

Abstract
Measuring biomarkers to identify and assess illness is a strategy growing in popularity and relevance. Although already in clinical use for treating and predicting cancer, no biological measurement is used clinically for any psychiatric disorder. Biomarkers could predict the course of a medical problem, and aid in determining how and when to treat. Several studies have indicated that of candidate psychiatric biomarkers detected using proteomic techniques, cholesterol and associated proteins, specifically apolipoproteins (Apos), may be of interest. Cholesterol is necessary for brain development and its synthesis continues at a lower rate in the adult brain. Apos are the protein component of lipoproteins responsible for lipid transport. There is extensive evidence that the levels of cholesterol and Apos may be disturbed in psychiatric disorders, including autistic spectrum disorders (ASD). Here, we describe putative serum biomarkers for psychiatric disorders, and the role of cholesterol and Apos in central nervous system (CNS) disorders.
Keywords: apolipoprotein, Apos, autism, biomarker, cholesterol, proteomics
Introduction
Measuring biomarkers to predict or identify illness is a strategy that is growing in popularity and relevance. Although the term biomarker has been used permissively in recent years, the United States Food and Drug Administration (FDA) strictly defines a biomarker as an objective measurement of a normal biological process, a pathological biological process or an objective measurement that indicates pharmacological response to a therapeutic (reviewed by [1]). Although biomarkers are already in clinical use for treating and predicting diseases such as cancer [[2],[3],,[4]], only candidate biomarkers are currently under study in the field of psychiatry. No true biomarker is currently used clinically to diagnose or identify any psychiatric disorder, despite proposals that biological changes underlie or contribute to many psychiatric syndromes. Biomarkers could lend a special significance to the field of psychiatry [5,6]. Many psychiatric disorders have an unclear cause and biomarkers could help to clarify the aetiology of psychiatric problems, or confirm that individuals categorized with the same disorder in fact have distinct syndromes with identical symptoms. They could aid in helping to predict the course of a disorder, and in determining how and when to treat. Perhaps based on this promise, interest in putative psychiatric biomarkers has recently spiked, with over 200 scientific articles published on this topic in 2008 according to one literature search, compared with no articles in 2002 and very few in previous years [6].
A potential molecular biomarker may consist of a difference in a measurable biochemical parameter, either intracellular or extracellular, between physiological and pathological states. Candidate biomarkers may be genes and their RNA transcription products, proteins and their post-translational modification or even higher order structures such as cholesterol lipid rafts and protein complexes or small molecular weight metabolites. Therefore, virtually almost any intracellular or extracellular molecule from the body may be a molecular biomarker candidate if changes in its levels are associated with a disease or disorder. Most researchers recognize biomarkers as an actual molecule or molecules, but biomarkers can consist of other characteristics. Although not under discussion in the present review, patterns of brain activity (such as those identified by functional magnetic resonance imaging, single photon emission computed tomography or electroencephalography), have also been explored as potential psychiatric biomarkers [7,8]. One molecular biomarker, if used for diagnostic and/or prognostic purposes, may not have the strongest statistical power by itself. Therefore, the current trend in molecular biomarker research leans towards identifying a signature of a set of biomarkers.
Candidate genomic biomarkers have been found for several psychiatric problems [9,10,11,12], often confirming the heritability of many disorders that were previously implicated based on familial patterns. The role of specific gene mutations and their interaction with environmental pressures to ultimately cause a psychiatric problem has been a topic of several studies [13,14,15,16]. However, although gene information is useful, genomic information does not necessarily reflect active protein levels [17] or tell the researcher about possible important post-translational modifications such as glycosylation, phosphorylation or formation/destruction of disulphide bridges to keep/disturb the protein's three-dimensional structure [18]. Such additional proteomic information may expand possibilities for a disorder's identification in clinical tests. Proteins represent the functional molecules in a biological system; therefore, study of proteins may take a researcher closer to identifying the cause of a disorder and could also suggest targets for therapeutics. Protein profiling of serum candidate biomarkers in psychiatry is therefore an area of great interest and one with great potential.
Methods for proteomic analysis
Sample fractionation/biochemical fractionation
Protein profiling in psychiatric research often involves examining proteins in blood sera or plasma, cerebrospinal fluid (CSF), saliva or urine. Sometimes, brain can be analysed if post-mortem or biopsied tissue is available. Blood sera are generally the most common fluid used. Advantages of protein profiling over genomic profiling include the possibility that levels of protein may alter along with the severity of the disorder or particular symptoms of the disorder. Protein levels could therefore be monitored not simply to determine disease presence but also severity and to gauge response to treatment. Several systems have been used to study proteins including Western blotting, ELISA, immunocytochemistry, protein chips and proteomics. Proteomics in particular may be an advantageous approach for understanding psychiatric disorders, as proteomic methods are relatively unbiased and reveal changed proteins without the need for a priori hypotheses [19,20]. In addition, proteomic techniques, when customized for particular needs, can theoretically identify any protein that is altered relative to healthy controls, whereas other techniques enable the measurement of only one or few pre-specified proteins [21,22,23,24,25]. Based on the complex nature of psychiatric disorders, one might expect the need to monitor multiple rather than single proteins [26].
A proteomics experiment usually involves biochemical fractionation of proteins and their identification by mass spectrometry (MS) [27,28,29,30,31,32,33]. The biochemical fractionation is performed by most researchers using electrophoresis and chromatography. These methods separate proteins according to their intrinsic (isoelectric point, molecular mass, hydrophobicity, etc.) and extrinsic (external charge induced by the buffer, detergent or dye) properties. Examples of electrophoresis methods include sodium dodecyl-sulphate polyacrylamide gel electrophoresis (SDS-PAGE), Tricine-PAGE, and Blue-Native PAGE. In addition to electrophoresis, chromatography is a choice for protein separation. Among them, ion exchange chromatography, size exclusion chromatography, hydrophobic interactions (or normal and reversed phase) chromatography and affinity chromatography are the most used methods. Given the wide range of choices for biochemical separation of proteins, protein complexes and peptides, countless possibilities for analyses exist. Once the proteins and peptides are biochemically fractionated, they are analysed by MS. In MS, the sample is ionized in the ionization source; then it travels through a mass analyser according to the mass over charge and then hits the detector, where the spectra are recorded.
MS
MS analyses of proteins and peptides are usually performed using Matrix Assisted Laser Desorption Ionization Mass Spectrometry (MALDI-MS) and/or electrospray-ionization-mass spectrometry (ESI/MS). In MALDI-MS, the protein or peptide sample is co-crystalized with a matrix (a compound that absorbs easily UV light and becomes ionized and then ionizes the protein or peptide sample), while in ESI/MS, the sample is ionized using the electric current (electrospray source). Depending on the type of the mass spectrometer, the sample may be analysed in either MS mode or in MS/MS mode. In MS mode, the compound to be analysed (protein or peptide) is ionized in the ion source, then analysed in the analyser and then detected in the detector. In MS/MS mode, the protein or peptide of interest is analysed twice: first, it is analysed in MS mode, as previously explained, then the peak that corresponds to this peptide or protein is selected and fragmented in pieces in MS/MS mode, and then the peaks that correspond to the fragments are analysed in the analyser and then detected in the detector.
A common combination that is widely used in proteomics is a mass spectrometer with ESI source and MS/MS capability combined with a liquid chromatography system (also called HPLC or high-performance liquid chromatography). A system capable of very low flow rates is named liquid chromatography tandem mass spectrometry (LC-MS/MS). Therefore, instead of analysing a considerable number of peptides simultaneously (and detecting only the most abundant peptides) by ESI-MS, pre-fractionation of these peptides on a HLPC prior to their analysis by MS allows sequential analysis of more peptides within the same experiment. In other words, if one has a sample with 100 peptides and analyses them simultaneously by ESI-MS, there would not be enough time to analyse all of them at once. However, if one uses LC-MS/MS, prefractionation performed by HPLC, this would allow the MS to analyse a higher number of peptides from the same sample.
Although the ionization methods are quite different (ESI source for ESI-MS and MALDI source for MALDI-MS), these two ionization methods do not exclude each other. On the contrary, these methods complement each other quite well. Regardless of the MS method chosen (MALDI-MS or LC-MS/MS), the end result is identification of a protein or a set of proteins. An example of a full proteomics experiment is shown in Figure. An example of a LC-MS/MS analysis of a peptide mixture, leading to the identification of a peptide that is part of ApoA1 protein, is shown in Figure.
Fig 1.
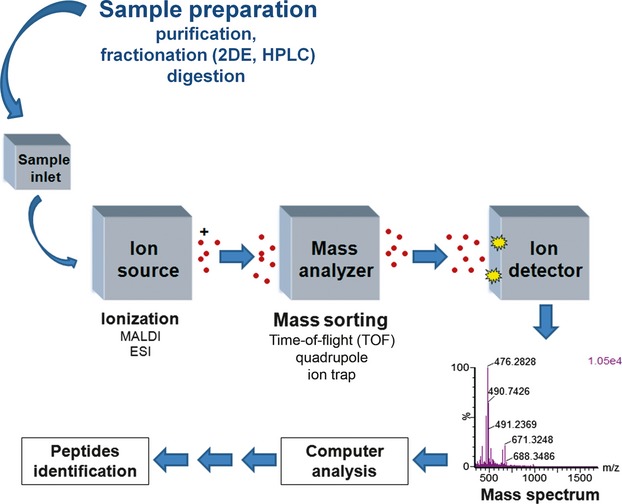
Example of a proteomics experiment. The sample is fractionated during the sample preparation. The fractionation may be electrophoresis (e.g. two dimensional electrophoresis, 2DE) or HPLC or other standard biochemical fractionation methods. The sample is then digested by an enzyme (usually trypsin) and then is ionized in the ion source of the MALDI-MS or ESI-MS mass spectrometer. The ions fly and are sorted through different types of mass analysers (Time of Flight, Quadrupole, Ion trap or a combination of them) and then are detected in the ion detector and recorded in a mass spectrum. If a protein is cleaved upon trypsin digestion in, for example, 10 peptides and only seven of them can fly into the mass spectrometer (e.g. due to their hydrophobicity or lack of basic residues or small size), then data analysis can lead to identification of the protein through a procedure named peptide mass fingerprinting (e.g. seven of ten peptides match with the peptides from a protein and ultimately lead to identification of that protein). In a different procedure shown in more detail in Figure, one of these seven peptides can be selected for fragmentation by the mass spectrometer and the fragmentation can lead to determination of the amino acid sequence of that peptide (which is part of the protein), which by itself can identify the full length protein.
Fig 2.
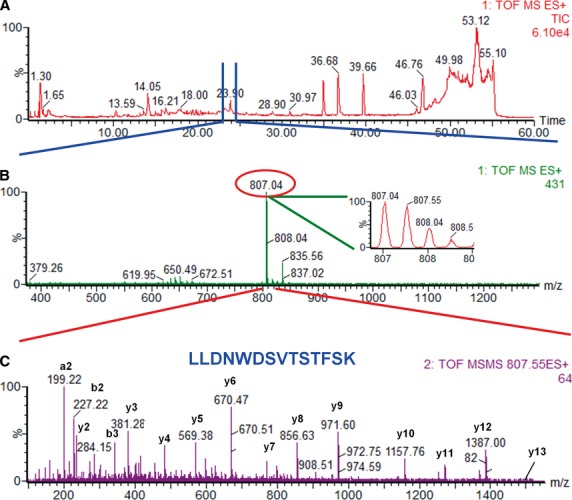
LC-MS/MS analysis of a peptide mixture for identification of ApoA1. The serum sample was separated by Tricine PAGE gel electrophoresis and the gel bands were cut according to their molecular mass and digested by trypsin. Here, the gel band that corresponds to about 25 kD was digested and analysed. The resulting peptide mixture was loaded onto a C18 reversed phase column and separated over a 60-min. linear gradient using an aqueous solution (solution A, which consisted of 0.1% (v/v) formic acid in HPLC water) and an organic solution [solution B, which consisted of 0.1% (v/v) formic acid in acetonitrile]. The gradient was achieved via a constant increase in the organic solvent from 2% to 55% in 40 min., followed by washing with 55% solution B for 2 min., a linear gradient from 55% solution B to 100% solution B over 5 min., washing with 100% solution B for 3 min. and then re-equilibration of the column with 2% solution B over the last 10 min. of run (A). The sample was fractionated using a Waters Alliance 2695 HPLC and analysed by a Micromass/Waters QTOF Micro mass spectrometer. During separation of the peptides by liquid chromatography (A), the mass spectrometer recorded a MS survey mass spectrum (B), in which one double-charged peak (2+) at m/z of 804.07 (expanded in the inbox) was fragmented by MS/MS and produced a MS/MS spectrum (C). Each MS/MS results from fragmentation of a peptide into peaks that correspond to the amino acid components of the peptide. Therefore, fragmentation of a peptide by MS/MS could lead to identification of that peptide by data analysis, which, in theory, could also lead to identification of one protein. The resulting peaks in the MS/MS spectrum correspond to a, b and y ions (mostly the peptidic bonds from the peptides are broken) from a peptide, which was part of ApoA1 protein. Data analysis of these peaks led to identification of the peptide with the amino acid sequence shown in (C). Data analysis of the MS/MS that corresponds to the peptide shown in (C), either alone or in combination with the data that resulted from the MS/MS of other peptides that are part of ApoA1 led to identification of the protein from the 25 kD band as ApoA1.
In addition to qualitative information, MS may provide quantitative information about a particular protein. Methods such as differential gel electrophoresis (DIGE) [34], isotope-coded affinity tag (ICAT) [35], stable isotope labelling by amino acids in cell culture (SILAC) [36], absolute quantification (AQUA) [37], multiple reaction monitoring (MRM) [38], or spectral counting [39] allow detection, identification and quantification of proteins or peptides.
The cholesterol system
Cholesterol and apolipoproteins
Several studies have indicated that of the candidate psychiatric biomarkers that can be detected using the previously mentioned proteomic techniques, cholesterol and proteins associated with the cholesterol system, specifically Apos, may be of particular interest [19,22,40,41].
Almost every bodily cell can synthesize cholesterol from acetyl-CoA and Apos of different types help transport cholesterol throughout the body and facilitate the uptake of cholesterol by cells. The composition of cholesterol plus an Apo (as well as other molecules such as phospholipids and triacylglycerols that can surround the lipid) is known as a lipoprotein [42]. Cholesterol the molecule is therefore distinct from common clinical terms regarding total cholesterol, which refer to the composition of low-density lipoprotein (LDL), high-density lipoprotein (HDL), and triglycerides [43]. LDL is well known as ‘bad cholesterol’ with the potential to build up on artery walls, form plaques and eventually atherosclerosis. HDL is the ‘good’ cholesterol, as higher HDL levels can prevent heart disease. HDL may actually remove cholesterol from plaques. Triglycerides are a form of bodily fat that also make up total cholesterol. High triglyceride levels can also contribute to high total cholesterol level and heart disease [43]. In the CNS, perturbations of the vascular system via cholesterol build-up can naturally contribute to stroke and possibly compromise the blood–brain barrier, causing or exacerbating a disorder.
Cholesterol
Cholesterol is necessary for brain development and its synthesis continues in the adult brain, although at a lower rate than when the brain is developing [44]. Cholesterol is an important part of cell plasma membranes [45,46], helping to determine membrane permeability to molecules as well as charged ions [44] and cholesterol is needed for synapse formation [47]. Naturally, this makes cholesterol important for normal neuron signalling. About 25% of all human bodily cholesterol is found in the brain, where cholesterol is made by both astrocytes and oligodendroglia. Almost all brain cholesterol is un-esterified and about 70% of brain cholesterol is found in myelin and the remaining cholesterol resides in neuronal and astrocytic plasma membranes [48]. Cholesterol in the brain is synthesized locally, due to the presence of the blood–brain barrier. This makes recycling of brain cholesterol by Apos particularly important, as little cholesterol is derived directly from lipoprotein cholesterol found in the circulation [48]. Apo interactions with cholesterol are therefore highly relevant to consider when studying CNS conditions.
Apos
Apos are the protein component of lipoproteins responsible for lipid transport. They combine with free cholesterol, cholesterol esters, phospholipids, and triacylglycerols, forming lipoproteins. Combinations of the Apo proteins with lipids can form different density particles, including low-density chylomicrons, VLDLs up to very high-density lipoproteins (VHDLs) [42]. This gene family includes APOA1, APOA2, APOA4, APOB-48, APOB-100, APOCI, APOCII, APOCIII, APOD, APOE, APOJ, APOL2, APOH [42,49]. Some atypical Apos do not form lipoproteins independently, but associate with other Apos or associate with other molecules via unconventional hydrophobic binding [49]. The function of Apos is principally to make lipoproteins stable and soluble in the blood. They therefore maintain the structure of lipoproteins, act as cofactors in enzymatic reactions and as lipoprotein receptor ligands [42]. The Apos found at the highest levels in CSF include ApoE and ApoA1. ApoJ, ApoD, ApoA2 and ApoA4 are also found in the CSF at lower levels [50,51]. Some notable Apos that have been implicated in psychiatric and neurological conditions include ApoE, ApoB, ApoA1 and ApoA4 [19,52,,53].
ApoE
ApoE is found in all plasma lipoproteins. About 10–20% of VLDLs are composed of apoE and 1–2% of HDLs. ApoE has three common potential isoforms, E2, E3 and E4 [54]. It is critical for cholesterol transport and lipoprotein particle metabolism and may also be involved in immune regulation, nerve regeneration, lipolytic enzyme activation, synaptogenesis, may be a ligand for various receptors and may promote neuronal homeostasis as well as tissue repair [53,55]. ApoE is synthesized in most organs and substantial amounts are produced in brain [56], which is the site in the body with the second greatest synthesis [57]. Brain-derived ApoE is separate from peripheral ApoE [58], primarily secreted and synthesized by astrocytes [59], although APOE is expressed by other brain cells, including neurons (reviewed in [49]). ApoE-containing lipoproteins bind LDL receptors found in neuronal membranes, acting to signal both axonal growth and neuron survival [59,60]. Both ApoE and cholesterol are essential for maintaining both myelin and neuronal synapses [55].
ApoB
ApoB is produced primarily in liver and intestine [61] and is found in two forms, named ApoB48 and ApoB100 [62,63]. It is the principal protein comprising plasma chylomicrons, VLDL and LDL [61]. ApoB100 is also found in intermediate density lipoproteins (IDL) and HDL [19]. The interaction between LDL and the LDL receptor is mediated by ApoB100 [64]. Modification of the APOB gene in mice can result in hydrocephaly or exocephaly, pointing towards a possible critical role in CNS development [65,66].
ApoA1 and ApoA4
ApoA1 is the major protein component of HDL [56,67]. It also facilitates reverse transport of cholesterol from tissues for excretion and by acting as a lecithin cholesterol acetyltransferase cofactor [68]. ApoA1 is present in neurons in the brain and spinal cord [69,70,,71] attesting to a possible role in cognition and mental processes. In contrast to ApoE, it is synthesized in the plasma and enters the brain by crossing the blood–brain barrier [72,73]. ApoA4 is a major component of chylomicrons and HDL, and to a limited extent VLDL [74]. ApoA4 may increase HDL formation and facilitate triglyceride clearance [75] and helps to remove cholesterol from peripheral cells, facilitating cholesterol deposition in the liver [74]. ApoA4 is mainly synthesized in the intestine and is secreted into plasma [56]. In the brain, it is also produced in the hypothalamus where it may act as a satiety signal [76].
The cholesterol system and specific disorders
Alzheimer's disease
The present review focuses primarily on psychiatric versus neurological illness; however, Alzheimer's disease (AD) merits particular mention due to the extensive literature attesting to cholesterol dysregulation in this CNS disorder [77]. Elevated total cholesterol and LDL levels correspond to increased neuritic plaque density in people with AD; this correlation is stronger for people with the APOE4 allele [78]. Having the ApoE4 isoform is the most established genetic risk factor for late-onset AD [53,79]. A protective role of the ApoE2 isoform has been proposed for late-onset AD [80]. Polymorphisms in APOC1, APOJ (clusterin) and APOD have been identified as AD risk factors as well [77,81]. ApoB has been colocalized to amyloid plaques in a mouse model of AD and in people with AD [82,83] and ApoB over-expression leads to neuronal degeneration in transgenic mice [84]. Increases in serum ApoB relative to age-matched controls have been observed in people with AD [85]. Supporting the possibility that Alzheimer's may at least in part be a vascular disease, increased ApoB levels and APOB polymorphisms are associated with increased stroke risk [86,87,,88]. ApoA1 declines have been associated with AD and AD animal models [52,89,,90], as well as mild cognitive impairment [90,91]. Increased ApoA1 benefits cognitive performance in an AD mouse model [92] and in humans with multiple sclerosis [93].
Schizophrenia
Much literature on schizophrenia focuses on the cholesterol perturbations and other metabolic side effects that occur as a result of antipsychotic treatment [94]. The present review does not consider the effects of drug treatment on cholesterol, but instead attempts to focus on cholesterol system disturbances that may occur intrinsic to the disease state. For example, increased levels of the ApoE4 isoform has been reported for schizophrenia [95], although other studies failed to support an association between ApoE4 alternations and schizophrenia [96,97]. Increased levels of brain and CNS ApoE protein [98,99,100,101] and LDL receptor-related protein [100] have also been observed, but also decreases in plasma ApoE [102]. ApoA1 declines have been associated with schizophrenia [40,103,104], although one study identified increases in CNS ApoA1 [101]. Variability in measurements of cholesterol-associated molecules may reflect schizophrenia heterogeneity. Although the direction of Apo disturbances is unclear in schizophrenia, aberrations in the system do seem consistent. Cholesterol metabolism in schizophrenia (independent of antipsychotic effects) warrants further study and elucidation.
Depression
The role of cholesterol levels in affective disorders has been frequently suggested, although whether or not cholesterol perturbations consistently occur in mood disorders such as depression remains a topic of debate [105]. For example, total cholesterol has been found to be lower in depressed people versus controls, with lower HDL, LDL and higher LDL triglyceride/ApoB ratio as well as more artherogenic LDL particle size in depressed individuals 106,107,108]. Low cholesterol has also been associated with increased suicidality [109]. Low LDL and LDL/HDL and LDL/ApoB ratios are reversed by antidepressant treatment, which is considered to be a favourable change in serum lipoproteins [106]. Low levels of HDL cholesterol are associated with long symptom duration [107]. In contrast, another study found that total cholesterol, HDL-cholesterol, LDL-cholesterol and ApoB levels were significantly higher and ApoA1 levels were significantly lower in people with major depression versus non-depressed controls [110].
Developmental disorders: ASDs
Autistic spectrum disorders are characterized by impaired social interaction, stereotyped behaviours/interests and communication deficits. They include ‘classic’ autism, Asperger's syndrome and pervasive developmental disorder not otherwise specified (PDD-NOS or atypical autism) [111]. Proteomic analysis in ASD is a sparsely explored research area with great potential. Much research indicates that cholesterol, in particular, may be altered in ASD [41]. A study examining ASD/non-ASD sibling pairs found dysregulation of cholesterol metabolism-associated genes in ASD [112]. Smith-Lemli-Opitz-Syndrome (SLOS) is an inborn decrease of cholesterol synthesis associated with ASD symptoms 113,114,115]. SLOS symptoms improve rapidly with cholesterol supplementation and ASD-affected children without SLOS have been identified with low total cholesterol [41]. However, SLOS is an uncommon cause of ASDs (about 1%). A recent study found that as high as 20% of an ASD sample of 100 children with ASD had hypocholesterolemia, suggesting that other syndromes associated with ASD may be characterized by low cholesterol [116]. For example, X-linked ichthyosis is another cholesterol-related disorder in which ASD symptoms are present [117]. Higher triglycerides (TG), lower HDL and LDL/HDL ratio were observed in autistic versus healthy boys [118]. Elevated total cholesterol and LDL have been observed in Asperger syndrome [119]. In addition, the levels of ApoB100 and ApoA4 were higher in children with high versus low-functioning autism [19]. Studies investigating ApoE expression and autism have failed to find any association 120,121]. In reflection of the possible important role of cholesterol in ASD, a large-scale clinical trial examining cholesterol supplementation on ASD symptoms has been initiated [122]. Recently, Taurines et al. (2010) found differences in the protein content of sera taken from 16 children with ASD versus 16 age-matched normal controls using matrix-assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-ToF-MS), but were not able to specify which proteins were altered [22,123]. They have suggested that one of the proteins may be an Apo [20]. Interestingly, in another developmental disorder, attention deficit hyperactivity disorder (ADHD), free cholesterol, HDL and ApoA1 were found to be higher in children with ADHD versus controls [124]. Cholesterol parameters in ADHD remain relatively unexplored.
Discussion
Proteomic considerations for analysis of Apos
The significance of cholesterol and of Apos in psychiatric disorders is still under-investigated and their roles wait to be revealed. Biochemically, Apo proteins are intensely post-translationally modified, which make it difficult to monitor these proteins in bodily fluids such as sera. For example, ApoE [54,125,126,127] has 317 amino acids (36 kD), but the first 18 are removed (signal sequence) and the remaining protein is post-translationally modified by phosphorylation at one serine residue and glycosylation of both N-linked- (one site) and O-linked (two sites) oligosaccharides. ApoB isoforms [62,128,129,130,131], ApoB100 and ApoB48, derive from the same initial APOB gene, but due to an editing event, a stop codon is introduced in the mRNA sequence and the ApoB48 is produced. ApoB100 has 4563 amino acids (515 kD) and ApoB48 has 2179 amino acids (243 kD). ApoBs are also intensely post-translationally modified. Both proteins have a 27 amino acid signal sequence that is removed upon secretion. ApoB100 has 19 potential sites for N-linked oligosaccharides, 8 disulphide bridges, two serine residues phosphorylated, one lysine modified at the N6 by an acetyl group, and one cysteine modified to S-palmitoyl-cysteine.
ApoA1 132,133,134,135,136] has 267 amino acids (31 kD). ApoA1 is processed from the pre-pro-protein into pro-protein through removal of the first 18 amino acids and then it is secreted into the blood stream, where is further processed into fully mature protein, by removal of six amino acids from the N-terminal part of the protein. Furthermore, this protein is also processed at the C-terminus, where one amino acid is removed, resulting in two protein isoforms: ApoA1 and ApoA1 (1-242). This protein is further modified by a phosphate group at one serine residue and by glycation at an asparagine residue. Furthermore, it has also been reported that ApoA1 is cleaved by serum proteases into a 14 kD fragment and the rest of the protein is degraded. ApoA4 133,137] has 396 amino acids (45 kD) and is one of the Apo proteins with the fewest post-translational modifications. The secreted form of the protein has the first 20 amino acids removed (signal sequence) and is then phosphorylated at one serine residue. A summary of the post-translational modifications in ApoE, ApoB, ApoA1 and ApoA4 proteins is shown in Figure.
Fig 3.
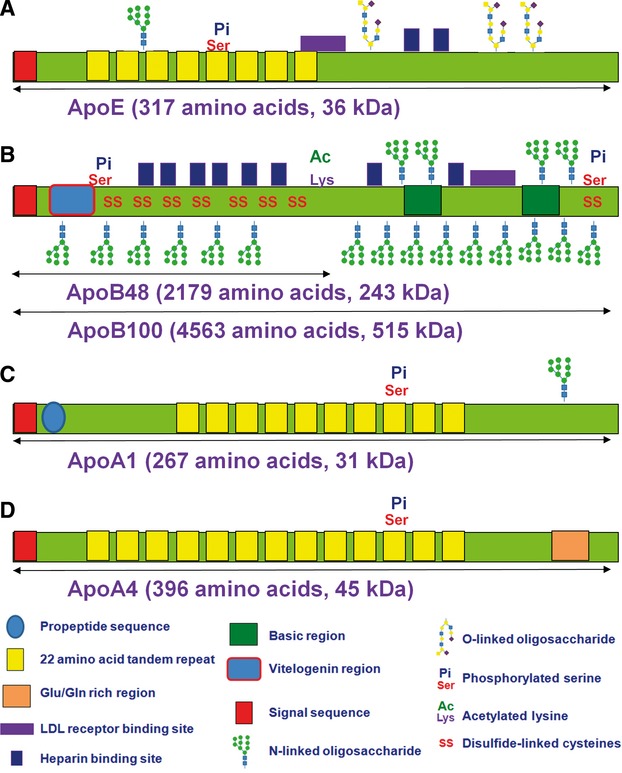
Schematic representing the post-translational modification of the ApoE, ApoB100, ApoA1 and ApoA4. ApoB48 is a truncated form of Apo100. The most important domains and motifs in these proteins are also shown, explained in the legend. The number of amino acids shown for each protein represents the unprocessed proteins; the actual length of the processed, mature proteins is shorter, but their size is larger, due to an additional modification such as glycosylation.
As observed from only a few examples, there are many places where a protein can go wrong. If one glycosylation site that is supposed to be occupied by an oligosaccharide is not occupied the solubility of the protein decreases and therefore its serum lifetime as well. If two cysteine residues are supposed to be disulphide bridges, but they are not, then the conformation of that protein may not be suitable for transport of cholesterol, or may not even be functional. Even worse, the free cysteine residues may form disulphide bonds with other proteins (similar or different proteins), leading to formation of aggregates in the blood that damage kidney function. Other insufficient or excessive modifications such as phosphorylation, palmitoylaton or acetylation may also responsible for changing the levels of proteins in general, and Apo proteins in particular, that may lead to diseases and/or disorders. To identify and thereby help prevent or treat a disease or disorder, MS and proteomics may be one option, although it is far too early to be validated as the method of choice by any scientist.
Considering diet and lifestyle effects on the cholesterol system
When conducting proteomic research, extrinsic effects, in particular diet, need to be accounted for. Disturbances in lipid metabolism may be secondary phenomena due to lifestyle factors, diet or medication. As already mentioned, atypical antipsychotics are frequently used to treat schizophrenia [138] and are increasingly being prescribed for depression as well, (generally as augmentation to another medication) [139]. Although no indicated medications exist for ASD, children with ASD are sometimes prescribed atypical antipsychotics for ASD symptoms or symptoms associated with comorbitities [140]. Atypical antipsychotics are well known to cause perturbations of the cholesterol system [138]. In addition, diet and other lifestyle factors such as smoking, physical activity and alcohol consumption can be less healthy in people with depression and schizophrenia, which in turn may affect cholesterol levels 141,142,143]. ASD specifically, is associated with stereotyped behaviour, which can include a limited diet and refusal to eat certain foods [144]. In addition, immunological disturbances have been identified in the gastrointestinal tract in subgroups of children with ASD [145]. It is therefore possible that in this subgroup, gastrointestinal processing contributes to cholesterol system disturbances. Studies measuring cholesterol-associated potential biomarkers in psychiatric disorders should ideally account for and measure these variables by identifying all relevant subject characteristics, such as medication use, lifestyle factors, dietary habits and gastrointestinal disorders.
Consequences of disturbed cholesterol and Apos in the CNS
In the event that a specific disturbance of the cholesterol system is identified as a consistent biomarker for a disorder or for disorder severity, several questions emerge, such as: Is the disturbance of the cholesterol system contributing to the disorder or is it a consequence? Does the magnitude of changes correspond to changes in specific symptoms? Can correction of cholesterol metabolism abnormalities alleviate symptoms? In ASD, a large-scale clinical trial examining cholesterol supplementation in children with ASD is currently underway to study this last question [122]. It is hoped that answers will rapidly emerge from this study. In schizophrenia and depression, there are several studies examining the effects of ingesting omega-3 fatty acids [43,146]. Potentially, ingestion of omega-3s or correction of an omega-6/omega-3 imbalance [43,147] can contribute to balancing the cholesterol system and alleviating psychiatric symptoms. Other possibilities include pharmacological interventions to correct cholesterol system abnormalities.
With ingestion of food supplements as well as oral medications, the permeability of the blood–brain barrier to serum cholesterol and carrier proteins also needs to be considered, based on the tendency of local brain cholesterol synthesis [48], as well as local brain synthesis of specific Apos and their different abilities to permeate the blood–brain barrier. For example, ApoE used in the brain tends to be synthesized there, whereas ApoA1 can cross the blood–brain barrier [58,59,72,73]. When considering Apos as therapeutics, the relevant Apo for a disorder as well as its permeability characteristics would need to be taken into account.
Conclusion
Putative protein biomarkers associated with the cholesterol system may be altered in psychiatric disorders. Proteomics has been a valuable tool for studying changes in the cholesterol system in CNS disease states. Perturbations of the cholesterol system seem clear in AD disease, depression and schizophrenia, although the direction of changes in cholesterol system molecules is somewhat inconsistent for depression and quite inconsistent in schizophrenia. Proteomics have been applied to the study of cholesterol system abnormalities in CNS disorders in adults, but are only recently being applied to the study of cholesterol systems in childhood. Untreated childhood disorders can have devastating and enduring consequences well into adulthood 148,149]; therefore, clues for diagnosis and early intervention are highly valuable. As cholesterol and associated molecules are critical to the formation of the CNS, the cholesterol system is a viable subject for studies of CNS development. There is considerable evidence that cholesterol and associated molecules may be disturbed in ASD. Further proteomic studies of cholesterol and associated molecules in ASD specifically are warranted.
Acknowledgments
This work was supported in part by a start-up grant from Clarkson University to CCD and by support from the U.S. Army research office through the Defense University Research Instrumentation Program (DURIP grant no. W911NF-11-1-0304). All authors contributed to writing this manuscript.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- Martins-de-Souza D. Is the word ‘biomarker’ being properly used by proteomics research in neuroscience? Eur Arch Psychiatry Clin Neurosci. 2010;260:561–2. doi: 10.1007/s00406-010-0105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallis AG, Fennell DA, Szutowicz E, et al. Biomarkers of clinical benefit from anti-epidermal growth factor receptor agents in patients with non-small-cell lung cancer. Br J Cancer. 2011;105:1–8. doi: 10.1038/bjc.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KA, Marshall DA, Haas JS, et al. Clinical practice patterns and cost effectiveness of human epidermal growth receptor 2 testing strategies in breast cancer patients. Cancer. 2009;115:5166–74. doi: 10.1002/cncr.24574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JS. Biomarker-based selection of therapy for colorectal cancer. Biomark Med. 2011;5:319–32. doi: 10.2217/bmm.11.38. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Vieira K, Hamlat E. Biomarkers in psychiatry: drawbacks and potential for misuse. Int Arch Med. 2010;3:1–6. doi: 10.1186/1755-7682-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Rose N. Biomarkers in psychiatry. Nature. 2009;460:202–7. doi: 10.1038/460202a. [DOI] [PubMed] [Google Scholar]
- Muller TJ, Thome J, Chiaramonti R, et al. A comparison of qEEG and HMPAO-SPECT in relation to the clinical severity of Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. 1997;247:259–63. doi: 10.1007/BF02900304. [DOI] [PubMed] [Google Scholar]
- Wolf C, Linden S, Jackson MC, et al. Brain activity supporting working memory accuracy in patients with paranoid schizophrenia: a functional magnetic resonance imaging study. Neuropsychobiology. 2011;64:93–101. doi: 10.1159/000323800. [DOI] [PubMed] [Google Scholar]
- Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience. 2009;164:331–43. doi: 10.1016/j.neuroscience.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Gogos JA. Molecules, signaling, and schizophrenia. Curr Top Behav Neurosci. 2010;4:629–56. doi: 10.1007/7854_2010_41. [DOI] [PubMed] [Google Scholar]
- Poelmans G, Pauls DL, Buitelaar JK, et al. Integrated genome-wide association study findings: identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am J Psychiatry. 2011;168:365–77. doi: 10.1176/appi.ajp.2010.10070948. [DOI] [PubMed] [Google Scholar]
- Weber H, Kittel-Schneider S, Gessner A, et al. Cross-Disorder analysis of bipolar risk genes: further evidence of DGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology. 2011;36:2076–85. doi: 10.1038/npp.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, et al. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010;167:509–27. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolassa IT, Kolassa S, Ertl V, et al. The risk of posttraumatic stress disorder after trauma depends on traumatic load and the catechol-o-methyltransferase Val(158)Met polymorphism. Biol Psychiatry. 2010;67:304–8. doi: 10.1016/j.biopsych.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Lahey BB, Rathouz PJ, Lee SS, et al. Interactions between early parenting and a polymorphism of the child's dopamine transporter gene in predicting future child conduct disorder symptoms. J Abnorm Psychol. 2011;120:33–45. doi: 10.1037/a0021133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Sarchiopone M, Carli V. Gene-environment interaction and suicidal behavior. J Psychiatr Pract. 2009;15:282–8. doi: 10.1097/01.pra.0000358314.88931.b5. [DOI] [PubMed] [Google Scholar]
- Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 1997;18:533–7. doi: 10.1002/elps.1150180333. [DOI] [PubMed] [Google Scholar]
- Junaid MA, Pullarkat RK. Proteomic approach for the elucidation of biological defects in autism. J Autism Dev Disord. 2001;31:557–60. doi: 10.1023/a:1013242910574. [DOI] [PubMed] [Google Scholar]
- Corbett BA, Kantor AB, Schulman H, et al. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry. 2007;12:292–306. doi: 10.1038/sj.mp.4001943. [DOI] [PubMed] [Google Scholar]
- Dudley E, Hassler F, Thome J. Profiling for novel proteomics biomarkers in neurodevelopmental disorders. Expert Rev Proteomics. 2011;8:127–36. doi: 10.1586/epr.10.97. [DOI] [PubMed] [Google Scholar]
- Spellman DS, Deinhardt K, Darie CC, et al. Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics. 2008;7:1067–76. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taurines R, Dudley E, Grassl J, et al. Proteomic research in psychiatry. J Psychopharmacol. 2011;25:151–96. doi: 10.1177/0269881109106931. [DOI] [PubMed] [Google Scholar]
- Turck CW, Iris F. Proteome-based pathway modelling of psychiatric disorders. Pharmacopsychiatry. 2011;44(Suppl. 1):S54–61. doi: 10.1055/s-0031-1271701. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Reckow S, Webhofer C, et al. Proteome scale turnover analysis in live animals using stable isotope metabolic labeling. Anal Chem. 2011;83:1665–72. doi: 10.1021/ac102755n. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Webhofer C, Reckow S, et al. A MS data search method for improved 15N-labeled protein identification. Proteomics. 2009;9:4265–70. doi: 10.1002/pmic.200900108. [DOI] [PubMed] [Google Scholar]
- Filiou MD, Turck CW, Martins-de-Souza D. Quantitative proteomics for investigating psychiatric disorders. Proteomics Clin Appl. 2011;5:38–49. doi: 10.1002/prca.201000060. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Darie CC, Shetty V, Spellman DS, et al. Blue Native PAGE and Mass Spectrometry Analysis of the Ephrin Stimulation-Dependent Protein-Protein Interactions in NG108-EphB2 Cells. Applications of Mass Spectrometry in Life Safety. Düsseldorf: Springer-Verlag; 2008. pp. 3–22. [Google Scholar]
- Darie CC, Biniossek ML, Gawinowicz MA, et al. Mass spectrometric evidence that proteolytic processing of rainbow trout egg vitelline envelope proteins takes place on the egg. J Biol Chem. 2005;280:37585–98. doi: 10.1074/jbc.M506709200. [DOI] [PubMed] [Google Scholar]
- Darie CC, Biniossek ML, Jovine L, et al. Structural characterization of fish egg vitelline envelope proteins by mass spectrometry. Biochemistry. 2004;43:7459–78. doi: 10.1021/bi0495937. [DOI] [PubMed] [Google Scholar]
- Darie CC, Biniossek ML, Winter V, et al. Isolation and structural characterization of the Ndh complex from mesophyll and bundle sheath chloroplasts of Zea mays. FEBS J. 2005;272:2705–16. doi: 10.1111/j.1742-4658.2005.04685.x. [DOI] [PubMed] [Google Scholar]
- Darie CC, Janssen WG, Litscher ES, et al. Purified trout egg vitelline envelope proteins VEbeta and VEgamma polymerize into homomeric fibrils from dimers in vitro. Biochim Biophys Acta. 2008;1784:385–92. doi: 10.1016/j.bbapap.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, et al. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Unlu M, Minden JS. Two-dimensional difference gel electrophoresis. Nat Protoc. 2006;1:1351–8. doi: 10.1038/nprot.2006.234. [DOI] [PubMed] [Google Scholar]
- Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Stemmann O, Zou H, Gerber SA, et al. Dual inhibition of sister chromatid separation at metaphase. Cell. 2001;107:715–26. doi: 10.1016/s0092-8674(01)00603-1. [DOI] [PubMed] [Google Scholar]
- Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7:2373–85. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Wang L, Prabakaran S, et al. Independent protein-profiling studies show a decrease in apolipoprotein A1 levels in schizophrenia CSF, brain and peripheral tissues. Mol Psychiatry. 2008;13:1118–28. doi: 10.1038/sj.mp.4002108. [DOI] [PubMed] [Google Scholar]
- Tierney E, Bukelis I, Thompson RE, et al. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:666–8. doi: 10.1002/ajmg.b.30368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 5. New York: W.H. Freeman and Company; 2008. [Google Scholar]
- Woods AG. Give a man a fish. Essential fatty acids in health and disease. Diabetes Self Manag. 2008;25:8–14. [PubMed] [Google Scholar]
- Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390:287–93. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange Y. Tracking cell cholesterol with cholesterol oxidase. J Lipid Res. 1992;33:315–21. [PubMed] [Google Scholar]
- Pal R, Barenholz Y, Wagner RR. Effect of cholesterol concentration on organization of viral and vesicle membranes. Probed by accessibility to cholesterol oxidase. J Biol Chem. 1980;255:5802–6. [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–7. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–15. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol. 2010;51:555–73. doi: 10.2217/CLP.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghini I, Barja F, Pometta D, et al. Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim Biophys Acta. 1995;1255:192–200. doi: 10.1016/0005-2760(94)00232-n. [DOI] [PubMed] [Google Scholar]
- Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–51. [PubMed] [Google Scholar]
- Liu HC, Hu CJ, Chang JG, et al. Proteomic identification of lower apolipoprotein A-I in Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;21:155–61. doi: 10.1159/000090676. [DOI] [PubMed] [Google Scholar]
- Takeda M, Martinez R, Kudo T, et al. Apolipoprotein E and central nervous system disorders: reviews of clinical findings. Psychiatry Clin Neurosci. 2010;64:592–607. doi: 10.1111/j.1440-1819.2010.02148.x. [DOI] [PubMed] [Google Scholar]
- Rall SC, Jr, Weisgraber KH, Innerarity TL, et al. Structural basis for receptor binding heterogeneity of apolipoprotein E from type III hyperlipoproteinemic subjects. Proc Natl Acad Sci USA. 1982;79:4696–700. doi: 10.1073/pnas.79.15.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Rodriguez F, Honer WG, Innis SM, et al. ApoE and cholesterol in schizophrenia and bipolar disorder: comparison of grey and white matter and relation with APOE genotype. J Psychiatry Neurosci. 2011;36:47–55. doi: 10.1503/jpn.090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ross MM, Tessitore A, et al. An initial characterization of the serum phosphoproteome. J Proteome Res. 2009;8:5523–31. doi: 10.1021/pr900603n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Linton MF, Gish R, Hubl ST, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88:270–81. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance A, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801:806–18. doi: 10.1016/j.bbalip.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Hayashi H. Lipid metabolism in the central nervous system and neurodegenerative diseases. Nippon Yakurigaku Zasshi. 2011;137:227–31. doi: 10.1254/fpj.137.227. [DOI] [PubMed] [Google Scholar]
- Demmer LA, Levin MS, Elovson J, et al. Tissue-specific expression and developmental regulation of the rat apolipoprotein B gene. Proc Natl Acad Sci USA. 1986;83:8102–6. doi: 10.1073/pnas.83.21.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman DA, Protter AA, Chen GC, et al. Structural comparison of human apolipoproteins B-48 and B-100. Biochemistry. 1987;26:5478–86. doi: 10.1021/bi00391a040. [DOI] [PubMed] [Google Scholar]
- Higuchi K, Hospattankar AV, Law SW, et al. Human apolipoprotein B (apoB) mRNA: identification of two distinct apoB mRNAs, an mRNA with the apoB-100 sequence and an apoB mRNA containing a premature in-frame translational stop codon, in both liver and intestine. Proc Natl Acad Sci USA. 1988;85:1772–6. doi: 10.1073/pnas.85.6.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullinger CR, Hennessy LK, Chatterton JE, et al. Familial ligand-defective apolipoprotein B. Identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest. 1995;95:1225–34. doi: 10.1172/JCI117772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homanics GE, Maeda N, Traber MG, et al. Exencephaly and hydrocephaly in mice with targeted modification of the apolipoprotein B (Apob) gene. Teratology. 1995;51:1–10. doi: 10.1002/tera.1420510102. [DOI] [PubMed] [Google Scholar]
- Homanics GE, Smith TJ, Zhang SH, et al. Targeted modification of the apolipoprotein B gene results in hypobetalipoproteinemia and developmental abnormalities in mice. Proc Natl Acad Sci USA. 1993;90:2389–93. doi: 10.1073/pnas.90.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas A. A review of plasma apolipoprotein A-I interactions with phosphatidylcholines. Exp Lung Res. 1984;6:255–70. doi: 10.3109/01902148409109252. [DOI] [PubMed] [Google Scholar]
- Akerloef E, Joernval H, Slotte H, et al. Identification of apolipoprotein A1 and immunoglobulin as components of a serum complex that mediates activation of human sperm motility. Biochemistry. 1991;30:8986–90. doi: 10.1021/bi00101a011. [DOI] [PubMed] [Google Scholar]
- Fujii H, Saito K, Hamakawa H, et al. Immunohistochemical localization and mRNA expression of apolipoprotein A-I in rat spinal cord. J Atheroscler Thromb. 2002;9:93–8. doi: 10.5551/jat.9.93. [DOI] [PubMed] [Google Scholar]
- Harr SD, Uint L, Hollister R, et al. Brain expression of apolipoproteins E, J, and A-I in Alzheimer's disease. J Neurochem. 1996;66:2429–35. doi: 10.1046/j.1471-4159.1996.66062429.x. [DOI] [PubMed] [Google Scholar]
- Panin LE, Russkikh GS, Polyakov LM. Detection of apolipoprotein A-I, B, and E immunoreactivity in the nuclei of various rat tissue cells. Biochemistry (Mosc) 2000;65:1419–23. doi: 10.1023/a:1002861008363. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, et al. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–60. [PubMed] [Google Scholar]
- Roheim PS, Carey M, Forte T, et al. Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci USA. 1979;76:4646–9. doi: 10.1073/pnas.76.9.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan S, Delvin E, Lambert M, et al. Apo A-IV: an update on regulation and physiologic functions. Biochim Biophys Acta. 2003;1631:177–87. doi: 10.1016/s1388-1981(03)00004-0. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ, Scheraldi CA, Yacoub LK, et al. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J Biol Chem. 1990;265:4266–72. [PubMed] [Google Scholar]
- Qin X, Tso P. The role of apolipoprotein AIV on the control of food intake. Curr Drug Targets. 2005;6:145–51. doi: 10.2174/1389450053174541. [DOI] [PubMed] [Google Scholar]
- Leduc V, Jasmin-Belanger S, Poirier J. APOE and cholesterol homeostasis in Alzheimer's disease. Trends Mol Med. 2010;16:469–77. doi: 10.1016/j.molmed.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Lesser GT, Beeri MS, Schmeidler J, et al. Cholesterol and LDL relate to neuritic plaques and to APOE4 presence but not to neurofibrillary tangles. Curr Alzheimer Res. 2011;8:303–12. doi: 10.2174/156720511795563755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins GA, Large CH, Rupniak HT, et al. Apolipoprotein E and Alzheimer's disease: a review of recent studies. Pharmacol Biochem Behav. 1997;56:675–85. doi: 10.1016/s0091-3057(96)00420-0. [DOI] [PubMed] [Google Scholar]
- Helisalmi S, Hiltunen M, Vepsalainen S, et al. Genetic variation in apolipoprotein D and Alzheimer's disease. J Neurol. 2004;251:951–7. doi: 10.1007/s00415-004-0470-8. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tsuchiya H, Ikeda K. Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer's disease. Neurosci Lett. 1992;134:264–6. doi: 10.1016/0304-3940(92)90531-b. [DOI] [PubMed] [Google Scholar]
- Takechi R, Galloway S, Pallebage-Gamarallage M, et al. Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem Cell Biol. 2009;131:661–6. doi: 10.1007/s00418-009-0567-3. [DOI] [PubMed] [Google Scholar]
- Bereczki E, Bernat G, Csont T, et al. Overexpression of human apolipoprotein B-100 induces severe neurodegeneration in transgenic mice. J Proteome Res. 2008;7:2246–52. doi: 10.1021/pr7006329. [DOI] [PubMed] [Google Scholar]
- Caramelli P, Nitrini R, Maranhao R, et al. Increased apolipoprotein B serum concentration in Alzheimer's disease. Acta Neurol Scand. 1999;100:61–3. doi: 10.1111/j.1600-0404.1999.tb00724.x. [DOI] [PubMed] [Google Scholar]
- Benn M, Nordestgaard BG, Jensen JS, et al. Mutation in apolipoprotein B associated with hypobetalipoproteinemia despite decreased binding to the low density lipoprotein receptor. J Biol Chem. 2005;280:21052–60. doi: 10.1074/jbc.M413877200. [DOI] [PubMed] [Google Scholar]
- Benn M, Nordestgaard BG, Skov Jensen J, et al. Polymorphisms in apolipoprotein B and risk of ischemic stroke. J Clin Endocrinol Metab. 2007;92:3611–7. doi: 10.1210/jc.2007-0221. [DOI] [PubMed] [Google Scholar]
- Walldius G, Aastveit AH, Jungner I. Stroke mortality and the apoB/apoA-I ratio: results of the AMORIS prospective study. J Intern Med. 2006;259:259–66. doi: 10.1111/j.1365-2796.2005.01610.x. [DOI] [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican AA, et al. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem. 2010;285:36945–57. doi: 10.1074/jbc.M110.127738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Poljak A, Smythe GA, et al. Plasma biomarkers for mild cognitive impairment and Alzheimer's disease. Brain Res Rev. 2009;61:69–80. doi: 10.1016/j.brainresrev.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Helbecque N, Codron V, Cottel D, et al. An apolipoprotein A-I gene promoter polymorphism associated with cognitive decline, but not with Alzheimer's disease. Dement Geriatr Cogn Disord. 2008;25:97–102. doi: 10.1159/000112176. [DOI] [PubMed] [Google Scholar]
- Lewis TL, Cao D, Lu H, et al. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:36958–68. doi: 10.1074/jbc.M110.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsis G, Panas M, Giogkaraki E, et al. An APOA1 promoter polymorphism is associated with cognitive performance in patients with multiple sclerosis. Mult Scler. 2009;15:174–9. doi: 10.1177/1352458508097217. [DOI] [PubMed] [Google Scholar]
- Tschoner A, Engl J, Laimer M, et al. Metabolic side effects of antipsychotic medication. Int J Clin Pract. 2007;61:1356–70. doi: 10.1111/j.1742-1241.2007.01416.x. [DOI] [PubMed] [Google Scholar]
- Harrington CR, Roth M, Xuereb JH, et al. Apolipoprotein E type epsilon 4 allele frequency is increased in patients with schizophrenia. Neurosci Lett. 1995;202:101–4. doi: 10.1016/0304-3940(95)12218-4. [DOI] [PubMed] [Google Scholar]
- Jonsson E, Lannfelt L, Engvall B, et al. Lack of association between schizophrenia and the apolipoprotein E epsilon 4 allele. Eur Arch Psychiatry Clin Neurosci. 1996;246:182–4. doi: 10.1007/BF02188951. [DOI] [PubMed] [Google Scholar]
- Town T, Fallin D, Crawford F, et al. Lack of association between the apolipoprotein E epsilon4 allele (APOE epsilon4) and chronic schizophrenia. Am J Med Genet. 1997;74:451–2. [PubMed] [Google Scholar]
- Dean B, Laws SM, Hone E, et al. Increased levels of apolipoprotein E in the frontal cortex of subjects with schizophrenia. Biol Psychiatry. 2003;54:616–22. doi: 10.1016/s0006-3223(03)00075-1. [DOI] [PubMed] [Google Scholar]
- Digney A, Keriakous D, Scarr E, et al. Differential changes in apolipoprotein E in schizophrenia and bipolar I disorder. Biol Psychiatry. 2005;57:711–5. doi: 10.1016/j.biopsych.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Gibbons AS, Thomas EA, Scarr E, et al. Low density lipoprotein receptor-related protein and apolipoprotein E expression is altered in schizophrenia. Front Psychiatry. 2010;1:19. doi: 10.3389/fpsyt.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-de-Souza D, Wobrock T, Zerr I, et al. Different apolipoprotein E, apolipoprotein A1 and prostaglandin-H2 D-isomerase levels in cerebrospinal fluid of schizophrenia patients and healthy controls. World J Biol Psychiatry. 2010;11:719–28. doi: 10.3109/15622971003758748. [DOI] [PubMed] [Google Scholar]
- Dean B, Digney A, Sundrum S, et al. Plasma apolipoprotein E is decreased in schizophrenia spectrum and bipolar disorder. Psychiatry Res. 2008;158:75–8. doi: 10.1016/j.psychres.2007.05.008. [DOI] [PubMed] [Google Scholar]
- La YJ, Wan CL, Zhu H, et al. Decreased levels of apolipoprotein A-I in plasma of schizophrenic patients. J Neural Transm. 2007;114:657–63. doi: 10.1007/s00702-006-0607-2. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wan C, Li H, et al. Altered levels of acute phase proteins in the plasma of patients with schizophrenia. Anal Chem. 2006;78:3571–6. doi: 10.1021/ac051916x. [DOI] [PubMed] [Google Scholar]
- De Berardis D, Conti CM, Serroni N, et al. The role of cholesterol levels in mood disorders and suicide. J Biol Regul Homeost Agents. 2009;23:133–40. [PubMed] [Google Scholar]
- Hummel J, Westphal S, Weber-Hamann B, et al. Serum lipoproteins improve after successful pharmacologic antidepressant treatment: a randomized open-label prospective trial. J Clin Psychiatry. 2011;72:885–91. doi: 10.4088/JCP.09m05853blu. [DOI] [PubMed] [Google Scholar]
- Lehto SM, Niskanen L, Tolmunen T, et al. Low serum HDL-cholesterol levels are associated with long symptom duration in patients with major depressive disorder. Psychiatry Clin Neurosci. 2010;64:279–83. doi: 10.1111/j.1440-1819.2010.02079.x. [DOI] [PubMed] [Google Scholar]
- Lehto SM, Ruusunen A, Niskanen L, et al. Elevated depressive symptoms and compositional changes in LDL particles in middle-aged men. Eur J Epidemiol. 2010;25:403–9. doi: 10.1007/s10654-010-9457-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olie E, Picot MC, Guillaume S, et al. Measurement of total serum cholesterol in the evaluation of suicidal risk. J Affect Disord. 2011;133:234–8. doi: 10.1016/j.jad.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Sarandol A, Sarandol E, Eker SS, et al. Oxidation of apolipoprotein B-containing lipoproteins and serum paraoxonase/arylesterase activities in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1103–8. doi: 10.1016/j.pnpbp.2006.04.012. [DOI] [PubMed] [Google Scholar]
- APA. Diagnostic and Statistical Manual of Mental Disorders. Revised 4th ed. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Hu VW, Nguyen A, Kim KS, et al. Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: altered pathways in neuronal development and steroid biosynthesis. PLoS One. 2009;4:e5775. doi: 10.1371/journal.pone.0005775. . doi: 10.1371/journal.pone.0005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneja A, Tierney E. Autism: the role of cholesterol in treatment. Int Rev Psychiatry. 2008;20:165–70. doi: 10.1080/09540260801889062. [DOI] [PubMed] [Google Scholar]
- Sikora DM, Pettit-Kekel K, Penfield J, et al. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2006;140:1511–8. doi: 10.1002/ajmg.a.31294. [DOI] [PubMed] [Google Scholar]
- Tierney E, Nwokoro NA, Porter FD, et al. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 2001;98:191–200. doi: 10.1002/1096-8628(20010115)98:2<191::aid-ajmg1030>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Tierney E, Bukelis I, Thompson RE, et al. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 2008;141B:666–8. doi: 10.1002/ajmg.b.30368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519–24. doi: 10.1136/jmg.2008.057729. [DOI] [PubMed] [Google Scholar]
- Kim EK, Neggers YH, Shin CS, et al. Alterations in lipid profile of autistic boys: a case control study. Nutr Res. 2010;30:255–60. doi: 10.1016/j.nutres.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Dziobek I, Gold SM, Wolf OT, et al. Hypercholesterolemia in Asperger syndrome: independence from lifestyle, obsessive-compulsive behavior, and social anxiety. Psychiatry Res. 2007;149:321–4. doi: 10.1016/j.psychres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Ashley-Koch AE, Jaworski J, Ma de Q, et al. Investigation of potential gene-gene interactions between APOE and RELN contributing to autism risk. Psychiatr Genet. 2007;17:221–6. doi: 10.1097/YPG.0b013e32809c2f75. [DOI] [PubMed] [Google Scholar]
- Raiford KL, Shao Y, Allen IC, et al. No association between the APOE gene and autism. Am J Med Genet B Neuropsychiatr Genet. 2004;125B:57–60. doi: 10.1002/ajmg.b.20104. [DOI] [PubMed] [Google Scholar]
- Clinicaltrials.gov. Cholesterol in ASD: Characterization and Treatment. ClinicalTrialsgov identifier: NCT00965068: US National Institutes of Health, 2011.
- Taurines R, Dudley E, Conner AC, et al. Serum protein profiling and proteomics in autistic spectrum disorder using magnetic bead-assisted mass spectrometry. Eur Arch Psychiatry Clin Neurosci. 2010;260:249–55. doi: 10.1007/s00406-009-0066-5. [DOI] [PubMed] [Google Scholar]
- Spahis S, Vanasse M, Belanger SA, et al. Lipid profile, fatty acid composition and pro- and anti-oxidant status in pediatric patients with attention-deficit/hyperactivity disorder. Prostaglandins Leukot Essent Fatty Acids. 2008;79:47–53. doi: 10.1016/j.plefa.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Paik YK, Chang DJ, Reardon CA, et al. Nucleotide sequence and structure of the human apolipoprotein E gene. Proc Natl Acad Sci USA. 1985;82:3445–9. doi: 10.1073/pnas.82.10.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall SC, Jr, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257:4171–8. [PubMed] [Google Scholar]
- Zannis VI, McPherson J, Goldberger G, et al. Synthesis, intracellular processing, and signal peptide of human apolipoprotein E. J Biol Chem. 1984;259:5495–9. [PubMed] [Google Scholar]
- Hardman DA, Protter AA, Schilling JW, et al. Carboxyl terminal analysis of human B-48 protein confirms the novel mechanism proposed for chain termination. Biochem Biophys Res Commun. 1987;149:1214–9. doi: 10.1016/0006-291x(87)90537-7. [DOI] [PubMed] [Google Scholar]
- Knott TJ, Pease RJ, Powell LM, et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature. 1986;323:734–8. doi: 10.1038/323734a0. [DOI] [PubMed] [Google Scholar]
- Knott TJ, Wallis SC, Powell LM, et al. Complete cDNA and derived protein sequence of human apolipoprotein B-100. Nucleic Acids Res. 1986;14:7501–3. doi: 10.1093/nar/14.18.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CY, Kim TW, Weng SA, et al. Isolation and characterization of sulfhydryl and disulfide peptides of human apolipoprotein B-100. Proc Natl Acad Sci USA. 1990;87:5523–7. doi: 10.1073/pnas.87.14.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer HB, Jr, Fairwell T, Kay L, et al. Human plasma proapoA-I: isolation and amino-terminal sequence. Biochem Biophys Res Commun. 1983;113:626–32. doi: 10.1016/0006-291x(83)91772-2. [DOI] [PubMed] [Google Scholar]
- Brewer HB, Jr, Ronan R, Meng M, et al. Isolation and characterization of apolipoproteins A-I, A-II, and A-IV. Methods Enzymol. 1986;128:223–46. doi: 10.1016/0076-6879(86)28070-2. [DOI] [PubMed] [Google Scholar]
- Cheung P, Chan L. Nucleotide sequence of cloned cDNA of human apolipoprotein A-I. Nucleic Acids Res. 1983;11:3703–15. doi: 10.1093/nar/11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Kao FT, Law ML, et al. Localization of the structural gene for human apolipoprotein A-I on the long arm of human chromosome 11. Proc Natl Acad Sci USA. 1984;81:508–11. doi: 10.1073/pnas.81.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders CC, Kornblihtt AR, Munro BS, et al. Gene structure of human apolipoprotein A1. Nucleic Acids Res. 1983;11:2827–37. doi: 10.1093/nar/11.9.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshourbagy NA, Walker DW, Paik YK, et al. Structure and expression of the human apolipoprotein A-IV gene. J Biol Chem. 1987;262:7973–81. [PubMed] [Google Scholar]
- Meyer JM. Antipsychotics and metabolics in the post-CATIE era. Curr Top Behav Neurosci. 2010;4:23–42. doi: 10.1007/7854_2010_45. [DOI] [PubMed] [Google Scholar]
- Konstantinidis A, Papageorgiou K, Grohmann R, et al. Increase of antipsychotic medication in depressive inpatients from 2000 to 2007: results from the AMSP International Pharmacovigilance Program. Int J Neuropsychopharmacol. 2011:1–9. doi: 10.1017/S1461145711000745. . doi: 10.1017/S1461145711000745. [DOI] [PubMed] [Google Scholar]
- McPheeters ML, Warren Z, Sathe N, et al. A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics. 2011;127:e1312–21. doi: 10.1542/peds.2011-0427. [DOI] [PubMed] [Google Scholar]
- Aubin HJ, Rollema H, Svensson TH, et al. Smoking, quitting, and psychiatric disease: a review. Neurosci Biobehav Rev. 2012;36:271–84. doi: 10.1016/j.neubiorev.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Buhagiar K, Parsonage L, Osborn DP. Physical health behaviours and health locus of control in people with schizophrenia-spectrum disorder and bipolar disorder: a cross-sectional comparative study with people with non-psychotic mental illness. BMC Psychiatry. 2011;11:104. doi: 10.1186/1471-244X-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacka FN, Mykletun A, Berk M, et al. The association between habitual diet quality and the common mental disorders in community-dwelling adults: the hordaland health study. Psychosom Med. 2011;73:483–90. doi: 10.1097/PSY.0b013e318222831a. [DOI] [PubMed] [Google Scholar]
- Emond A, Emmett P, Steer C, et al. Feeding symptoms, dietary patterns, and growth in young children with autism spectrum disorders. Pediatrics. 2010;126:337–42. doi: 10.1542/peds.2009-2391. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Wakefield AJ. Immune activation of peripheral blood and mucosal CD3+ lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms. J Neuroimmunol. 2006;173:126–34. doi: 10.1016/j.jneuroim.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Akter K, Gallo DA, Martin SA, et al. A review of the possible role of the essential fatty acids and fish oils in the aetiology, prevention or pharmacotherapy of schizophrenia. J Clin Pharm Ther. 2011 doi: 10.1111/j.1365-2710.2011.01265.x. ; in press: doi: 10.1111/j.365-2710.011.01265.x. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP. Evolutionary aspects of diet: the omega-6/omega-3 ratio and the brain. Mol Neurobiol. 2011;44:203–15. doi: 10.1007/s12035-010-8162-0. [DOI] [PubMed] [Google Scholar]
- McEachin JJ, Smith T, Lovaas OI. Long-term outcome for children with autism who received early intensive behavioral treatment. Am J Ment Retard. 1993;97:359–91. [PubMed] [Google Scholar]
- Shaw M, Caci H, Hodgkins P, et al. Review of studies of ADHD: long-term outcomes with and without treatment. Eur Psychiatry. 2011;26(Suppl. 1):579. [Google Scholar]