

Abstract
Manufacturing procedures for cellular therapies are continuously improved with particular emphasis on product safety. We previously developed a dendritic cell (DC) cancer vaccine technology platform that uses clinical grade lipopolysaccharide (LPS) and interferon (IFN)-y for the maturation of monocyte derived DCs. DCs are frozen after 6 hrs exposure at a semi-mature stage (smDCs) retaining the capacity to secret interleukin (IL)-12 and thus support cytolytic T-cell responses, which is lost at full maturation. We compared closed systems for monocyte enrichment from leucocyte apheresis products from healthy individuals using plastic adherence, CD14 selection, or CD2/19 depletion with magnetic beads, or counter flow centrifugation (elutriation) using a clinical grade in comparison to a research grade culture medium for the following DC generation. We found that elutriation was superior compared to the other methods showing 36 ± 4% recovery, which was approximately 5-fold higher as the most frequently used adherence protocol (8 ± 1%), and a very good purity (92 ± 5%) of smDCs. Immune phenotype and IL-12 secretion (adherence: 1.4 ± 0.4; selection: 20 ± 0.6; depletion: 1 ±0.5; elutriation: 3.6 ± 1.5 ng/ml) as well as the potency of all DCs to stimulate T cells in an allogeneic mixed leucocyte reaction did not show statistically significant differences. Research grade and clinical grade DC culture media were equally potent and freezing did not impair the functions of smDCs. Finally, we assessed the functional capacity of DC cancer vaccines manufactured for three patients using this optimized procedure thereby demonstrating the feasibility of manufacturing DC cancer vaccines that secret IL-12 (9.4 ± 6.4 ng/ml). We conclude that significant steps were taken here towards clinical grade DC cancer vaccine manufacturing.
Keywords: cancer vaccination, good manufacturing practice, in-line monocyte enrichment, dendritic cell manufacturing, quality control
Introduction
Dendritic cell (DC)-based cancer vaccines are now widely explored for the treatment of cancer [1]. Regulatory authorities around the world are beginning to catch up with the biotechnology of such cellular therapies making them increasingly the subject of government regulation. Consequently, good manufacturing practice (GMP) standards have become mandatory for the clinical development of DC-based cancer vaccines. Advances made in the development of clinical grade technologies for collecting cells or manipulating them in vitro contribute toward the compliance of GMP standards in the manufacturing of DCs for cancer vaccination.
Although DCs for cancer vaccination may be generated form haematopoietic stem cells [2], they are more commonly differentiated from peripheral blood monocytes by cultivation in interleukin (IL)-4 and granulocyte-macrophage colony-stimulation factor (GM-CSF) supplemented medium [3]. The initial step of DC manufacturing is the collection of peripheral blood mononuclear cells (PBMC) from a leucocyte apheresis product. From the PBMCs, monocytes may be enriched by several techniques. Classically, the capacity of monocytes to adhere to plastic surfaces was employed to separate them from non-adherent lymphocytes [4]. More sophisticated technologies utilize magnetic beads coated with either anti-CD14 for the selection of monocytes [5, 6] or a combination of anti-CD2 and anti-CD19 antibodies for the depletion of T and B lymphocyte from monocytes [5, 7, 8]. More recently an elutriation procedure was introduced for monocyte enrichment from leucocyte apheresis products, which is based on a counter-flow centrifugation principle [9–12]. All three technologies are semi-automated in-line procedures using clinical grade material. It seems therefore a reasonable and timely measure for manufacturers of DC cancer vaccines to start implementing one of these advanced monocyte enrichment technologies.
Other aspects of DC manufacturing for clinical application concern the choice of a clinical grade culture medium that omits the use of bovine serum or the selection of a suitable maturation stimulus. In the majority of the recently conducted DC cancer vaccine trials, the DCs were exposed to a cytokine cocktail comprised of tumour necrosis factor (TNF)-α, prostaglandin (PG) -E2, IL-1 β and IL-6 [3]. Such DCs lack the capacity for IL-12 secretion and thus for effective type I polarization of an immune response that ultimately leads to the support of cytolytic immunity [13]. We, thus have developed a DC cancer vaccine technology platform that uses clinical grade lipopolysaccharide (LPS) and interferon (IFN)-γ for DC maturation and has the capacity to trigger the release of IL-12 [14, 15]. However, IL-12 secretion ceases 24 hrs after exposure to LPS/IFN-γ[16]. Thus, a hallmark of our DC cancer vaccine technology is that we apply DCs as soon as 6 hrs after LPS/IFN-γ activation enabling the release of IL-12 during DC/T-cell interaction [17].
In a series of four clinical pilot trials for DC cancer vaccination we have so far treated more than 50 patients with paediatric and adult malignancies using a conventional protocol [17]. The improvement in the enrichment of monocytes using elutriation, the successful implementation of a clinical grade DC culture medium, as well as optimizations of the LPS/IFN-γ maturation using a clinical grade LPS preparation certified by the US Pharmacopeia for the use in human beings [18] prompted us to change our DC manufacturing standard protocol also in our clinical trials. Analysing the first three DC cancer vaccines for patients indicated that the modified procedure was also suitable for patients resulting in the generation of DCs that fulfilled all quality as well as potency criteria.
Material and methods
Leucocyte apheresis
Leucocytes were collected using an Amicus leucocyte apheresis device (Baxter, Deerfield, IL, USA) from healthy volunteers and patients suffering from various neoplasias treated in the context of clinical trials that were approved by the responsible institutions review boards. All individuals gave their informed consent to these studies according to the World Medical Association Declaration of Helsinki. Cell numbers and subsets were determined on a Sysmex cell counter (Sysmex, Bornbarch, Germany) and/or by flow cytometry.
Monocyte enrichment
Monocytes were enriched by plastic adherence as described previously [17] using AIM-V (Invitrogen, Carlsbad, CA, USA) supplemented with 1% human pooled AB plasma (Octaplas, Octapharma, Vienna, Austria) or CellGro medium (CellGenix, Freiburg, Germany). For the in-line procedures, we followed the instructions provided by the manufacturers. Using the Elutra cell separator (Gambro BCT, Lakewood, CO, USA), monocytes were enriched from the leucocyte apheresis product by loading into the elutriation chamber while maintaining the centrifuge speed at 2400 rpm. Thereafter, the centrifuge speed and the flow of elutriation media (PBS/HSA Baxter, New Jersey, NJ, USA) were held constant for cell fractionation. Selection of monocytes was done with the CliniMACS cell selection system (Miltenyi, Bergisch Gladbach, Germany) that uses CD14-coated magnetic beads to retain monocytes in a magnetic column. Depletion of T and B lymphocytes for the enrichment of monocytes was done using the Isolex 300i Magnetic Cell Selector (Nexell, Irvine, CA, USA). Lymphocytes were retained in a magnetic column by connecting them to CD2 and CD19 coated magnetic beads and collecting the flow through. All semi-automated monocyte enrichment procedures are designed to process a full leucocyte apheresis product. Each monocyte enrichment procedure was performed with freshly isolated leucocytes.
Flow cytometry
Leucocyte apheresis and monocyte enrichment products were analysed for total leucocytes, T lymphocytes, B lymphocytes, monocytes and granulocytes by antibody labelling with anti-CD45-FITC, anti-CD3-PerCP, anti-CD19-APC, anti-CD14-APC and anti-CD15-FITC (BD Pharmingen San Diego, CA, USA), respectively, using the Trucount system (Becton Dickinson, New Jersey, NJ, USA). Labelled cells were analysed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA).
DC manufacturing
We followed a previously optimized protocol for the generation of DCs [5] but exchanged research grade material against clinical grade culture reagents and supplements. Monocytes isolated by the respective enrichment procedures described above were cultured at a density of 1 × 106 monocytes/cm2 either in AIM-V medium supplemented with 2% pooled human AB plasma or in CellGro DC medium at 37°C in a humidified incubator for 6 days. Based on optimization and validation experiments, the culture media AIM-V/2%OP or CellGro were supplemented with 1000 U/ml human GM-CSF and 300 U/ml human IL-4 (both from CellGenix Freiburg, Germany) and replaced with the same volume and units of medium plus GM-CSF and IL-4 on day 3. On day 6, the DC differentiation culture was supplemented with 50 ng/ml IFN-γ (Boehringer Ingelheim Vienna, Austria) and lipopolysaccharide (LPS, E. coli strain O111: B4 Calbiochem, San Diego, CA, USA), ranging from 1 to 1000 ng/ml, and cultivated for 6 hrs to generate semi-mature (sm) DCs that were subsequently frozen; patient's DC vaccines were manufactured with clinical grade LPS (US Pharmacopeia, Bethesda, MD, USA). Excess LPS was removed using three washing steps and subsequent freezing of the DC cancer vaccine. The biological effect of LPS on the DCs was quality controlled by DC immune phenotyping, IL-12 secretion and an alloMLR (see below). After discussion and in agreement with European (EMEA) and Austrian (AGES PharmMed) regulators, no further assessment of the amount of LPS or recombinant cytokines from the differentiation culture as well as the multitude of molecules other than IL-12 released by the DCs into the final product was done.
DC immune phenotyping
The maturation status of the DCs was determined using the following antibodies: anti-CD86-APC (BD Pharmingen, San Diego, CA, USA), anti-CD80-PE (Immunotech, Beckman Coulter, Fullerton, CA, USA), anti-CD83-APC (all three from BD Pharmingen, San Diego, CA, USA), anti-MHC I-PE, anti-MHC II-FITC (both from Dako Cytomation, Carpinteria, CA, USA) and anti-CD45-PerCP (BD Pharmingen, San Diego, CA, USA). The viability of the DCs was measured by propidium iodide staining (Sigma, St. Louis, MO, USA). Cells were analysed using a FACS Calibur flow cytometer. The appropriate isotype control antibodies were included in the analysis.
IL-12 detection by ELISA
IL-12 concentrations in the supernatant of the DC cultures were measured as described previously [17].
Allogeneic mixed leucocyte reactions
Stimulating DCs (10,000, 2000 or 400) were placed in triplicates with 105 allogeneic responder PBMCs in 200 μl AIM-V medium supplemented with 2% pooled human plasma on a 96-well round bottom plate. For a positive reference, 105 responder cells were stimulated with 100 ng/ml staphylococcal enterotoxin A/B (SEA/SEB, Toxin Technologies Inc., Sarasota, FL USA). On day 4, the co-culture was incubated for another 18 hrs with 1 μCi of tritium thymidine solution (NEN Life Science Products, Boston, MA, USA). Finally, the cells were harvested with a Skatron harvester (Lier Norway). The incorporated tritium thymidine was counted using a Trilux β-plate reader (Wallac Oy, Turku, Finland). Alternatively, allogeneic PBMCs were labelled with Calboxy-fluorescein diacetate, succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR, USA) and mixed with DCs in a ratio of 1/5, 1/10, 1/20, 1/40 and 1/80. For the controls, no DCs or SEA/SEB was added. Finally, the PBMCs were labelled with anti-CD3-PerCP and analysed using a FACS Calibur flow cytometer. The percentage of CD3 positive CFSE negative T lymphocytes was determined.
Statistics
Statistical significance of all results was calculated by use of an unpaired Student's t-test. Results are given in mean ± standard error of mean (SEM).
Results
Manufacturing of LPS/IFN-/-activated dendritic cell-based cancer vaccines
We have established a standard operating procedure (SOP) for the manufacturing of a cancer vaccine based on monocyte-derived DCs that are pulsed with tumour antigen followed by activation with LPS in the presence of IFN-γ (Fig. 1A). This SOP is the result of the experiments described in this paper that have been performed over a time period of 3 years including preliminary optimization and validation experiments. Most extensively we optimized the isolation of monocytes by comparing adherence with the semi-automated methods, elutriation, selection and depletion procedures using research and clinical grade media (Fig. 1B).
1.
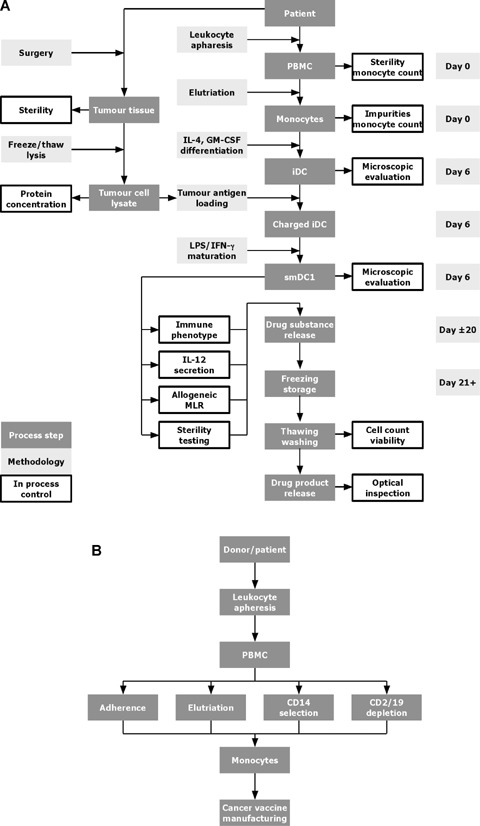
Dendritic cell (DC) manufacturing. (A) Flow chart of the standard operating procedure (SOP) for the manufacturing of a cancer vaccine. From patients undergoing tumour surgery a piece of tumour tissue is delivered to the manufacturing facility. The tumour tissue is disrupted mechanically, and the tumour cells are lysed to enrich soluble protein containing tumour antigens. After recovery from surgery, leucocyte apheresis is performed to collect peripheral blood mononuclear cells (PBMCs) from the patients, the monocytes are enriched and cultivated for 6 days in the presence of interleukin (IL)-4 and granulocyte-macrophage colony-stimulation factor (GM-CSF) in order to obtain iDCs. The iDCs are charged with tumour antigens, exposed to LPS/IFN-γ to trigger maturation and cryopreserved until treatment. An aliquot of the DC cancer vaccine is subjected to quality control, a potency assay and sterility control. If all criteria are met the DC cancer vaccine is released for treatment. (B) Flow chart of DC manufacturing using different monocyte enrichment protocols. Monocytes are isolated from PBMCs of a healthy donor or cancer patient using leucocyte apheresis. The PBMCs are further subjected to monocyte enrichment using adherence, or semi-automated elutriation, CD14 selection, or CD2/19 depletion. The differentiation into DCs is done in the same way independently of the enrichment procedure used (see Fig. 1A).
Leucocyte apheresis
We analysed leucocyte apheresis products from 11 selected healthy donors and three patients suffering from neoplastic diseases (Fig. 2). The three patients received a DC cancer vaccine, manufactured according to Fig. 1, in the course of a clinical trial that was approved by the institutional review board of the Medical University Vienna. The leucocyte apheresis products contained 4–10 × 109 PBMCs that included 0.6–3 × 109 monocytes for further enrichment. The products of healthy volunteers contained 71 ± 3% lymphocytes, 19 ±3% monocytes and 5.6 ±1.4% granulocytes, of patient donors 57 ± 3%, 29 ± 3% and 7.5 ± 1.8%, respectively.
2.
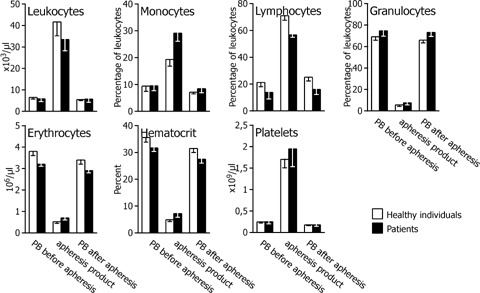
Cell composition of leucocyte apheresis products. The leucocyte apheresis product as well as peripheral blood (PB) before and immediately after leucocyte apheresis from healthy individuals (n= 11) and patients (n= 3) was analysed for the leucocyte number and distribution (monocytes, lymphocytes, granulocytes), erythrocyte number, haematocrit and platelet number. Percentage and cell number are given as mean ± SD. Comparisons of whole leucocyte apheresis products as well as peripheral blood before and after leucocyte apheresis from healthy and patient donors show no statistically significant differences.
Recovery and purity of monocytes after enrichment
The percentage of monocytes in the leucocyte apheresis products from healthy donors before enrichment was similar for all enrichment procedures. Among the enrichment procedures, illustrated in Fig. 3, we isolated monocytes with a mean recovery for selection, depletion and elutriation of 59 ± 4%, 41 ± 3% and 87 ± 7%, respectively. The purity was found to be 96 ± 2% of leucocytes for selection, and 61 ± 4% and 82 ± 3% for depletion and elutriation, respectively. Product analysis could not be performed for monocytes enriched by adhesion without disrupting the culture.
3.
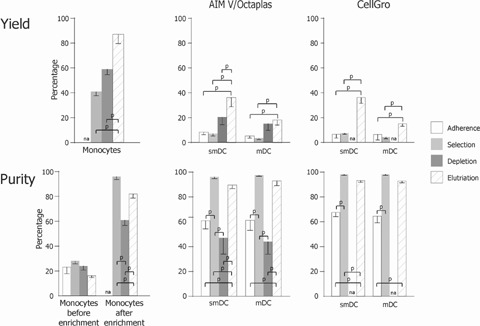
Recovery and Purity of LPS/IFN-γ activated-DCs manufactured by using different monocyte enrichment protocols. Six hours (smDCs) and 48 hrs (mDCs) LPS/IFN-γ-activated iDCs were generated using monocytes from healthy individuals. Monocytes were enriched from leucocyte apheresis products by plastic adherence, CD14 selection, CD2/CD19 depletion or elutriation (as indicated) using AIM V/Octaplas or clinical grade CellGro DC Medium. Upper: The mean percentage ± SEM of recovery of monocytes after enrichment, smDCs or mDCs, as indicated, is given relative to monocytes in the leucocyte apheresis product. Lower: The mean percentage ± SEM of monocytes before and after enrichment, smDCs and mDCs, as indicated, is given relative to the total number of leucocytes. Number of independent DC preparations in AIM-V/Octaplas medium: adherence, n= 10; selection, n= 5; depletion, n= 9; elutriation, n= 7; and CellGro DC medium: adherence, n= 5, selection, n= 3; depletion, n= 2; elutriation, n= 10. na, no data available; p, P < 0.01.
Recovery and purity of semi-mature dendritic cells after 6 hrs of LPS/IFN-γ activation
Next, we analysed smDCs differentiated from monocytes followed by 6 hrs DC maturation using LPS/IFN-γ. In our cancer vaccine design, smDCs are frozen in liquid nitrogen at this stage and applied to the patients immediately after thawing and washing (Fig. 1A). We therefore froze the smDCs and measured recovery and purity after thawing (Fig. 3). Mean recovery of adherence, selection, depletion and elutriation was 7 ± 1%, 8 ± 1%, 22 ± 4% and 36 ± 4%, respectively. Mean purity in the same sequence as above was 63 ± 14%, 97 ± 1%, 49 ± 9% and 92 ± 5% smDCs of leucocytes. No differences in yield and purity of smDCs were found when comparing research or clinical grade medium for all monocyte enrichment procedures.
Recovery and purity of mature dendritic cells after 48 hrs of LPS/IFN-γ activation
We re-cultivated smDCs recovered from freezing for 2 days and determined recovery and purity (Fig. 3). Mean recovery of adherence, selection, depletion and elutriation was 6 ± 2%, 4 ± 1%, 15 ± 3% and 16 ± 2%, respectively. Mean purity in the same sequence as above was 62 ± 5%, 97 ± 0%, 42 ± 8% and 93 ± 2% mDCs of leucocytes, respectively. Research and clinical grade medium was equally suitable for manufacturing of mDCs from any of the monocyte enrichment procedures.
DC quality and potency control
According to the quality control protocol (Fig. 1A), we determined the immune phenotype by analysing the expression density of typical DC membrane molecules, IL-12 secretion and the support of an alloMLR during differentiation from smDCs into mDCs. MHC I and MHC II molecules, CD80 and CD86 co-stimulatory molecules of the B7-family and CD83, a DC differentiation marker, were up-regulated, and IL-12 secretion was induced independently of the culture medium or selection procedure (adherence: 1.4 ± 0.4; selection: 2.0 ± 0.6; depletion: 1 ± 0.5; elutriation: 3.6 ± 15 ng/ml; Fig. 4A and B). All DCs manufactured from monocytes enriched by the various procedures had a comparable capacity for T lymphocyte activation in an alloMLR (Fig. 4C).
4.
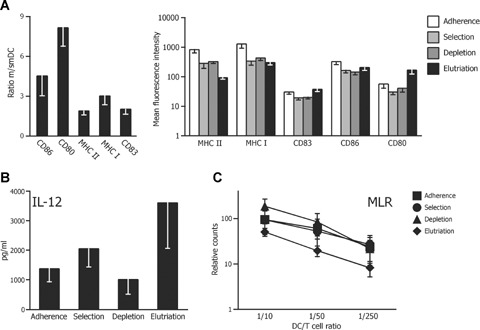
DC quality control. Monocytes from healthy individuals were enriched from leucocyte apheresis products by different enrichment procedures, as indicated, differentiated into DCs followed by activation with LPS/IFN-γ. DC preparations using AIM-V/Octaplas or CellGro DC medium are combined. (A) The left-hand panel shows the ratio of increase in the expression density ± SEM of the indicated DC maturation markers measured 6 hrs (smDCs) and 48 hrs (mDC) after activation. DCs generated from elutriated monocytes (number of preparations, n= 35) are analysed. The right-hand panel compares the expression density mean fluorescence intensity (MFI) ± SEM of maturation markers measured on mDCs that are generated from monocytes isolated by the indicated enrichment procedures. MFIs of the isotype controls are below 5 (data not shown). (B) Secretion of IL-12 ± SEM secreted from mDCs analysed in (A) is illustrated. (C) Proliferation of allogeneic PBMCs in co-cultures with the DCs analysed in (A) is given relative to PBMCs stimulated with the super-antigen staphylococcal enterotoxin A/B (SEA/SEB) (normalized to 100%) and the background proliferation of unstimulated PBMCs (normalized to 0%). Number of preparations in (B) and (C): adherence, n= 10; selection, n= 8; depletion, n= 11, elutriation, n= 35. Comparisons of the enrichment procedures in (A) to (C) show no statistically significant differences.
Optimization of conditions for LPS/IFN-γ maturation
Based on this comparative evaluation of the different monocyte enrichment procedures, we implemented elutriation and the clinical grade medium in the SOP for the manufacturing of our DC cancer vaccine (Fig. 1A). In order to confirm that this procedure is suitable for the manufacturing of an effective cancer vaccine, we further optimized the maturation with LPS/IFN-γ together with the DC freezing step after 6 hrs (Fig. 5). This represents the cancer vaccine, which allows the secretion of IL-12 after administration to the patients. We observed up-regulation of maturation-associated DC membrane molecules as well as IL-12 secretion at LPS concentrations of 10–1000 ng/ml. The viability of DCs at those concentrations was higher than 80%. Freezing after 6 hrs of maturation did not affect the surface expression of DC maturation markers, IL-12 secretion and the viability of the DCs.
5.
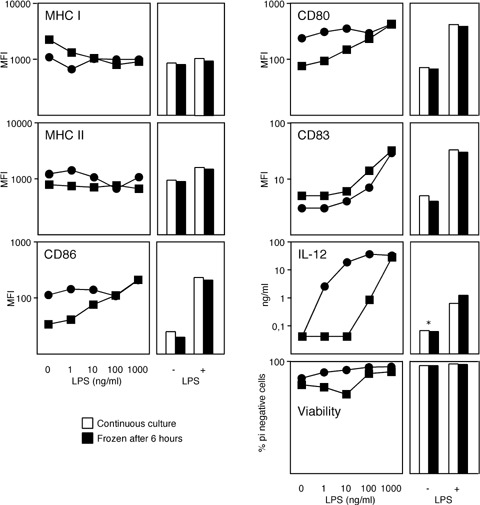
Optimization of DC maturation. Left-hand panels: Expression density (MFI) of the indicated DC membrane molecules and viability of mDCs as well as IL-12 secretion during the differentiation of smDCs into mDCs is shown after exposure to increasing concentrations of lipopolysaccharide (LPS) at constant IFN-γ concentration. Data from two healthy donors are shown. Right-hand panel: LPS at a concentration of 30 ng/ml was applied for 6 hrs (+) or the DCs were left immature (–) in the presence of IFN-β. According to the DC-manufacturing protocol for cancer vaccines, DC cultures were frozen after 6 hrs in liquid nitrogen, recovered from freezing and re-cultured; or DCs were continuously cultured until hour = 48 as indicated. * Below the detection limit of the IL-12 ELISA.
Feasibility of DC cancer vaccine manufacturing for patients
We have so far treated more than 50 patients in four DC cancer vaccine pilot trials. In these trials we used the conventional adherence procedure to enrich patient's monocytes from leucocyte apheresis products. Based on the data above, we manufactured cancer vaccines for three patients following the novel SOP implementing elutriation and clinical grade DC medium (Fig. 1A). We measured a recovery of 29 ± 7% of smDCs with a mean purity of 88 ± 1% (Fig. 6). The viability of smDCs and mDCs differentiated during the quality control was above 80%. From all three patients we obtained mDCs with a typical immune phenotype, IL-12 (9.4 ± 6.4 ng/ml) secretion and satisfactory stimulatory capacity for allogeneic T lymphocytes meeting our quality criteria.
6.
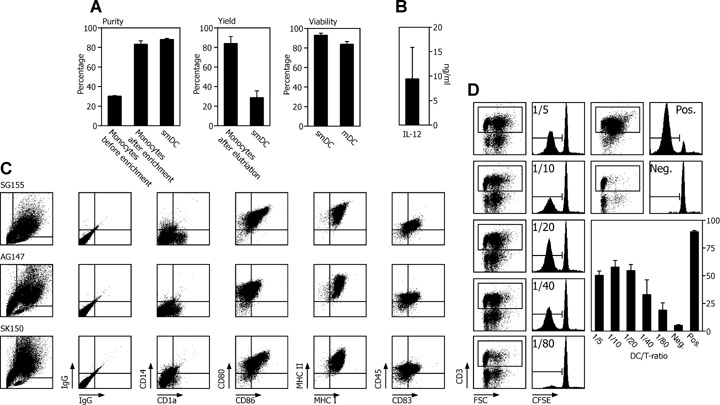
Validation of the DC manufacturing process for three patients. DC quality control of cancer vaccines from three cancer patients using monocytes enriched by elutriation was performed according to the flow chart shown in Fig. 1A. For DC activation, 30 ng/ml LPS was used. (A) Purity, yield and viability ± SEM was assessed for monocytes, smDCs and mDCs as indicated. (B) Mean ± SEM of IL-12 secreted from three vaccines. (C) Immune phenotype measuring the expression density of the depicted DC membrane molecules for three patients. (D) AlloMLR using CFSE dilution was performed as potency assay for the stimulatory capacity of smDCs at the indicated DC/PBMC ratios. The dot plots illustrate the gating for proliferating CD3 expressing T lymphocytes that lost CFSE due to proliferation as shown in the histograms. As a negative control we used un-stimulated PBMCs, as positive control PBMCs exposed to the super-antigen SEA/SEB (one representative experiment of three is given). The bar graph shows the mean ± SEM of the percentage of proliferating allogeneic T lymphocytes co-cultured with DCs from three patients.
Discussion
DC-based cancer vaccines have the potential to become an independent module in the treatment of tumours supplementing the standard treatment regime that includes surgery, chemotherapy and radiation therapy [1]. At the verge of widespread clinical application, it is mandatory that the safety of such pharmaceuticals is beyond doubt and that the manufacturing protocols guarantee the quality of the DC cancer vaccines. During the last years, we have continuously worked on the improvement of DC manufacturing using LPS/IFN-γ as the activation stimulus [5, 14, 15, 17, 19]. By comparing different monocyte enrichment procedures in this study using leucocyte apheresis products from healthy individuals, we designed a new SOP for the manufacturing of clinically applied DCs implementing elutriation as the most effective enrichment procedure and a clinical grade culture medium that did not impair DC quality. The feasibility of our newly established protocol was confirmed by the manufacturing of LPS/IFN-γ-activated smDCs for the treatment of three cancer patients. The DCs complied with quality and potency criteria that have been defined according to previous in vitro studies [14, 15]. The culture conditions for monocyte differentiation into DCs are well established and allow only minor modifications [20]. A more critical step in the manufacturing of DC cancer vaccines is the enrichment of monocytes from leucocyte apheresis products [5]. Classically adherence to plastic surfaces was used to separate monocytes from other PBMCs [4]. This method has clear advantages by being easy to perform and by not requiring additional laboratory equipment. However, it is an open method that makes it a weak link in the chain of GMP manufacturing. Hence, technologies for in-line monocyte enrichment were developed based on two major principles: magnetic cell sorting using monoclonal antibodies and counter flow elutriation based on cell density [9–12]. Using monoclonal antibodies two strategies are feasible: enrichment of monocytes using magnetic bead-coupled monoclonal antibodies selecting CD14 expressing monocytes [5, 6]; or depleting CD2 or CD19 expressing T- and B lymphocytes from PBMCs [5, 7, 8]. All three techniques may be performed as an in-line procedure, which is a significant step towards GMP compliance and hence product safety. The immune phenotype, IL-12 secretion, and the stimulatory capacity of the DCs obtained by all methods were similar (Fig. 4). There may be a tendency of lower IL-12 secretion when DCs where manufactured from monocytes enriched by adherence, selection or depletion, that because of considerable individual differences did not reach statistical significance. However, DCs manufactured from monocytes enriched by any of the four enrichment procedures conferred full functional capacity to DCs. Significant differences were observed in the purity and recovery of DCs (Fig. 3), identifying elutriation as the most favourable method for the manufacturing of DCs according to our technical requirements. Elutriation yielded the highest recovery of monocytes with purity only slightly lower as obtained with CD14 selection, the method that resulted in the purest monocyte population but had very poor recovery. Of note, contaminating cells after elutriation were mainly granulocytes, which do not survive the culture period or the freezing step (data not shown). This mainly explains the increase in purity from elutriated monocytes to differentiated mDCs. Using the other methods the main contaminating cells were lymphocytes, which survive in the culture and thus remain as contaminants in the final cancer vaccine product. CD2/CD19 depletion showed a considerably lower recovery of mDCs compared to elutriation. CD2 is expressed by monocyte/DC subsets in the peripheral blood, which might contribute to the poor results of the depletion method [21]. In addition, DCs generated with depleted monocytes showed lymphocyte contamination similar to DCs manufactured from monocytes enriched by adherence (data not shown).
For the manufacturing of the DC cancer vaccine described in this paper, we elected to measure the immune phenotype and the secretion of IL-12 as quality criteria 2 days after re-cultivation of a thawed aliquot of 6 hrs LPS/IFN-γ matured smDCs. The stimulatory capacity of the smDCs in an alloMLR is used as potency assay. The 2 days’ re-cultivation is intended to mimic the situation after inoculation of the smDCs into the patient's lymph node where the final differentiation step from smDCs to mDCs is assumed to occur. We could demonstrate that freezing of smDCs generated with elutriated monocytes did not impair the immune phenotype, IL-12 secretion or the viability of DCs also at high LPS concentrations up to 1000 ng/ml. Having the option of giving high LPS concentrations is critical as we observed high variations concerning LPS responsiveness in different donors (Fig. 5). For the manufacturing of a DC cancer vaccine for three patients according to a revised SOP (Fig. 1A), we used a mean LPS concentration of 30 ng/ml which induced a mature phenotype meeting our quality criteria (Fig. 6).
After discussion and in agreement with European (EMEA) and Austrian (AGES PharmMed) regulators we implied additional quality tests showing the absence of bacteria, mycoplasma and selected viruses. Obviously, there are many more features that are critically important for the quality and function of DCs such as the ability to present specific antigens, the ability to cross-present exogenous antigens on MHC class I, the nature of the cytokines produced by T lymphocytes co-cultured with the DCs or the migratory ability of DCs. However, we have shown previously that DCs matured by exposure to LPS/IFN-γ are characterized by a clearly defined phenotype, most importantly IL-12 secretion, and have the capacity for the presentation of exogenous antigens to MHC class I restricted cytotoxic T lymphocytes. Also, T lymphocytes co-cultivated with LPS/IFN-γ-activated DCs enabled for IL-12 production, secrete IFN-γ, which is a critical indicator of type I polarization [14, 22]. The migratory capacity of DCs receiving a strong maturation stimulus in vitro is considerably limited without additional treatment with PG E2 [14, 22–24]. Thus, we chose the intranodal application route in order to complement for this deficiency of LPS/IFN-γ-activated DCs [17]. In general, limitations of patient's material as well as time constraints in a clinical cancer vaccination setting preclude extensive quality control emphasizing the necessity of controlling carefully selected DC features that reflect their stimulatory potential aimed for in clinical applications.
The majority of clinical DC cancer vaccine trials were performed using monocyte-derived DCs. However, other methods are available, one of which is the manufacturing of DCs from haematopoietic stem cells [2]. A direct comparison of the specific features of monocyte versus stem cell-derived DCs as well as their respective potential to induce anti-tumour immunity is not available but would benefit the design of DC cancer vaccines. Mouse models are not suitable to answer this question, as it is not reasonably possible to collect sufficient amounts of peripheral blood monocytes for DC manufacturing and thus mouse models typically use stem cell-derived DCs. Another possibility for the design of a DC cancer vaccine is the direct collection of DCs from peripheral blood [25, 26]. Such peripheral blood DCs expanded with Flt3 ligand represent potent antigen presenting cells to prime CD8+ T cells [27]. Although DCs may be readily isolated from mouse spleen, it is not clear how such DCs compare to human peripheral blood DCs. Thus, the utility of peripheral blood DCs in the design of a cancer vaccine is currently uncertain.
More and more cell-based medicinal products enter the mainstream of clinical research and development. Thus, it becomes increasingly important that safety and quality issues are addressed in the manufacturing of such medicines. For DC manufacturing, a multitude of protocols has been used [20]. Here, only selected steps in the highly complex procedure of DC cancer vaccine manufacturing, which, however, we consider of critical importance, were analysed in detail. Other important aspects include DC maturation using toll-like receptor ligands, cytokine cocktails or T lymphocyte-derived signals. A maturation stimulus more commonly used than LPS/IFN-γ for the manufacturing of DC-based cancer vaccines is a cocktail comprised of TNF-α, PG-E2, IL-1 β and IL-6. Recovery as well as purity of DCs was not significantly different when we compared this cytokine cocktail and LPS/IFN-γ to trigger maturation (data not shown). Further aspects of cancer vaccine manufacturing include the cellular source: haematopoietic stem cells or peripheral blood monocytes; antigen loading: synthetic peptides, recombinant proteins, tumour cell material, RNA or DNA vectors; the route of administration: intradermally subcutaneously, intravenously, intranodally or combinations thereof. In order to reach the ultimate goal of demonstrating clinical benefit of DC-based cancer vaccination, many of these variables may need to be studied and their respective impact on the treatment procedure evaluated. Also, in order to conduct meta-analysis of different DC cancer vaccine trials, it may be important to define standards for DC manufacturing to facilitate comparisons between clinical studies. With this paper, we attempt a first step towards that goal by showing the superiority of elutriation over other procedures for the enrichment of monocytes in the manufacturing of DC-based cancer vaccines; the evaluation of a clinical grade culture medium; the optimization of a DC maturation procedure; and the development of quality and potency assays for DC cancer vaccines.
Acknowledgments
This work was supported by grants from the Vienna Business Agency and the Austrian Research Promotion Fund. Trimed Biotech GmbH is a subsidiary of AOP Orphan Pharmaceuticals AG that also provides private equity funding for Trimed.
Author information
AM Dohnal, C Eichstill, D Wagner, D Wimmer and T Felzmann are employees of Trimed and thus declare competing financial interests.
References
- 1.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, Pineiro L, Steinman R, Fay J. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61:6451–8. [PubMed] [Google Scholar]
- 3.Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, Grabbe S, Rittgen W, Edler L, Sucker A, Zimpfer-Rechner C, Berger T, Kamarashev J, Burg G, Jonuleit H, Tuttenberg A, Becker JC, Keikavoussi P, Kampgen E, Schuler G. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17:563–70. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]
- 4.Vakkila J, Lehtonen E, Koskimies S, Hurme M. Dendritic cells in human peripheral blood: effective enrichment from the nonadherent cells. Immunol Lett. 1987;15:229–36. doi: 10.1016/0165-2478(87)90029-0. [DOI] [PubMed] [Google Scholar]
- 5.Felzmann T, Witt V, Wimmer D, Ressmann G, Wagner D, Paul P, Huttner K, Fritsch G. Monocyte enrichment from leukapharesis products for the generation of DCs by plastic adherence, or by positive or negative selection. Cytotherapy. 2003;5:391–8. doi: 10.1080/14653240310003053. [DOI] [PubMed] [Google Scholar]
- 6.Padley DJ, Dietz AB, Gastineau DA, Vuk-Pavlovic S. Mature myeloid dendritic cells for clinical use prepared from CD14+ cells isolated by immunomagnetic adsorption. J Hematother Stem Cell Res. 2001;10:427–9. doi: 10.1089/152581601750289037. [DOI] [PubMed] [Google Scholar]
- 7.Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from non-proliferating progenitors in human blood. J Immunol Methods. 1996;196:121–35. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 8.Suen Y, Lee SM, Aono F, Hou S, Loudovaris M, Ofstein G, Bender JG. Comparison of monocyte enrichment by immuno-magnetic depletion or adherence for the clinical-scale generation of DC. Cytotherapy. 2001;3:365–75. doi: 10.1080/146532401753277184. [DOI] [PubMed] [Google Scholar]
- 9.Berger TG, Strasser E, Smith R, Carste C, Schuler-Thurner B, Kaempgen E, Schuler G. Efficient elutriation of monocytes within a closed system (Elutra) for clinical-scale generation of dendritic cells. J Immunol Methods. 2005;298:61–72. doi: 10.1016/j.jim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Erdmann M, Dorrie J, Schaft N, Strasser E, Hendelmeier M, Kampgen E, Schuler G, Schuler-Thurner B. Effective clinical-scale production of dendritic cell vaccines by monocyte elutriation directly in medium, subsequent culture in bags and final antigen loading using peptides or RNA transfection. J Immunother. 2007;30:663–74. doi: 10.1097/CJI.0b013e3180ca7cd6. [DOI] [PubMed] [Google Scholar]
- 11.Lemarie C, Sugaye R, Kaur I, Taga T, Chabannon C, Schuyler R, McMannis J. Purification of monocytes from cryopre-served mobilized apheresis products by elutriation with the Elutra device. J Immunol Methods. 2007;318:30–6. doi: 10.1016/j.jim.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 12.Ten Brinke A, Karsten ML, Dieker MC, Zwaginga JJ, Vrielink H, Marieke van Ham S. Generation of dendritic cells for immunotherapy is minimally impaired by granulocytes in the monocyte preparation. Immunobiology. 2006;211:633–40. doi: 10.1016/j.imbio.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 13.De Jong EC, Smits HH, Kapsenberg ML. Dendritic cell-mediated T cell polarization. Springer Semin Immunopathol. 2005;26:289–307. doi: 10.1007/s00281-004-0167-1. [DOI] [PubMed] [Google Scholar]
- 14.Felzmann T, Huttner KG, Breuer SK, Wimmer D, Ressmann G, Wagner D, Paul P, Lehner M, Heitger A, Holter W. Semi-mature IL-12 secreting dendritic cells present exogenous antigen to trigger cytolytic immune responses. Cancer Immunol Immunother. 2005;54:769–80. doi: 10.1007/s00262-004-0637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hüttner KG, Breuer SK, Paul P, Majdic O, Heitger A, Felzmann T. Generation of potent anti-tumor immunity in mice by interleukin-12-secreting dendritic cells. Cancer Immunol Immunother. 2005;54:67–77. doi: 10.1007/s00262-004-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1:3116. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 17.Dohnal A, Witt V, Hugel H, Holter W, Gadner H, Felzmann T. Phase I study of tumor Ag-loaded IL-12 secreting semi-mature DC for the treatment of pediatric cancer. Cytotherapy. 2007;9:755–70. doi: 10.1080/14653240701589221. [DOI] [PubMed] [Google Scholar]
- 18.Suffredini AF, Hochstein HD, McMahon FG. Dose-related inflammatory effects of intravenous endotoxin in humans: evaluation of a new clinical lot of Escherichia coli O: 113 endotoxin. J Infect Dis. 1999;179:1278–82. doi: 10.1086/314717. [DOI] [PubMed] [Google Scholar]
- 19.Felzmann T, Buchberger M, Lehner M, Printz D, Kircheis R, Wagner E, Gadner H, Holter W. Functional maturation of dendritic cells by exposure to CD40L transgenic tumor cells, fibroblasts or keratinocytes. Cancer Letters. 2001;168:145–54. doi: 10.1016/s0304-3835(01)00526-2. [DOI] [PubMed] [Google Scholar]
- 20.Felzmann T, Gadner H, Holter W. Dendritic cells as adjuvants in antitumor immune therapy. Onkologie. 2002;25:456–64. doi: 10.1159/000067441. [DOI] [PubMed] [Google Scholar]
- 21.Crawford K, Gabuzda D, Pantazopoulos V, Xu J, Clement C, Reinherz E, Alper CA. Circulating CD2+ monocytes are dendritic cells. J Immunol. 1999;163:5920–8. [PubMed] [Google Scholar]
- 22.Dohnal AM, Inthal A, Felzmann T, Glatt S, Sommergruber W, Mann G, Gadner H, Panzer-Grumayer ER. Leukemia-associated antigenic isoforms induce a specific immune response in children with T-ALL. Int J Cancer. 2006;119:2870–7. doi: 10.1002/ijc.22224. [DOI] [PubMed] [Google Scholar]
- 23.Luft T, Maraskovsky E, Schnurr M, Knebel K, Kirsch M, Gorner M, Skoda R, Ho AD, Nawroth P, Bierhaus A. Tuning the volume of the immune response: strength and persistence of stimulation determine migration and cytokine secretion of dendritic cells. Blood. 2004;104:1066–74. doi: 10.1182/blood-2003-12-4146. [DOI] [PubMed] [Google Scholar]
- 24.Jefford M, Schnurr M, Toy T, Masterman KA, Shin A, Beecroft T, Tai TY, Shortman K, Shackleton M, Davis ID, Parente P, Luft T, Chen W, Cebon J, Maraskovsky E. Functional comparison of DCs generated in vivo with Flt3 ligand or in vitro from blood monocytes: differential regulation of function by specific classes of physiologic stimuli. Blood. 2003;102:1753–63. doi: 10.1182/blood-2002-12-3854. [DOI] [PubMed] [Google Scholar]
- 25.Thomas R, Lipsky PE. Human peripheral blood dendritic cell subsets Isolation and characterization of precursor and mature antigen-presenting cells. J Immunol. 1994;153:4016–28. [PubMed] [Google Scholar]
- 26.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, Van Beckhoven A, Liles TM, Engleman EG, Levy R. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99:1517–26. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 27.Davis ID, Chen Q, Morris L, Quirk J, Stanley M, Tavarnesi ML, Parente P, Cavicchiolo T, Hopkins W, Jackson H, Dimopoulos N, Tai TY, MacGregor D, Browning J, Svobodova S, Caron D, Maraskovsky E, Old LJ, Chen W, Cebon J. Blood dendritic cells generated with Flt3 lig-and and CD40 ligand prime CD8+ T cells efficiently in cancer patients. J Immunother. 2006;29:499–511. doi: 10.1097/01.cji.0000211299.29632.8c. [DOI] [PubMed] [Google Scholar]