pharmacotherapies targeting Aβ
Although the exact mechanism is still unclear, it is widely believed that dysfunctional Aβ metabolism is the underlying cause for the neurodegeneration and dementia observed in AD. Therefore, a leading strategy for the development of AD pharmacotherapies is modulation of Aβ production, aggregation and/or clearance. It is assumed that altering these processes will stop and/or reverse the pathological neuronal loss and the clinical cognitive decline. We will briefly summarize key findings of the major AD pharmacological strategies being development to target various aspects of Aβ metabolism (see Fig. 2).
2.
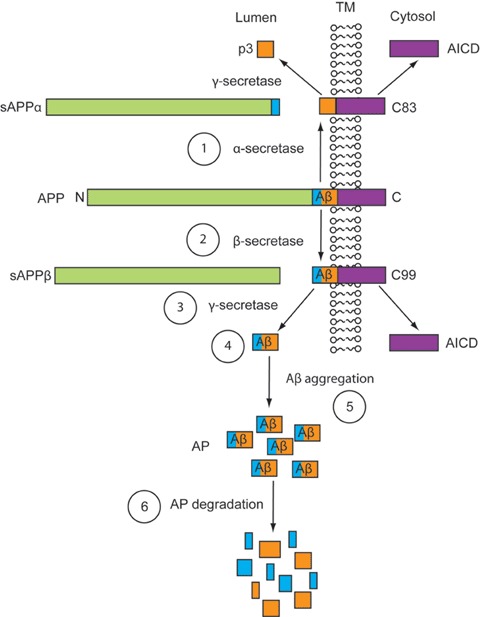
Pharmacotherapeutic strategies for the treatment of Alzheimer's disease targeting Aβ. Schematic representation of the anti-amyloidogenic targets for potential pharmacotherapies: (i) α-secretase activators, (ii) β-secretase modulators/inhibitors, (iii) 7-secretase modulators/inhibitors, (iv) Aβ immunotherapy, (v) Aβ aggregation inhibitors, (vi) Amyloid-plaque degredation enhancers. Abbreviations: Aβ (amyloid-β), AICD (APP intracellular domain); AP (amyloid plaque); APP (amyloid precursor protein); sAPPα (soluble APP-α); sAPPβ (soluble APP-β); TM (trans-membrane).
Inhibitors and/or modulators of the secretases
The amyloid precursor protein (APP) is an evolutionary conserved type I transmembrane glycoprotein [79] that belongs to a family of proteins, including amyloid protein precursor-like protein1 (APLP1) and APLP2 [80, 81]. Both the amino and carboxyl terminals of APP can be divided into several regions, each with its own characteristics and functions [82]. The overall function of APP is unclear; however, it is believed to be important during the development of the CNS and in response to stress or injury [83]. APP has been suggested to act as a cell-surface receptor and may also be involved in cell adhesion and/or neurite outgrowth [84, 85]. APP is synthesized in the endoplasmic reticulum, undergoes N-and O-glycosylation in the Golgi, and is translocated from the trans-Golgi network to the cell surface via the secretory pathway [86]. During and/or after trafficking, APP undergoes degradation via the ubiquitin-proteasome system [87] and/or various forms of autophagy [88, 89]. Neuronal macroautophagy induction and impaired clearance of several autophagy intermediates is evident in the AD brain, leading to an overproduction and accumulation of intracellular Aβ in autophagic vacuoles [90, 91].
APP also undergoes proteolytic processing through either the non-amyloidogenic or the amyloidogenic pathways [92]. During the non-amyloidogenic pathway, the membrane-bound enzyme α-secretase cleaves APP within its Aβ domain, resulting in the extracellular secretion of soluble APP-α (sAPP-α) and the production of a short membrane-bound COOH-terminal fragment (CTF), α-CTF or C83 [93]. Subsequent 7-secretase cleavage of C83 results in the secretion of a 3-kD peptide termed p3 out of the cell [94], and release of the APP intracellular domain (AICD) into the cytoplasm [95]. Enzymes that have been suggested to have α-secretase activity include members of a disintegrin and metalloprotease family of proteins, ADAM 10 and ADAM 17 or TACE (tumour necrosis factor-α converting enzyme) [96–98]. The amyloidogenic pathway is initiated when β-secretase, identified as the aspartyl protease β-site APP cleaving enzyme (BACE1, Asp-2 or memapsin-2) [99, 100], cleaves APP at the N-terminal part of the Aβ domain. This cleavage leads to the extracellular release of sAPPβ, while the β-CTF or C99 fragment remains membrane bound. Sequential γ-secretase cleavage of C99, at the C-terminal of Aβ, allows the shedding of the AICD and the secretion of Aβ species of variable length, into the lumen or extracellular space [101]. γ-Secretase is thought to be an intramembranous-cleaving polytopic aspartyl protease [102], comprised a complex of presenilin1 (PS1), presenilin2 (PS2), nicastrin, aph-1 and pen-2 [103–105]. The presenilins (PSs) are transmembrane homologue proteins [106], which have been shown to be essential for the γ-secretase cleavage of APP [107, 108] as well as other type I proteins [109]. Mutations in PSs have been shown to alter APP processing and Aβ levels in mice [110] and are associated with the inheritance of early onset familial AD in human beings [111].
Following their discovery and characterization, the APP secretases became attractive targets in the quest for an AD treatment. The logic behind modulating the APP secretases is two fold: stimulating α-secretase cleavage in order to direct APP processing towards the non-amyloidogenic pathway or suppressing β- and/or γ-secretase cleavage in order to reduce the amount of Aβ produced. It has been shown that muscarinic AChE-receptor agonists can foster α-secretase processing of APP to subsequently result in a reduction in Aβ levels [112, 113]. This has been further demonstrated in animal models of AD, where the treatment of triple transgenic mice [114] with the M1 AChR agonist NGX267 (TorreyPines Therapeutics, La Jolla, CA, USA) resulted in reduced Aβ1–42, reduced amyloid load and decreased τ-phosphorylation as well as improved behaviour [115]. Numerous β- and γ-secretase inhibitors and/or modulators have also been designed; however the majority of these agents are not specific for the secretase cleavage of APP and thus may prevent the cleavage and processing of additional substrates, which could result in various adverse effects [116, 117]. At the moment, the β-secretase inhibitor TAK-070 (Takeda Pharmaceutical Co. Ltd., Osaka, Japan) is undergoing a phase I clinical trial. A number of γ-secretase-targeting compounds are in early clinical development, including a selective γ-secretase inhibitor (BMS-708163; Bristol-Myers Squibb, New York, NY, USA) and a γ-secretase modulator (E2012; Eisai Inc., Woodcliff Lake, NJ, USA). The most advanced compound, however, is the γ-secretase inhibitor hydroxyl-valeryl monobenzocaprolactam/LY450139 dihydrate (Eli Lilly, Indianapolis, IN, USA). A 40-week, multi-centre, randomized, double-blinded, dose escalation, placebo-controlled, parallel assignment phase II study (safety, tolerability and biomarker assessment) with LY450139 dihydrate in individuals with mild-to-moderate AD showed that individuals who received either the low (100 mg/day) or high (140 mg/day) dose of the drug had a significant (∼60%) decrease in plasma Aβ1–40 compared to placebo; however, Aβ1–40 changes in cerebrospinal fluid (CSF) were not statistically significant [118]. Recruitment of approximately 1,500 individuals for a phase III trial to study the effects of LY450139 dihydrate (100 or 140 mg per day) on the rate of cognitive and functional decline versus placebo over a 2-year period has begun, with the clinical trial estimated to be complete in the first quarter of 2012.
A focal point at ICAD 2008 was the announcement by Myriad Genetics (Salt Lake City, UT, USA) that the most extensive (1,649 patients treated over 18 months in a phase III) AD clinical trial ever to be completed (tarenflurbil/Flurizan™ 800 mg/twice daily or placebo) had failed to demonstrate significant differences in any of its outcome measures, including ADAS-Cog and Alzheimer's Disease Cooperative Study Activities of Daily Living (ADCS-ADL) scores. Thus, the γ-secretase modulator Flurizan™ was ineffective in slowing disease progression. The failure of this trial has raised many issues within the AD research community with the main question being whether or not β- and/or γ-secretase modulators should still be considered as a therapeutic target. Many scientists believe that a wiser strategy to targeting Aβ production is to target Aβ after it has been synthesized.
Aβ aggregation inhibitors
As described above, Aβ is constitutively synthesized at the membrane surface by proteolytic cleavage and is then secreted [119]. Aβ typically ranges between 38 and 43 amino acid residues in length with Aβ1–40 and Aβ1–42 being the most prominent types in AD [120]. Following its secretion, extracellular Aβ can later be internalized back into the cell by poorly understood molecular mechanisms. Recently, it was reported that in the absence of apolipoprotein E (ApoE), Aβ1–42 is internalized in axons of primary neurons via a clathrin-independent endocytic pathway involving lipid rafts [121]. The rapid turnover of Aβ in the brain [122, 123] suggests efficient clearance and/or degradation mechanism(s) of the peptide are in place. Detection of Aβ in plasma and CSF [124], implies that Aβ can be transported from the CNS across the BBB into the periphery. In this regard, a few receptors (involved in cholesterol and/or lipid metabolism) have been suggested to mediate Aβ efflux from the brain, including MDR1-P-glycoprotein (P-gp/ABCB1) [125], receptor for advanced glycation end products (RAGE) [126] and the extensively studied low-density lipoprotein receptor-related protein (LRP). Aβ has been shown to bind directly to LRP-1 and LRP-2/megalin or indirectly, by binding to their ligands: apolipoprotein J and E (ApoJ and ApoE, respectively) and α2-macroglobulin (α2 M) [127–129]. Aβ-LRP1/2 complexes can be internalized and delivered to the endosomal/lysosomal compartments, where they either undergo autophagy in a similar manner to APP, or they may undergo transcytosis into the CSF or plasma [130, 131]. Aβ is finally eliminated through the kidney and liver via LRP [132, 133] or by liver X receptor [134–136]. Alternatively, Aβ can be catabolized via enzymatic degradation [137]. To this end, several classes of enzymes have been identified, including the serine proteases plasmin and tissue plasminogen activator [138–140], and the metalloproteases neprilysin [141–144], insulin degrading enzyme [145–148], as well as the zinc-dependent endothelin-converting enzyme 1 [149, 150] and matrix metalloproteinases 2 and 9 (MMP2 and MMP9, respectively) [151–153].
The fact that Aβ is normally produced in the body throughout life, is present in various organs and bodily fluids, and that the body has evolved sophisticated mechanisms for its metabolism (as detailed above) suggest that Aβ has a physiological role [154]. Although the function of Aβ is yet to be elucidated, Aβ has been proposed as an acute-phase apolipoprotein with metal-binding and antioxidant activities [155–160]. The idea that Aβ has a functional role leads us to the conclusion that with old age, and more specifically with the late onset of AD, Aβ either losses its physiological function or gains a pathological function [155, 156]. There are several theories as to factor(s) that may turn Aβ from being a physiological to a pathological agent; however, none of these hypotheses are definite and all of them still have many caveats. However, it has been consistently demonstrated that Aβ exerts neurotoxic and synaptotoxic affects both in vitro[161] and in vivo[162]. Researchers have turned to the study of Aβ structure in search of clues as to its toxic effects. It was found that soluble Aβ monomers assume a random coil or α-helix conformation; however, in AD they undergo a structural change into a pleated β-sheet [163]. This induces the peptide to form low molecular weight oligomers, higher molecular weight complexes (protofibrils and amyloid-β derived diffusible ligands or ADDLs), mature fibrils and amyloid plaques (APs) in the neuropil and the vasculature [164–166]. In vitro studies have shown that amyloidogenesis and fibrillogenesis can be affected not only by the type of Aβ produced and its conformation, but also by factors such as time, concentration, temperature, pH and metal ion concentration [167]. For many years it was believed that the toxic effects of Aβ were a result of the mature Aβ fibrils; however, recent studies suggest that low molecular weight, soluble, oligomeric forms of Aβ1–42 rather than Aβ1–40[168] are more neurotoxic than the mature Aβ fibrils [169–173]. Indeed, the severity of AD correlates more closely with cerebral concentrations of soluble Aβ rather than insoluble Aβ load (reviewed by Lesne and Kotilinek [174]). As our understanding of Aβ structure improves and with the advent of more advanced techniques, the development of inhibitors of Aβ oligomers will improve [175]. Candidate drugs in this category are synthetic peptides based on the Aβ17–21 sequence, with the five-amino-acid β-sheet breaker peptide Ac-LPFFD-NH2 (iAβ5p) as its lead compound [176, 177], the discontinued tramiprosate/Alzhemed™ (Neurochem Inc.) and ELND-005/AZD-103 (developed as a joint venture by Elan Pharma International Ltd., Dublin, Ireland and Transition Therapeutics, Toronto, ON, Canada). Tramiprosate/Alzhemed™ is in fact a variant of the amino acid taurine (3-amino-1-propanylsulfonic acid [3-APS]) [178], which prevents sulphated glycosaminoglycans from promoting the oligomerization of soluble Aβ[179], but at the same time also enhances non-toxic τ-aggregation in vitro[180, 181]. Unfortunately, pre-clinical studies of tramiprosate/ Alzhemed™ in TgCRND8 mice did not include an investigation of τ-pathology or any behavioural testing. Phase II trial results showed the only significant effect of tramiprosate/Alzhemed™ treatment was a dose-dependent reduction in CSF Aβ1–42, but had no significant impact on CSF Aβ1–40 and τ, or on psychometric scores [182, 183]. Despite these disappointing results, the investigational drug progressed into a phase III trial in Northern America, which was recently declared by the FDA to have failed. As a result, the European Phase III study of tramiprosate/Alzhemed™ has been abandoned and the compound is being marketed as a nutraceutical, although a phase II trial for its use as a preventative of hemorrhagic stroke in patients with cerebral amyloid angiopathy (CAA) is ongoing. Another investigational drug, ELND-005/AZD-103 (Transition Therapeutics, Toronto, ON, Canada and Elan, Dublin, Ireland), is an orally administrated compound that crosses the BBB and is believed to break-down Aβ aggregates and prevent further Aβ oligomerization from taking place. In transgenic mouse models of AD, ELND-005/AZD-103 treatment improved their spatial memory performance in the Morris Water Maze. In several phase I studies, single and multiple ascending doses of ELND-005/AZD-103 were shown to have good safety, tolerability and pharmacokinetic profiles. At present, ELND-005/AZD-103 is undergoing an 18-month phase II trial in 340 patients with mild-to-moderate AD in order to confirm its safety and to evaluate its efficacy on cognition and functionality.
Another approach has been to try and characterize the mecha-nism(s) involved in the neurotoxicity of Aβ as a basis for developing pharmacotherapeutics that modulate these processes. Aβ-associated neurotoxicity may be attributed to various factors [184], including: Aβ interactions with intracellular target(s) and/or extracellular Aβ interaction with membrane surface receptor(s), cholesterol, lipids and lipoproteins [185, 186]. Activation of microglia and inflammatory factors [187] and induction of apoptosis by Aβ-mediated activation of cysteine aspartyl proteases termed caspases [114, 188, 189] have also been proposed to have neurotoxic effects. Berman and colleagues recently demonstrated that Aβ oligomer-induced neurotoxicity is due to the destabilization of phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) metabolism [190]. Another proposed mechanism of Aβ toxicity is the promotion of ion-channel formation and calcium ion (Ca2+) influx [191]. This theory gained support from pre-clinical and early clinical trials with different neuronal L-type calcium channel blockers, such as S-312-d, nimodipine and MEM 1003 (Memory Pharmaceuticals, Montvale, NJ, USA) [192–195]. However, meta-analysis of clinical studies revealed that nimodipine only slows down the disease progression and may be effective only in certain types of dementia [196]. As for MEM 1003, late last year Memory Pharmaceuticals announced that the drug failed to show changes in ADAS-Cog scores between treated and control mild-to-moderate AD patients in a phase IIa trial, yet the company is still testing the efficacy of MEM 1003 in individuals with bipolar disorder (www.memorypharma.com).
Passive or active immunization
A novel and controversial approach to treating AD is based on vaccine therapy. Transgenic mouse models of AD actively immunized with Aβ[197–200] or passively immunized with humanized anti-Aβ antibodies [201–208] showed reduced Aβ and τ-pathology, neutralized soluble Aβ oligomers, attenuated synaptic degeneration and improved synaptic plasticity, all of which were accompanied by improved learning. Immunization against Aβ thus appeared to be the much-anticipated breakthrough in the development of AD therapeutics, in addition to being the primary test of the amyloid cascade hypothesis. An active immunization strategy was rapidly advanced into clinical trials by Elan, and following successful completion of the phase I trial, a phase IIa trial with AN-1792/Betabloc was initiated by Elan/Wyeth. This study was terminated after four patients presented with symptoms consistent with autoimmune meningoencephalitis [209, 210] and by the end of 2002 there were 18 known cases [211]. A subsequent autopsy analysis of a phase I study patient, who died 20 months after the first inoculation, indicated evidence of encephalitis [212]. This, together with three later autopsy cases of AN-1792-immunized AD patients, highlighted the persistence of CAA despite the removal of Aβ from plaques [213], consistent with observations from studies in PDAPP mice [214, 215] and monkeys [216]. A follow-up study of a further 36 patients demonstrated that many developed anti-Aβ antibodies, which was consistent with a slowing in the rate of cognitive decline 12 months after completion of the trial [217]. Patients with the highest titres also displayed the greatest slowing in cognitive decline [218]. While encouraging, MRI scans of the antibody responders revealed a reduction in total brain volume and the rates of cognitive decline in the non-responders appeared more rapid than typical [219]. However, a composite neuropsychological performance study has shown that the patients developing Aβ antibodies showed improvements in memory, attention and concentration, along with decreases in the level of τ-protein in CSF [220]. The most recent data to emerge from the original immunization trial, however, appear to confound some of these earlier reports. The long-term clinical follow-up of 80 patients demonstrated that, despite a varied degree of Aβ plaque removal, there was no prevention of progressive neurodegeneration and no evidence for improved survival [221]. Of note, seven of the eight immunized patients that underwent autopsy, including two patients with near complete removal of plaques, had severe end-stage dementia prior to death [221]. Despite its tragic outcome, valuable lessons learnt from this failed trial have lead researchers to develop more selective, advanced immunotherapies [222–225], including another active Aβ vaccine developed by Elan and Wyeth (Madison, NJ, USA) (ACC-001). Affiris GmbH (Vienna, Austria) is also developing an active immunization program with AFFITOPE AD01 (phase I study due to be completed in November 2008) and AFFITOPE AD02 (recruitment stage for a phase I trial due to be completed in early 2009).
The development of intravenous recombinant humanized anti-Aβ monoclonal immunoglobulins (IVIg), which avoid the induction of an immune response, continues in parallel. Two small, independent phase I investigations of AD patients with IVIg over six months proved to be safe, stopped the cognitive deterioration and in most cases even resulted in a slight improvement of ADAS-Cog scores [226]. Examples of passive vaccines against Aβ in various stages of research and development are: phase I (V950, Merck, Whitehouse Station, NJ, USA; PF-04360365, Pfizer, New York, NY, USA), completed phase II (LY2062430, Eli Lilly, Indianapolis, IN, USA), and ongoing parallel phase II and III (AAB-001/Bapineuzumab, Elan with Wyeth, Madison, NJ, USA). Data from a phase II study with LY2062430 indicate that the monoclonal antibodies lead to elevated levels of Aβ1–40 and Aβ1–42, both in serum and CSF; however, SPECT analysis did not reveal any reduction in APs and no improvement in cognition was detected. Despite this, the company has announced its intention to commence a phase III study with LY2062430 in the coming year. With regards to AAB-001/bapineuzumab, modified intent-to-treat (MITT) interim analysis of phase II studies showed no significant changes in ADAS-Cog and Disability Assessment Scale for Dementia in the total study population and no statistically significant changes in any of the cognitive or functional efficacy endpoints in the ApoE4 carrier sub-group. In fact, a significant elevation in ventricular volume was observed in ApoE4 carriers treated with the drug. However, post hoc MITT analysis of the results did show statistically significant differences from baseline in ADAS-Cog, the Neuropsychological Test Battery and the Clinical Dementia Rating Sum of Boxes, as well as the Brain Boundary Shift Integral in the non-ApoE4 carrier sub-group treated with AAB-001/bapineuzumab compared to placebo. It should be noted that individuals treated with the drug experienced significantly more cases of cataracts, deep vein thrombosis, syncope, seizures and pulmonary embolism, as compared to placebo control patients. Importantly, vasogenic edema was observed only in drug-treated patients and mostly in ApoE4 carriers treated with the highest dose of the drug (2.0 mg/kg). The significance of the results, however, will only be made clear once a final analysis is done after the completion of all phase II and III trials.
The metal hypothesis of AD
It is evident that both Aβ and τ are involved in the development and progression of AD; however, pharmacological strategies directed at these targets have not yet proven to be disease modifying in human studies. In particular, several investigational drugs that target Aβ have failed to show any correlation between a reduction in amyloid burden and improvement in cognitive functions in large-scale clinical trials (as mentioned above). While such data might indicate that the ‘amyloid hypothesis’ of AD is not necessarily the correct one, there remains considerable debate as to whether it has yet to be truly tested in the clinic. Numerous factors have been proposed to account for the poor performance of several frontline drugs, including: patient confounds (e.g. ApoE genotype, overall rates of cognitive decline in placebo groups), trial design (e.g. is a ‘treatment’ protocol, as opposed to a ‘prevention’ protocol, the best way to test the hypothesis) and drug penetration (e.g. it is suggested that Flurizan may have failed because of a poor pharmacodynamic profile). While the debate over the validity of the amyloid cascade hypothesis will no doubt continue, it remains likely that there are other critical factors playing a role in AD pathogenesis.
Metal ions are one such possibility, as cerebral concentrations of zinc (Zn), copper (Cu) and iron (Fe) ions are significantly elevated in AD, compared to age-matched controls [227–230], and metals have been implicated in several other neurodegenerative diseases [231–234]. Here, we will review the various events in AD pathogenesis in which metal ions are involved, and then discuss the pharmacotherapeutics being developed to modulate metal ions in AD.
There is an increasing amount of evidence suggesting that τ and NFTs may in some way be involved in, or regulated by, metal metabolism. Zinc ions (Zn2+) [235] and the iron regulatory protein-2 [236], for example, have been found to co-localize with NFT-containing neurons. Addition of Zn2+ to mouse and human neuroblastoma cells (N2a and SH-SY5Y, respectively) induces τ-hyperphosphorylation [237], whereas the opposite result is seen in hippocampal neurons with the addition of pyrolidium dithiocarbamate (PDTC) [238] or iron citrate (FeC6H5O7) [239]. Ferric ions (Fe3+) and cupric ions (Cu2+) can bind to various ‘repeat’ motifs on τ, thus altering the protein's conformation, promoting its phosphorylation [238] and inducing its aggregation [240–242]. In the case of iron, this effect can be reversed by reducing Fe3+ to Fe2+ (ferrous ions) [243]. As for APs, they have been shown to be enriched with Cu (400 μM), Zn (1 mM) and Fe (1 mM) [114, 176, 192–194], suggesting that there may be an interaction between metals, APP and Aβ that may influence Aβ aggregation and Aβ-associated toxicity.
It has been demonstrated that APP contains putative zinc and copper-binding domains (CuBD) both in its ectodomain and in its Aβ sequence (see Fig. 3). Little is known about the APP Zn-binding domain; however, it has been established that its CuBD consists of a tyrosine (Tyr168), a methionine (Met170) and two histidine (His147, 151) residues that are able to coordinate Cu2+ and reduce it to Cu+[244]. The similarities between the CuBD on APP and Cu chaperone proteins suggest that APP may play a role in metal homeostasis [245]. This notion has recently gained support from findings that the translation of APP mRNA is governed by the binding of an iron-regulatory element to its 5′-untranslated region such that in an Fe-enriched environment APP translation is up-regulated, whereas it is down-regulated in response to an Fe-deficient milieu [246, 247]. Moreover, increasing Cu levels in vitro can shift APP processing towards the non-amyloidogenic pathway and result in decreased Aβ production [222–225]. This may result from an increase in GSK-3β phosphorylation, which activates phosphatidylinositol-3-kinase (PI3K) to result in the secretion of MMPs that can degrade Aβ[225]. In addition, genetically modified animal models of AD provide vital clues as to the affects of APP and Aβ on metal-ions and vice versa. Tg2576 mice that over-express the Swedish double mutant APP695 (K-670-N and M-671-L) exhibit AD-related behavioural and cognitive changes (memory and spatial learning impairments) [248] and AD-related pathology (substantially elevated levels of full-length APP, CTFs and cerebral extracellular Aβ) [249]. However, their cerebral Cu (but not Fe) levels are significantly reduced [224, 250]. C100 mice over-express Aβ and the C-terminal of APP, yet have significantly lower levels of both Cu and Fe in the brain [250]. Conversely, APP (and APLP2) knockout mice have raised brain and liver Cu levels [251] and develop reactive cerebral gliosis and locomotor-behavioural changes with age [252]. These studies all suggest a role for APP in metal homeostasis. As a further demonstration that metal homeostasis is important in the pathogenesis of AD, when APPswe/PS1P-264-L-expressing mice, which also have ∼15% lower brain Cu levels compared to non-transgenic controls, are crossed with TxJ ‘toxic milk’ mice (that have a mutated ATPase7b transporter and a consequent elevation in Cu levels), the resulting progeny have markedly reduced AP load and Aβ levels [224]. Similarly, increasing dietary copper intake in APP23 mice (carrying the Swedish mutation of human APP751, regulated by the murine Thy-1.2 promoter [253]) resulted in reduced Aβ levels and a prolonged lifespan [222]. Conversely, increasing dietary Cu intake in normal rabbits resulted in elevated Aβ levels and impaired learning [134, 254]. Thus, metal homeostasis appears to be intimately involved in Aβ metabolism.
3.
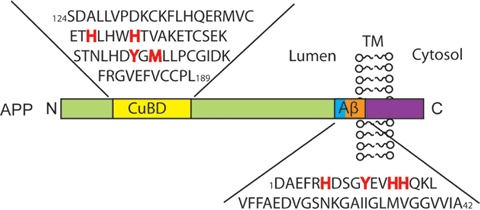
Copper binding domains on APP. APP contains two high-affinity copper binding domains: one on its N-terminus and the other on the Aβ sequence. Highlighted in red are the copper binding ligands in the CuBD and in the Aβ1–42 sequence. Abbreviations: Aβ (amyloid-β); APP (amyloid precursor protein); CuBD (copper binding domains); TM (trans-membrane).
These in vivo studies are supported by a wealth of in vitro data demonstrating that low concentrations of Zn2+ promote the rapid aggregation of Aβ at physiological pH [255–259]. At mildly acidic pH, Cu2+ (and Fe3+) have also been shown to induce Aβ precipitation [227, 230, 260–262]. These data suggest that the synaptic cleft is an ideal location for Aβ metallation and aggregation, as neurotransmission results in peak concentrations of ∼300 μM Zn2+[263, 264] and up to 100 μM Cu2+[265–267]. This is supported by observations of a significant reduction in plaque formation in a transgenic mouse model of AD (Tg2576) lacking the zinc transporter 3 (ZnT3) protein (Tg2576/ZnT3−/−) [268, 269], which is responsible for zinc enrichment and transport into pre-synaptic vesicles [270, 271]. The complicated process of Aβ aggregation makes it is difficult to characterize the binding of metals to Aβ, and while there have been numerous reports on the affinity and stoichiometry of Aβ-metal binding, results have varied depending on: the Aβ source (mouse, rat or human), Aβ sequence or length (Aβx-16/28/40/42), Aβ species (monomers, oligomers, etc.), as well as the reaction conditions (sample preparation, type and concentration of buffer, pH, incubation time and/or technique used). Most researchers are in agreement that Aβ binds Cu2+ and Zn2+ in a 1:1 ratio [272–276]; however, there have also been reports of Zn2+ binding to Aβ in a 2:1 [277] and 3:1 stoichiometry [278], and of Cu2+ binding to Aβ in a 2:1 ratio when copper is in excess [279, 280]. Mounting evidence indicates that the Aβ:metal ions ratio modulates not only Aβ conformation (random coil, α-helix or β-sheet) and aggregation [281–283], but also the morphology of the Aβ aggregates (amorphous, non-fibrillar or fibrillar) [280, 284, 285]. There is also an ongoing debate as to the binding affinity and kinetics of Aβ to Cu2+ and Zn2+, with dissociation constants (Kd) ranging from nM to μM for Cu2+-Aβ[272, 286, 287] and for Zn2+-Aβ[255, 276, 287–291]. A novel study has even suggested an initial, weak Zn2+-Aβ40 complex, which quickly turns into a high-affinity complex, possibly due to a conformational change of the peptide [287]. In order to resolve the issues above, it is imperative that the metal-binding site(s) of Aβ and APP are defined and that the relationship between the structural features of the protein and its function in health and disease can be elucidated. Recent studies [287, 292] utilizing the electrospray-ionization mass spectrometry, Raman spectroscopy, electron paramagnetic resonance, circular dichroism, nuclear magnetic resonance, X-ray diffraction and extended X-ray absorption fine structure spectroscopies have determined the coordination of Cu and Zn by His6, His13, His14[163, 255, 262, 272, 284, 286, 293–301] and a fourth ligand. The fourth donor could be Tyr10[293, 301] and/or Glu11[288, 302] for Zn2+, or Tyr10[293, 296] and/or Asp1[272, 298, 299] for Cu2+. Interestingly, mouse and rat Aβ contains three amino acid substitutions (R-5-G, Y-10-F and H-13-R), which prevent the formation of intermolecular histidine bridges [293, 303, 304] and therefore do not allow metal-induced Aβ aggregation in vitro[256, 260] and cerebral Aβ deposits in vivo[305].
In summary, the above findings demonstrate APP and/or Aβ play a major physiological role in regulating metal-ion levels. This cumulative data has lead Bush, Tanzi and colleagues to propose ‘the metal theory of AD’[306], which stipulates that age-related endogenous metal dyshomeostasis in the brain allows binding of redox-active metal ions (Cu2+ and Fe3+) to Aβ. This can lead to neurotoxicity as Cu2+ stabilizes the neurotoxic, oligomeric Aβ species [307–309], induces the covalent di-tyrosine crosslink of Aβ[274, 286, 287, 310–317] and promotes the generation of SDS-resistant copper-derived diffusible ligands [278, 286, 316]. Metallated-Aβ also has an increased affinity for the phospholipid heads of the membrane bilayer [318, 319], which acts as a reduc-tant in the production of reactive oxygen species (ROS) via Fenton and Haber-Weiss chemistry [320, 321]. The resulting radicals, such as hydrogen peroxide (H2O2) and superoxide (OH_), induce oxidative stress damage of lipids, proteins and DNA, ultimately leading to synaptic and neuronal loss [230, 231, 320–326]. Based on this hypothesis, pharmacotherapeutics that aim to restore metal homeostasis, inhibit Aβ-metal interactions and/or inhibit metallated Aβ-catalysed oxidation are being developed.
AD pharmacotherapies targeting metal ions
The equilibrium (concentrations, distribution, stability and bio-availability) of metal ions is critical for many physiological functions. This is particularly true for the CNS, where metals are essential for development and maintenance of enzymatic activities, mitochondrial function [327, 328], myelination [329], neurotransmission [330], learning and memory [331, 332]. Due to their importance, cells have evolved complex machinery for controlling metal-ion homeostasis. However, when these mechanisms fail, the altered homeostasis of metal ions can result in a disease state, including several neurodegenerative disorders [333, 334]. Understanding the complex structural and functional interactions of metal ions with the various intracellular and extracellular components of the CNS, under normal conditions and during neurodegeneration, is essential for the development of effective therapies [335]. Accordingly, modulation of metal ions has been proposed as a disease-modifying therapeutic strategy for AD [336–338] and other neurodegenerative diseases [339, 340]. Antioxidants and metal-modulators represent two such therapeutic strategies.
Antioxidants
Antioxidant molecules are capable of neutralizing free or incorrectly bound metals, thereby interfering with the ‘down-stream’ generation of ROS and other radicals. Therefore, antioxidants may be used mainly as a preventative approach [341]. Numerous molecules with antioxidant properties, such as oestrogen, melatonin, vitamin C and E (L-ascorbate and α- topopherol, respectively), ginkgo bilboa extract, curcumin and flavonoids, have been shown to have neuroprotective effects against Aβ-induced toxicity in cell-based experiments [342, 343] and animal models [344–348], but have had conflicting results in a clinical setting [349–351].
Metal chelators
By definition, metal chelators bind strongly to two or more metal ions and form a cyclic ring, which converts the metal ions into an inert form and depletes the total pool of bioavailable metals. Desferrioxamine (DFO), an Fe chelator with high binding affinities for Zn, Cu and aluminium (Al) [352], was the first such agent to enter clinical investigations for the treatment of AD. Results of a 2-year-long, blinded phase II trial with a cohort of 48 AD patients demonstrated that 125 mg intramuscular injections twice daily for 5 days a week significantly slowed down the decline of some cognitive functions, compared to the two control arms (an oral placebo or no treatment) [353]. DFO, however, is a large hydrophilic molecule, which is not orally bio-available and does not normally penetrate the BBB. Hence, it is unknown whether the beneficial effect seen with the DFO treatment was due to the drug's interaction and/or chelation of metals, or due to a different mechanism all together [354]. Another hexadentate chelator, DP-109 (DPharm, Rehovot, Israel), is a large synthetic pro-drug that becomes activated following the cleavage of its two long-chain esters. Daily administration of DP-109 by oral gavage to female Tg2576 mice over a 3-month period reduced the formation and deposition of CAA and APs, as well as re-solubilized Aβ[355]. Like DFO, DP-109 is not expected to cross the BBB, therefore the way it exerts its anti-amyloidogenic effect is still not clear. Recently, DP-109 and DP-460 (another Ca, Cu and Zn lipophilic chelator) were reported to have neuroprotective effects in a G93A transgenic mouse model of amyotrophic lateral sclerosis [356], another neurodegenerative disease associated with metal imbalance [357, 358]. Other chelating agents have been reported to have different effects in vitro, including reduced Aβ42-induced oxidative stress [359], and the solubilization of hypophosphory-lated τ[360] and Aβ from AD brain [361]. Further in vivo studies with these chelators is required to further advance this therapeutic route and to rule out any systemic effects.
An alternative approach to chelation is to modulate metals with metallo-complexes. Such an approach serves to remove metals from biologically deleterious sites and potentially deliver them to areas of deficiency, thereby maintaining overall metal homeostasis.
Metal complexes
Metallo-complexes are emerging as a new potential therapeutic for AD. The rational guiding this strategy is the delivery of Cu, for example, to cellular compartment which are Cu-deficient, using metallo-complexes of pyrrolidine dithiocarbamate (M2+-PDTC) or bis(thiosemicarbazone) (M2+-BTSC), or preventing the harmful binding of Cu to Aβ, using platinum (Pt) complexed to 1,10 phenanthroline derivatives (L-PtCl2).
PDTC is traditionally considered an inhibitor of the transcription-factor regulator nuclear factor-κB (NF-κB) with anti-inflammatory, antioxidant and anti-apoptotic properties [362–364]– all of which have been attributed to the synergistic interaction between PDTC, Cu and/or Zn [365–371]. As well as preventing the nuclear translocation of NF-κB in a neonatal hypoxia-ischaemia model, PDTC also activates Akt and inhibits GSK-3β[372]. In vivo, oral PDTC treatment of APP/PS1 double transgenic mice resulted in increased cerebral Cu levels, as compared to non-treated APP/PS1 mice, as well as down-regulation of the GSK-3β signalling cascade, which lead to a decrease in τ-phosphorylation and an improvement in spatial memory, but had no effect on amyloid burden, glial activation or oxidative stress [238]. The latest data to emerge indicate that PDTC complexed to either Cu2+ or Zn2+ can act as proteasome inhibitors to induce apoptosis in numerous human cancer cells [373–375]. It would be of interest to examine if the same effects occur in cellular and/or animal models of AD.
The metallo-complexes of diacetylbis(N4-methylthiosemicarbazone) (M2+-ATSM) and glyoxalbis(NR4– methylthiosemicarbazone) (M2+-GTSM) have both been shown to have anti-bacterial, anti-fungal and anti-neoplastic/cytotoxic activities, by selectively delivering exogenous metal ions into metal-deficient cells [376, 377]. Cu2+-ATSM is membrane permeable, selective for oxygen-deprived (hypoxic) cells, and is redox inactive therefore the ligand retains its Cu molecule [378, 379]. These properties are being exploited for its development as a radiotherapeutic agent [380–382] and as a radiopharmaceutical for positron emission tomography imaging [383, 384]. Cu2+-GTSM can also cross the BBB; however, once inside the cell it is reduced by various cellular reductants and releases its Cu molecule, which is made available for the cell [378, 385, 386]. Treatment of hAPP695-overexpressing CHO cells with Cu2+/Zn2+-BTSC ligands resulted in increased intracellular metal levels that, in turn, activated Akt/PI3K, c-Jun N-terminal kinase and GSK-3 [387]. Phosphorylation of the above kinases lead to the up-regulation of MMPs, which reduced extracellular levels of Aβ[387]. Examination of the effects of Cu2+/Zn2+-BTSC ligands on τ and translation of these studies to animal models of AD is currently underway.
Other radiopharmaceutical-based compounds being evaluated for treatment of AD are 1,10 phenanthroline derivatives complexed to platinum (Pt2+). These ligand-PtCl2 complexes have been designed to bind and alkylate the imidazole side chains on histidine residues 6, 13 and 14 on Aβ, thereby preventing the detrimental binding of Cu2+ to this Aβ metal binding site and subsequent Aβ-Cu2+ binding to the cell membrane [388]. This study identified the Pt(4,7-diphenyl-[1,10] phenanthroline)Cl2 as a compound that binds to Aβ, changes the conformation of Aβ and inhibits Aβ aggregation [388]. In addition, this complex is able to inhibit Aβ-related neurotoxicity (restore the cell viability of primary mouse cortical neurons and suppresses the Cu2+-Aβ-dependent H2O2 generation), and reverse Aβ-inhibited long-term potentiation (LTP) of mouse hippocampal slices as a measure of synaptotoxicity [388]. Future evaluation of the compound's ability to cross the BBB and exert beneficial effects in animal models for AD need to be performed prior to its advanced development as an AD pharmacotherapeutic.
The Aβ-metal interaction can be targeted not only to the Aβ sequence that binds metals, but also to the metals themselves.
Metal-protein attenuating compounds (MPACs)
MPACs have weak, reversible affinity towards metals, which enables them to compete with endogenous ligands for metal ions, target the harmful ‘up stream’ metal-protein reactions and restore normal metal levels in specific cellular compartments [389]. The first-generation series of MPACs were based on clioquinol (CQ; 5-chloro-7-iodo-8-hydroxyquinoline). CQ is highly lipophilic, absorbed quickly, can convert to glucuronated and sulphate metabolites, is able to cross the BBB and is excreted in urine and faeces [390–395]. CQ had been used as a therapeutic in cattle and human beings with Zn-deficiency diseases and for many decades was prescribed as an oral anti-amebic in addition to being used for the treatment of dysentery and diarrhoea [396, 397]. However, its oral preparation was withdrawn from the market during the 1960s to 1970s, as it was suspected to be involved in the development of subacute myelo-optico-neuropathy (SMON) [398–401]. SMON is characterized by sensory and motor disorders in the lower limbs, peripheral neuropathy and visual impairment due to demyelation of the spinal cord, optic nerve and peripheral nerves [402]. SMON affected people worldwide; however, it reached near-epidemic proportions in Japan, where a few related deaths were reported [403]. At the time, a mechanistic link between CQ and SMON was not established [404]. Later, it was suggested that CQ may transport metals into the CNS, which leads to neurotoxicity. Early studies demonstrated that CQ-Fe3+, but not CQ or Fe3+ alone, induced degeneration of cultured retinal neuroblasts [405] by increasing cellular Fe concentrations and promoting lipid per-oxidation [406]. However, it is now believed that intake of CQ at doses far exceeding the recommended ones and for prolonged periods, together with a post-World War II iron-deficient diet, are the reasons for a vitamin B12 deficiency that presented as SMON in Japan [407, 408].
CQ binds Cu2+ and Zn2+ (2:1 ratio) in a square, planar arrangement [409, 410] and exerts different effects on Cu and Zn, depending on its route of administration and the system in which it is tested [411–413]. The known interaction of CQ with Cu2+ and Zn2+ thus prompted an investigation into the effects of CQ on AD-related pathology. CQ was initially shown to dissolve synthetic Aβ-Cu2+/Zn2+ aggregates and amyloid deposits from post-mortem AD brain [414]. This then prompted a study of the oral administration of CQ to Tg2576 mice over 9 weeks, which resulted in the normalization of cerebral Cu and Zn levels, a reduction in H2O2 synthesis, and a significant decrease in cortical amyloid deposition by ∼49%, compared to control littermates [415]. Subsequently, CQ was shown to reverse Cu-suppressed, but not Zn-suppressed Aβ1–40 fibril formation [416], and to rescue Ca2+-mediated Aβ toxicity in neuronal cell culture [417]. Other studies, however, have suggested that CQ increases oxidative neurotoxicity [418]. As previously mentioned, CQ treatment caused a reduction in Aβ levels in CHO-APP cells, accompanied by increased phosphorylation of GSK-3 and MMP2/3 activity [225]. The cumulative data led to CQ being entered into clinical trials for the treatment of AD (PBT-1, Prana Biotechnology, Melbourne, Victoria, Australia), in which CQ slowed the cognitive decline of moderate to severe AD patients, with no signs of severe adverse effects. It also influenced CSF-τ, lowered plasma Aβ1–42 with no change to CSF-Ap1–42 levels [419]. Subsequent phase II/III studies, however, were stalled by the difficulties encountered in preventing di-iodo-8-hydroxy quinoline contamination during the required larger scale chemical synthesis for such trials [420]. The subsequent drug discovery program identified PBT2 (Prana Biotechnology) as an 8-hydroxy quinoline that lacks iodine, thereby enabling easier chemical synthesis, and which also has higher solubility and increased BBB permeability than CQ. This compound was then extensively screened in a variety of pre-clinical assays. In APP/PS1 Tg mice, PBT2 was shown to decrease soluble interstitial Aβ within hours, and to improve cognitive performance to levels equivalent to or greater than wild-type controls within days of treatment [421]. In addition, there was a significant decrease in insoluble Aβ load and the phosphorylation of τ, as well as a significant increase in synaptophysin levels – suggesting that a number of primary indices that characterize the AD brain had been successfully modulated by this orally bioavailable MPAC [421]. PBT2 then progressed into human clinical trial, and following a successful phase I study, it entered into a randomized, double blind, placebo-controlled, multi-centred, 12-week-long phase IIa trial with 78 mild AD patients (Prana Biotechnology). This study demonstrated safety and tolerability, reduced CSF levels of Aβ1–42 and improved cognition in patients taking PBT2 as compared to placebo [422]. Taken together, these data support the notion that the modulation of metals may be sufficient to significantly alter the onset and progression of AD, and that targeting metals may represent a more potent disease intervention than systemically targeting the production or degradation of the Aβ protein; however, these concepts need to be further explored in a larger phase III trial.
While CQ is continuing to be examined as a therapeutic for other diseases, such as Parkinson's disease, Prion diseases, Huntington's disease, diabetes and cancer [373, 411, 423–431], a finer dissection of the mechanism of action of drugs such as CQ and PBT2 will enable researchers to better design additional pharmacotherapies for the treatment of AD and other diseases.
Conclusion
It is evident that AD pathogenesis is a complex process involving both genetic and environmental factors; therefore development of effective disease-modifying drugs is proving to be a difficult task. Aβ, τ and metals are some of the therapeutic targets identified and compounds that modulate them represent promising drug candidates. With ongoing basic science and clinical research, we look forward to a greater understanding of the pathogenesis of AD, the completion of several comprehensive clinical trials and the development of new potential pharmacotherapeutic agents for the treatment and/or prevention of AD.
Disclosers and Acknowledgements
The authors would like to acknowledge Dr. Robert Cherny for discussing the ideas presented. Y.B. is supported by the Commonwealth Scientific and Industrial Research Organization (CSIRO) Preventative-Health Flagship. P.A.A., C.L.M., K.J.B. and A.I.B. are consultants to Prana Biotechnology. We gratefully acknowledge the support of the National Health and Medical Research Council of Australia, Australian Research Council and the Alzheimer's Association (USA).
References
- 1.Mount C, Downton C. Alzheimer disease: progress or profit? Nat Med. 2006;12:780–4. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- 2.Nagy Z. The last neuronal division: a unifying hypothesis for the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2005;9:531–41. doi: 10.1111/j.1582-4934.2005.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lleo A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer's disease. Annu Rev Med. 2006;57:513–33. doi: 10.1146/annurev.med.57.121304.131442. [DOI] [PubMed] [Google Scholar]
- 4.Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45:583–95. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Sonkusare SK, Kaul CL, Ramarao P. Dementia of Alzheimer's disease and other neurodegenerative disorders–memantine, a new hope. Pharmacol Res. 2005;51:1–17. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system – too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–9. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 8.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–24. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 9.Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipopro-tein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer's disease: results of the Alzheimer's Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurol Scand Suppl. 2006;114:3–7. doi: 10.1111/j.1600-0404.2006.00690.x. [DOI] [PubMed] [Google Scholar]
- 10.Sparks DL, Petanceska S, Sabbagh M, Connor D, Soares H, Adler C, Lopez J, Ziolkowski C, Lochhead J, Browne P. Cholesterol, copper and Aβ in controls, MCI, AD and the AD Cholesterol-Lowering Treatment Trial (ADCLT) Curr Alzheimer Res. 2005;2:527–39. doi: 10.2174/156720505774932296. [DOI] [PubMed] [Google Scholar]
- 11.Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, Wasser D, Johnson-Traver S, Lochhead J, Ziolwolski C. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62:753–7. doi: 10.1001/archneur.62.5.753. [DOI] [PubMed] [Google Scholar]
- 12.Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Petanceska S, Browne P, Wassar D, Johnson-Traver S, Lochhead J, Ziolkowski C. Atorvastatin therapy lowers circulating cholesterol but not free radical activity in advance of identifiable clinical benefit in the treatment of mild-to-moderate AD. Curr Alzheimer Res. 2005;2:343–53. doi: 10.2174/1567205054367900. [DOI] [PubMed] [Google Scholar]
- 13.Landreth G. Therapeutic use of agonists of the nuclear receptor PPAR-γ in Alzheimer's disease. Curr Alzheimer Res. 2007;4:159–64. doi: 10.2174/156720507780362092. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–73. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 15.Risner ME, Saunders AM, Altman JFB, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 16.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–8. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- 17.Doraiswamy PM, Xiong GL. Pharmacological strategies for the prevention of Alzheimer's disease. Expert Opin Pharmacother. 2006;7:1–10. doi: 10.1517/14656566.7.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Szekelya CA, Thornea JE, Zandia PP, Eka M, Messiasa E, Breitnerc JCS, Goodmana SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159–69. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 19.Aisen PS. The potential of anti-inflammatory drugs for the treatment of Alzheimer's disease. Lancet Neurol. 2002;1:279–84. doi: 10.1016/s1474-4422(02)00133-3. [DOI] [PubMed] [Google Scholar]
- 20.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 21.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Tolnay M, Probst A. Review: tau protein pathology in Alzheimer's disease and related disorders. Neuropathol Appl Neurobiol. 1999;25:171–87. doi: 10.1046/j.1365-2990.1999.00182.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 24.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the micro-tubuleassociated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alonso ADC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–6. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandelkow E-M, Mandelkow E. Tau in Alzheimer's disease. Trends Cell Biol. 1998;8:425–7. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- 28.Brandt R, Lee G. The balance between tau protein's microtubule growth and nucle-ation activities: implications for the formation of axonal microtubules. J Neurochem. 1993;61:997–1005. doi: 10.1111/j.1471-4159.1993.tb03613.x. [DOI] [PubMed] [Google Scholar]
- 29.Trojanowski JQ, Schmidt ML, Shin RW, Bramblett GT, Rao D, Lee VM. Altered tau and neurofilament proteins in neuro-degenerative diseases: diagnostic implications for Alzheimer's disease and Lewy body dementias. Brain Pathol. 1993;3:45–54. doi: 10.1111/j.1750-3639.1993.tb00725.x. [DOI] [PubMed] [Google Scholar]
- 30.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gail VWJ, Judith AH. Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimers Dis. 1999;1:329–51. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe A, Hasegawa M, Suzuki M, Takio K, Morishima-Kawashima M, Titani K, Arai T, Kosik KS, Ihara Y. In vivo phos-phorylation sites in fetal and adult rat tau. J Biol Chem. 1993;268:25712–7. [PubMed] [Google Scholar]
- 33.Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16:355–62. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- 34.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and Non-proline-directed Phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–9. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 35.Betts JC, Loviny TLF, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelec-trospray mass spectrometry. J Neurochem. 1998;71:2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 36.Iqbal K, Grundke-Iqbal I. Metabolic/signal transduction hypothesis of Alzheimer's disease and other tauopathies. Acta Neuropathol. 2005;109:25–31. doi: 10.1007/s00401-004-0951-y. [DOI] [PubMed] [Google Scholar]
- 37.Iqbal K, Del C. Alonso A, Chen S, Chohan MO, EI-Akkad E, Gong C-X, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–12. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VMY. PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol. 2001;168:402–12. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- 40.Cheng-Xin Gong TJSIG-IKI. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–7. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 41.Fei Liu IG-IKIC-XG. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–50. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 42.Gong CX, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer's disease abnormally phosphorylated tau by protein phosphatase-2A. Neuroscience. 1994;61:765–72. doi: 10.1016/0306-4522(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 43.Pei J-J, Gong C-X, An W-L, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer's disease. Am J Pathol. 2003;163:845–58. doi: 10.1016/S0002-9440(10)63445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iqbal K, Grundke-Iqbal I. Tau phosphatase activity as a therapeutic target for AD. Drug News Perspect. 1998;11:10–4. doi: 10.1358/dnp.1998.11.1.863668. [DOI] [PubMed] [Google Scholar]
- 45.Ferrer I, Gomez-Isla T, Puig B, Freixes M, RibÈ E, Dalfó E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 46.Savage MJ, Lin Y-G, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-ter-minal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–85. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y. Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J Biol Chem. 2004;279:44795–801. doi: 10.1074/jbc.M408127200. [DOI] [PubMed] [Google Scholar]
- 48.Lee K-Y, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN. Elevated neu-ronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. 1999;34:21–9. doi: 10.1016/s0168-0102(99)00026-7. [DOI] [PubMed] [Google Scholar]
- 49.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreac-tivity and microtubule binding. Neuron. 1993;11:153–63. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 50.Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow E-M, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase. J Biol Chem. 1995;270:7679–88. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 51.Ishiguro K, Ihara Y, Uchida T, Imahori K. A novel tubulin-dependent protein kinase forming a paired helical filament epitope on tau. J Biochem. 1988;104:319–21. doi: 10.1093/oxfordjournals.jbchem.a122465. [DOI] [PubMed] [Google Scholar]
- 52.Ishiguro K, Omori A, Sato K, Tomizawa K, Imahori K, Uchida T. Aserine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci Lett. 1991;128:195–8. doi: 10.1016/0304-3940(91)90259-v. [DOI] [PubMed] [Google Scholar]
- 53.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3β is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–72. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 54.Bhat RV, Haeberlein SLB, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–7. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- 55.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–87. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 56.Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration cancer, and inflammation. Med Res Rev. 2002;22:373–84. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 57.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 59.Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312:922–9. doi: 10.1016/j.bbrc.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 60.Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-β precursor protein processing. Biochemistry. 2004;43:6899–908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 61.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–5. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rapoport M, Ferreira A. PD98059 prevents neurite degeneration induced by fibrillar β-amyloid in mature hippocampal neurons. J Neurochem. 2000;74:125–33. doi: 10.1046/j.1471-4159.2000.0740125.x. [DOI] [PubMed] [Google Scholar]
- 63.Sarno S, Ruzzene M, Frascella P, Pagano MA, Meggio F, Zambon A, Mazzorana M, Maira GD, Lucchini V, Pinna LA. Development and exploitation of CK2 inhibitors. Mol Cell Biochem. 2005;274:69–76. doi: 10.1007/s11010-005-3079-z. [DOI] [PubMed] [Google Scholar]
- 64.Pedersen LM, Lien GF, Bollerud I, Gjerstad J. Induction of long-term potentiation in single nociceptive dorsal horn neurons is blocked by the CaMKII inhibitor AIP. Brain Res. 2005;1041:66–71. doi: 10.1016/j.brainres.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 65.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–25. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 66.Pallas M, Canudas AM, Verdaguer E, Allgaier C, De Arriba SG, Alvira D, Sureda FX, Camins A. Inhibitors of cyclin-dependent kinases: potential drugs for the treatment of neurodegenerative disorders? Curr Med Chem Cent Nerv Syst Agents. 2005;5:101–9. [Google Scholar]
- 67.Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006;103:9673–8. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr Top Med Chem. 2006;6:579–95. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- 69.Iqbal K, Grundke-lqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med. 2008;12:38–55. doi: 10.1111/j.1582-4934.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stoothoff WH, Johnson GVW. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 71.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–8. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pickhardt M, Gazova Z, Von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow E-M, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–35. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 73.Pickhardt M, Biernat J, Khlistunova I, Wang YP, Gazova Z, Mandelkow EM, Mandelkow E. N-Phenylamine derivatives as aggregation inhibitors in cell models of tauopathy. Curr Alzheimer Res. 2007;4:397–402. doi: 10.2174/156720507781788765. [DOI] [PubMed] [Google Scholar]
- 74.Khlistunova I, Pickhardt M, Biernat J, Yipeng W, Mandelkow E-M, Mandelkow E. Inhibition of tau aggregation in cell models of tauopathy. Curr Alzheimer Res. 2007;4:544–6. doi: 10.2174/156720507783018307. [DOI] [PubMed] [Google Scholar]
- 75.Gozes I, Zaltzman R, Hauser J, Brenneman DE, Shohami E, Hill JM. The expression of activity-dependent neuro-protective protein (ADNP) is regulated by brain damage and treatment of mice with the ADNP derived peptide, NAP, reduces the severity of traumatic head injury. Curr Alzheimer Res. 2005;2:149–53. doi: 10.2174/1567205053585873. [DOI] [PubMed] [Google Scholar]
- 76.Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323:438–49. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- 77.Gozes I, Spivak-Pohis I. Neurotrophic effects of the peptide NAP: a novel neuro-protective drug candidate. Curr Alzheimer Res. 2006;3:197–9. doi: 10.2174/156720506777632790. [DOI] [PubMed] [Google Scholar]
- 78.Geerts H. AL-108 and AL-208, formulations of the neuroprotective NAP fragment of activity-dependent neuroprotective protein, for cognitive disorders. Curr Opin Investig Drugs. 2008. 2008;9:800–11. [PubMed] [Google Scholar]
- 79.Rosen DR, Martin-Morris L, Luo LQ, White K. A Drosophila gene encoding a protein resembling the human β-amyloid protein precursor. Proc Natl Acad Sci USA. 1989;86:2478–82. doi: 10.1073/pnas.86.7.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sprecher CA, Grant FJ, Grimm G, O'Hara PJ, Norris F, Norris K, Foster DC. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry. 1993;32:4481–6. doi: 10.1021/bi00068a002. [DOI] [PubMed] [Google Scholar]
- 81.Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid β protein precursor. Nat Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- 82.Reinhard C, Hebert SS, De Strooper B. The amyloid-β precursor protein: integrating structure with biological function. EMBO J. 2005;24:3996–4006. doi: 10.1038/sj.emboj.7600860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Panegyres PK. The functions of the amyloid precursor protein gene. Rev Neurosci. 2001;12:1–39. doi: 10.1515/revneuro.2001.12.1.1. [DOI] [PubMed] [Google Scholar]
- 84.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–70. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 85.Zheng H, Koo E. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:1–12. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamazaki T, Koo EH, Selkoe DJ. Trafficking of cell-surface amyloid β-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J Cell Sci. 1996;109:999–1008. doi: 10.1242/jcs.109.5.999. [DOI] [PubMed] [Google Scholar]
- 87.Forloni G, Terreni L, Bertani I, Fogliarino S, Invernizzi R, Assini A, Ribizzi G, Negro A, Calabrese E, Volonte MA, Mariani C, Franceschi M, Tabaton M, Bertoli A. Protein misfolding in Alzheimer's and Parkinson's disease: genetics and molecular mechanisms. Neurobiol Aging. 2002;23:957–76. doi: 10.1016/s0197-4580(02)00076-3. [DOI] [PubMed] [Google Scholar]
- 88.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–35. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 89.Nixon RA. Autophagy amyloidogenesis and Alzheimer's disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 90.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 91.Yu WH, Cuervo AM, Kumar A, Peterhoft CM, Schmidt SD, Lee J-H, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy – a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nunan J, Small DH. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett. 2000;483:6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 93.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid-β peptide during constitutive processing of its precursor. Science. 1990;248:1122–4. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 94.Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ. β-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem. 1993;268:3021–4. [PubMed] [Google Scholar]
- 95.Cao X, Sudhof TC. Dissection of amyloid-β precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–11. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 96.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 97.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–7. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 98.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–7. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hussain I, Powell D, Hewlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol Cell Neurosci. 1999;14:419–27. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 100.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature. 1999;402:533–7. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 101.Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomquist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thai LJ. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature. 1993;361:260–3. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- 102.Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Rahmati T, Donkor IO, Selkoe DJ. Peptidomimetic probes and molecular modeling suggest that Alzheimer's γ-secretase is an intramembrane-cleaving aspartyl protease. Biochemistry. 1999;38:4720–7. doi: 10.1021/bi982562p. [DOI] [PubMed] [Google Scholar]
- 103.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of γ-secretase activity. Nat Cell Biol. 2003;5:486–8. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 104.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D. aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 105.Periz G, Fortini ME. Functional reconstitution of γ-secretase through coordinated expression of presenilin, nicastrin, Aph-1 and Pen-2. J Neurosci Res. 2004;77:309–22. doi: 10.1002/jnr.20203. [DOI] [PubMed] [Google Scholar]
- 106.Li X, Greenwald I. Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci USA. 1998;95:7109–14. doi: 10.1073/pnas.95.12.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398:513–7. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 108.Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000;2:463–5. doi: 10.1038/35017108. [DOI] [PubMed] [Google Scholar]
- 109.Kopan R, IIagan MXG. γ-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 110.Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–53. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 111.St George-Hyslop PH, Petit A. Molecular biology and genetics of Alzheimer's disease. C R Biol. 2005;328:119–30. doi: 10.1016/j.crvi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 112.Fisher A, Michaelson DM, Brandeis R, Haring R, Chapman S, Pittel Z. M1 muscarinic agonists as potential disease-modifying agents in Alzheimer's disease: rationale and perspectives. Ann N Y Acad Sci. 2000;920:315–20. doi: 10.1111/j.1749-6632.2000.tb06941.x. [DOI] [PubMed] [Google Scholar]
- 113.Wolf BA, Wertkin AM, Jolly YC, Yasuda RP, Wolfe BB, Konrad RJ, Manning D, Ravi S, Williamson JR, Lee VMY. Muscarinic regulation of Alzheimer's disease amyloid precursor protein secretion and amyloid β-protein production in human neuronal NT2N cells. J Biol Chem. 1995;270:4916–22. doi: 10.1074/jbc.270.9.4916. [DOI] [PubMed] [Google Scholar]
- 114.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbarl Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 115.Caccamo A, Oddo S, Billings LM, Green KN, Martlnez-Corla H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–82. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 116.Evln G, Sernee MF, Masters CL. Inhibition of γ-secretase as a therapeutic intervention for Alzheimer's disease: prospects, limitations and strategies. CNS Drugs. 2006;20:351. doi: 10.2165/00023210-200620050-00002. [DOI] [PubMed] [Google Scholar]
- 117.Tschape J-A, Hartmann T. Therapeutic perspectives in Alzheimers disease. Recent Patents CNS Drug Discov. 2006;1:119–27. doi: 10.2174/157488906775245354. [DOI] [PubMed] [Google Scholar]
- 118.Flelsher AS, Raman R, Slemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvln JE, Pesklnd ER, Qulnn JF, Sherzal A, Sowell BB, Alsen PS, Thai LJ. Phase 2 safety trial targeting amyloid-β production with a γ-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65:1031–8. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Haass C, Hung AY, Schlossmacher MG, Oltersdorf T, Teplow DB, Selkoe DJ. Normal cellular processing of the β-amyloid precursor protein results in the secretion of the amyloid-β peptide and related molecules. Ann N Y Acad Sci. 1993;695:109–16. doi: 10.1111/j.1749-6632.1993.tb23037.x. [DOI] [PubMed] [Google Scholar]
- 120.Morgan C, Colombres M, Nunez MI, Inestrosa NC. Structure and function of amyloid in Alzheimer's disease. Prog Neurobiol. 2004;74:323–49. doi: 10.1016/j.pneurobio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 121.Saavedra L, Mohamed A, Ma V, Kar S, De Chaves EP. Internalization of β-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282:35722–32. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]
- 122.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheskl KE, Holtzman DM. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Savage MJ, Trusko SP, Howland DS, Plnsker LR, Mlstretta S, Reaume AG, Greenberg BD, Slman R, Scott RW. Turnover of amyloid β-protein in mouse brain and acute reduction of its level by phorbol ester. J Neurosci. 1998;18:1743–52. doi: 10.1523/JNEUROSCI.18-05-01743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Slnha S, Schlossmacher M, Whaley J, Swlndlehurst C. Isolation and quantification of soluble Alzheimer's β-peptide from biological fluids. Nature. 1992;359:325–7. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 125.Kuhnke D, Jedlltschky G, Grube M, Krohn M, Jucker M, Mosyagln I, Cascorbl I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer's amyloid-β peptides – implications for the mechanisms of Aβ clearance at the bloodbrain barrier. Brain Pathol. 2007;17:347–53. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovlc S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghlso J, Franglone B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Llllenslek B, Nawroth P, Hofman F, Klndy M, Stern D, Zlokovic B. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 127.Tanzi RE, Molr RD, Wagner SL. Clearance of Alzheimer's Aβ peptide: the many roads to perdition. Neuron. 2004;43:605–8. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 128.Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2006;27:909–18. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hammad SM, Ranganathan S, Louklnova E, Twal WO, Argraves WS. Interaction of apolipoprotein j-amyloid β-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid β petptide. J Biol Chem. 1997;272:18644–9. doi: 10.1074/jbc.272.30.18644. [DOI] [PubMed] [Google Scholar]
- 130.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parlsl M, LaRue B, Hu HW, Spljkers P, Guo H, Song X, Lentlng PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004;43:333–44. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 131.Herz J. LRP: a bright beacon at the blood-brain barrier. J Clin Invest. 2003;112:1483–5. doi: 10.1172/JCI20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tamakl C, Ohtsukl S, Iwatsubo T, Hashimoto T, Yamada K, Yabukl C, Terasakl T. Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid β-peptide by the liver. Pharm Res. 2006;23:1407–16. doi: 10.1007/s11095-006-0208-7. [DOI] [PubMed] [Google Scholar]
- 133.Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lentlng PJ, Zhenhua W, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhlhul Z, Zlokovic BV. Clearance of amyloid-β by circulating lipoprotein receptors. Nat Med. 2007;13:1029–31. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sparks DL. Cholesterol metabolism and brain amyloidosis: evidence for a role of copper in the clearance of Aβ through the liver. Curr Alzheimer Res. 2007;4:165–9. doi: 10.2174/156720507780362119. [DOI] [PubMed] [Google Scholar]
- 135.Koldamova R, Lefterov I. Role of LXR and ABCA1 in the pathogenesis of Alzheimer's disease – implications for a new therapeutic approach. Curr Alzheimer Res. 2007;4:171–8. doi: 10.2174/156720507780362227. [DOI] [PubMed] [Google Scholar]
- 136.Cao G, Bales KR, DeMattos RB, Paul SM. Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: relevance to Alzheimer's disease therapeutics. Curr Alzheimer Res. 2007;4:179–84. doi: 10.2174/156720507780362173. [DOI] [PubMed] [Google Scholar]
- 137.Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–80. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 138.Klnston IB, Castro MJM, Anderson S. In vitro stimulation of tissue-type plasmino-gen activator by Alzheimer amyloid β-peptide analogues. Nat Med. 1995;1:138–42. doi: 10.1038/nm0295-138. [DOI] [PubMed] [Google Scholar]
- 139.Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S. Tissue plasminogen activator requires plasminogen to modulate amyloid-β neurotoxicity and deposition. J Neurochem. 2000;75:2172–7. doi: 10.1046/j.1471-4159.2000.0752172.x. [DOI] [PubMed] [Google Scholar]
- 140.Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashl T, Price D, Walker D, Scheff S, McGlllls JP, Rydel RE, Estus S. The plasmin system is induced by and degrades amyloid-β aggregates. J Neurosci. 2000;20:3937–46. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yasojlma K, Aklyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- 142.Iwata N, Tsubukl S, Takakl Y, Watanabe K, Seklguchl M, Hosokl E, Kawashlma-Morlshlma M, Lee H-J, Hama E, Sekine-Alzawa Y, Saldo TC. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 143.Iwata N, Satoshi T, Yoshie T, Keiro S, Bao L, Gerard NP, Gerard C, Emi H, Lee H-J, Saido TC. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 144.Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of Aβ-degrading endopeptidase, neprilysin in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- 145.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998;273:32730–8. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 146.Kurochkin IV, Goto S. Alzheimer's β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–7. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 147.Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004;24:11120–6. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–65. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–8. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 150.Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–4. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 151.Roher AE, Kasunic TC, Woods AS, Cotter RJ, Ball MJ, Fridman R. Proteolysis of Aβ peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun. 1994;205:1755–61. doi: 10.1006/bbrc.1994.2872. [DOI] [PubMed] [Google Scholar]
- 152.Backstrom JR, Lim GP, Cullen MJ, Tokes ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40) J Neurosci. 1996;16:7910–9. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yin K-J, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu F-F, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee J-M. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J Neurosci. 2006;26:10939–48. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Esteban JA. Living with the enemy: a physiological role for the β-amyloid peptide. Trends Neurosci. 2004;27:1–3. doi: 10.1016/j.tins.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 155.Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN. Amyloid-β: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-β. Brain Res Rev. 2003;43:1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 156.Kontush A. Amyloid-β: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer's disease. Free Radic Biol Med. 2001;31:1120–31. doi: 10.1016/s0891-5849(01)00688-8. [DOI] [PubMed] [Google Scholar]
- 157.Kontush A. Amyloid-β: Acute-phase apolipoprotein with metal-binding activity. J Alzheimers Dis. 2005;8:129–37. doi: 10.3233/jad-2005-8205. [DOI] [PubMed] [Google Scholar]
- 158.Kontush A, Atwood CS. Amyloid-β: phylogenesis of a chameleon. Brain Res Rev. 2004;46:118–20. doi: 10.1016/j.brainresrev.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 159.Berthon G. Does human βA4 exert a protective function against oxidative stress in Alzheimer's disease? Med Hypotheses. 2000;54:672–7. doi: 10.1054/mehy.1999.0924. [DOI] [PubMed] [Google Scholar]
- 160.Zou K, Gong J-S, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002;22:4833–41. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–20. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 162.Frautschy SA, Baird A, Cole GM. Effects of injected Alzheimer β-amyloid cores in rat brain. Proc Natl Acad Sci USA. 1991;88:8362–6. doi: 10.1073/pnas.88.19.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu ST, Hewlett G, Barrow CJ. Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the Aβ peptide of Alzheimer's disease. Biochemistry. 1999;38:9373–8. doi: 10.1021/bi990205o. [DOI] [PubMed] [Google Scholar]
- 164.Serpell LC. Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 165.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–42. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 166.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–52. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 167.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of con-ditions for amyloid-β peptide oligomeriza-tion and fibrillogenesis. J Biol Chem. 2003;278:11612–22. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 168.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electro-physiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–84. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–28. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 172.Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–8. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 174.Lesne S, Kotilinek L. Amyloid plaques and amyloid-β oligomers: an ongoing debate. J Neurosci. 2005;25:9319–20. doi: 10.1523/JNEUROSCI.3246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Look GC, Jerecic J, Cherbavaz DB, Pray TR, Breach J-CR, Crosier WJ, Igoudin L, Hironaka CM, Lowe RM, McEntee M, Ruslim-Litrus L, Hsiu-Mei W, Sue Z, Catalano SM, Goure WF, Summa D, Krafft GA. Discovery of ADDL-targeting small molecule drugs for Alzheimer's disease. Curr Alzheimer Res. 2007;4:562–7. doi: 10.2174/156720507783018271. [DOI] [PubMed] [Google Scholar]
- 176.Chacon MA, Barria MI, Soto C, Inestrosa NC. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol Psychiatry. 2004;9:953–61. doi: 10.1038/sj.mp.4001516. [DOI] [PubMed] [Google Scholar]
- 177.Giordano C, Masi A, Pizzini A, Sansone A, Consalvi V, Chiaraluce R, Lucente G. Synthesis and activity of fibrillogenesis peptide inhibitors related to the 17–21 β-amyloid sequence. Eur J Med Chem. doi: 10.1016/j.ejmech.2008.03.036. In Press. [DOI] [PubMed] [Google Scholar]
- 178.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 179.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J, Szarek WA, Tremblay P. Targeting soluble Aβ peptide with tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–47. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 180.Santa-Maria I, Hernandez F, Del Rio J, Moreno F, Avila J. Tramiprosate, a drug of potential interest for the treatment of Alzheimer's disease, promotes an abnormal aggregation of tau. Mol Neurodegener. 2007;2:1–12. doi: 10.1186/1750-1326-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Santa-María I, Hernández F, Moreno FJ, Avila J. Taurine, an inducer for tau polymerization and a weak inhibitor for amyloid-β-peptide aggregation. Neurosci Lett. 2007;429:91–4. doi: 10.1016/j.neulet.2007.09.068. [DOI] [PubMed] [Google Scholar]
- 182.Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, Garceau D. A phase II study targeting amyloid-β with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67:1757–63. doi: 10.1212/01.wnl.0000244346.08950.64. [DOI] [PubMed] [Google Scholar]
- 183.Geerts H. NC-531 (Neurochem) Curr Opin Investig Drugs. 2004;5:95–100. [PubMed] [Google Scholar]
- 184.Crouch PJ, Harding S-ME, White AR, Camakaris J, Bush AI, Masters CL. Mechanisms of Aβ mediated neurodegen-eration in Alzheimer's disease. Int J Biochem Cell Biol. 2008;40:181–98. doi: 10.1016/j.biocel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 185.Talaga P, Quere L. The plasma membrane: a target and hurdle for the development of anti-Aβ drugs? Curr Drug Targets CNS Neurol Disord. 2002;1:567–74. doi: 10.2174/1568007023338897. [DOI] [PubMed] [Google Scholar]
- 186.Verdier Y, Zarandi M, Penke B. Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J Pept Sci. 2004;10:229–48. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- 187.Roller AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996;271:20631–5. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 188.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–5. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001;898:350–7. doi: 10.1016/s0006-8993(01)02018-2. [DOI] [PubMed] [Google Scholar]
- 190.Berman DE, Dall'armi C, Voronov SV, Mclntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G. Oligomeric amyloid-β peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci. 2008;11:547–54. doi: 10.1038/nn.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–89. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fritze J, Walden J. Clinical findings with nimodipine in dementia: test of the calcium hypothesis. J Neural Transm Suppl. 1995;46:439–53. [PubMed] [Google Scholar]
- 193.Pierrot N, Ghisdal P, Caumont A-S, Octave J-N. Intraneuronal amyloid-β1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–50. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- 194.Rose GM, Ong VS, Woodruff-Pak DS. Efficacy of MEM 1003, a novel calcium channel blocker, in delay and trace eye-blink conditioning in older rabbits. Neurobiol Aging. 2007;28:766–73. doi: 10.1016/j.neurobiolaging.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 195.Yagami T, Ueda K, Sakaeda T, Itoh N, Sakaguchi G, Okamura N, Hori Y, Fujimoto M. Protective effects of a selective L-type voltage-sensitive calcium channel blocker, S-312-d, on neuronal cell death. Biochem Pharmacol. 2004;67:1153–65. doi: 10.1016/j.bcp.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 196.López-Arrieta J, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002;3:1–56. doi: 10.1002/14651858.CD000147. [DOI] [PubMed] [Google Scholar]
- 197.Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. β-Amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Home P, Heslin D, French J, Mount HTJ, Nixon RA, Mercken M, Bergeron C, Fraser PE, St. George-Hyslop P, Westaway D. Amyloid β peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 199.Town T, Tan J, Sansone N, Obregon D, Klein T, Mullan M. Characterization of murine immunoglobulin G antibodies against human amyloid-β1–42. Neurosci Lett. 2001;307:101–4. doi: 10.1016/s0304-3940(01)01951-6. [DOI] [PubMed] [Google Scholar]
- 200.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Amyloid β peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 201.Bard F, Cannon C, Barbour R, Burke R-L, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 202.Brendza R, Bacskai B, Cirrito J, Simmons K, Skoch J, Klunk W, Mathis C, Bales K, Paul S, Hyman B, Holtzman D. Anti-abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–33. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.DeMattos RB, Bales KR, Cummins DJ, Dodart J-C, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dodart J-C, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 205.Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-amyloid antibodies block β-amyloid fibril formation and prevent β-amyloid-induced neurotoxicity. Brain. 2003;126:1935–9. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- 206.Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-β antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:6213–20. doi: 10.1523/JNEUROSCI.0664-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid β protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–7. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–83. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Check E. Nerve inflammation halts trial for Alzheimer's drug. Nature. 2002;415:462. doi: 10.1038/415462a. [DOI] [PubMed] [Google Scholar]
- 210.Senior K. Dosing in phase II trial of Alzheimer's vaccine suspended. Lancet Neurol. 2002;1:3. doi: 10.1016/s1474-4422(02)00023-6. [DOI] [PubMed] [Google Scholar]
- 211.Orgogozo JMM, Gilman SMF, Dartigues JFMDP, Laurent BM, Puel MM, Kirby LCM, Jouanny PMDP, Dubois BM, Eisner LM, Flitman SM, Michel BFM, Boada MM, Frank AMDP, Hock CM. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 212.Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-b peptide: a case report. Nat Med 04. 2003;9:448. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 213.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Aβ species removal after Aβ42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 214.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 215.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-β deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–72. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 216.Lemere CA, Beierschmitt A, Iglesias M, Spooner ET, Bloom JK, Leverone JF, Zheng JB, Seabrook TJ, Louard D, Li D, Selkoe DJ, Palmour RM, Ervin FR. Alzheimer's disease Aβ vaccine reduces central nervous system Aβ levels in a non-human primate, the Caribbean vervet. Am J Pathol. 2004;165:283–97. doi: 10.1016/s0002-9440(10)63296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, Von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–5. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 218.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Miiller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, De Quervain DJF, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–54. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 219.Fox NCMF, Black RSM, Gilman SMF, Rossor MNMF, Griffith SGMDPM, Jenkins LP, Koller MMM. for the ANST. Effects of Aβ immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–72. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 220.Gilman S, Koller M, Black RS, Jenkins L, Griffith SGMD, Fox NC, Eisner L, Kirby L, Rovira M, BM D, Forette F, Orgogozo JM. Team ANS. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 221.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JAR. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 222.Bayer TA, Schafer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schussel K, Eikenberg O, Sturchler-Pierrat C, Abramowski D, Staufenbiel M, Multhaup G. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Aβ production in APP23 transgenic mice. Proc Natl Acad Sci USA. 2003;100:14187–92. doi: 10.1073/pnas.2332818100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Borchardt T, Camakaris J, Cappai R, Masters CL, Beyreuther K, Multhaup G. Copper inhibits β-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem J. 1999;344:461–7. [PMC free article] [PubMed] [Google Scholar]
- 224.Phinney AL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, Home P, Yang J, Sekoulidis J, Coomaraswamy J, Chishti MA, Cox DW, Mathews PM, Nixon RA, Carlson GA, George-Hyslop PS, Westaway D. In vivo reduction of amyloid-β by a mutant copper transporter. Proc Natl Acad Sci USA. 2003;100:14193–8. doi: 10.1073/pnas.2332851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.White AR, Du T, Laughton KM, Volitakis I, Sharpies RA, Xilinas ME, Hoke DE, Holsinger RMD, Evin G, Cherny RA, Hill AF, Barnham KJ, Li Q-X, Bush AI, Masters CL. Degradation of the Alzheimer disease amyloid β-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006;281:17670–80. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- 226.Dodel RC, Du Y, Depboylu C, Hampel H, Frolich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nolker C, Moller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against β-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–4. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Adlard PA, Bush AI. Metals and Alzheimer's disease. J Alzheimers Dis. 2006;10:145–63. doi: 10.3233/jad-2006-102-303. [DOI] [PubMed] [Google Scholar]
- 228.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 229.Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal ageing and Alzheimer's disease. J Neural Transm Park Dis Dement Sect. 1991;3:231–58. [PubMed] [Google Scholar]
- 230.Bush AI. The metallobiology of Alzheimer's disease. Trends Neurosci. 2003;26:207–14. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 231.Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2000;4:184–91. doi: 10.1016/s1367-5931(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 232.Koh J-Y. Zinc and disease of the brain. Mol Neurobiol. 2001;24:99–106. doi: 10.1385/MN:24:1-3:099. [DOI] [PubMed] [Google Scholar]
- 233.Madsen E, Gitlin JD. Copper and iron disorders of the brain. Annu Rev Neurosci. 2007;30:317–37. doi: 10.1146/annurev.neuro.30.051606.094232. [DOI] [PubMed] [Google Scholar]
- 234.Barnham KJ, Bush AI. Metals in Alzheimer's and Parkinson's diseases. Curr Opin Chem Biol. 2008;12:222–8. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 235.Suh SW, Jensen KB, Jensen MS, Suva DS, Kesslak PJ, Danscher G, Frederickson CJ. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Res. 2000;852:274–8. doi: 10.1016/s0006-8993(99)02096-x. [DOI] [PubMed] [Google Scholar]
- 236.Smith MA, Wehr K, Harris PLR, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer's disease. Brain Res. 1998;788:232–6. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 237.Bjorkdahl C, Sjogren MJ, Winblad B, Pei J-JCA. Zinc induces neurofilament phosphorylation independent of p70 S6 kinase in N2a cells. Neuroreport. 2005;16:591–5. doi: 10.1097/00001756-200504250-00015. [DOI] [PubMed] [Google Scholar]
- 238.Malm TM, Livonen H, Goldsteins G, Keksa-Goldsteine V, Ahtonleml T, Kannlnen K, Salminen A, Auriola S, Van Groen T, Tanlla H, Koistinaho J. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting β-amyloid burden. J Neurosci. 2007;27:3712–21. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Egana JT, Zambrano C, Nunez MT, Gonzalez-Billault C, Maccioni RB. Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals. 2003;16:215–23. doi: 10.1023/a:1020727218493. [DOI] [PubMed] [Google Scholar]
- 240.Ma Q, Li Y, Du J, Kanazawa K, Nemoto T, Nakanishi H, Zhao Y. Binding of copper (II) ion to an Alzheimer's tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers. 2005;79:74–85. doi: 10.1002/bip.20335. [DOI] [PubMed] [Google Scholar]
- 241.Ma Q, Li Y, Du J, Liu H, Kanazawa K, Nemoto T, Nakanishi H, Zhao Y. Copper binding properties of a tau peptide associated with Alzheimer's disease studied by CD, NMR, and MALDI-TOF MS. Peptides. 2006;27:841–9. doi: 10.1016/j.peptides.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 242.Zhou L-X, Du J-T, Zeng Z-Y, Wu W-H, Zhao Y-F, Kanazawa K, Ishizuka Y, Nemoto T, Nakanishi H, Li Y-M. Copper (II) modulates in vitro aggregation of a tau peptide. Peptides. 2007;28:2229–34. doi: 10.1016/j.peptides.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 243.Yamamoto A, Shin RW, Hasegawa K, Naiki H, Sato H, Yoshimasu F, Kitamoto T. Iron (III) induces aggregation of hyper-phosphorylated τ and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer's disease. J Neurochem. 2002;82:1137–47. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x. [DOI] [PubMed] [Google Scholar]
- 244.Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R. Structure of the Alzheimer's disease amyloid precursor protein copper binding domain A regulator of neuronal copper homeostasis. J Biol Chem. 2003;278:17401–7. doi: 10.1074/jbc.M300629200. [DOI] [PubMed] [Google Scholar]
- 245.Bayer TA, Multhaup G. Involvement of amyloid β precursor protein (AβPP) modulated copper homeostasis in Alzheimer's disease. J Alzheimers Dis. 2005;8:201–6. doi: 10.3233/jad-2005-8212. [DOI] [PubMed] [Google Scholar]
- 246.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–28. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 247.Venti A, Giordano T, Eder P, Bush AL, Lahiri DK, Greig NH, Rogers JT. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5’-untranslated region. Ann N Y Acad Sci. 2004;1035:34–48. doi: 10.1196/annals.1332.003. [DOI] [PubMed] [Google Scholar]
- 248.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–92. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 249.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 250.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li Q-X. Overexpression of Alzheimer's disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–6. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 251.White AR, Reyes R, Mercer JFB, Camakaris J, Zheng H, Bush AL, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999;842:439–44. doi: 10.1016/s0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- 252.Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van Der Ploeg LH. β-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–31. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 253.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold K-H, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–92. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Sparks DL, Schreurs BG. Trace amounts of copper in water induce β-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:11065–9. doi: 10.1073/pnas.1832769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bush AI, Pettingell WH, Jr, Paradis MD, Tanzi RE. Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem. 1994;269:12152–8. [PubMed] [Google Scholar]
- 256.Bush AI, Pettingell WH, Multhaup G, D Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994;265:1464–7. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 257.Esler WP, Stimson ER, Jennings JM, Ghilardi JR, Mantyh PW, Maggio JE. Zinc-induced aggregation of human and rat β-amyloid peptides in vitro. J Neurochem. 1996;66:723–32. doi: 10.1046/j.1471-4159.1996.66020723.x. [DOI] [PubMed] [Google Scholar]
- 258.Brown AM, Tummolo DM, Rhodes KJ, Hofmann JR, Jacobsen JS, Sonnenberg-Reines J. Selective aggregation of endogenous β-amyloid peptide and soluble amyloid precursor protein in cerebrospinal fluid by zinc. J Neurochem. 1997;69:1204–12. doi: 10.1046/j.1471-4159.1997.69031204.x. [DOI] [PubMed] [Google Scholar]
- 259.Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of β-amyloid peptide. J Neurochem. 1993;61:1171–4. doi: 10.1111/j.1471-4159.1993.tb03639.x. [DOI] [PubMed] [Google Scholar]
- 260.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NME, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–26. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 261.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Copper homeostasis and neurodegenerative disorders (Alzheimer's, Prion, and Parkinson's diseases and amyotrophic lateral sclerosis) Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 262.Miura T, Suzuki K, Kohata N, Takeuchi H. Metal binding modes of Alzheimer's amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry. 2000;39:7024–31. doi: 10.1021/bi0002479. [DOI] [PubMed] [Google Scholar]
- 263.Assaf SY, Chung S-H. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–6. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 264.Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–8. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 265.Hartter DE, Barnea A. Brain tissue accumulates 67copper by two ligand-dependent saturable processes. A high affinity, low capacity and a low affinity, high capacity process. J Biol Chem. 1988;263:799–805. [PubMed] [Google Scholar]
- 266.Kardos J, Kovacs I, Hajos F, Kalman M, Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett. 1989;103:139–44. doi: 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- 267.Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25:239–46. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Friedlich AL, Lee J-Y, Van Groen T, Cherny RA, Volitakis I, Cole TB, Palmiter RD, Koh J-Y, Bush AI. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer's disease. J Neurosci. 2004;24:3453–9. doi: 10.1523/JNEUROSCI.0297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA. 2002;99:7705–10. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Linkous DH, Flinn JM, Koh JY, Lanzirotti A, Bertsch PM, Jones BF, Giblin LJ, Frederickson CJ. Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J Histochem Cytochem. 2008;56:3–6. doi: 10.1369/jhc.6A7035.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Frederickson CJ, Bush AI. Synaptically released zinc: physiological functions and pathological effects. Biometals. 2001;14:353–66. doi: 10.1023/a:1012934207456. [DOI] [PubMed] [Google Scholar]
- 272.Karr JW, Akintoye H, Kaupp LJ, Szalai VA. N-Terminal deletions modify the Cu2+ binding site in amyloid-β. Biochemistry. 2005;44:5478–87. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 273.Syme CD, Viles JH. Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-β peptide (Aβ) of Alzheimer's disease. Biochim Biophys Acta. 2006;1764:246–56. doi: 10.1016/j.bbapap.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 274.Jiang D, Men L, Wang J, Zhang Y, Chickenyen S, Wang Y, Zhou F. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathological relevance. Biochemistry. 2007;46:9270–82. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Clements A, Allsop D, Walsh DM, Williams CH. Aggregation and metal-binding properties of mutant forms of the amyloid β peptide of Alzheimer's disease. J Neurochem. 1996;66:740–7. doi: 10.1046/j.1471-4159.1996.66020740.x. [DOI] [PubMed] [Google Scholar]
- 276.Mekmouche Y, Coppel Y, Hochgräfe K, Guilloreau L, Talmard C, Mazarguil H, Faller P. Characterization of the Zn(II) binding to the peptide amyloid-β1–16 linked to Alzheimer's disease. Chembiochem. 2005;6:1663–71. doi: 10.1002/cbic.200500057. [DOI] [PubMed] [Google Scholar]
- 277.Kozin SA, Zirah S, Rebuffat S, Hui Bon Hoa G, Debey P. Zinc binding to Alzheimer's Aβ(1–16) peptide results in stable soluble complex. Biochem Biophys Res Commun. 2001;285:959–64. doi: 10.1006/bbrc.2001.5284. [DOI] [PubMed] [Google Scholar]
- 278.Atwood CS, Huang X, Khatri A, Scarpa RC, Kim YS, Moir RD, Tanzi RE, Roher AE, Bush AI. Copper catalyzed oxidation of Alzheimer Aβ. Cell Mol Biol. 2000;46:777–83. [PubMed] [Google Scholar]
- 279.Guilloreau L, Damian L, Coppel Y, Mazarguil H, Winterhalter M, Faller P. Structural and thermodynamical properties of CuII amyloid-β16/28 complexes associated with Alzheimer's disease. J Biol Inorg Chem. 2006;11:1024–38. doi: 10.1007/s00775-006-0154-1. [DOI] [PubMed] [Google Scholar]
- 280.Ha C, Ryu J, Park CB. Metal ions differentially influence the aggregation and deposition of Alzheimer's β-amyloid on a solid template. Biochemistry. 2007;46:6118–25. doi: 10.1021/bi7000032. [DOI] [PubMed] [Google Scholar]
- 281.Han D, Wang H, Yang P. Molecular modeling of zinc and copper binding with Alzheimer's amyloid β-peptide. Biometals. 2008;21:189–96. doi: 10.1007/s10534-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 282.Li W, Zhang J, Su Y, Wang J, Qin M, Wang W. Effects of zinc binding on the conformational distribution of the amyloid-β peptide based on molecular dynamics simulations. J Phys Chem. 2007;111:13814–21. doi: 10.1021/jp076213t. [DOI] [PubMed] [Google Scholar]
- 283.Morgan DM, Dong J, Jacob J, Lu K, Apkarian RP, Thiyagarajan P, Lynn DG. Metal switch for amyloid formation: insight into the structure of the nucleus. J Am Chem Soc. 2002;124:12644–5. doi: 10.1021/ja0273086. [DOI] [PubMed] [Google Scholar]
- 284.Dong J, Canfield JM, Mehta AK, Shokes JE, Tian B, Childers WS, Simmons JA, Mao Z, Scott RA, Warncke K, Lynn DG. Engineering metal ion coordination to regulate amyloid fibril assembly and toxicity. Proc Natl Acad Sci USA. 2007;104:13313–8. doi: 10.1073/pnas.0702669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Sangmi J, Sunil S. The aggregated state of amyloid-β peptide in vitro depends on Cu2+ ion concentration. Angew Chem Int Ed Engl. 2007;46:3959–61. doi: 10.1002/anie.200700318. [DOI] [PubMed] [Google Scholar]
- 286.Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI. Characterization of copper interactions with Alzheimer amyloid β peptides. Identification of an attomolar-affinity copper binding site on amyloid β1–42. J Neurochem. 2000;75:1219–33. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 287.Tougu V, Karafin A, Palumaa P. Binding of zinc(II) and copper(II) to the full-length Alzheimer's amyloid-β peptide. J Neurochem. 2008;104:1249–59. doi: 10.1111/j.1471-4159.2007.05061.x. [DOI] [PubMed] [Google Scholar]
- 288.Gaggelli E, Janicka-Klos A, Jankowska E, Kozlowski H, Migliorini C, Molteni E, Valensin D, Valensin G, Wieczerzak E. NMR studies of the Zn2+ interactions with rat and human β-amyloid (1–28) peptides in water-micelle environment. J Phys Chem. 2008;112:100–9. doi: 10.1021/jp075168m. [DOI] [PubMed] [Google Scholar]
- 289.Danielsson J, Pierattelli R, Banci L, Graslund A. High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid β-peptide. FEBS J. 2007;274:46–59. doi: 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- 290.Garzon-Rodriguez W, Yatsimirsky AK, Glabe CG. Binding of Zn(II), Cu(II), and Fe(II) ions to Alzheimer's Aβ peptide studied by fluorescence. Bioorg Med Chem Lett. 1999;9:2243–8. doi: 10.1016/s0960-894x(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 291.Clements A, Allsop D, Walsh DM, Williams CH. Aggregation and metal-binding properties of mutant forms of the amyloid Aβ peptide of Alzheimer's disease. J Neurochem. 1996;66:740–7. doi: 10.1046/j.1471-4159.1996.66020740.x. [DOI] [PubMed] [Google Scholar]
- 292.Streltsov V. X-ray absorption and diffraction studies of the metal binding sites in amyloid β-peptide. Eur Biophys J. 2008;37:257–63. doi: 10.1007/s00249-007-0232-5. [DOI] [PubMed] [Google Scholar]
- 293.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer's disease amyloid-β binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–73. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 294.Yang D-S, McLaurin J, Qin K, Westaway D, Fraser PE. Examining the zinc binding site of the amyloid-β peptide. Eur J Biochem. 2000;267:6692–8. doi: 10.1046/j.1432-1327.2000.01767.x. [DOI] [PubMed] [Google Scholar]
- 295.Zirah S, Rebuffat S, Kozin SA, Debey P, Fournier F, Lesage D, Tabet J-C. Zinc binding properties of the amyloid fragment Aβ(1–16) studied by electrospray-ionization mass spectrometry. Int J Mass Spectrom. 2003;228:999–1016. [Google Scholar]
- 296.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J Biol Chem. 2003;278:2977–82. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 297.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amy-loid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–73. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 298.Karr JW, Kaupp LJ, Szalai VA. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. J Am Chem Soc. 2004;126:13534–8. doi: 10.1021/ja0488028. [DOI] [PubMed] [Google Scholar]
- 299.Karr JW, Szalai VA. Role of aspartate-1 in Cu(II) binding to the amyloid-β peptide of Alzheimer's disease. J Am Chem Soc. 2007;129:3796–7. doi: 10.1021/ja068952d. [DOI] [PubMed] [Google Scholar]
- 300.Kowalik-Jankowska T, Ruta M, Wisniewska K, Lankiewicz L. Coordination abilities of the 1–16 and 1–28 fragments of β-amyloid peptide towards copper(II) ions: a combined potentiometric and spectroscopic study. J Inorg Biochem. 2003;95:270–82. doi: 10.1016/s0162-0134(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 301.Stellato F, Menestrina G, Serra M, Potrich C, Tomazzolli R, Meyer-Klaucke W, Morante S. Metal binding in amyloid β-peptides shows intra- and inter-peptide coordination modes. Eur Biophys J. 2006;35:340–51. doi: 10.1007/s00249-005-0041-7. [DOI] [PubMed] [Google Scholar]
- 302.Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, Debey P, Rebuffat S. Structural changes of region 1–16 of the Alzheimer disease amyloid β-peptide upon zinc binding and in vitro aging. J Biol Chem. 2006;281:2151–61. doi: 10.1074/jbc.M504454200. [DOI] [PubMed] [Google Scholar]
- 303.Liu ST, Hewlett G, Barrow CJ. Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the abeta peptide of Alzheimer's disease. Biochemistry. 1999;38:9373–8. doi: 10.1021/bi990205o. [DOI] [PubMed] [Google Scholar]
- 304.Morgan DM, Dong J, Jacob J, Lu K, Apkarian RP, Thiyagarajan P, Lynn DG. Metal switch for amyloid formation: insight into the structure of the nucleus. J Am Chem Soc. 2002;124:12644–5. doi: 10.1021/ja0273086. [DOI] [PubMed] [Google Scholar]
- 305.Vaughan D, Peters A. The structure of neuritic plaque in the cerebral cortex of aged rats. J Neuropathol Exp Neurol. 1981;40:472–87. doi: 10.1097/00005072-198107000-00009. [DOI] [PubMed] [Google Scholar]
- 306.Bush AI, Tanzi RE. Therapeutics for Alzheimer's disease based on the metal hypothesis. Neurotherapeutics. 2008;5:421–32. doi: 10.1016/j.nurt.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S. Zinc lowers amyloid-β toxicity by selectively precipitating aggregation intermediates. Biochemistry. 2007;46:10655–63. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 308.Tew DJ, Bottomley SP, Smith DP, Ciccotosto GD, Babon J, Hinds MG, Masters CL, Cappai R, Barnham KJ. Stabilization of neurotoxic soluble β-sheet-rich conformations of the Alzheimer's disease amyloid-β peptide. Biophys J. 2008;94:2752–66. doi: 10.1529/biophysj.107.119909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Yoshiike Y, Tanemura K, Murayama O, Akagi T, Murayama M, Sato S, Sun X, Tanaka N, Takashima A. New insights on how metals disrupt amyloid β -aggregation and their effects on amyloid-β cytotoxicity. J Biol Chem. 2001;276:32293–9. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- 310.Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer's disease β-amyloid. FASEB J. 2004;18:1427–9. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- 311.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Copper mediates dityrosine cross-linking of Alzheimer's amyloid-beta. Biochemistry. 2004;43:560–8. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 312.Smith DP, Ciccotosto GD, Tew DJ, Fodero-Tavoletti MT, Johanssen T, Masters CL, Barnham KJ, Cappai R. Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer's disease amyloid-β peptide. Biochemistry. 2007;46:2881–91. doi: 10.1021/bi0620961. [DOI] [PubMed] [Google Scholar]
- 313.Barnham KJ, Ciccotosto GD, Tickler AK, Ali FE, Smith DG, Williamson NA, Lam Y-H, Carrington D, Tew D, Kocak G, Volitakis I, Separovic F, Barrow CJ, Wade JD, Masters CL, Cherny RA, Curtain CC, Bush AI, Cappai R. Neurotoxic, redox-competent Alzheimer's β-amyloid is released from lipid membrane by methionine oxidation. J Biol Chem. 2003;278:42959–65. doi: 10.1074/jbc.M305494200. [DOI] [PubMed] [Google Scholar]
- 314.Crouch PJ, Barnham KJ, Duce JA, Blake RE, Masters CL, Trounce IA. Copper-dependent inhibition of cytochrome c oxi-dase by Aβ1 42 requires reduced methionine at residue 35 of the Aβ peptide. J Neurochem. 2006;99:226–36. doi: 10.1111/j.1471-4159.2006.04050.x. [DOI] [PubMed] [Google Scholar]
- 315.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon ML, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the Aβ(1–40) and Aβ(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 316.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Copper mediates dityrosine cross-linking of Alzheimer's amyloid-β. Biochemistry. 2004;43:560–8. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 317.Naylor R, Hill A, Barnham K. Neurotoxicity in Alzheimer's disease: is covalently crosslinked Aβ responsible? Eur Biophys J. 2008;37:265–8. doi: 10.1007/s00249-007-0243-2. [DOI] [PubMed] [Google Scholar]
- 318.Lau T-L, Ambroggio EE, Tew DJ, Cappai R, Masters CL, Fidelio GD, Barnham KJ, Separovic F. Amyloid-β peptide disruption of lipid membranes and the effect of metal ions. J Mol Biol. 2006;356:759–70. doi: 10.1016/j.jmb.2005.11.091. [DOI] [PubMed] [Google Scholar]
- 319.Ciccotosto GD, Tew D, Curtain CC, Smith D, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Barnham KJ. Enhanced toxicity and cellular binding of a modified amyloid β peptide with a methionine to valine substitution. J Biol Chem. 2004;279:42528–34. doi: 10.1074/jbc.M406465200. [DOI] [PubMed] [Google Scholar]
- 320.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–14. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 321.Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim Biophys Acta. 2007;1768:1976–90. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 322.Atwood CS, Huang X, Moir RD, Tanzi RE, Bush AI. Role of free radicals and metal ions in the pathogenesis of Alzheimer's disease. Met Ions Biol Syst. 1999;36:309–64. [PubMed] [Google Scholar]
- 323.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–16. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 324.Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Afl peptides. J Biol Inorg Chem. 2004;9:954–60. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 325.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–6. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 326.Sayre LM, Perry G, Harris PLR, Liu Y, Schubert KA, Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease. J Neurochem. 2000;74:270–9. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 327.Rossi L, Lombardo M, Ciriolo M, Rotilio G. Mitochondrial dysfunction in neurodegenerative diseases associated with copper imbalance. Neurochem Res. 2004;29:493–504. doi: 10.1023/b:nere.0000014820.99232.8a. [DOI] [PubMed] [Google Scholar]
- 328.Masayoshi Y, Masatsugu K, Shoji O. Role of zinc as an activator of mitochondrial function in rat liver. Biochem Pharmacol. 1982;31:1289–93. doi: 10.1016/0006-2952(82)90018-1. [DOI] [PubMed] [Google Scholar]
- 329.Ortiz E, Pasquini JM, Thompson K, Felt B, Butkus G, Beard J, Connor JR. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res. 2004;77:681–9. doi: 10.1002/jnr.20207. [DOI] [PubMed] [Google Scholar]
- 330.Smart TG, Hosie AM, Miller PS. Zn2+ Ions: Modulators of Excitatory and Inhibitory Synaptic Activity. Neuroscientist. 2004;10:432–42. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- 331.Takeda A. Zinc homeostasis and functions of zinc in the brain. Biometals. 2001;14:343–51. doi: 10.1023/a:1012982123386. [DOI] [PubMed] [Google Scholar]
- 332.Bhatnagara S, Tanejaa S. Zinc and cognitive development. Br J Nutr. 2001;85:S139–S45. doi: 10.1079/bjn2000306. [DOI] [PubMed] [Google Scholar]
- 333.Gaeta A, Hider RC. The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br J Pharmacol. 2005;146:1041–59. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Que EL, Domaille DW, Chang CJ. Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem Rev. 2008;108:1517–49. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]
- 335.Bush AI, Curtain C. Twenty years of metallo-neurobiology: where to now? Eur Biophys J. 2008;37:241–5. doi: 10.1007/s00249-007-0228-1. [DOI] [PubMed] [Google Scholar]
- 336.Crouch PJ, White AR, Bush AI. The modulation of metal bio-availability as a therapeutic strategy for the treatment of Alzheimer's disease. FEBS J. 2007;274:3775–83. doi: 10.1111/j.1742-4658.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- 337.Finefrock AE, Bush AI, Doraiswamy PM. Current Status of Metals as Therapeutic Targets in Alzheimer's Disease. J Am Geriatr Soc. 2003;51:1143–8. doi: 10.1046/j.1532-5415.2003.51368.x. [DOI] [PubMed] [Google Scholar]
- 338.Bush AI. Metal complexing agents as therapies for Alzheimer's disease. Neurobiol Aging. 2002;23:1031–8. doi: 10.1016/s0197-4580(02)00120-3. [DOI] [PubMed] [Google Scholar]
- 339.Brown DR. Interactions between metals and α-synuclein – function or artefact? FEBS J. 2007;274:3766–74. doi: 10.1111/j.1742-4658.2007.05917.x. [DOI] [PubMed] [Google Scholar]
- 340.Doraiswamy PM, Finefrock AE. Metals in our minds: therapeutic implications for neurodegenerative disorders. Lancet Neurol. 2004;3:431–4. doi: 10.1016/S1474-4422(04)00809-9. [DOI] [PubMed] [Google Scholar]
- 341.Behl C, Moosmann B. Antioxidant neuro-protection in Alzheimer's disease as preventive and therapeutic approach. Free Radic Biol Med. 2002;33:182–91. doi: 10.1016/s0891-5849(02)00883-3. [DOI] [PubMed] [Google Scholar]
- 342.Zatta P, Tognon G, Carampin P. Melatonin prevents free radical formation due to the interaction between β-amyloid peptides and metal ions [Al(III), Zn(II), Cu(II) Mn(II), Fe(II)] J Pineal Res. 2003;35:98–103. doi: 10.1034/j.1600-079x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 343.Shishodia S, Sethi G, Aggarwal BB. Curcumin: Getting Back to the Roots. Ann N Y Acad Sci. 2005;1056:206–17. doi: 10.1196/annals.1352.010. [DOI] [PubMed] [Google Scholar]
- 344.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 345.Feng Z, Chang Y, Cheng Y, Zhang B-I, Qu Z-W, Qin C, Zhang J-T. Melatonin alleviates behavioral deficits associated with apopto-sis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer's disease. J Pineal Res. 2004;37:129–36. doi: 10.1111/j.1600-079X.2004.00144.x. [DOI] [PubMed] [Google Scholar]
- 346.Matsubara E, Bryant-Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, Herbert D, Cruz-Sanchez F, Chyan Y-J, Smith MA, Perry G, Shoji M, Abe K, Leone A, Grundke-lkbal I, Wilson GL, Ghiso J, Williams C, Refolo LM, Pappolla MA. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer's disease. J Neurochem. 2003;85:1101–8. doi: 10.1046/j.1471-4159.2003.01654.x. [DOI] [PubMed] [Google Scholar]
- 347.Cheng Y, Feng Z, Zhang Q-Z, Zhang J-T. Beneficial effects of melatonin in experimental models of Alzheimer disease1. Acta Pharmacol Sin. 2006;27:129–39. doi: 10.1111/j.1745-7254.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 348.Defeudis FV. Bilobalide and neuroprotection. Pharmacol Res. 2002;46:565–8. doi: 10.1016/s1043-6618(02)00233-5. [DOI] [PubMed] [Google Scholar]
- 349.Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JCS. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch Neurol. 2004;61:82–8. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 350.Oken BS, Storzbach DM, Kaye JA. The Efficacy of Ginkgo biloba on cognitive function in Alzheimer disease. Arch Neurol. 1998;55:1409–15. doi: 10.1001/archneur.55.11.1409. [DOI] [PubMed] [Google Scholar]
- 351.Schneider LS, DeKosky ST, Farlow MR, Tariot PN, Hoerr R, Kieser M. A randomized, double-blind, placebo-controlled trial of two doses of ginkgo biloba extract in dementia of the Alzheimer's type. Curr Alzheimer Res. 2005;2:541–51. doi: 10.2174/156720505774932287. [DOI] [PubMed] [Google Scholar]
- 352.Keberle H. The biochemistry of desferrioxamine and its relation to iron metabolism. Ann N Y Acad Sci. 1964;119:758–68. doi: 10.1111/j.1749-6632.1965.tb54077.x. [DOI] [PubMed] [Google Scholar]
- 353.McLachlan DRC, Dalton AJ. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–8. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 354.Cuajungco MP, Faget KY, Huang X, Tanzi RE, Bush AI. Metal chelation as a potential therapy for Alzheimer's disease. Ann N Y Acad Sci. 2000;920:292–304. doi: 10.1111/j.1749-6632.2000.tb06938.x. [DOI] [PubMed] [Google Scholar]
- 355.Lee J-Y, Friedman JE, Angel I, Kozak A, Koh J-Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human β-amyloid precursor protein transgenic mice. Neurobiol Aging. 2004;25:1315–21. doi: 10.1016/j.neurobiolaging.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 356.Petri S, Calingasan NY, Alsaied OA, Wille E, Kiaei M, Friedman JE, Baranova O, Chavez JC, Beal MF. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2007;102:991–1000. doi: 10.1111/j.1471-4159.2007.04604.x. [DOI] [PubMed] [Google Scholar]
- 357.Carrì MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull. 2003;61:365–74. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 358.Roos PM, Vesterberg O, Nordberg M. Metals in motor neuron diseases. Exp Biol Med. 2006;231:1481–7. doi: 10.1177/153537020623100906. [DOI] [PubMed] [Google Scholar]
- 359.Perluigi M, Joshi G, Sultana R, Calabrese V, De Marco C, Coccia R, Butterfield DA. In vivo protection by the xanthate tricyclodecan-9-yl-xanthogenate against amyloid β-peptide (1–42)-induced oxidative stress. Neuroscience. 2006;138:1161–70. doi: 10.1016/j.neuroscience.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 360.Shin R-W, Kruck TPA, Murayama H, Kitamoto T. A novel trivalent cation chela-tor Feralex dissociates binding of aluminum and iron associated with hyper-phosphorylated τ of Alzheimer's disease. Brain Res. 2003;961:139–46. doi: 10.1016/s0006-8993(02)03893-3. [DOI] [PubMed] [Google Scholar]
- 361.Cherny RA, Barnham KJ, Lynch T, Volitakis I, Li Q-X, McLean CA, Multhaup G, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Chelation and intercalation: complementary properties in a compound for the treatment of Alzheimer's disease. J Struct Biol. 2000;130:209–16. doi: 10.1006/jsbi.2000.4285. [DOI] [PubMed] [Google Scholar]
- 362.Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor κ B activation in intact cells. J Exp Med. 1992;175:1181–94. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Liu SF, Ye X, Malik AB. Inhibition of NF-κB activation by pyrrolidine dithiocarbamate prevents in vivo expression of proinflammatory genes. Circulation. 1999;100:1330–7. doi: 10.1161/01.cir.100.12.1330. [DOI] [PubMed] [Google Scholar]
- 364.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 2003;22:3356–66. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Burkitt MJ, Bishop HS, Milne L, Tsang SY, Provan GJ, Nobel CSI, Orrenius S, Slater AFG. Dithiocarbamate toxicity toward thymocytes involves their copper-catalyzed conversion to thiuram disulfides, which oxidize glutathione in a redox cycle without the release of reactive oxygen species. Arch Biochem Biophys. 1998;353:73–84. doi: 10.1006/abbi.1998.0618. [DOI] [PubMed] [Google Scholar]
- 366.Nobel CSI, Burgess DH, Zhivotovsky B, Burkitt MJ, Orrenius S, Slater AFG. Mechanism of dithiocarbamate inhibition of apoptosis: thiol oxidation by dithiocarbamate disulfides directly inhibits processing of the caspase-3 proenzyme. Chem Res Toxicol. 1997;10:636–43. doi: 10.1021/tx970006a. [DOI] [PubMed] [Google Scholar]
- 367.Nobel CSI, Kimland M, Lind B, Orrenius S, Slater AFG. Dithiocarbamates induce apoptosis in thymocytes by raising the intracellular level of redox-active copper. J Biol Chem. 1995;270:26202–8. doi: 10.1074/jbc.270.44.26202. [DOI] [PubMed] [Google Scholar]
- 368.Verhaegh GW, Richard MJ, Halnaut P. Regulation of p53 by metal ions and by antioxidants: dithiocarbamate down-regulates p53 DNA-binding activity by increasing the intracellular level of copper. Mol Cell Biol. 1997;17:5699–706. doi: 10.1128/mcb.17.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Kim CH, Kim JH, Hsu CY, Ahn YS. Zinc is required in pyrrolidine dithiocarbamate inhibition of NF-κB activation. FEBS Lett. 1999;449:28–32. doi: 10.1016/s0014-5793(99)00390-7. [DOI] [PubMed] [Google Scholar]
- 370.Iseki A, Kambe F, Okumura K, Niwata S, Yamamoto R, Hayakawa T, Seo H. Pyrrolidine dithiocarbamate inhibits TNF-α-dependent activation of NF-κB by increasing intracellular copper level in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2000;276:88–92. doi: 10.1006/bbrc.2000.3452. [DOI] [PubMed] [Google Scholar]
- 371.Furuta S, Ortiz F, Zhu Sun X, Wu H-H, Mason A, Momand J. Copper uptake is required for pyrrolidine dithiocarbamate-mediated oxidation and protein level increase of p53 in cells. Biochem J. 2002;365:639–48. doi: 10.1042/BJ20011251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Nurmi A, Goldsteins G, Närväinen J, Pihlaja R, Ahtoniemi T, Gröhn O, Koistinaho J. Antioxidant pyrrolidine dithiocarbamate activates Akt-GSK signaling and is neuroprotective in neonatal hypoxia-ischemia. Free Radic Biol Med. 2006;40:1776–84. doi: 10.1016/j.freeradbiomed.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 373.Daniel K, Chen D, Orlu S, Cui Q, Miller F, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7:R897–908. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP. Pyrrolidine dithiocarbamate-zinc(II) and -copper(II) complexes induce apoptosis in tumor cells by inhibiting the proteasomal activity. Toxicol Appl Pharmacol. 2008;231:24–33. doi: 10.1016/j.taap.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Ding W-Q, Yu H-J, Lind SE. Zinc-binding compounds induce cancer cell death via distinct modes of action. Cancer Lett. 2008;271:251–9. doi: 10.1016/j.canlet.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 376.Beraldo H, Gambino D. The wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini Rev Med Chem. 2004;4:31–9. doi: 10.2174/1389557043487484. [DOI] [PubMed] [Google Scholar]
- 377.Petering DH. Concerning the role of zinc in the antitumor activity of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazonato) zinc(II) and related chelates. Biochem Pharmacol. 1974;23:567–76. doi: 10.1016/0006-2952(74)90621-2. [DOI] [PubMed] [Google Scholar]
- 378.Dearling J, Lewis J, Mullen G, Welch M, Blower P. Copper bis(thiosemicarbazone) complexes as hypoxia imaging agents: structure-activity relationships. J Biol Inorg Chem. 2002;7:249–59. doi: 10.1007/s007750100291. [DOI] [PubMed] [Google Scholar]
- 379.Maurer RI, Blower PJ, Dilworth JR, Reynolds CA, Zheng Y, Mullen GED. Studies on the mechanism of hypoxic selectivity in copper bis(thiosemicar -bazone) radiopharmaceuticals. J Med Chem. 2002;45:1420–31. doi: 10.1021/jm0104217. [DOI] [PubMed] [Google Scholar]
- 380.Lewis JS, Laforest R, Buettner TL, Song S-K, Fujibayashi Y, Connett JM, Welch MJ. Copper-64-diacetyl-bis(N 4 -methylth-iosemicarbazone): An agent for radiotherapy. Proc Natl Acad Sci USA. 2001;98:1206–11. doi: 10.1073/pnas.98.3.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Cowley AR, Davis J, Dilworth JR, Donnelly PS, Dobson R, Nightingale A, Peach JM, Shore B, Kerr D, Seymour L. Fluorescence studies of the intra-cellular distribution of zinc bis(thiosemicarbazone) complexes in human cancer cells. Chem Commun. 2005;7:845–7. doi: 10.1039/b417206j. [DOI] [PubMed] [Google Scholar]
- 382.Holland JP, Aigbirhio FI, Betts HM, Bonnitcha PD, Burke P, Christlieb M, Churchill GC, Cowley AR, Dilworth JR, Donnelly PS, Green JC, Peach JM, Vasudevan SR, Warren JE. Functionalized bis(thiosemicarbazonato) complexes of zinc and copper: synthetic platforms toward site-specific radiopharmaceuticals. Inorg Chem. 2007;46:465–85. doi: 10.1021/ic0615628. [DOI] [PubMed] [Google Scholar]
- 383.Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A. Copper-62-ATSM: a new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med. 1997;38:1155–60. [PubMed] [Google Scholar]
- 384.Vavere AL, Lewis JS. Cu-ATSM: A radio-pharmaceutical for the PET imaging of hypoxia. Dalton Trans. 2007;43:4893–902. doi: 10.1039/b705989b. [DOI] [PubMed] [Google Scholar]
- 385.Xiao Z, Donnelly PS, Zimmermann M, Wedd AG. Transfer of copper between bis(thiosemicarbazone) ligands and intracellular copper-binding proteins. insights into mechanisms of copper uptake and hypoxia selectivity. Inorg Chem. 2008;47:4338–47. doi: 10.1021/ic702440e. [DOI] [PubMed] [Google Scholar]
- 386.Kraker A, Krezoski S, Schneider J, Minkel D, Petering DH. Reaction of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazonato) Cu(II) with Ehrlich cells. Binding of copper to metallothionein and its relationship to zinc metabolism and cell proliferation. J Biol Chem. 1985;260:13710–8. [PubMed] [Google Scholar]
- 387.Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, Sharples RA, Hill AF, Li Q-X, Masters CL, Barnham KJ, White AR. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-β peptide. J Biol Chem. 2008;283:4568–77. doi: 10.1074/jbc.M705957200. [DOI] [PubMed] [Google Scholar]
- 388.Barnham KJ, Kenche VB, Ciccotosto GD, Smith DP, Tew DJ, Liu X, Perez K, Cranston GA, Johanssen TJ, Volitakis I, Bush AI, Masters CL, White AR, Smith JP, Cherny RA, Cappai R. Platinum-based inhibitors of amyloid-β as therapeutic agents for Alzheimer's disease. Proc Natl Acad Sci USA. 2008;105:6813–8. doi: 10.1073/pnas.0800712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Masters CL, Beyreuther K. Alzheimer's centennial legacy: prospects for rational therapeutic intervention targeting the Aβ amyloid pathway. Brain. 2006;129:2823–39. doi: 10.1093/brain/awl251. [DOI] [PubMed] [Google Scholar]
- 390.Bondiolotti G, Sala M, Pollera C, Gervasoni M, Puricelli M, Ponti W, Bareggi SR. Pharmacokinetics and distribution of clioquinol in golden hamsters. J Pharm Pharmacol. 2007;59:387–93. doi: 10.1211/jpp.59.3.0008. [DOI] [PubMed] [Google Scholar]
- 391.Bondiolotti GP, Pollera C, Pirola R, Bareggi SR. Determination of 5-chloro-7-iodo-8-quinolinol (clioquinol) in plasma and tissues of hamsters by high-performance liquid chromatography and electrochemical detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;837:87–91. doi: 10.1016/j.jchromb.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 392.Hayashi M, Fuwa T, Awazu S, Hanano M. Differences in species of iodochlorhydroxyquin absorption, metabolism, and excretion. Chem Pharm Bull. 1976;24:2589–96. doi: 10.1248/cpb.24.2589. [DOI] [PubMed] [Google Scholar]
- 393.Kotaki H, Yamamura Y, Tanimura Y, Saitoh Y, Nakagawa F, Tamura Z. Intestinal absorption and metabolism of clioquinol in the rat. J Pharmacobiodyn. 1983;6:881–7. doi: 10.1248/bpb1978.6.881. [DOI] [PubMed] [Google Scholar]
- 394.Kotaki H, Yamamura Y, Tanimura Y, Saitoh Y, Nakagawa F, Tamura Z. Determination of chinoform and its metabolites in plasma by gas chromatography-mass spectrometry. Chem Pharm Bull. 1983;31:299–304. doi: 10.1248/cpb.31.299. [DOI] [PubMed] [Google Scholar]
- 395.Liewendahl K, Kivikangas V, Lamberg BA. Metabolism of 125-iodochloroxyquinoline in man. II. Metabolites in plasma, urine and faeces. Nuclear-Medizin. 1967;6:32–43. [PubMed] [Google Scholar]
- 396.Neldner KH, Hambidge KM. Zinc therapy of acrodermatitis enteropathica. N Engl J Med. 1975;292:879–82. doi: 10.1056/NEJM197504242921702. [DOI] [PubMed] [Google Scholar]
- 397.Flagstad T. Intestinal absorption of 65 Zinc in A46 (Adema disease) after treatment with oxychinolines. Nordisk Veterinar Medicine. 1977;29:96–100. [PubMed] [Google Scholar]
- 398.Tsubaki T, Honma Y, Hoshi M. Neurological syndrome associated with clioquinol. Lancet. 1971;297:696–7. doi: 10.1016/s0140-6736(71)92699-7. [DOI] [PubMed] [Google Scholar]
- 399.Clioquinol and neurological disease. Br Med J. 1971;2:291–2. [PMC free article] [PubMed] [Google Scholar]
- 400.Osterman PO. Myelopathy after clioquinol treatment. Lancet. 1971;2:544. doi: 10.1016/s0140-6736(71)90460-0. [DOI] [PubMed] [Google Scholar]
- 401.Shimada Y, Tsuji T, Igata A, Steinitz H. Halogenated oxyquinoline derivatives and neurological syndromes. Lancet. 1971;2:41–3. doi: 10.1016/s0140-6736(71)90023-7. [DOI] [PubMed] [Google Scholar]
- 402.Tateishi J. Subacute myelo-optico-neuropathy: Clioquinol intoxication in humans and animals. Neuropathology. 2000;20:S20–S24. doi: 10.1046/j.1440-1789.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- 403.Konagaya M, Matsumoto A, Takase S, Mizutani T, Sobue G, Konishi T, Hayabara T, Iwashita H, Ujihira T, Miyata K, Matsuoka Y. Clinical analysis of longstanding subacute myelo-optico-neuropathy: sequelae of clioquinol at 32 years after its ban. J Neurol Sci. 2004;218:85–90. doi: 10.1016/j.jns.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 404.Nakae K, Yamamoto S-I, Shigematsu I, Kono R. RElation between subacute myelooptic neuropathy (S.M.O.N.) and clioquinol: nationwide survey. Lancet. 1973;301:171–3. doi: 10.1016/s0140-6736(73)90004-4. [DOI] [PubMed] [Google Scholar]
- 405.Ohtsuka K, Ohishi N, Eguchi G, Yagi K. Degeneration of retinal neuroblasts by chinoform-ferric chelate. Cell Mol Life Sci. 1982;38:120–2. doi: 10.1007/BF01944564. [DOI] [PubMed] [Google Scholar]
- 406.Yagi K, Ohtsuka K, Ohishi N. Lipid peroxidation caused by chinoform-ferric chelate in cultured neural retinal cells. Cell Mol Life Sci. 1985;41:1561–3. doi: 10.1007/BF01964807. [DOI] [PubMed] [Google Scholar]
- 407.Yassin MS, Ekblom J, Fberg C, Oreland L. Transmethylation Reactions and Autoradiographic Distribution of Vitamin B12: Effects of Clioquinol Treatment in Mice. Jpn J Pharmacol. 1998;78:55–61. doi: 10.1254/jjp.78.55. [DOI] [PubMed] [Google Scholar]
- 408.Yassin MS, Ekblom J, Xilinas M, Gottfries CG, Oreland L. Changes in uptake of vitamin B12 and trace metals in brains of mice treated with clioquinol. J Neurol Sci. 2000;173:40–4. doi: 10.1016/s0022-510x(99)00297-x. [DOI] [PubMed] [Google Scholar]
- 409.DiVaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P. Clioquinol, a drug for Alzheimer's disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg Chem. 2004;43:3795–7. doi: 10.1021/ic0494051. [DOI] [PubMed] [Google Scholar]
- 410.Wagner CC, Calvo S, Torre MH, Baran EJ. Vibrational spectra of clioquinol and its Cu(II) complex. J Raman Spec. 2007;38:373–6. [Google Scholar]
- 411.Ding W-Q, Liu B, Vaught JL, Palmiter RD, Lind SE. Clioquinol and docosa-hexaenoic acid act synergistically to kill tumor cells. Mol Cancer Ther. 2006;5:1864–72. doi: 10.1158/1535-7163.MCT-06-0067. [DOI] [PubMed] [Google Scholar]
- 412.Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, Multhaup G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer's disease. J Biol Chem. 2004;279:51958–64. doi: 10.1074/jbc.M407410200. [DOI] [PubMed] [Google Scholar]
- 413.Nitzan Y, Sekler I, Frederickson C, Coulter D, Balaji R, Liang S-L, Margulis A, Hershfinkel M, Silverman W. Clioquinol effects on tissue chelatable zinc in mice. J Mol Med. 2003;81:637–44. doi: 10.1007/s00109-003-0462-7. [DOI] [PubMed] [Google Scholar]
- 414.Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Aqueous dissolution of Alzheimer's disease Aβ amyloid deposits by biometal depletion. J Biol Chem. 1999;274:23223–8. doi: 10.1074/jbc.274.33.23223. [DOI] [PubMed] [Google Scholar]
- 415.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y-S, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–76. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 416.Raman B, Ban T, Yamaguchi K-i, Sakai M, Kawai T, Naiki H, Goto Y. Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid β peptide. J Biol Chem. 2005;280:16157–62. doi: 10.1074/jbc.M500309200. [DOI] [PubMed] [Google Scholar]
- 417.Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23:5088–95. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Benvenisti-Zarom L, Chen J, Regan RF. The oxidative neurotoxicity of clioquinol. Neuropharmacology. 2005;49:687–94. doi: 10.1016/j.neuropharm.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 419.Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sj'gren M, Wallin A, Xilinas M, Gottfries CG. Treatment of Alzheimer's disease with clioquinol. Dement Geriatr Cogn Disord. 2001;12:408–14. doi: 10.1159/000051288. [DOI] [PubMed] [Google Scholar]
- 420.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li Q-X, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 421.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li Q-X, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 422.Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW. Safety, efficacy and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–86. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 423.Arbiser JL, Kraeft SK, Van Leeuwen R, Hurwitz SJ, Selig M, Dickersin GR, Flint A, Byers HR, Chen LB. Clioquinol-zinc chelate: a candidate causative agent of subacute myelooptic neuropathy. Mol Med. 1998;4:665–70. [PMC free article] [PubMed] [Google Scholar]
- 424.Chen WH, Wang M, Yu SS, Su L, Zhu DM, She JQ, Cao XJ, Ruan DY. Clioquinol and vitamin B12 (cobalamin) synergistically rescue the lead-induced impairments of synaptic plasticity in hippocampal dentate gyrus area of the anesthetized rats in vivo. Neuroscience. 2007;147:853–64. doi: 10.1016/j.neuroscience.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 425.Choi SM, Choi K-O, Park Y-K, Cho H, Yang EG, Park H. Clioquinol, a Cu(II)/Zn(II) chelator, inhibits both ubiq-uitination and asparagine hydroxylation of hypoxia-inducible factor-1α, leading to expression of vascular endothelial growth factor and erythropoietin in normoxic cells. J Biol Chem. 2006;281:34056–63. doi: 10.1074/jbc.M603913200. [DOI] [PubMed] [Google Scholar]
- 426.Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Genetic or pharmacological iron chelation prevents mptp-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- 427.Masuda T, Hida H, Kanda Y, Aihara N, Ohta K, Yamada K, Nishino H. Oral administration of metal chelator ameliorates motor dysfunction after a small hemorrhage near the internal capsule in rat. J Neurosci Res. 2007;85:213–22. doi: 10.1002/jnr.21089. [DOI] [PubMed] [Google Scholar]
- 428.Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc Natl Acad Sci USA. 2005;102:11840–5. doi: 10.1073/pnas.0502177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Pollera C, Lucchini B, Formentin E, Bareggi S, Poli G, Ponti W. Evaluation of anti-prionic activity of clioquinol in an in vivo model (Mesocricetus auratus) Vet Res Commun. 2005;29:253–5. doi: 10.1007/s11259-005-0055-8. [DOI] [PubMed] [Google Scholar]
- 430.Ponti W, Sala M, Pollera C, Braida D, Poli G, Bareggi S. In vivo model for the evaluation of molecules active towards transmissible spongiform encephalopathies. Vet Res Commun. 2004;28:307–10. doi: 10.1023/b:verc.0000045433.45346.1c. [DOI] [PubMed] [Google Scholar]
- 431.Priel T, Aricha-Tamir B, Sekler I. Clioquinol attenuates zinc-dependent beta-cell death and the onset of insulitis and hyperglycemia associated with experimental type I diabetes in mice. Eur J Pharmacol. 2007;565:232–9. doi: 10.1016/j.ejphar.2007.02.064. [DOI] [PubMed] [Google Scholar]