

Abstract
In individuals with inflammatory bowel diseases, intestinal fibrosis is a serious clinical complication with no specific therapies. Patients develop bowel fistulae and strictures that usually require surgery and often reoccur. The main driver of gut fibrogenesis is believed to be chronic inflammation, which leads to mesenchymal cell recruitment and activation. Recent findings suggest that the environment—in particular the microbiome—plays a critical role in this process.
Crohn’s disease (CD) and ulcerative colitis (UC) are lifelong relapsing and remitting inflammatory conditions of the gastrointestinal tract. Both forms of inflammatory bowel disease (IBD) are highly heterogeneous. The vast majority of CD patients present with inflammation of the gut at diagnosis. The disease eventually progresses to include complications, such as fistulae or strictures, in about 70% of patients (1). More than 30% of CD patients develop fibrosis-induced intestinal obstruction, with debilitating symptoms such as severe abdominal pain, nausea, vomiting and inability to pass stool (1). These individuals often ultimately require surgery, but inflammation and re-stenosis commonly reoccur (2).
Although immunosuppressants such as azathioprine (AZA)/6-mercaptopurine and biologics [antibodies to tumor necrosis factor (TNF)-α, for example] can control intestinal inflammation (3), there are no specific antifibrotic therapies. Although new preliminary data suggest that early, prolonged use of immunosuppressive or biologic agents can reduce the need for surgery and hospitalizations (4), we need a mechanistic understanding of intestinal fibrosis to develop preventive and therapeutic approaches.
Intestinal fibrogenesis
Fibrosis is defined as an excessive accumulation of extracellular matrix (ECM) that ultimately leads to organ dysfunction, resulting from chronic tissue damage, impaired wound healing, and an underlying expansion of mesenchymal cells (5). These events occur in both forms of IBD but in distinct locations: in UC the colonic mucosa and submucosa are affected, while in CD fibrosis extends through the entire thickness of the small or large bowel wall, including the muscularis propria and serosa (6). Intestinal fibrosis in IBD is a considered a classic chronic inflammation-driven process, with the inflammation being much more severe than in fibrosis of the kidney, liver, or lung (5, 7). The ultimate outcome of the disease depends on the severity, duration and location of inflammation: The intestine has a remarkable ability to fully regenerate after short-lived insults—such as infectious enteritis, acute peptic ulcer disease, or mild diverticulitis—but it cannot do so in the face of unrelenting, severe inflammation. When repair mechanisms are continuously activated, scar tissue forms a stricture and eventually the bowel is obstructed.
The main effector cell for fibrosis in all organs, including the intestine, is the mesenchymal cell, which exists in three distinct but interrelated forms: the fibroblast, the myofibroblast, and the smooth muscle cell (5) (collectively called fibroblasts here). In health, fibroblasts in the intestinal wall produce small amounts of ECM (collagen and fibronectin). In response to injury or inflammation, fibroblasts become highly versatile repair units that rapidly increase in numbers, migrating up a gradient of growth factors from a multitude of sources inside and outside of the intestine (Fig. 1). These cells are exposed to a complex mix of immune cells, non-immune cells and their products, most of which directly or indirectly activate fibroblasts to produce excess ECM and, under pathological conditions, fibrosis (Fig. 2; Table 1).
Figure 1.
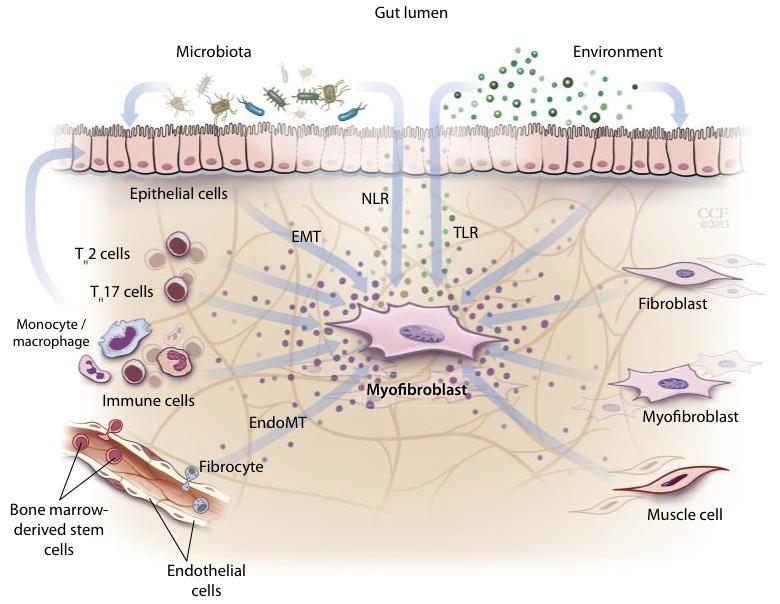
Sources of fibroblasts in inflammatory bowel disease-associated fibrosis. Fibroblast precursors can derive from intestinal stellate cells (6) or from fully differentiated epithelial or endothelial cells under inflammatory pressure [epithelial- (42) or endothelial-to-mesenchymal transition (EMT) (43)]. Fibrocytes, circulating fibroblast precursors, which express both hematopoietic (CD14, CD45) and mesenchymal (CD34, Col I) cell markers (44), as well as bone marrow stem cells (which replenish intestinal fibroblasts) are recruited to inflamed or injured intestine (6). The role of pericytes, the main origin of fibroblasts in kidney fibrosis (5), has not been investigated in intestinal stricture formation.
Table 1.
Profibrotic Immune- and Non-Immune Cell Factors in the Intestine
| Growth factors | Transforming growth factor (TGF)-β1 Platelet derived growth factor (PDGF) Insulin-like growth factor (IGF)-1 Epidermal growth factor (EGF) Basic fibroblast growth factor (bFGF) |
(45–51) |
| Cytokines | Interleukin (IL)-1 IL-4 IL-13 IL-17 |
(52–55) |
| Chemokines | Monocyte chemotactic protein (MCP)-1 Macrophage inflammatory protein (MIP)-1α MIP-1β MIP-3α |
(56, 57) |
| Bacterial products | Toll-like receptor ligands Nod-like receptor ligands |
(17, 18, 58) |
| Matrix molecules | Hyaluronan Laminins Collagens |
(59, 60) |
Environmental factors in intestinal fibrosis
Environmental factors inside and outside the gut can trigger or ameliorate IBD pathogenesis (8), although the relevant agents remain poorly characterized. Probable exogenous agents include pollution, diet, lifestyle, drugs, psychosocial stress, infections, and xenobiotics. Endogenous influences include the gut microbiome and secondary products of inflammation and metabolism.
Microbiome-host interaction
Most human-associated microbes are found in the gut, where they influence health and disease. Alterations in the immune system, the barrier function of the intestine or the composition of the microflora can upset the host-microbiome interaction and lead to inflammation. Essentially all intestinal immune (dendritic cells, macrophages, lymphocyte etc.) and non-immune cells (epithelial cells, fibroblasts, endothelial cells, etc.) sense microbe-derived, pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors, including Toll-like receptors (TLRs) and Nod-like receptors (NLRs) (9). Recognition of microbial molecules by these receptors induces innate and adaptive immune responses. Each intestinal cell type can have a distinct response to the same pattern recognition receptors, and different receptors can elicit the same response in a given cell [a common response is the activation of nuclear factor kB (NFkB)] (9).
Pattern recognition receptors and mesenchymal cells
Fibroblasts express multiple TLRs and NLRs (10–12) that, once activated, drive the cell towards a proinflammatory or pro-fibrotic phenotype (13, 14). Although these pathways have been extensively studied in the intestine, the link between the microbiota and fibrogenesis has been predominantly explored in the liver.
Via TLR4, the microbe-derived ligand lipopolysaccaride (LPS) in the portal vein or translocation of bacteria or their products to the liver promotes fibrogenesis (15, 16). Blocking TLR4 signaling in mice or reducing liver exposure to intestinal microbes by decontaminating the gut with antibiotics inhibits experimental liver fibrosis (12). Hepatic stellate cells (HSC), which are closely related to intestinal fibroblasts, carry all known human TLRs, are activated by TLR4 ligands, and thereby mediate these fibrotic effects (11, 12). TLR4 ligands also indirectly contribute to fibrogenesis by rendering HSC more susceptible to TGF-β1 through downregulation of BAMBI, a TGF-β1 decoy receptor (12). By analogy, the fibroblasts of the intestine likely initiate TLR-mediated pro-fibrogenic events in the inflamed gut.
Translocation of PAMPs through the gut epithelium brings them in direct contact with the so-called subepithelial myofibroblasts located immediately below the basement membrane. Intestinal fibroblasts express TLR1 to 9 and Nod1 and 2 (17, 18). Exposure of human intestinal fibroblasts to live bacteria (E. coli or S. aureus) activates NFkB, causes subsequent secretion of cytokines and chemokines, and enhances fibroblast contraction, another fibroblast function possibly important for the luminal narrowing of the intestine in stricture formation. Several TLRs could mediate this effect (13, 17): Ligands to TLR4 (predominantly from gram-negative bacteria) or TLR2 (predominantly from gram-positive bacteria) activate NFkB and cause cytokine and chemokine secretion in intestinal fibroblasts (14, 17), and the TLR5 ligand flagellin (present in all flagellated bacteria – gram-positive and -negative) induces a pro-inflammatory and pro-fibrotic phenotype in primary human intestinal fibroblasts (18). In addition, PAMPs can also expand and recruit intestinal fibroblasts: TLR5 ligands promote fibroblast cell cycle entry and proliferation (19), and colonic epithelial cells undergo epithelial-to-mesenchymal transition after ligation of the TLR4 variant D299G (20).
Microbe-induced intestinal fibrosis in animal models
In several animal models, microbes initiate or perpetuate gut inflammation and fibrosis: (i) The spontaneous, senescence-accelerated SAMP1/YitFc mouse exhibits chronic CD-like ileitis in the absence of chemical, immunologic, or genetic manipulations, but intestinal inflammation is attenuated under pathogen-free conditions. (ii) IL-10 knock out mice show no inflammation under pathogen-free conditions, but develop intestinal inflammation upon transfer to regular housing conditions. (iii) In chemically induced, colitis in mice, the epithelial barrier is breached and colonic proteins modified, activating a delayed-type hypersensitivity reaction; inflammation is reduced after treatment with antibiotics (21). Additionally, microbial products can be directly pro-fibrogenic: Injection of a bacterial cell wall component into rat bowel wall induces inflammation that eventually evolves to fibrosis (22). Injection of fecal material or anaerobic bacteria into autologous animals (23) has the same effect. In the above described models, fibrogenesis is accompanied by an increase in classical pro-fibrotic mediators, such as TGF-β1, in the mucosa and can be inhibited by antibiotic treatment. Blockade of TGF-β1 reduces intestinal collagen deposition, showing that microbial components bypassing the intestinal barrier (e.g. by barrier dysfunction or injection) triggers inflammation and fibrosis, and that TGF-β1 is a key pro-fibrotic mediator.
The microbiota in human gut fibrogenesis
These in vitro findings and animal studies indicate that microbes and their products can be pro-fibrogenic in the gut. But what do these results mean for understanding and treatment of fibrosis in IBD?
Human genetic studies can be informative about gut microbiota participation in fibrogenesis. Gene variants that affect innate immunity, located in or near genes involved in bacterial recognition and processing, are genetically linked to IBD and CD (24). These variants are best exemplified by polymorphisms of the nucleotide-binding oligomerization domain-containing protein 2/caspase recruitment domain-containing protein 15 (NOD2/CARD15) gene, an intracellular pattern recognition receptor that binds muramyl dipeptide, a cell wall component of many gram-positive and -negative bacteria. Loss of function of NOD2 enhances inflammatory responses. CD patients carrying the most prevalent of these variants (Arg702Trp, Gly908Arg and the frameshift mutation Leu1007insC) are at increased risk for complications such as bowel obstruction and the need for surgery (25). The risk of these complications increases proportionally with the number of variants carried by the patient (26). The low prevalence and incomplete penetrance of these variants make them unsuitable as biomarkers of disease outcome, but understanding how they lead to exacerbated disease will be informative.
CD is associated with shifts in the composition of the enteric microbiota, so-called dysbiosis. These shifts include an overall reduced diversity of the microflora plus a relative depletion of the phyla Firmicutes and Bacteroidetes and increased frequencies of Actinobacteria and Proteobacteria, as compared to healthy individuals (27, 28). The loss of Firmicutes is largely a result of fewer species of the order clostridiales (27). Possibly related is the fact that host genetic variants, such as NOD2/CARD15 or autophagy-related protein 16-like 1 (ATG16L1), are also linked to shifts in microbial composition. The clinical CD phenotype, too, including age at surgical intervention, is associated with changes in Faecalibacterium and Escherichia abundance (27). These hints point toward a relationship between the composition of the microbial community and the course of IBD, but it remains to be determined which is the cause and which the consequence.
Circulating antibodies against microbial peptides are found in some patients with IBD and are believed to arise from an immune response towards the luminal microbiota. These antibodies are qualitatively and quantitatively associated with and predictive of a more complicated disease phenotype; a higher percentage of patients with more severe CD carry these antibodies than do those with milder forms (29).
Food and other environmental agents
Non-microbial factors, such as gadolinium, cigarette smoking, alcohol or iron induce or exacerbate organ fibrosis in, for example, the skin or liver (30, 31). Because the intestine is in constant contact with numerous exogenous factors, this is likely also to be the case for the intestine. A link between the environment and gut biology may be the aryl-hydrocarbon receptor (AhR), a promiscuous ligand-dependent transcription factor that controls the expression of cytochrome P450 genes (32). AhR agonists are found among a variety of environmental factors, such as food components---extracts from vegetables (broccoli, cauliflower, and cabbage), fruits (apple), herbs (ginseng, ginkgo biloba, and black cohosh) and pesticides used in agriculture (cyprodinil), but also include aromatic hydrocarbons such as the toxin dioxin. Endogenous ligands to the AhR have been described as well (32).
The AhR controls diverse processes (cell proliferation, differentiation, migration, etc), and has been suggested to have anti-fibrotic properties: Among these is inhibition of TGF-β1-induced myofibroblast differentiation and ECM production in primary human lung, orbital and corneal fibroblasts in vitro (33). AhR activation also exerts an anti-fibrotic effect in experimental bile duct, liver and heart fibrosis (34, 35). Despite the fact that the AhR ligand dioxin is a component of cigarette smoke and smoking has been linked to more severe CD courses, AhR activation reduces the clinical and histologic severity of experimental colitis (whether induced by chemical agents or T-cell transfer) in rodents (36, 37). It remains to be determined, if specific AhR ligands exert distinct effects, such as suppression of inflammation and fibrosis. Diet, intestinal microbiota and AhR appear to be functionally linked: AhR KO animals harbor a distinct gut flora with increased colonization by Bacteroides spp., a finding that can be reproduced by depriving wild type mice of AhR-activating food components (37). Together, data from in vitro and in vivo experiments (36, 37) suggest that activation of the AhR can protect against intestinal inflammation and fibrogenesis, making this environment-sensing receptor a potential target for anti-fibrotic therapy in IBD.
Obstacles and unknowns
Several key fundamental questions remain, the most of which important is: What are the early triggers that lead to intestinal fibrosis in IBD? No other organ is exposed to as many endogenous and exogenous factors as the gut and in no other organ is the relation between the environment and the immune system as intimate. The multitude of substances bathing the intestinal walls makes it very possible that several distinct agents or mechanisms can cause IBD and subsequent fibrosis and that intestinal fibrosis should not be seen as driven only by inflammation, but as the result of a complex interplay between exogenous agents, immune and non-immune cells.
A second open issue is that we do not understand the natural history of fibrostenosing IBD, especially with respect to the gut microbiome. Is the abnormal bacterial composition of the gut microbiota in patients with IBD a primary or secondary phenomenon? The microbes found in a patient with established IBD may not be the same as the organisms that triggered the initial events.
Indeed, intestinal fibrosis, once initiated, may be self-perpetuating. In mice with intestinal fibrosis triggered by Salmonella, the early blockade of inflammation prevents fibrosis, but with continuing inflammation fibrosis becomes progressive – even if inflammation is brought under control (38). This observation at the bench fits well with human clinical trials indicating that early suppression of intestinal inflammation can better prevent the later development of complications than treatment of established disease (4, 39). This may be because the quality of IBD inflammation changes over time, from an acute proinflammatory type to a chronic profibrotic type (40). Additionally, ECM accumulation leads to an increased tissue stiffness that can, by itself, drive fibrogenesis through integrin-mediated fibroblast activation (41). Further studies will clarify whether the timing of therapeutic intervention is likely to be as important as the precise type of the anti-fibrotic intervention.
A further difficulty is that no commonly accepted histopathological or clinical definition of intestinal fibrosis exists. This makes testing of new anti-fibrotic therapies difficult, as it limits efficient inclusion of patients in clinical trials as well as the choice of relevant endpoints. Further, there are no reliable biomarkers to monitor fibrostenosing IBD.
Clinicians still see the development of fibrotic strictures as unavoidable and accept endoscopic or surgical interventions as part of the routine management of patients with complicated IBD. Even so, the notion that fibrosis is inevitable and irreversible has been challenged by demonstrations that established fibrosis in the liver can regress (5). By unraveling the steps leading to fibrotic pathology in the intestines of IBD patients, we may reveal targets for therapeutic agents that can cause reversal and healing of intestinal fibrosis in IBD patients. Interventions could include restoration of intestinal barrier integrity or the use of selective TLR and NLR antagonists to reduce the effective exposure of fibroblasts to microbial and environmental factors.
Highlights.
Intestinal fibrosis is a frequent, serious sequela of IBD that has no specific treatment available.
Intestinal fibrosis shares many of its molecular features with other forms of fibrosis, but the gut microbiota and contents may play a particularly prominent and unique role.
In intestinal fibrosis, molecules with pathogen-associated molecular patterns cross the gut epithelial barrier and trigger an inflammatory and ultimately fibrotic response.
The early triggers for intestinal fibrosis in IBD and its natural history are not well understood and require further research.
Acknowledgments
Funding: F.R. is supported by NIH-T32 1T32DK083251-01A1, Digestive Disease Institute, Cleveland Clinic Foundation, Cleveland, USA.
Footnotes
Competing interests: The author receives consulting fees from GA Generic Assays GmbH, Germany
References
- 1.Cosnes J, Nion-Larmurier I, Beaugerie L, Afchain P, Tiret E, Gendre JP. Impact of the increasing use of immunosuppressants in Crohn’s disease on the need for intestinal surgery. Gut. 2005;54:237–241. doi: 10.1136/gut.2004.045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farmer RG, Whelan G, Fazio VW. Long-term follow-up of patients with Crohn’s disease. Relationship between the clinical pattern and prognosis. Gastroenterology. 1985;88:1818–1825. doi: 10.1016/0016-5085(85)90006-x. [DOI] [PubMed] [Google Scholar]
- 3.Armuzzi A, Van Assche G, Reinisch W, Pineton de Chambrun G, Griffiths A, Sladek M, Preiss JC, Lukas M, D’Haens G. Results of the 2nd scientific workshop of the ECCO (IV): therapeutic strategies to enhance intestinal healing in inflammatory bowel disease. Journal of Crohn’s & colitis. 2012;6:492–502. doi: 10.1016/j.crohns.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Peyrin-Biroulet L, Oussalah A, Williet N, Pillot C, Bresler L, Bigard MA. Impact of azathioprine and tumour necrosis factor antagonists on the need for surgery in newly diagnosed Crohn’s disease. Gut. 2011;60:930–936. doi: 10.1136/gut.2010.227884. [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Science translational medicine. 2013;5:167sr161. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 6.Rieder F, Fiocchi C. Intestinal fibrosis in IBD--a dynamic, multifactorial process. Nature reviews. 2009;6:228–235. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 7.Rieder F, Brenmoehl J, Leeb S, Scholmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130–139. doi: 10.1136/gut.2006.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rappaport SM. Discovering environmental causes of disease. Journal of epidemiology and community health. 2012;66:99–102. doi: 10.1136/jech-2011-200726. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Uehara A, Takada H. Functional TLRs and NODs in human gingival fibroblasts. J Dent Res. 2007;86:249–254. doi: 10.1177/154405910708600310. [DOI] [PubMed] [Google Scholar]
- 11.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 12.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature medicine. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki H, Kobayashi R, Ishikawa H, Awano N, Yamagoe S, Miyazaki Y, Matsumoto T. Activation of COL1A2 promoter in human fibroblasts by Escherichia coli. FEMS Immunol Med Microbiol. 2012;65:481–487. doi: 10.1111/j.1574-695X.2012.00979.x. [DOI] [PubMed] [Google Scholar]
- 14.Burke JP, Cunningham MF, Watson RW, Docherty NG, Coffey JC, O’Connell PR. Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br J Surg. 2010;97:1126–1134. doi: 10.1002/bjs.7045. [DOI] [PubMed] [Google Scholar]
- 15.Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, Hsu WC, Huang CC, Wang SS, Lo KJ. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22:165–172. doi: 10.1016/0168-8278(95)80424-2. [DOI] [PubMed] [Google Scholar]
- 16.Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY, Li CP, Chu CJ, Lu RH, Lee SD. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scandinavian journal of gastroenterology. 1997;32:942–946. doi: 10.3109/00365529709011206. [DOI] [PubMed] [Google Scholar]
- 17.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 18.Rieder F, Bhilocha S, Schirbel A, Ouyang Z, West G, Atreja A, Rho H, De La Motte C, Fiocchi C. Activation of Toll-Like Receptor (TLR) 5 Induces a Pro-Fibrogenic Phenotype on Human Intestinal Myofibroblasts (HIF) - A Novel Pathway Mediated by Caspase 1 (Abstract) Gastroenterology. 2011;142:S116. [Google Scholar]
- 19.Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. The Journal of biological chemistry. 2005;280:20620–20627. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 20.Eyking A, Ey B, Runzi M, Roig AI, Reis H, Schmid KW, Gerken G, Podolsky DK, Cario E. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology. 2012;141:2154–2165. doi: 10.1053/j.gastro.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rieder F, Kessler S, Sans M, Fiocchi C. Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. American journal of physiology. 2012;303:G786–801. doi: 10.1152/ajpgi.00059.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pucilowska JB, Williams KL, Lund PK. Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. American journal of physiology. 2000;279:G653–659. doi: 10.1152/ajpgi.2000.279.4.G653. [DOI] [PubMed] [Google Scholar]
- 23.Mourelle M, Salas A, Guarner F, Crespo E, Garcia-Lafuente A, Malagelada JR. Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology. 1998;114:519–526. doi: 10.1016/s0016-5085(98)70535-9. [DOI] [PubMed] [Google Scholar]
- 24.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D’Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak-Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D’Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol. 2011;106:699–712. doi: 10.1038/ajg.2011.19. [DOI] [PubMed] [Google Scholar]
- 26.Weersma RK, Stokkers PC, Cleynen I, Wolfkamp SC, Henckaerts L, Schreiber S, Dijkstra G, Franke A, Nolte IM, Rutgeerts P, Wijmenga C, Vermeire S. Confirmation of multiple Crohn’s disease susceptibility loci in a large Dutch-Belgian cohort. Am J Gastroenterol. 2009;104:630–638. doi: 10.1038/ajg.2008.112. [DOI] [PubMed] [Google Scholar]
- 27.Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, Pace NR, Li E. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis. 2008;14:858–867. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 29.Rieder F, Lawrance IC, Leite A, Sans M. Predictors of fibrostenotic Crohn’s disease. Inflamm Bowel Dis. 2011;17:2000–2007. doi: 10.1002/ibd.21627. [DOI] [PubMed] [Google Scholar]
- 30.Swaminathan S, Shah SV. New insights into nephrogenic systemic fibrosis. J Am Soc Nephrol. 2007;18:2636–2643. doi: 10.1681/ASN.2007060645. [DOI] [PubMed] [Google Scholar]
- 31.Wood MJ, Powell LW, Ramm GA. Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood. 2008;111:4456–4462. doi: 10.1182/blood-2007-11-122374. [DOI] [PubMed] [Google Scholar]
- 32.Barouki R, Coumoul X, Fernandez-Salguero PM. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007;581:3608–3615. doi: 10.1016/j.febslet.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 33.Lehmann GM, Xi X, Kulkarni AA, Olsen KC, Pollock SJ, Baglole CJ, Gupta S, Casey AE, Huxlin KR, Sime PJ, Feldon SE, Phipps RP. The aryl hydrocarbon receptor ligand ITE inhibits TGFbeta1-induced human myofibroblast differentiation. The American journal of pathology. 2011;178:1556–1567. doi: 10.1016/j.ajpath.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson TC, Hodgson P, Fernandez-Salguero P, Neumeister M, Gonzalez FJ. Hepatic fibrosis and cytochrome P450: experimental models of fibrosis compared to AHR knockout mice. Hepatol Res. 2000;17:112–125. doi: 10.1016/s1386-6346(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 35.Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–268. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- 36.Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F, Monteleone G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237–248. 248, e231. doi: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–640. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 38.Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. 2012;18:460–471. doi: 10.1002/ibd.21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feagan BG, Panaccione R, Sandborn WJ, D’Haens GR, Schreiber S, Rutgeerts PJ, Loftus EV, Jr, Lomax KG, Yu AP, Wu EQ, Chao J, Mulani P. Effects of adalimumab therapy on incidence of hospitalization and surgery in Crohn’s disease: results from the CHARM study. Gastroenterology. 2008;135:1493–1499. doi: 10.1053/j.gastro.2008.07.069. [DOI] [PubMed] [Google Scholar]
- 40.Kugathasan S, Saubermann LJ, Smith L, Kou D, Itoh J, Binion DG, Levine AD, Blumberg RS, Fiocchi C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 42.Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. The Journal of biological chemistry. 2010;285:20202–20212. doi: 10.1074/jbc.M110.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. The American journal of pathology. 2012;179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uehara H, Nakagawa T, Katsuno T, Sato T, Isono A, Noguchi Y, Saito Y. Emergence of fibrocytes showing morphological changes in the inflamed colonic mucosa. Dig Dis Sci. 2010;55:253–260. doi: 10.1007/s10620-009-0730-7. [DOI] [PubMed] [Google Scholar]
- 45.Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis. 2001;7:226–236. doi: 10.1097/00054725-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Dammeier J, Brauchle M, Falk W, Grotendorst GR, Werner S. Connective tissue growth factor: a novel regulator of mucosal repair and fibrosis in inflammatory bowel disease? The international journal of biochemistry & cell biology. 1998;30:909–922. doi: 10.1016/s1357-2725(98)00046-6. [DOI] [PubMed] [Google Scholar]
- 47.Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology. 1996;110:975–984. doi: 10.1053/gast.1996.v110.pm8613031. [DOI] [PubMed] [Google Scholar]
- 48.Lawrance IC, Maxwell L, Doe W. Inflammation location, but not type, determines the increase in TGF-beta1 and IGF-1 expression and collagen deposition in IBD intestine. Inflamm Bowel Dis. 2001;7:16–26. doi: 10.1097/00054725-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Kumagai S, Ohtani H, Nagai T, Funa K, Hiwatashi NO, Shimosegawa, Nagura H. Platelet-derived growth factor and its receptors are expressed in areas of both active inflammation and active fibrosis in inflammatory bowel disease. Tohoku J Exp Med. 2001;195:21–33. doi: 10.1620/tjem.195.21. [DOI] [PubMed] [Google Scholar]
- 50.Eivindson M, Gronbaek H, Flyvbjerg A, Frystyk J, Zimmermann-Nielsen E, Dahlerup JF. The insulin-like growth factor (IGF)-system in active ulcerative colitis and Crohn’s disease: relations to disease activity and corticosteroid treatment. Growth Horm IGF Res. 2007;17:33–40. doi: 10.1016/j.ghir.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Hoffmann P, Reinshagen M, Zeeh JM, Lakshmanan J, Wu VS, Goebell H, Gerken G, Eysselein VE. Increased expression of epidermal growth factor-receptor in an experimental model of colitis in rats. Scandinavian journal of gastroenterology. 2000;35:1174–1180. doi: 10.1080/003655200750056655. [DOI] [PubMed] [Google Scholar]
- 52.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nature medicine. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 53.Spencer DM, Veldman GM, Banerjee S, Willis J, Levine AD. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology. 2002;122:94–105. doi: 10.1053/gast.2002.30308. [DOI] [PubMed] [Google Scholar]
- 54.Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, Thompson RC, Dinarello CA. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. The Journal of clinical investigation. 1990;86:972–980. doi: 10.1172/JCI114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fichtner-Feigl S, Strober W, Geissler EK, Schlitt HJ. Cytokines mediating the induction of chronic colitis and colitis-associated fibrosis. Mucosal immunology. 2008;1(Suppl 1):S24–27. doi: 10.1038/mi.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motomura Y, Khan WI, El-Sharkawy RT, Verma-Gandhu M, Verdu EF, Gauldie J, Collins SM. Induction of a fibrogenic response in mouse colon by overexpression of monocyte chemoattractant protein 1. Gut. 2006;55:662–670. doi: 10.1136/gut.2005.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papadakis KA, Targan SR. The role of chemokines and chemokine receptors in mucosal inflammation. Inflamm Bowel Dis. 2000;6:303–313. doi: 10.1002/ibd.3780060408. [DOI] [PubMed] [Google Scholar]
- 58.Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. 2008;134:768–780. doi: 10.1053/j.gastro.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 59.Vaday GG, Lider O. Extracellular matrix moieties, cytokines, and enzymes: dynamic effects on immune cell behavior and inflammation. Journal of leukocyte biology. 2000;67:149–159. doi: 10.1002/jlb.67.2.149. [DOI] [PubMed] [Google Scholar]
- 60.Schor H, Vaday GG, Lider O. Modulation of leukocyte behavior by an inflamed extracellular matrix. Developmental immunology. 2000;7:227–238. doi: 10.1155/2000/51902. [DOI] [PMC free article] [PubMed] [Google Scholar]