

Abstract
Mesenchymal stem cells (MSCs) play a crucial role in tissue repair by secretion of tissue nutrient factors such as hepatocyte growth factor (HGF). However, studies examining the effects of HGF on the proliferation and differentiation of MSCs used different concentrations of HGF and reported conflicting conclusions. This study aimed to determine the mechanisms by which different concentrations of HGF regulate MSC proliferation and osteogenic differentiation, and validate the mechanism in an animal model of early stage avascular necrosis of femoral head (ANFH). Our results demonstrate that a low concentration of HGF (20 ng/ml) preferentially promotes MSC osteogenic differentiation through increased c-Met expression and phosphorylation, Akt pathway activation, and increased expression of p27, Runx2 and Osterix. In contrast, a high concentration of HGF (100 ng/ml) strongly induced proliferation by inducing strong activation of the ERK1/2 signalling pathway. As validated by animal experiments, high localized expression of HGF achieved by transplantation of HGF transgenic MSCs into ANFH rabbits increased the number of MSCs. Subsequently, 2 weeks after transplantation, HGF levels decreased and MSCs differentiated into osteoblasts and resulted in efficient tissue repair. Our results demonstrate that sequential concentration changes in HGF control the proliferation and osteogenic differentiation of MSCs in vivo. This phenomenon can be exploited therapeutically to induce bone regeneration and, in turn, improve the efficacy of pharmacological intervention for ANFH treatment.
Keywords: hepatocyte growth factor, mesenchymal stem cells, proliferation, osteogenic differentiation, avascular necrosis of femoral head
Introduction
Mesenchymal stem cells (MSCs) are mesoderm-like adult stem cells present in the stroma of various organs and can differentiate into many terminal-differentiated cell types. Such pluripotency, the convenience of isolation and the ability to proliferate extensively in vitro make MSCs the best candidate cells for tissue engineering [1]. MSC-based treatments have already been applied in the clinic for treatment of ischaemic conditions, including avascular necrosis of the femoral head (ANFH) [2, 3] and myocardial infarction [4, 5], and show great promise as a therapeutic. Although the exact mechanism of MSC-mediated tissue repair is still not fully understood, there is evidence indicating that secretion of nutrient factors, such as hepatocyte growth factor (HGF), plays an important role [6].
Hepatocyte growth factor is a pleitropic cytokine that is mainly secreted by mesenchymal cells and exerts potent mitogenic effects and promotes nutrient absorption and utilization to promote tissue repair in liver [7, 8], heart [9, 10] and muscle [11] through autocrine and paracrine action modalities. MSCs express both HGF and its receptor, c-Met [12, 13], suggesting that HGF secreted by MSCs may also perform an autocrine function. Such characteristics make MSC- and HGF-based therapies highly attractive. The rate of tissue repair mediated by HGF-expressing MSCs following myocardial infarction is far higher than that by non-infected MSCs in vivo [14]. In addition, HGF-null MSCs failed to improve angiogenesis in a model of ischaemic limb [15]. In a previous study, we achieved excellent efficacy in treating early stage ANFH with HGF-expressing bone marrow stroma cells (BMSCs) [16]. These results demonstrate that HGF plays a key role in MSC-mediated ischaemic tissue repair, but the mechanism is still not clear.
Other previous studies observing the effects of HGF on MSC proliferation and differentiation, including osteogenesis, reported different conclusions with various HGF concentrations. Treatment with 100 ng/ml HGF greatly decreased BMP-induced osteogenic differentiation of human MSCs and mouse C2C12 cells [17]. Overexpression of HGF in chicken skeletal muscle satellite cells inhibited myogenesis through induction of Twist and down-regulation of p27 [18]. In contrast, treatment with 10 ng/ml HGF acted cooperatively with 1,25-dihydroxyvitamin D3 to simultaneously promote human MSC proliferation and osteogenic differentiation [19]. Treatment of mouse MSCs with 20 ng/ml HGF for 48 hrs induced expression of myocyte-specific transcription factor and structural genes [12]. Collectively, these results suggest that different concentrations of HGF may affect the proliferation and differentiation of MSCs in various ways, therefore leading to confusion in understanding the precise effects of HGF treatment on MSCs under physiological conditions and delay or impair the clinical application of HGF-based treatments.
In this study, we compared the effects of different concentrations of HGF on the proliferation and osteogenic differentiation of MSCs and dissected the underlying mechanisms in vitro and in vivo. The results show that treatment of MSCs with a high concentration of HGF (100 ng/ml) promotes proliferation through activation of ERK1/2 and suppresses osteogenic differentiation through inhibition of c-Met expression and phosphorylation, the Akt signalling pathway, expression of cell cycle inhibitor p27 and osteogenic-associated transcription factors Runx2 and Osterix. In contrast, treatment of MSCs with a low concentration of HGF (20 ng/ml) promotes osteogenic differentiation through opposite effects on the above molecules. Furthermore, transplantation of HGF-expressing MSCs in animals with ANFH demonstrates the therapeutic potential of HGF-based treatments for ischaemic conditions depends on the appropriate sequential concentration change.
Materials and methods
Isolation and culture of MSCs
Rabbit MSCs were isolated as reported previously [16] and cultured in growth medium (GM), DMEM-low glucose (Hyclone Ltd., Logan, UT, USA) containing 10% foetal bovine serum (FBS; Hyclone Ltd.), and cell confluence was kept below 80% at all times in culture. All experiments were carried out with cells passaged no more than five times. MSCs were identified according to their capacity to differentiation into osteoblasts, chondrocytes and adipoblasts under the specific conditions described later.
Differentiation assays
Chondrogenic and adipogenic differentiation
Mesenchymal stem cells were seeded as a monolayer in six-well plates (Nunc, Thermo Fisher Scientific, Waltham, MA, USA). The chondrogenic (Cat. no. RBXMX-04041) and adipogenic (Cat. nos. RBXMX-04031 and RBXMX-04032) differentiation assays were performed according to the manufacturer’s instructions (Cyagen Biosciences Inc., Goleta, CA, USA). When the differentiation process was complete, the cells were fixed with 4% paraformaldehyde for 30 min., stained with Alcian Blue (Cat. no. 101105c01) or Oil Red O storage solution (Cat. no.: 100801c01; Cyagen Biosciences Inc.) for 30 min., followed by washes with double-distilled H2O (ddH2O). The pictures were taken at 100χ magnification with a Nikon Eclipse Ti–U inverted microscope and Nikon Digital Sight DS-Ril camera (Nikon Corporation, Tokyo, Japan).
Osteogenic differentiation
Mesenchymal stem cells were plated at 2.1 χ 104 cells/cm2 and cultured to 100% confluence. The medium was replaced with osteogenic differentiation medium (DM), GM supplemented with 0.1 μM dexamethasone, 0.2 mM ascorbate-2-phosphate and 10 mM β-glycerophosphate (Sigma-Aldrich, St. Louis, MO, USA). The medium was changed every 2–3 days and the culture was maintained for 21 days. From this point forward ‘differentiation’ is used to indicate ‘osteogenic differentiation’.
Alkaline phosphatase (ALP) assay
Mesenchymal stem cells were washed three times with 0.01M phosphate buffer solution (PBS) at the indicated times after treatment, and then stained using the NBT-BCIP Alkaline Phosphatase Color Development Kit (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions and captured, and Image-Pro Plus software version 6.0 (IPP 6.0, Media Cybernetics, Inc., MD, USA) was used to calculate the ALP activity indicated as the integrated optical density (IOD) of staining in unit culture area.
Calcium accumulation assay
Alizarin red sulfate (AR-S) staining was used to measure the formation of mineralized extracellular matrix (ECM) in vitro. After incubation in DM for 21 days, cells were fixed with 75% ethanol for 10 min. and stained with 0.1% (w/v) AR-S (Weiga Scientific Co. Ltd., Guangzhou, China) for 30 min. Cells were washed with PBS to remove non-specific staining and pictures were taken. Cultures were then disdained by incubation in 10% (w/v) cetylpyridinium chloride (CPC)–PBS for 1 hr. The AR-S concentration in the supernatant was determined by reading the absorbance at 562 nm using a Varioskan Flash microplate reader (Thermo Fisher Scientific) and comparing results with the standard curve obtained by serially diluting AR-S solution in CPC–PBS.
Cell proliferation assay
Mesenchymal stem cells (2.5 χ 103 cells/well) were seeded in triplicate in 96-well plates (Nunc) and incubated overnight. Cells were starved in GM without FBS for 24 hrs and then incubated in DM containing 2% FBS as well as 20 or 100 ng/ml HGF (PeproTech Inc., Rocky Hill, NJ, USA) for 24 hrs. Then the EdU (5′-ethynyl-2′-deoxyuridine) incorporation assay was performed to quantify cell proliferation [20, 21] using the Cell-Light™ EdU DNA Cell Proliferation Kit (Guangzhou Ribobio Co., Ltd, Guangzhou, China) according to the manufacturer’s instructions. As a negative control, cells in the same HGF treatment group were not subsequently treated with EdU. More than five random fields per well were captured at 100χ magnification, and IPP 6.0 was used to calculate the percentage of EdU-positive cells (identified by Apollo® 643 fluorescence) in total cells (identified by Hoechst33342 nuclei staining). In some experiments cells were pre-treated with 30 μM PD98059 [12] or 100 nM wortmannin [12] (Beyotime) for 1 hr prior to HGF stimulation.
Cell viability
Mesenchymal stem cells were seeded in quadruplicate in 96-well plates (Nunc) at a density of 5 χ 103 cells/well in DM supplemented with 20 or 100 ng/ml HGF. The medium was changed every 3 days. On day 1, 4 and 7, the medium was replaced with 100 μl/well fresh DM to avoid volume discrepancy due to evaporation, and then cell viability was detected using the Cell Counting Kit-8 (CCK-8, Dojindo Laboratorise, Tokyo, Japan) according to the manufacturer’s instruction. The absorbance was detected at 450 nm and referenced with 630 nm. After deducting the background absorbance, the emitted light intensity is linearly related to the cell activity [22], thus quantifying the number of live cells. In the indicated experiments, specific chemical inhibitors were added 1 hr before HGF stimulation and every time the medium was changed.
RNA isolation and real-time quantitative RT-PCR (qRT-PCR) analysis
Total RNA was extracted using the E.Z.N.A.® Total RNA Kit (OMEGA Bio-tek, Inc., Norcross, GA, USA) according to the manufacturer’s instruction, and the concentration was determined using a spectrophotometer (Biophotometer Plus, Eppendorf, Hamburg, Germany). RT-PCR analysis was performed by pre-treating 1 μg total RNA with 1 U RNase-free-DNase I (Fermentas, Life Sciences, Ontario, Canada), and then reverse transcribing using the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas) according to manufacturer’s instruction. The FastStart Universal SYBR Green Master (ROX) kit (Roche Applied Science, Mannheim, Germany) and ABI PRISM 7900HT Sequence Detection System (Applied Biosystems Inc., Foster City, CA, USA) were used to perform real-time qRT-PCR. Quantification of target mRNA abundance was analysed with the SDS software Version 2.3 (Applied Biosystems) using the comparative threshold cycle (Ct) method [23]. Relative quantification relates the PCR signal of the sample transcript in the treated MSCs to that of the control MSCs at each time. The fold change in cDNAs (target gene) after GAPDH (reference gene) normalization was determined by the following formula: fold change ∇ 2ΔΔCt, where ΔΔCt ∇ (CtTarget − CtGAPDH)sample − (CtTarget − CtGAPDH)control. The PCR primer sequences used are listed in Table 1.
Table 1.
Primers employed in quantitative real-time RT-PCR
| Gene | Primer pairs | Amplicon | Accession number |
|---|---|---|---|
| Vimentin | F ∇ 5′- AGGCAAAGCAGGAGTCAAATGAG -3′ R ∇ 5′- TCCAGAGCCATCTTGACATTGAG -3′ | 250 bp | XM_002717420.1 |
| Runx2 | F ∇ 5′- GTCATGGCGGGTAACGATGA -3′ R ∇ 5′- CCCACAAATCTCAGATCGTTGAA -3′ | 101 bp | XM_002714704.1 |
| SOX9 | F ∇ 5′- AGTACCCGCACCTGCACAAC -3′ R ∇ 5′- TACTTGTAGTCCGGGTGGTCTTTC -3′ | 145 bp | XM_002719499.2 |
| PPAR-γ3 | F ∇ 5′- ACGACCAGGTGACTCTGCTCAA -3′ R ∇ 5′- CCACTGAGAATGATGACGGCTAT -3′ | 250 bp | NM_001082148.1 |
| GAPDH | F ∇ 5′- GCACGGTCAAGGCTGAGAAC -3′ R ∇ 5′- GCCTTCTCCATGGTGGTGAA -3′ | 151 bp | NM_001082253.1 |
| c-Met | F ∇ 5′- GCTGAAACCCAATAGTTGAGTTTGG -3′ R ∇ 5′- AGAGCGGATTTCCCTATGTTGAC -3′ | 201 bp | NM_001171040.1 |
| p27 | F ∇ 5′- CCTCCGAAGACGTCAGACGTA -3′ R ∇ 5′- GTGCTTATACAGGATGTCCATTCCA -3′ | 151 bp | DQ845181.1 |
| Osterix | F ∇ 5′- CAAAGCAGGCACGAAGAAG -3′ R ∇ 5′- GCCCATTGGTGCTTGAAAAG -3′ | 101 bp | XM_002711016.1 |
Western blot analysis
Total cell protein was extracted using RIPA buffer (ShangHai Biocolor BioScience Technology Company, Shanghai, China) containing 5 μl of PhosSTOP Phosphatase inhibitor Cocktail (Roche), and Western blot analysis was performed as previously described [12]. The following primary antibodies were used: phosphorylated-Akt (p-Akt) (D9E; 1:2000), Akt (C67E7; 1:2000), phosphorylated-c-Met (D26; 1:2000), c-Met (25H2; 1:2000) (Cell Signaling Technology, Inc., Beverly, MA, USA), ERK2 (C-14; 1:2000), phosphorylated-ERK1/2 (p-ERK1/2) (E-4; 1:1000), Runx2 (27-K; 1:2000), p27 (F-8; 1:2000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and GAPDH (1:2000; Zhongshan Goldenbridge Biotechnology Co., Ltd. Beijing, China). After extensive washing, immunocomplexes on PVDF membrane were detected with appropriate horseradish peroxidase–conjugated secondary antibodies (1:5000; Zhongshan Goldenbridge). The membranes were developed with the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to manufacturer’s instructions and exposed to Fuji Medical X-Ray Film (FUJIFILM Corporation, Tokyo, Japan). Quantification of protein expression detected by Western blot analysis was performed using Quantity-One software version 4.6.7 (Bio-Rad, Hercules, CA, USA).
Animal surgery and MSC injection
Fifty-five New Zealand rabbits (The Experimental Animal Center of Nanfang Hospital, Guangzhou, China) were maintained under specific pathogen-free conditions and were randomly assigned to four treatment groups. All animals received humane care according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996). The experiment protocol was approved by the Animal Ethics Committee at Southern Medical University. Five animals were left untreated as normal controls (Normal), while ANFH was induced in the remaining 50 animals by injection of horse serum (10 ml/kg; Hyclone) followed by prednisolone acetate (7.5 mg/kg; Pharmacia & Upjohn Co., Kalamazoo, MI, USA) through ear vein without anaesthesia as previously described [24]. After injecting the hormone, 200,000 U of penicillin was administrated intramuscularly into the buttock of each animal. Five rabbits with ANFH were left untreated as model controls (ANFH). Fifteen animals were treated with transplantation of HGF-expressing MSCs through the tunnel of core decompression (ANFH+MSC+HGF) as described previously [16]. Another 15 rabbits received blank Ad vector-infected MSCs as vector infection controls (ANFH+MSC+vector). The remaining 15 animals received transplantation of uninfected MSCs as treatment controls (ANFH+MSC). For each treatment group, 106 cells in 100 μl GM without FBS were transferred into the necrotic femoral head. During the period, no animal died unintentionally. Animals were killed by air injection through ear vein at 2 days, 2 weeks and 4 weeks post-treatment and femoral head tissue obtained. Unilateral tissue sample was paraffin embedded and sections were prepared for haematoxylin and eosin staining and immunolabelling with the following antibodies: anti-HGF antibody (BOSTER Bioengineering Co. Ltd., Wuhan, China), anti-p-ERK1/2 (E-4) and anti-p-Akt (D9E) as previously described [16]. RNA was also extracted from the other side of medullary cavity tissue and expression of p27, Runx2 and Osterix mRNA was determined using qRT-PCR. Pictures of the HE staining and immunohistochemistry (IHC) were taken during microscopic analysis (Nikon) and IPP 6.0 was used to count the number of empty lacunae, the number of haematopoietic medullary cells, and the IOD of staining in unit cavity area using at least ten randomly selected fields per section.
Statistical analysis
Data from the cellular experiments are expressed as the mean ± S.D. Data from animal experiments are expressed as the mean ± S.E. One-way ANOVA was applied to determine the statistical significance. Least significant difference or Dunnett’s T3 was performed for post hoc multiple comparisons. Differences among groups were considered to be statistically significant when P < 0.05. SPSS statistical software version 16.0 (SPSS, Chicago, IL, USA) was used to perform all statistical analyses.
Results
Isolation and characterization of rabbit MSCs
Pluripotency was determined by differentiating MSCs into osteoblasts, chondroblasts or adipocytes (Fig. 1). The osteoblasts appeared elongated and slim (Fig. 1A, panel f), and stained positively for both ALP activity and calcium deposition (Fig. 1A, panels g and h) and negatively for lipid accumulation and acidic mucosubstance (Fig. 1A, panels i and j). Chondroblasts stained positively for acidic mucosubstance with Alcian Blue (Fig. 1A, panel n), but not for adipocyte and osteoblast markers (Fig. 1A, panels l, m and o). Adipocytes contained lipid droplets in the cytoplasm and only stained positively with Oil Red O (Fig. 1A, panel t), and were negative for osteoblast and chondroblast markers (Fig. 1A, panels q–s). MSCs were negative for all the differentiation markers (Fig. 1A, panels a–e).
Fig 1.
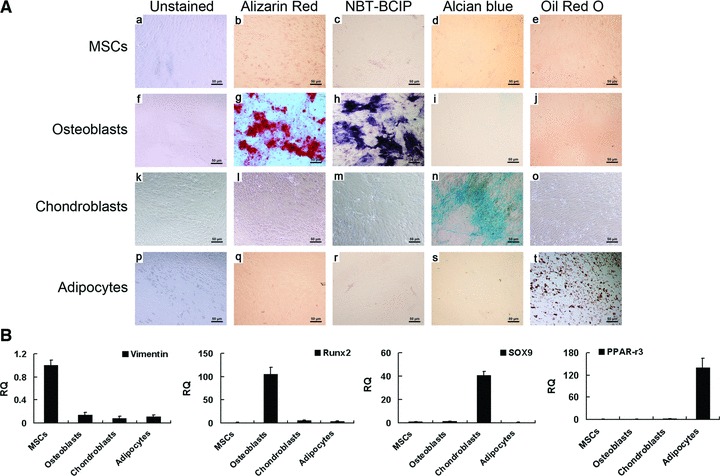
Differentiation of rabbit MSCs into specific cell lineages. (A) MSCs (a–e), osteoblasts (f–j), chondroblasts (k–o) and adipocytes (p–t) were left unstained (a, f, k and p), or stained with Alizarin Red (day 21, b, g, l and q), NBT-BCIP (day 14, c, h, m and r), Alcian blue (day 27, d, i, n and s) or Oil Red O (day 27, e, j, o and t). Scale bar ∇ 50 μm. (B) Quantitative RT-PCR analysis of mRNA expression of the indicated lineage-associated markers. MSCs, osteoblasts, chondroblasts and adipocytes specifically expressed Vimentin, Runx2, SOX9 and PPAR-gamma3, respectively. RQ: relative mRNA expression normalized to GAPDH.
Furthermore, osteoblasts expressed Runx2, chondroblasts expressed SOX9 and adipocytes expressed PPAR-gamma3 (Fig. 1B). In contrast, MSCs expressed these differentiation marker genes at low levels, but specifically expressed the MSC marker, Vimentin (Fig. 1B).
Effect of HGF concentration on the proliferation of MSCs
Previous studies have reported that HGF treatment effects proliferation and differentiation of MSCs with contradictory conclusions from different research groups. So we chose two concentrations, 20 ng/ml (H20, low concentration) and 100 ng/ml (H100, high concentration), to investigate the effects of HGF treatment on MSCs.
To analyse the effects of HGF concentration on proliferation of MSCs, cells were maintained in DM in the presence or absence of HGF for up to 7 days. Results of the EdU incorporation assay show that proliferation of MSCs positively correlates with the concentration of HGF (Fig. 2A and B). The cell viability assay show no significant difference between untreated cells or cells treated with 20 ng/ml of HGF (H20 treatment) from day 1 to 7, but, 100 ng/ml of HGF in DM (H100 treatment) showed an increase in cell viability at days 4 and 7 (Fig. 2C). Notably, this difference was more pronounced at day 4 than at day 7, indicating that H100 treatment has stronger effects on the proliferation of MSCs in the early stage of differentiation than in the late stage. In summary, the above results show that a high concentration of HGF (100 ng/ml) has a much stronger effect in promoting proliferation of MSCs than a low concentration of HGF (20 ng/ml).
Fig 2.
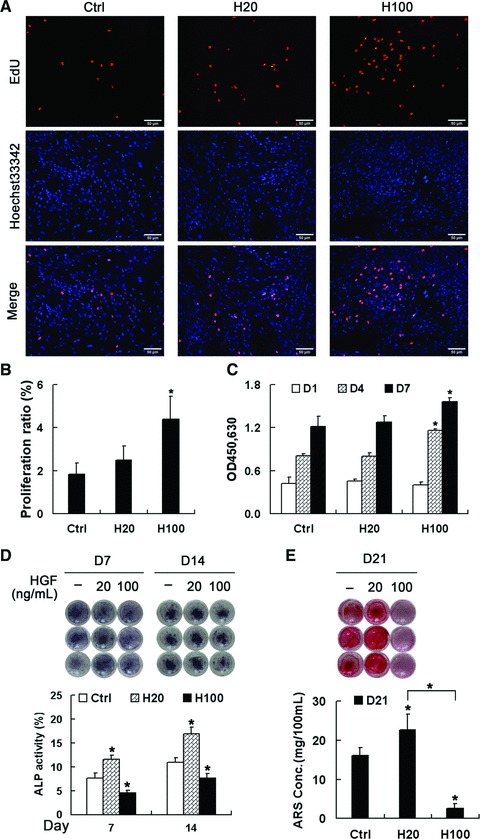
Effects of HGF concentration on MSC proliferation and osteogenic differentiation. (A, B) EdU incorporation assays in MSCs after treatment with HGF for 24 hrs. Scale bar ∇ 50 μm. (C) Cell viability assays after treatment with HGF for 1, 4 and 7 days. Both assays indicate that H100 treatment more strongly induces proliferation compared with H20 treatment. (D, E) MSCs were maintained in DM containing HGF for 21 days and the level of osteogenesis was assessed at the indicated times by staining with NBT-BCIP to show ALP (D, upper panel) or with alizarin red sulfate (AR-S) to show calcium deposition (E, upper panel). (D and E, lower panels) Quantification analysis showed that H20 promotes differentiation, which was strongly suppressed by H100. *P < 0.05, compared with control group at each time point.
Effect of HGF concentration on the differentiation of MSCs
For analysing the effects of HGF concentration on differentiation of MSCs, cells were differentiated in DM containing or not HGF for up to 21 days. The degree of differentiation of MSCs was evaluated by ALP activity at days 7 and 14, and calcium deposition at day 21. Results show that H20 treatment highly increased ALP activity compared with DM alone, which was partially suppressed by H100 treatment on both days 7 and 14 (Fig. 2D). Consistently, AR-S staining and quantification show that calcium deposition at the end of differentiation was much more in the H20-treatment group than the group cultured in DM alone (Fig. 2E). However, H100 treatment almost completely eliminated ECM mineralization. In summary, compared with the non-treatment group, treatment with 20 ng/ml HGF promotes both the early and late stages of differentiation of MSCs, which is inhibited by treatment with 100 ng/ml HGF.
Phosphorylation of c-Met is correlated with the concentration of HGF and may affect the differentiation of MSCs
Previous reports have demonstrated the importance of HGF/c-Met signalling for differentiation [25]. In this study, we first investigated the effect of short-term treatment of HGF on the activation of c-Met. Both H20 and H100 treatment for 15 min. activated c-Met compared with the non-treatment group, however, H20 treatment showed a much stronger effect than H100 (Fig. 3A).
Fig 3.
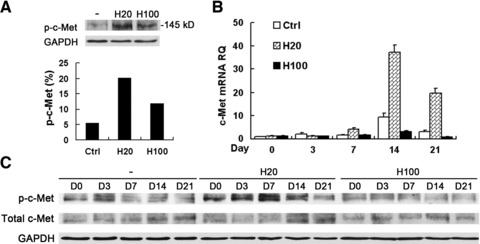
Activation of c-Met was highly enhanced by H20, but suppressed by H100 during osteogenic induction. (A) Western blot analysis of MSCs left untreated (–) or treated with HGF using anti-phosphorylated-c-Met (top panel) or anti-GAPDH (as a loading control, bottom panel) antibodies. H20 treatment led to a much stronger activation of p-c-Met than H100 treatment beginning from 15 min. (B) Quantitative RT-PCR of c-Met mRNA expression during differentiation with long-term treatment of HGF. H20 treatment stimulated significantly higher levels of c-Met, which were partially suppressed by H100 treatment compared with control. (C) C-Met activation during differentiation by Western blot analysis is in consistency with short-term treatment with HGF. RQ: relative mRNA expression normalized to GAPDH.
Next, the long-term effects of HGF treatment on c-Met expression and activation were examined. There was a general trend of increasing c-Met expression levels in all three groups after osteogenic induction to a peak level at day 14. However, the level of c-Met was highly up-regulated in the H20-treatment group, but greatly suppressed in the H100-treatment group, compared with the non-treatment group (control), especially at days 14 and 21 (Fig. 3B). These expression patterns were confirmed by Western blot analysis (Fig. 3C). H20 treatment produced the highest level of phosphorylated c-Met (p-c-Met), whereas H100 treatment gradually decreased the p-c-Met levels, compared with the control (Fig. 3C). Therefore, treatment with 20 ng/ml HGF not only induced the highest level of p-c-Met but also increased the expression level of total c-Met, which is further increased and maintained during the whole process of differentiation. In contrast, activation of c-Met by short-term treatment with 100 ng/ml HGF gradually decreases during long-term differentiation. Thus, low and high concentrations of HGF have dramatically different effects on c-Met activation and expression.
Requirement of ERK1/2 and Akt pathways for MSC proliferation and differentiation
ERK1/2 and Akt mediate important signalling pathways downstream of HGF/c-Met [26]. In this study, we first investigated the short-term effects of HGF treatment on the phosphorylation of ERK1/2 and Akt. H20 treatment showed a greater increase in Akt phosphorylation compared with H100 treatment and the effect was prolonged by at least 60 min. However, H100 treatment activated ERK1/2 more strongly than H20 treatment (Fig. 4A). Both ERK1/2 and Akt activation was completely abolished by 1 hr pre-treatment with 30 μM PD98059 and 100 nM Wortmannin, respectively. PD98059 acts in vivo as a highly specific inhibitor of MEK1 activation and the MAP kinase cascade including ERK1/2 [27-30] and prevents activation by upstream activators [29]. Wortmannin is a very potent, specific and direct inhibitor of PI3 kinase [31, 32] with irreversible and non-competitive effects, which in turn inhibits activation of downstream Akt. Thus, short-term treatment with different concentrations of HGF diversely affects activations of Akt and ERK1/2 signalling pathway.
Fig 4.
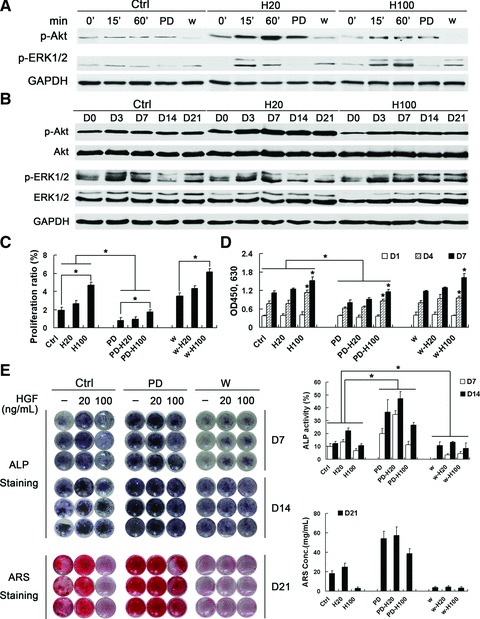
Effects of ERK1/2 and Akt pathways on MSC proliferation and differentiation. (A) Western blot analysis of ERK and Akt activation in MSCs after short-term treatment with HGF. H20 treatment resulted in greater Akt and weaker ERK1/2 phosphorylation in comparison with H100 treatment. Activation of ERK1/2 and Akt were inhibited by pre-treatment for 1 h with PD98059 (30 μM PD) or wortmannin (10 nM w), respectively. (B) Western blot analysis of ERK1/2 and Akt activation during differentiation with long-term treatment of HGF. Both ERK1/2 and Akt were continuously phosphorylated in similar pattern with short-term HGF treatment, which was maintained to the end of differentiation. (C) EdU incorporation assays in MSCs pre-treated with PD98059 and wortmannin for 1 hr prior to incubation with HGF for 24 hrs. (D) Cell viability assays after treatment with HGF for 1, 4 and 7 days. Assays indicate that MSC proliferation was significantly suppressed by PD98059, but not wortmannin pre-treatment. (E, F) MSCs were maintained in DM containing HGF for 21 days. The level of osteogenesis was assessed by staining for ALP activity and calcium deposition with NBT-BCIP and ARS, respectively. Quantification analysis showed that PD98059 pre-treatment strongly promoted differentiation which was greatly suppressed by wortmannin pre-treatment; *P < 0.05.
The role of HGF-induced ERK1/2 and Akt activation in MSC differentiation was further investigated. Both ERK1/2 and Akt were continuously phosphorylated during MSC differentiation. Notably, long-term H20 treatment still led to higher phosphorylation levels of Akt than H100 treatment, but decreased ERK1/2 activation between days 14 and 21 similarly with in the control (Fig. 4B). In contrast, H100 treatment maintained and even increased the phosphorylation level of ERK1/2 to the end of differentiation. Thus, short- and long-term effects on ERK1/2 and Akt phosphorylation are similar for treatment with the same concentration of HGF, but vary between treatments with different concentrations of HGF.
To determine whether ERK1/2 and Akt activation is required for HGF-induced MSC proliferation, we pre-treated cells with PD98059 and wortmannin and performed EdU incorporation assays (Fig. 4C). Inhibition of MEK1 prevented HGF-induced proliferation, indicating that ERK1/2 activation is required. In contrast, wortmannin pre-treatment did not significantly affect HGF-induced proliferation. Furthermore, cell viability assays over 7 days showed similar results (Fig. 4D). These data further indicate that H100 treatment affects proliferation more strongly than H20 treatment through ERK1/2 activation.
Cells were maintained in DM for 21 days in the presence of specific inhibitors and H20 or H100. Unexpectedly, inhibition of ERK1/2 signalling strikingly enhanced MSC ALP activity and calcium deposition, which was further enhanced by co-treatment with H20 (Fig. 4E). Interestingly, PD98059-induced differentiation was partially suppressed by H100 co-treatment. In contrast, pre-treatment with wortmannin significantly decreased ALP activity (Fig. 4E, left- and right-upper panels) and almost completely abolished ECM mineralization (Fig. 4E, left- and right-lower panels). Although HGF co-treatment increased wortmannin-induced ALP expression (Fig. 4E, left- and right-upper panels), it had no effect on calcium deposition (Fig. 4E, left- and right-lower panels).
Collectively, these results suggest that the ERK1/2 pathway, preferentially activated by 100 ng/ml of HGF, is required for MSC proliferation and partially inhibits differentiation. Furthermore, Akt activation, preferentially stimulated by 20 ng/ml of HGF, is indispensable for differentiation especially for ECM mineralization.
Effects of HGF concentration on expression of the cell cycle inhibitor, p27 and transcription factors required for osteogenic differentiation
It has been shown that HGF modulates cell proliferation and differentiation by regulating the expression of p27, which can trigger differentiation of stem cells [18]. Runx2 and Osterix are both osteoblast-specific transcription factors and their expression promotes osteoblast differentiation [33-36]. Quantitative PCR analysis showed that after osteogenic induction peak expression levels were reached at day 7 for p27 or at day 14 for Runx2 and Osterix, and then decreased until the end of differentiation (Fig. 5A–C). Compared with the control, H20 treatment significantly promoted their expression (P < 0.05), which was greatly suppressed by H100 treatment (P < 0.05) in contrast. The expression differences of p27 and Runx2 between H20 and H100 treatment were also confirmed by Western blot analysis (Fig. 5D).
Fig 5.
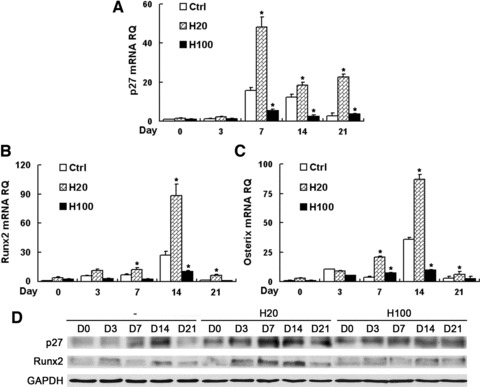
HGF regulates p27 and transcription factor expression during MSC differentiation. Quantitative RT-PCR was performed to detect mRNA expression of p27 (A), Runx2 (B) and Osterix (C) in MSCs maintained under osteogenic conditions containing HGF for 21 days. (D) Western blot analysis of p27 and Runx2 expression. H20 treatment induced a much higher expression levels of p27, Runx2 and Osterix, which were suppressed by H100-treatment compared with control group. RQ: relative mRNA expression normalized to GAPDH. *P < 0.05, compared with control group at each time point.
HGF-expressing MSCs can promote the recovery of ANFH
To determine whether the differential effects of low and high HGF concentrations on MSCs affects treatment efficacy in bone injury in vivo, an ANFH model was established in rabbit. Animals were treated by transplantation of HGF-expressing MSCs. We verified changes in phenotype by HE staining, protein expression by IHC and gene expression by qPCR analysis at different time points after treatment with MSCs expressing HGF.
The HE staining results show that treatment with uninfected MSCs or empty vector-infected MSCs increased the amount of haematopoietic tissue and reduced empty bone lacunae compared with the non-treatment ANFH group, which showed no significant tissue changes during the treatment period (Fig. 6A–C). However, the HGF-expressing MSC-treatment group showed a much more organized arrangement of the trabeculae, increased amount of bone matrix, increased number of osteoblasts at the edges of the trabeculae and newly generated capillaries on the bone plates (Fig. 6A–C). These results demonstrate that the HGF-expressing MSC-treated group achieved more therapeutic efficacy for ANFH compared with other treatment groups, which is similar with results from our previous study [16].
Fig 6.
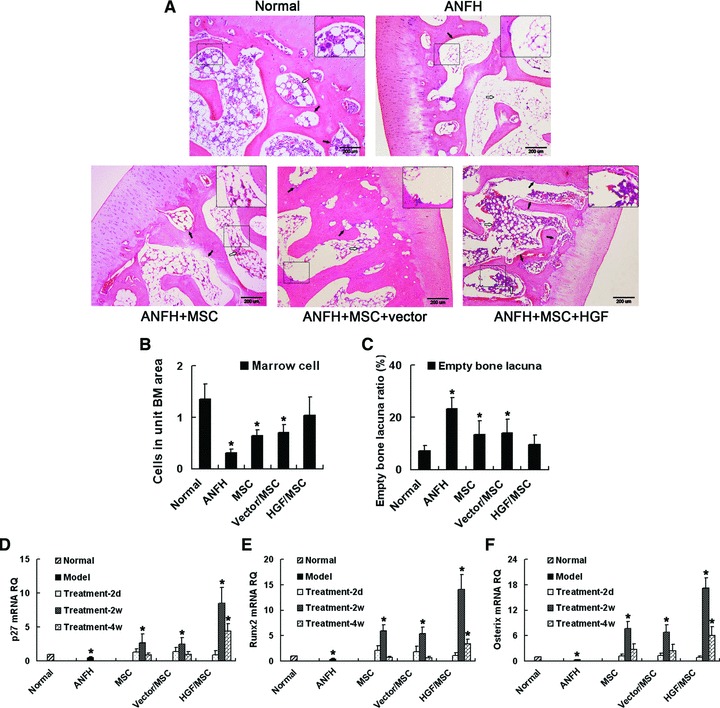
Transplantation of HGF-expressing MSCs promotes the recovery from ANFH. (A–C) Histopathological examination of treatment efficacy for hormone-induced ANFH by HE staining. ANFH was induced and treated with non-infected MSCs (ANFH+MSC), vector-infected MSCs (ANFH+MSC+vector) or HGF-expressing MSCs (ANFH+MSC+HGF). (A) Black arrows indicate osteoblasts along the trabeculae. White arrows indicate haematopoietic tissue in the medullary cavity (B) which and empty lacunae in the trabeculae (C) were semi-quantitatively analysed. The increased haematopoietic tissue and decreased empty lacunae were most significant in the HGF-expressing MSC-treatment group compared to the non-treatment group. Expression of p27 (D), Runx2 (E) and Osterix (F) was determined by qRT-PCR and all the highest expression levels occurred in the HGF-expressing MSC-treatment group. RQ: relative mRNA expression normalized to GAPDH. *P < 0.05, compared with the normal group. Scale bar ∇ 200 μm.
Results of IHC showed that after induction of ANFH, the HGF expression levels decreased to approximately 20% of the levels of the Normal group in vivo. Transplantation of HGF-expressing MSCs treatment sharply increased HGF levels at the local site and was maintained for about 2 weeks (Fig. 7A). After treatment for 4 weeks, the HGF level significantly decreased again, but was still higher than the normal level. The expression pattern of HGF after treatment with HGF-expressing MSCs in vivo is in line with our previous in vitro results [16]. However, treatment with non-infected or vector-infected MSCs did not significantly enhance the local HGF protein levels (Fig. 7A), while increased ERK1/2 activation compared with the non-treated ANFH group. But, the strongest activation of p-ERK1/2 was observed in the HGF-expressing MSC-treated group in which the p-ERK1/2 level was close to the Normal level and decreased after 2 weeks (Fig. 7B). Activation of Akt was increased 2 weeks after transplantation of all MSCs, and attained the highest level in the HGF-expressing MSC-treatment group compared with the other treatment groups (Fig. 7C). Consistently, the expression levels of p27, Runx2 and Osterix mRNA in the ANFH group were almost completely suppressed, but were increased 2 weeks after transplantation of HGF-expressing MSCs, which were significantly higher than in the other transplantation groups (P < 0.05; Figs. 6D–F).
Fig 7.
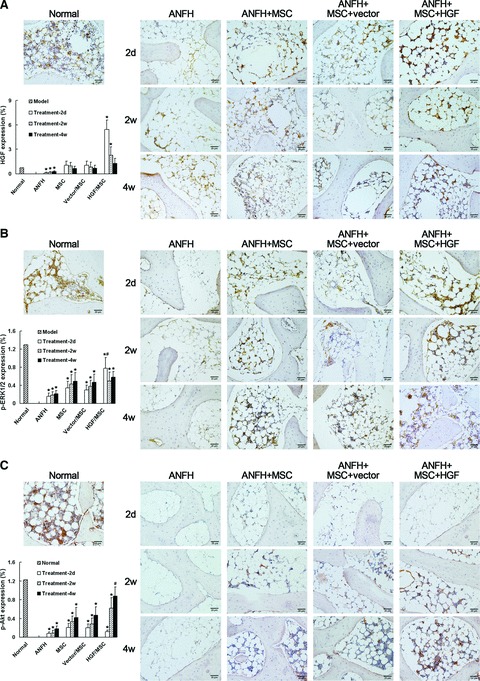
Immunohistochemical detection and semi-quantitative analysis of HGF expression (A), phosphorylation of ERK1/2 (p-ERK1/2) (B) and Akt (p-Akt) (C) in vivo after induction of hormone-induced ANFH and treatment with transplantation of MSCs. The level of HGF greatly decreased after induction of ANFH, but significantly increased 2 days after transplantation of MSCs, accompanied by increased p-ERK1/2. Two weeks post-transplantation HGF levels decreased, which was followed by a significant increase in p-Akt levels. The effects were most remarkable in the HGF-expressing MSC-treatment group. *P < 0.05, compared with the normal group. #P < 0.05, compared with the non-infected MSC-treated group. Scale bar ∇ 20 μm.
Discussion
Our in vitro studies demonstrate that HGF has diverse effects on rabbit MSC proliferation and differentiation depending on the concentration. H100 treatment induces proliferation of MSCs more strongly than H20 treatment. Furthermore, differentiation of MSCs is enhanced by H20 treatment, but partially inhibited by H100 treatment. Such concentration-dependent effects of HGF likely plays an important role in MSC-mediated tissue repair in vivo and can be exploited for treatment in the clinic.
Previous studies have shown that after myocardial infarction, liver injury and muscle injury, the inactive, single chain HGF precursor is cleaved by serine proteases to produce an active heterodimer [37-39]. Its concentration in serum increases quickly after injury before gradually decreasing and, in turn, leading to ischaemic disease progression [40, 41]. It was reported that HGF concentrations increased by 20-fold in rat serum within 1 hr after hepatectomy, then the concentration slowly decreased over the next 24 hr and was back to basal levels after 3 days [42]. The rapid increase in HGF concentration was suggested to be one of the signals for DNA synthesis in hepatocytes [42]. HGF was also shown to regulate cell fate in vitro, enhance proliferation, and inhibit differentiation of myogenic cells in the early stage of muscle tissue development until generating a sufficient number of cells to achieve proper tissue morphology, and myogenesis begins [18]. Thus, the rapid increase in HGF concentration after injury is likely to promote MSC proliferation to generate the appropriate number of cells required for tissue regeneration, which is then followed by a gradual decrease in HGF concentration to induce MSC differentiation for tissue repair. These studies show that the in vivo expression levels are tightly related with the dose-dependent functions of HGF in both proliferation and differentiation. However, although ‘cytokine supplement therapy’ for peripheral arterial disease has been presented over the last 10 years [43] and the efficacy for administration of HGF in improving the state of ischaemic illness has been demonstrated [44-46], no one has linked the therapeutic effect of HGF concentration changes on the treatment of ischaemic disease.
In our previous studies we showed that in vitro transfection of BMSCs with replication-deficient recombinant adenoviral vectors expressing the human HGF gene (Ad-HGF) lead to high levels of HGF expression (133 ng/ml) in the first week post-infection, which then decreased to 19 ng/ml about 2 weeks later [16]. Similarly, in this study we transplanted such HGF-expressing MSCs into the diseased region during the early stage of ANFH in a rabbit model and achieved excellent results. The animal experiments demonstrated that a high level of HGF expression immediately after transplantation, which may promote MSC proliferation for more than 1 week, during which the number of MSCs would continuously increase to meet the cell requirement for tissue reconstruction. The subsequent decrease in HGF levels promoted MSC differentiation in the osteogenic microenvironment for re-establishment of bone tissue, which is demonstrated by the increased number of osteoblasts. These results suggest that changes in HGF concentration enhance the ability of MSCs to induce femoral head tissue repair in hormone-induced ANFH. Thus, localized expression of HGF in vivo by transplantation of MSCs transfected with Ad-HGF is a promising treatment for ANFH.
In contrast to traumatic ANFH, significant down-regulation of HGF expression and the absence of tissue repair were observed in hormone-induced ANFH without therapy, which may be due to the decreased number and impaired activity of MSCs [47]. However, the significantly up-regulated expression of HGF following transplantation still efficiently promotes MSC-mediated tissue repair. At the end of observation (21 days), the HGF level in vivo was still higher than the Normal level, which is likely a result of transplanted HGF-expressing MSCs remaining in the tissue. Longer treatment times may show that the differentiation of MSCs and the HGF levels will eventually return to Normal levels when the transplanted MSCs are differentiated.
Activation of c-Met, the specific receptor of HGF, is required for critical processes during adulthood, including liver regeneration [48], wound healing [48] and MSC differentiation [25]. Unexpectedly, increased HGF expression after tissue injury is accompanied by a simultaneous decrease in c-Met mRNA expression [49]. In this study, H20 treatment but not H100 treatment increased c-Met receptor expression and induced stronger activation, and, in turn, may further affect the downstream signalling pathway to diversely regulate differentiation. Thus, c-Met expression may control the ability of MSCs to mobilize after HGF stimulation; high levels of c-Met promote differentiation and low levels induce proliferation.
Many studies demonstrated specificity in downstream signals of HGF/c-Met corresponding to distinct biological effects [26]. Among them, the ras/ERK1/2 pathway is necessary and sufficient for HGF-induced mitogenic effects. In contrast, the PI3K/Akt pathway seems to be mostly associated with motility, inhibition of apoptosis and induction of differentiation [12, 50]. In this study, we found that after inhibiting ERK1/2 phosphorylation, the differentiation of MSCs was significantly induced and proliferation was suppressed. Conversely, inhibition of Akt activation almost completely abolished extracellular mineralization. H20 treatment preferentially induces activation of the Akt pathway and, subsequently, high levels of p27, Runx2 and Osterix, thus promoting MSC differentiation. In contrast, H100 treatment enhances cell proliferation by strongly activating ERK1/2 signalling and suppressing expression of other factors. These findings suggest that different concentrations of HGF have divergent effects on cell cycle and transcriptional control and, in turn, modulate differentiation. In animal experiments increased activation of ERK1/2 and proliferation was observed shortly after transplantation of HGF-expressing MSCs. HGF expression decreased 2 weeks after transplantation, and the levels of p-Akt and osteogenic transcription factors increased and were sustained. Our results demonstrate that increased expression of HGF in MSCs promotes tissue repair.
Our findings demonstrate that the underlying mechanisms effecting proliferation and differentiation of MSCs differ between low and high concentration HGF environments. These results may help explain how HGF concentration changes after ischaemic injury helps tissue repair and how we can manipulate the concentration of HGF in vivo to improve the therapeutic efficacy.
Acknowledgments
This work was financially supported by State 863 Projects (2007AA02Z458), Program for New Century Excellent Talents in University (NCET-07-0410) & Science and Technology Project of Guangzhou (2009Z1-E441).
Authors’ contributions
QW carried out the cell experiments, participated in the immunoassay and animal experiment and performed the statistical analysis.
LZ participated in the design of the study, the immunoassay and molecular biology studies.
CZ carried out the analysis of immunohistochemistry results and participated in the animal experiment.
MZ participated in the animal experiment and the immunoassay.
WL participated in the molecular biology studies.
LM conceived of the study, and participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Satija N, Gurudutta G, Sharma S, et al. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev. 2007;16:7–23. doi: 10.1089/scd.2006.9998. [DOI] [PubMed] [Google Scholar]
- 2.Gangji V, Hauzeur J, Matos C, et al. Treatment of osteonecrosis of the femoral head with implantation of autologous bone-marrow cells. A pilot study. J Bone Joint Surg Am. 2004;86:1153–60. doi: 10.2106/00004623-200406000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Daltro G, Fortuna V, Araújo M, et al. Femoral head necrosis treatment with autologous stem cells in sickle cell disease. Acta Ortop Bras. 2008;16:23–7. [Google Scholar]
- 4.Stamm C, Westphal B, Kleine H, et al. Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet. 2003;361:45–6. doi: 10.1016/S0140-6736(03)12110-1. [DOI] [PubMed] [Google Scholar]
- 5.Sch chingerV, Erbs S, Els sser A, et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355:1210–21. doi: 10.1056/NEJMoa060186. [DOI] [PubMed] [Google Scholar]
- 6.Kinnaird T, Stabile E, Burnett M, et al. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res. 2004;94:678–85. doi: 10.1161/01.RES.0000118601.37875.AC. [DOI] [PubMed] [Google Scholar]
- 7.Noji S, Tashiro K, Koyama E, et al. Expression of hepatocyte growth factor gene in endothelial and Kupffer cells of damaged rat livers, as revealed by in situ hybridization. Biochem Biophys Res Commun. 1990;173:42–7. doi: 10.1016/s0006-291x(05)81018-6. [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto J. Gene therapy for liver cirrhosis. J Gastroenterol Hepatol. 2000;15:D33–6. doi: 10.1046/j.1440-1746.2000.02146.x. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda S, Goto Y, Baba T, et al. Enhanced secretion of cardiac hepatocyte growth factor from an infarct region is associated with less severe ventricular enlargement and improved cardiac function. J Am Coll Cardiol. 2000;36:115–21. doi: 10.1016/s0735-1097(00)00675-6. [DOI] [PubMed] [Google Scholar]
- 10.Xin X, Yang S, Ingle G, et al. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158:1111–20. doi: 10.1016/S0002-9440(10)64058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheehan S, Tatsumi R, Temm-Grove C, et al. HGF is an autocrine growth factor for skeletal muscle satellite cells in vitro. Muscle Nerve. 2000;23:239–45. doi: 10.1002/(sici)1097-4598(200002)23:2<239::aid-mus15>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 12.Forte G, Minieri M, Cossa P, et al. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23–33. doi: 10.1634/stemcells.2004-0176. [DOI] [PubMed] [Google Scholar]
- 13.Neuss S, Becher E, W ltje M, et al. Functional expression of HGF and HGF receptor/c-met in adult human mesenchymal stem cells suggests a role in cell mobilization, tissue repair, and wound healing. Stem Cells. 2004;22:405–14. doi: 10.1634/stemcells.22-3-405. [DOI] [PubMed] [Google Scholar]
- 14.Duan H, Wu C, Wu D, et al. Treatment of myocardial ischemia with bone marrow-derived mesenchymal stem cells overexpressing hepatocyte growth factor. Molecular Therapy. 2003;8:467–74. doi: 10.1016/s1525-0016(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 15.Cai L, Johnstone B, Cook T, et al. Suppression of hepatocyte growth factor production impairs the ability of adipose-derived stem cells to promote ischemic tissue revascularization. Stem Cells. 2007;25:3234–43. doi: 10.1634/stemcells.2007-0388. [DOI] [PubMed] [Google Scholar]
- 16.Wen Q, Ma L, Chen Y, et al. Treatment of avascular necrosis of the femoral head by hepatocyte growth factor-transgenic bone marrow stromal stem cells. Gene Therapy. 2008;15:1523–35. doi: 10.1038/gt.2008.110. [DOI] [PubMed] [Google Scholar]
- 17.Standal T, Abildgaard N, Fagerli U, et al. HGF inhibits BMP-induced osteoblastogenesis: possible implications for the bone disease of multiple myeloma. Blood. 2007;109:3024–30. doi: 10.1182/blood-2006-07-034884. [DOI] [PubMed] [Google Scholar]
- 18.Leshem Y, Spicer D, Gal-Levi R, et al. Hepatocyte growth factor (HGF) inhibits skeletal muscle cell differentiation: a role for the bHLH protein twist and the cdk inhibitor p27. J Cell Physiol. 2000;184:101–9. doi: 10.1002/(SICI)1097-4652(200007)184:1<101::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 19.D’Ippolito G, Schiller P, Perez-stable C, et al. Cooperative actions of hepatocyte growth factor and 1, 25-dihydroxyvitamin D3 in osteoblastic differentiation of human vertebral bone marrow stromal cells. Bone. 2002;31:269–75. doi: 10.1016/s8756-3282(02)00820-7. [DOI] [PubMed] [Google Scholar]
- 20.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105:2415–20. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Momcilovi O, Choi S, Varum S, et al. Ionizing radiation induces ataxia telangiectasia mutated-dependent checkpoint signaling and G (2) but not G (1) cell cycle arrest in pluripotent human embryonic stem cells. Stem Cells. 2009;27:1822–35. doi: 10.1002/stem.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takasu H, Sugita A, Uchiyama Y, et al. c-Fos protein as a target of anti-osteoclastogenic action of vitamin D, and synthesis of new analogs. Clin Res. 2006;116:528–35. doi: 10.1172/JCI24742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali-Benali M, Alary R, Joudrier P, et al. Comparative expression of five Lea Genes during wheat seed development and in response to abiotic stresses by real-time quantitative RT-PCR. Biochim. Biophys Acta. 2005;1730:56–65. doi: 10.1016/j.bbaexp.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Wen Q, Ma L, Chen Y, et al. A rabbit model of hormone-induced early avascular necrosis of the femoral head. Biomed Environ Sci. 2008;21:398–403. doi: 10.1016/S0895-3988(08)60060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosová I, Link D, Nolta J. shRNA-mediated decreases in c-Met levels affect the differentiation potential of human mesenchymal stem cells and reduce their capacity for tissue repair. Tissue Eng Part A. 2010;16:2627–39. doi: 10.1089/ten.tea.2009.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comoglio P. Pathway specificity for Met signalling. Nat Cell Biol. 2001;3:E161–2. doi: 10.1038/35083116. [DOI] [PubMed] [Google Scholar]
- 27.Crews C, Alessandrini A, Erikson R. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–80. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 28.Leevers S, Paterson H, Marshall C. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–4. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 29.Rosen L, Ginty D, Weber M, et al. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–21. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 30.Cowley S, Paterson H, Kemp P, et al. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–52. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi S, Kakita S, Takahashi I, et al. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267:2157–63. [PubMed] [Google Scholar]
- 32.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3, 4, 5-trisphosphate in neutrophil responses. Biochem J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 34.Otto F, Thornell A, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 35.Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 36.Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 37.Rosová I, Dao M, Capoccia B, et al. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells. 2008;26:2173–82. doi: 10.1634/stemcells.2007-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato T, Fujieda H, Murao S, et al. Sequential changes of hepatocyte growth factor in the serum and enhanced c-Met expression in the myocardium in acute myocardial infarction. Jpn Circ J. 1999;63:906–8. doi: 10.1253/jcj.63.906. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–9. doi: 10.1016/0006-291x(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–3. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 41.Sonnenberg E, Meyer D, Weidner K, et al. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol. 1993;123:223–35. doi: 10.1083/jcb.123.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paku S, Schnur J, Nagy P, et al. Origin and structural evolution of the early proliferating oval cells in rat liver. Am J Pathol. 2001;158:1313–23. doi: 10.1016/S0002-9440(10)64082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nabel EG. Gene therapy for cardiovascular disease. Circulation. 1995;91:541–8. doi: 10.1161/01.cir.91.2.541. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi S, Morishita R, Nakamura S, et al. Potential role of hepatocyte growth factor, a novel angiogenic growth factor, in peripheral arterial disease: downregulation of HGF in response to hypoxia in vascular cells. Circulation. 1999;100:II301–8. doi: 10.1161/circ.100.suppl_2.Ii-301. [DOI] [PubMed] [Google Scholar]
- 45.Morishita R, Makino H, Aoki M, et al. Phase I/iia clinical trial of therapeutic angiogenesis using hepatocyte growth factor gene transfer to treat critical limb ischemia. Arterioscler Thromb Vasc Biol. 2011;31:713–20. doi: 10.1161/ATVBAHA.110.219550. [DOI] [PubMed] [Google Scholar]
- 46.Rastogi S, Guerrero M, Wang M, et al. Myocardial transfection with naked DNA plasmid encoding hepatocyte growth factor prevents the progression of heart failure in dogs. Am J Physiol Heart Circ Physiol. 2011;300:H1501–9. doi: 10.1152/ajpheart.00636.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein R, Jilka R, Parfitt A, et al. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gentile A, Trusolino L, Comoglio P. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27:85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 49.Yanagita K, Matsumoto K, Sekiguchi K, et al. Hepatocyte growth factor may act as a pulmotrophic factor on lung regeneration after acute lung injury. J Biol Chem. 1993;268:21212–7. [PubMed] [Google Scholar]
- 50.Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]