

Abstract
Oncostatin M (OSM) is a pleiotropic cytokine of the IL-6 family and displays both pro-inflammatory and anti-inflammatory activities. We studied the impact of OSM on the gene activation profile of human synovial cells, which play a central role in the progression of inflammatory responses in joints. In synovial cells stimulated with lipopolysaccharide and recombinant human granulocyte-macrophage colony-stimulating factor, recombinant human OSM and native OSM secreted by human granulocytes both reduced the gene expression and secretion of IL-1β and CXCL8, but increased that of IL-6 and CCL2. This impact on synovial cell activation was not obtained using IL-6 or leukaemia inhibitory factor. Signal transducer and activator of transcription-1 appeared to mediate the effects of OSM on stimulated human synovial fibroblasts. In the murine dorsal air pouch model of inflammation, OSM reduced the expression of the pro-inflammatory cytokines IL-1β and TNF-α in lining tissues, and their presence in the cavity. These results as a whole suggest an anti-inflammatory role for OSM, guiding inflammatory processes towards resolution.
Keywords: oncostatin M, synovial fibroblasts, neutrophils, cytokines/chemokines, inflammation, murine dorsal air pouch
Introduction
Oncostatin M is a pleiotropic cytokine associated with haematopoiesis, development, tissue remodelling and inflammation [1]. Oncostatin M is structurally and functionally related to other members of the IL-6 family, such as IL-6, IL-11 and leukaemia inhibitory factor (LIF), all of which perform their signalling function via the common transducing receptor chain glycoprotein gp130 [2, 3]. Although somewhat redundant, each ensures a distinct array of biological functions via receptor combinations formed by initial binding to a specific receptor subunit (IL-6R, IL-11R, LIFR, OSMR), followed by pairing with gp130 to engage intracellular signalling [4]. Human OSM has the exceptional capability of engaging two different receptor complexes combining gp130 either with the LIF receptor subunit LIFR, a type-I OSM receptor, or with the type-II OSM receptor OSMRβ [5], eliciting distinct signal transduction events and cellular responses [6, 7]. As well, OSM seems to promote either pro-inflammatory or pro-resolution events, depending on the situation [8, 9-16]. In a number of inflammatory murine models, OSM is a potent suppressor of inflammation and tissue destruction [13, 17, 18]. Some of the disparities reported in the literature may also be due to differences in the source (bacteria, yeast, adenovirus) and the purity of the recombinant form. Alternative approaches have allowed to circumvent this problem; studies in OSM-deficient mice have established key roles for OSM in haematopoiesis [19] and confirmed its protective properties in autoimmune diseases, by regulating apoptotic thymocyte clearance [20].
Numerous cell types participate in the pathogenesis of inflammatory diseases. Polymorphonuclear neutrophils (PMNs), which play important roles in the early stages of inflammation, are often the first and most abundant cell type to migrate to injury sites and recognized as a major source of OSM [14, 21]. Polymorphonuclear neutrophils and OSM are both found in increased amounts in the synovial fluids of rheumatoid arthritis patients [22-24]. It is also clear that synovial fibroblasts play a central role in the inflammatory process [25, 26]. These cells respond to and stimulate inflammation, participate in autoimmune responses and promote bone destruction [27]. Synovial fibroblasts also interact with immune cells in the joint, like T cells [28], B cells [29], neutrophils and macrophages (reviewed by Ref. [26]). In view of the reported impact of synovial fibroblasts on cell engagement patterns in joint tissues, their activation status is regarded as an important regulatory checkpoint in the progression of inflammatory responses. In contrast, impact of OSM on the activation status of synovial cells remains elusive.
In this study, we sought to elucidate the impact that recombinant, as well as endogenous PMN-secreted OSM, might have on the gene activation profile of human synovial fibroblasts, and on a model of local inflammation. Together, the evidence obtained suggests that OSM may limit the onset of inflammation and steer progression of inflammatory processes towards resolution.
Materials and methods
All experiments involving human tissues received approval from the Université Laval ethics committee.
Materials
Normal human primary synovial fibroblasts (HSFs) were purchased from Asterand (Detroit, MI, USA). Lipopolysaccharide (LPS) from Escherichia coli O111:B4 was obtained from Sigma-Aldrich (Oakville, ON, Canada). Recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-1β were obtained from Peprotech Inc (Rocky Hill, NJ, USA). Recombinant human and murine OSM and the human neutralizing antibody were obtained from R&D Systems (Minneapolis, MN, USA). Non-specific goat IgG from R&D Systems was used as matched isotype control. Recombinant human LIF and IL-6 were purchased from Cedarlane (Burlington, ON, Canada). DFP (diisopropylfluorophosphate) was from EMD Biosciences (San Diego, CA, USA) and sodium orthovanadate from Sigma-Aldrich. Leupeptin and aprotinin were obtained from ICN Biomedicals (Irvin, CA, USA). The MEK inhibitor U0126 was from Cayman chemical (Ann Arbor, MI, USA) and the universal JAK inhibitor I was from Calbiochem (EMD chemicals, Gibbstown, NJ, USA). Cellular culture products DMEM/F12, serum, essential amino acids and lymphocyte separation medium were purchased from Wisent (St-Bruno, QC, Canada). ELISA kits for human IL-1β, human CXCL8, murine TNF-α, murine IL-1β were obtained from Biolegend (San Diego, CA, USA) kits for human and murine IL-6 were obtained from Biosource Invitrogen (Carlsbad, CA, USA) and the kit for human OSM was obtained from Antigenix America Inc. (Huntington Station, NY, USA). Antibodies used for immunoblots were: rabbit anti-phospho-signal transducer and activator of transcription-1 (STAT-1) (Tyr701) and rabbit anti-STAT-1 from Santa Cruz Technology (Santa Cruz, CA, USA), rabbit anti-phospho-ERK1/2 from EMD Millipore (Billerica, MA, USA) and the HRP-linked donkey anti-rabbit antibody from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Polymorphonuclear neutrophil isolation and stimulation
Informed consent was obtained in writing from all providers of human tissues. Data collection and analyses were performed anonymously. Polymorphonuclear neutrophils were isolated as originally described [30] with modifications [31]. Briefly, venous blood collected on isocitrate anticoagulant solution from healthy volunteers was centrifuged (250 χ g, 10 min.) and the resulting platelet-rich plasma was discarded. Leucocytes were obtained following sedimentation of erythrocytes in 2% Dextran T-500 (Sigma-Aldrich). Polymorphonuclear neutrophils were then separated from other leucocytes by centrifugation on a 10-ml lymphocyte separation medium. Contaminating erythrocytes were removed using 20 sec. of hypotonic lysis. Purified granulocytes (>95% neutrophils, <5% eosinophils) contained less than 0.2% monocytes, as determined by esterase staining. Viability was greater than 98%, as determined by trypan blue dye exclusion. The entire cell isolation procedure was carried out under sterile conditions at room temperature.
Polymorphonuclear neutrophils were re-suspended at a concentration of 5 χ 106 cells/ml in DMEM/F12 (50/50) supplemented with 2% heat-inactivated foetal calf serum (FCS), non-essential amino acids, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 0.2 μg/ml diisopropylfluorophosphate and 0.1 U/ml adenosine deaminase (ADA), the latter to prevent accumulation of endogenous adenosine in the medium, thus minimizing the previously demonstrated modulating effects of adenosine on inflammatory factor production by PMNs [32-34]. Stimulation consisted of exposing PMNs for 4 hrs to LPS (100 ng/ml) and GM-CSF (1.4 nM) at 37°C. Cell-free supernatants of stimulated PMNs were used to stimulate HSFs or stored at −20°C for cytokine measurement.
Primary human synovial fibroblast culture and stimulation
Human primary synovial fibroblasts were cultured in DMEM/F12 (50/50) containing 10% FCS at 37°C in a humidified 5% CO2 atmosphere and passaged with trypsin/EDTA treatments. Prior to stimulation, confluent cell layers in 25-cm2 flasks (for gene expression analysis) or 6-well plates (for immunoblots) were starved for 24 hrs in serum-free culture medium. Stimulations consisted of a 24 hrs exposure to cell-free supernatants of PMN culture, or to fresh medium containing LPS (100 ng/ml) and GM-CSF (1.4 nM), in the absence or presence of recombinant OSM, LIF or IL-6 (10 ng/ml each), as indicated. When mentioned, an anti-OSM neutralizing antibody (10 μg/ml) was added to the cell-free supernatant 30 min. before HSF stimulation. When used, the MEK inhibitor U0126 (10 μM) and JAK inhibitor I (0.5 μM) were added to the culture medium 15 min. before the cells stimulation. All HSF experiments were performed between cell passages 9 and 14. Cell layers were rinsed twice with PBS before RNA isolation.
Mouse dorsal air pouches
The Université Laval research animal protection committee approved the experimental design involving mice. Air pouches were raised on the dorsum of 6- to 8-week-old CD1 mice by subcutaneous injection of 5 ml of sterile air on day 0 and 3 ml on day 3. Experiments were conducted on day 6. Individual air pouches (one per mouse) were injected either with LPS alone (500 ng in 1 ml of endotoxin-free PBS) or in combination with recombinant murine OSM (25 μg/ml). Mice were sacrificed 1, 4 or 8 hrs after these injections and the air pouch was washed twice with ice-cold PBS (total of 2 ml). Leucocyte suspensions were enumerated with an automated cell counter (Cellometer auto T4; ESBE, Markham, ON, USA) and cell-free supernatants (exudates) were used for cytokine measurements. The subcutaneous connective tissue limiting the pouches was biopsied and processed for mRNA expression analysis. At least eight mice per group were used in each of two separate experiments.
RNA isolation
Total RNA from HSFs and pouch connective tissue were isolated using Trizol (Gibco, Burlington, VT, USA) according to the manufacturer’s protocol, with modifications. Briefly, cells or tissue were mixed into 1 ml Trizol and 200 μl of chloroform were added. After mixing, samples were centrifuged at 10,000 χ g for 15 min. at 4°C. The aqueous phase was mixed with an equal volume of isopropanol, held at room temperature for 10 min. and then centrifuged at 12,000 χ g for 10 min. at 4°C. Supernatants were discarded and the precipitated RNA pellets were washed twice using 500 μl of 75% ethanol. Pellets were allowed to air-dry for 5–10 min. and resuspended in RNAse-free water. Total mRNA was quantitated using a Qubit™ fluorometer (Invitrogen Life Technology, Carlsbad, CA, USA).
cDNA reverse transcription and real-time PCR
First-strand cDNA synthesis was performed with 1 μg of total RNA with Superscript II® reverse transcriptase (Invitrogen) under manufacturer-recommended conditions, using 500 ng of random hexamers. Amplification of cDNA was performed in a real-time PCR rotor-Gene 3000 analyser operated with Rotor-Gene software version 6.0.19 (Corbett Research, Mort lake, New South Wales, Australia). Each sample consisted of 1 μl of cDNA, 1.3 mM MgCl2, 0.2 mM dNTP, 500 nM of primers, 0.1 U of TaqDNA polymerase (Roche Diagnostics, Indianapolis, IN, USA) and SYBR Green I dye (Invitrogen; 1:30,000 dilution) in a reaction volume of 20 μl. Amplification was achieved using 35 cycles of 95°C, 58°C and 72°C steps of 20 sec. each. Reaction specificity was ascertained after each amplification using the Melt® procedure (58–99°C, 1°C/5 sec.), according to the manufacturer’s protocol. The difference in expression level was determined by normalization to the expression of GAPDH as previously described [35]. The human and murine primers used are listed in Table 1.
Table 1.
Sequences of primers for real-time PCR
| Species | Gene | Forward | Reverse |
|---|---|---|---|
| Human | GAPDH | 5′-CGAGATCCCTCCAAAATCAA-3′ | 5′-TTCACACCCATGACGAACAT-3′ |
| IL-1β | 5′-GGACAAGCTGAGGAAGATGC-3′ | 5′-TCGTTATCCCATGTGTCGAA-3′ | |
| IL-6 | 5′-CACAGACAGCCACTCACCTC-3′ | 5′-TTTTCTGCCAGTGCCTCTTT-3′ | |
| CXCL8 | 5′-GTGCAGTTTTGCCAAGGAGT-3′ | 5′-CTCTGCACCCAGTTTTCCTT-3′ | |
| CCL2 | 5′-AGCAGCAAGTGTCCCAAAGA-3′ | 5′-TTGGGTTTGCTTGTCCAGGT-3 | |
| Mouse | GAPDH | 5′-aactttggcattgtagaagg-3′ | 5′-acacattggggttaggaaca-3′ |
| IL-1β | 5′-tcacagcagcacatcaacaa-3′ | 5′-tgtcctcatcctggaaggtc-3′ | |
| IL-1α | 5′- gttctgccattgaccatctc-3′ | 5′-ctcagccgtctcttcttcag-3′ | |
| IL-1RA | 5′-ttgtgccaagtctggagatg-3′ | 5′-ttctcagagcggatgaaggt-3′ | |
| IL-6 | 5′-agttgccttcttgggactga-3′ | 5′-tccacgatttcccagagaac-3′ | |
| TNF-α | 5′-tagccaggagggagaacaga-3′ | 5′-ttttctggagggagatgtgg-3′ | |
| CCL3 | 5′-accatgacactctgcaacca-3′ | 5′-cccaggtctctttggagtca-3′ | |
| CCL4 | 5′-tgtctgccctctctctcctc-3′ | 5′-gagcaaggacgcttctcagt-3′ | |
| CCL8 | 5′-taaggctccagtcacctgct -3′ | 5′-tctggaaaaccacagcttcc-3′ | |
| CXCL2 | 5′-agtgaactgcgctgtcaatg-3′ | 5’-ttagccttgcctttgttcag-3′ |
Immunoblots and densitometry
Where mentioned, HSFs were pre-incubated for 15 min. with the MEK inhibitor U0126 (10 μM) or Jak inhibitor I (0.5 μM) before stimulation with LPS and GM-CSF ± OSM as described earlier. Following 24 hrs of appropriate treatments, cells were put on ice, washed with cold PBS and scraped in lysis buffer (10 mM tris buffer pH 7.4, 1% sodium dodecyl sulfate (SDS), containing the following antiprotease cocktail: 0.2 μg/ml diisopropylfluorophosphate (DFP), 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1 mM sodium orthovanadate). Samples were boiled for 5 min., centrifuged at 12,000 r.p.m. for 15 min. and supernatants were stored at −20°C for use in immunoblots. Protein concentration was measured using the Quant-iT™ Protein Assay Kit (Invitrogen Corporation, Carlsbad, CA, USA) and 25 μg of total proteins for each sample were subjected to 10% SDS-PAGE and transferred to immobilon membranes (Millipore, Bedford, MA, USA). Membranes were soaked for 30 min. at room temperature in tris buffered saline (TBS; 25 mM Tris-HCl pH 7.6, 0.2M NaCl, 0.15% Tween 20) containing 5% (w/v) bovine serum albumin, washed three times in TBS and incubate overnight at 4°C with anti-phospho-STAT-1 (1/200) or anti-STAT-1 (1/200). For the detection of phospho-ERK1/2, membranes were blocked at 37°C for 30 min. with gelatin and incubated 1 hr at 37°C with the anti-phospho-ERK1/2 (1/1,000). Membranes were then washed three times in TBS and incubated for 45 min. with the HRP-linked anti-rabbit antibody (1/10,000). Enzyme expression was revealed on thermo scientific CL-XPosure films (Fisher Scientific, Toronto, Canada) with ECL-Plus (PerkinElmer Life Sciences, Boston, MA, USA). Immunoblot autoradiograms were digitalized and densitometric analyses (integrated density of bands) were performed with the NIH Image software (http://rsb.info.nih.gov).
Measurement of secreted cytokines/chemokines
All cytokine/chemokine measurements were performed with ELISA kits, following the manufacturer’s instructions. OSM in PMN supernatants, as well as IL-6, CXCL8, CCL2 and IL-1β from HSF supernatants (control or stimulated for 24 hrs as described earlier) were thus quantified. For IL-1β measurement, stimulated HSFs were washed with PBS, scraped into PBS containing protease inhibitors (leupeptin; 10 μg/ml, aprotinin; 10 μg/ml and DFP; 0,2 μg/ml) and centrifuged at 12,000 χ g for 15 min. at 4°C. Murine IL-1β, TNF-α, IL-6 and CCL2 in the cell-free exudates of dorsal air pouches were also quantified.
Statistical analysis
Where applicable, statistical analysis was performed with the nonparametric ANOVA (Kruskal–Wallis test). Pairwise comparisons were performed with the Mann–Whitney U-test (Prism 5.0c software; Graphpad Inc., La Jolla, CA, USA). Differences were considered significant when P < 0.05.
Results
Recombinant OSM decreases levels of IL-1β mRNA and secreted protein in stimulated HSFs
A 24-hr stimulation with the 100 ng/ml LPS and 1.4 nM GM-CSF combination up-regulated IL-1β, IL-6, CXCL8 (IL-8) and CCL2 (MCP-1) mRNAs respectively, by factors of 224 ± 58, 433 ± 29, 407 ± 66 and 26 ± 7 (mean ± S.E.M., n ∇ 6, not illustrated) compared to resting cells. The impact of recombinant OSM on the expression of these genes in stimulated primary HSFs, is shown in Figure 1. Oncostatin M reduced this stimulation by 78% in the case of IL-1β (Fig. 1A, black bar), an effect lessened in the presence of specific OSM-neutralizing antibody (OSM + anti-OSM; dark grey bar) but not by the isotype-matched non-specific antibody (light grey bar). The specific neutralizing antibody by itself had no significant effect on IL-1β expression (white bar). Oncostatin M had a similar effect on CXCL8 up-regulation, while it increased the up-regulation of IL-6 and CCL2 (Fig. 1B). Neither IL-1α nor TNF-α mRNAs were detected in HSFs under any of the conditions tested. Oncostatin M had no significant impact on the expression of any of these genes in unstimulated cells (data not shown). In HSFs stimulated with the classical agonist IL-1β (30 nM), OSM had similar effects to those mentioned earlier. Up-regulation of IL-1β and CXCL8 mRNA expression were reduced respectively, by 64.1% ± 11.8 and 84.4% ± 5.4 while IL-6 and CCL2 up-regulation were enhanced by 2.35 ± 0.64 and 2.32 ± 0.81 folds (mean ± S.E.M., n ∇ 3).
Fig 1.
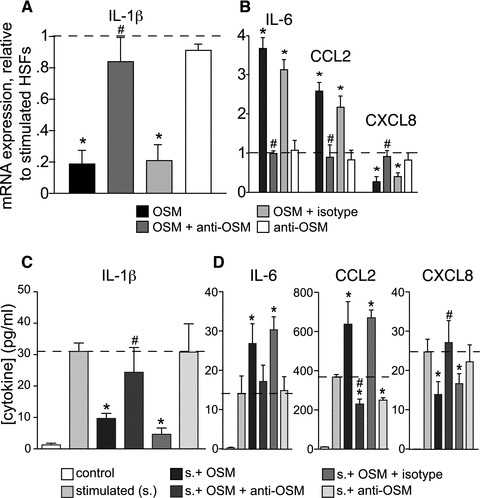
Impact of OSM on gene expression (A, B) and protein secretion (C, D) in stimulated HSFs. HSFs were stimulated for 24 hrs with a combination of LPS (100 ng/ml) and GM-CSF (1.4 nM). Human recombinant OSM (10 ng/ml) was added with or without 10 μg/ml of neutralizing anti-OSM, or isotype-matched non-specific antibody. Levels of IL-1β (A), IL-6, CXCL8 and CCL2 mRNA (B) were measured by real-time PCR. Results (mean ± S.E.M., n ∇ 6) are expressed as multiples relative to stimulated cells (dotted line). Protein levels of IL-1β (C), IL-6, CCL2 and CXCL8 (D) in supernatants were determined by ELISA (mean ± S.E.M., n ∇ 6). *Significantly different (P < 0.05) from stimulated cells. #Significantly different from the stimulated cell in the presence of OSM. Control: unstimulated cells.
We next studied the impact of recombinant OSM on the secretion of cytokines by stimulated HSFs. Secretion of IL-1β was markedly reduced by OSM (P < 0.01; Fig. 1C). Oncostatin M also decreased the secretion of CXCL8, while increasing that of IL-6 and CCL2 (Fig. 1D), consistent with patterns observed for mRNA. In each case, the influence of OSM on protein secretion could be reversed by the presence of its neutralizing antibody, while the isotype control antibody had no significant effect (Fig. 1C and D, pale grey bars).
Polymorphonuclear neutrophil-secreted OSM decreases IL-1β mRNA and protein levels in stimulated HSFs
To ascertain that the activities of recombinant OSM were not merely a result of its source, we sought to determine the impact of native, endogenous OSM on the cytokine expression profile of HSFs. We stimulated freshly isolated human PMNs with LPS and GM-CSF, a condition known to induce OSM secretion in these cells [21]. Stimulations were performed in presence of ADA (0.1 U/ml), a condition which prevents accumulation of endogenous adenosine in cell suspensions, thus eliminating the well-documented modulating effects of adenosine on neutrophil activation [32-34]. We thus obtained OSM concentrations of about 4 ng/ml (based on ELISA) in supernatants of PMN cell suspensions (Fig. 2A) and used these supernatants to stimulate HSFs in culture. Supernatants from resting PMNs were used as control. Fold increases in gene expression were as follows: 14.4 ± 3 for IL-1β, 38.9 ± 7.9 for IL-6, 35.7 ± 9.3 for CXCL8 and 6.2 ± 0.4 for CCL2 (mean ± S.E.M., n ∇ 6, not shown). Adding neutralizing anti-OSM antibody to the supernatant caused levels of IL-1β mRNA to double, while significantly decreasing those of IL-6 and CCL2 (Fig. 2B) and slightly increasing CXCL8 mRNA. At the level of cytokine secretion, the anti-OSM antibody had a similar impact, causing IL-1β concentrations to double (Fig. 2C), while decreasing that of IL-6 and CCL2 by more than 50%. The antibody also increased CXCL8 secretion. These results corroborate those obtained with recombinant OSM, thereby confirming that both OSM sources share comparable pro-resolution properties, notably by modulating the expression of inflammatory genes, including IL-1β, in HSFs.
Fig 2.
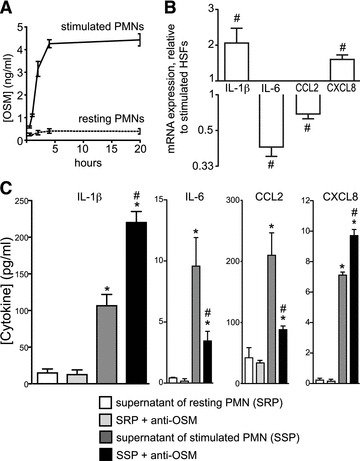
Impact of PMN-derived OSM on gene expression and protein secretion in stimulated HSFs. PMNs were stimulated with a combination of LPS (100 ng/ml) and GM-CSF (1.4 nM) for 4 hrs. (A) OSM concentrations in supernatants of resting or LPS/GM-CSF-stimulated PMNs were measured by ELISA (mean ± S.E.M., n ∇ 3). (B) HSFs were incubated for 24 hrs with supernatants of resting or stimulated PMNs with or without anti-OSM neutralizing antibody (10 μg/ml). Expression levels are presented relative to the levels for cells stimulated in the absence of anti-OSM (as reciprocals in the case of decreases), as measured using real-time PCR (mean ± S.E.M., n ∇ 4). (C) IL-1β, IL-6, CCL2 and CXCL8 concentrations were quantified in cell-free supernatants by ELISA. Results are expressed as mean ± S.E.M. (n ∇ 4). *Significantly different from SRP. #Significantly different (P < 0.05) from SSP.
Neither LIF nor IL-6 produces the same effects as OSM
We compared the response of HSFs to exogenous OSM with those of two other members of this family of cytokines, LIF and IL-6, to address the specificity of the former. Responses of stimulated HSFs to OSM, LIF or IL-6 are shown in Figure 3. Neither IL-6 (10 ng/ml) nor LIF (10 ng/ml) produced effects similar to those of OSM on IL-1β, IL-6, CXCL8 or CCL2 expression. LIF slightly increased IL-6 mRNA expression as previously reported [15] but had no effect on expression of the other three genes. IL-6 had no significant impact on the expression of any of these genes. Even at concentrations up to 100 ng/ml, neither LIF nor IL-6 modulated the mRNA response to any significant degree (data not shown).
Fig 3.
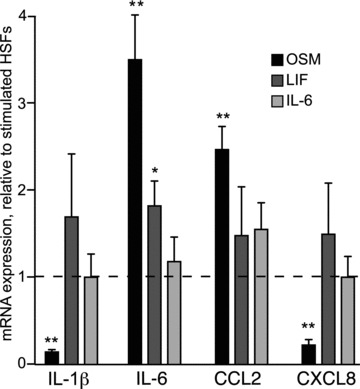
Response of HSF mRNA levels to LIF and IL-6. HSFs were stimulated with a combination of LPS and GM-CSF for 24 hrs in the absence or presence of OSM, LIF or IL-6 (10 ng/ml each). Levels of IL-1β, IL-6, CXCL8 and CCL2 mRNA were quantified by real-time PCR. Results (mean ± S.E.M., n ∇ 3) are expressed as multiples relative to stimulated cells (dotted line). *P < 0.05, **P < 0.01 from the stimulated condition.
Implication of the JAK/STAT pathway
Reported intracellular pathways positively linked with OSM in other cell types include the JAK/STAT (STAT-1 and 3) and MAPK (ERK1/2 and p38) pathways [1, 4, 36]. Impact of OSM on the activation of ERK1/2 and STAT-1 in stimulated HSFs, as assessed by their level of phosphorylation, are presented in Figure 4A. While phosphorylation of ERK1/2 and STAT-1 were marginal in response to stimulations with LPS and GM-CSF, levels significantly increased in the presence of OSM. For both ERK1/2 and STAT-1, this increase could be reversed by the OSM-neutralizing antibody, but not by the matched isotype non-specific antibody. MEK inhibition completely abolished ERK1/2 phosphorylation, and STAT-1 phosphorylation partially as well. Reciprocally, JAK inhibitor I inhibited the STAT-1 phosphorylation and decreased that of ERK 1/2. Signal for STAT-3 was weak compared to that of STAT-1 in HSFs, but its phosphorylation levels consistently followed that of STAT-1 (data not shown). Moreover, phosphorylation of p38 MAPK was never observed in our experiments. ERK1/2 appeared slightly phosphorylated in resting HSFs. By itself, OSM induced STAT-1 phosphorylation in resting HSFs but this did not result in significant effects on any of the expression of the studied genes (vide supra). These results confirm a link between OSM and the JAK/STAT pathway in HSFs and provide additional evidence for the cross-talk existing between the JAK/STAT and ERK signalling pathways [37, 38]. At the mRNA level, JAK inhibition totally abolished the effects of OSM on IL-1β, CXCL8, IL-6 and CCL2 (Fig. 4B). In contrast, presence of the MEK inhibitor only affected the increase of IL-6 mRNA, indicating that the JAK/STAT pathway is predominant in the OSMRβ transduction in stimulated HSFs. A summary of observations relating to OSMRβ activation in stimulated HSFs is depicted in Figure 4C.
Fig 4.
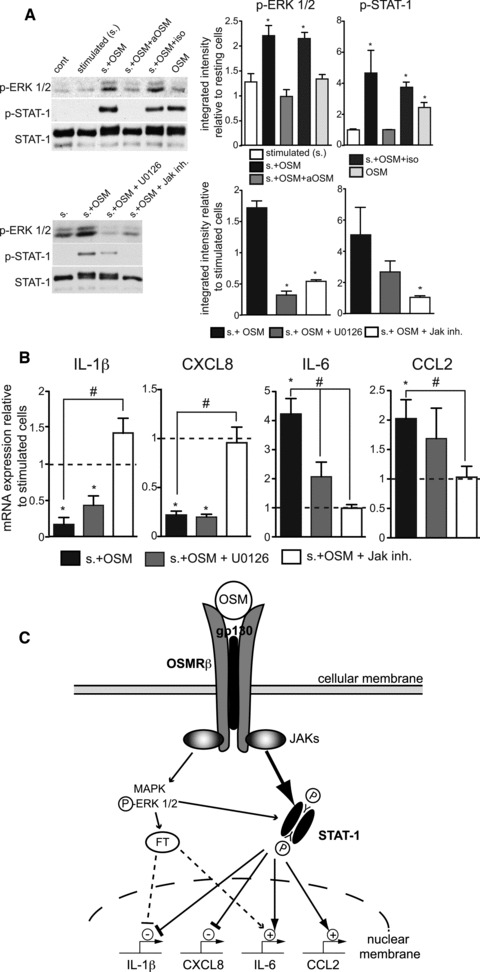
Signalling pathways mediating the OSM-effects on the IL-1β, CXCL8, IL-6 and CCL2 expression in stimulated HSFs. (A) HSFs were stimulated or not with a combination of LPS and GM-CSF for 10 min. in the absence or presence of OSM (10 ng/ml) and its neutralizing antibody (10 μg/ml), as indicated, prior to lysis and subsequent immunoblot analysis using primary antibodies raised against phospho-ERK1/2, phospho-STAT-1 (Tyr 701) and total STAT-1 (as a loading control). Immunoblots shown are representative of three independent experiments; bands were quantified by densitometric analysis and results presented as mean ± S.E.M. When indicated, cells were pre-incubated for 15 min. with the MEK inhibitor U0126 (10 μM) or the Jak inhibitor (0.5 μM) before stimulation in the presence of OSM. *P < 0.05 from the control condition (fixed at 1). (B) Otherwise identical conditions were used to study the consequences of a 24-hr stimulation in the presence of OSM and the metabolic inhibitors, on the mRNA expression of IL-1β, CXCL8, IL-6 and CCL2 by real-time PCR. Results (mean ± S.E.M., n ∇ 4) are expressed as multiples relative to stimulated cells (dotted line). *P < 0.05 from the stimulated condition, #P < 0.05 from the s. + OSM condition. (C) Schematic representation of the OSMRβ signalling in LPS and GM-CSF stimulated HSFs. OSM receptor engagement induces activation/phosphorylation of STAT-1 and also increases ERK1/2 phosphorylation. This leads to a STAT-1-dependent negative regulation of the IL-1β and CXCL8 genes transcription and to the up-regulation of the IL-6 and CCL2 genes.
Oncostatin M reduces inflammatory responses to LPS in the murine air pouch model
In view of clear anti-inflammatory effects of both recombinant and native OSM on cultured HSFs, we evaluated the potential of OSM as a modulator of inflammatory reactions in vivo, using an inflammatory model sharing key structural features with the synovial environment, namely the murine dorsal air pouch [39]. Injecting LPS alone or in combination with OSM into the air pouches and then sacrificing the mice 1, 4 or 8 hrs later, we assessed the impact of OSM on leucocyte migration, on the expression of ten genes (recognized as being solicited in this model according to; Ref. [33]) in the pouch connective tissue lining (composed primarily of fibroblasts, macrophages and endothelial cells) and on the accumulation of cytokines in pouch exudate. OSM had no significant effect on the number of leucocytes migrating into LPS-injected air pouches (Fig. 5). This cell population is composed of 90% ± 1.2 PMNs; 5% ± 0.08 lymphocytes–monocytes at 4 hrs and 80% ± 1.6; 15% ± 0.3 respectively at 8 hrs. Ratios were not different in the presence of OSM (data not shown). Based on real-time PCR measurement of mRNA expression, OSM significantly inhibited IL-1β, IL-1α, TNF-α, CCL3, CCL4, CCL8 and CXCL2 expression in the pouch lining tissues, albeit with different kinetics (Fig. 6). IL-1β mRNA levels were reduced by half in the presence of OSM at the 1-hr point, while IL-1α expression was reduced by nearly 70%, while the expression of IL-1RA remained essentially unchanged. OSM delayed the onset of TNF-α mRNA synthesis, with marked inhibition at the 4-hr point, and increased IL-6 mRNA levels at t ∇ 8 hrs. CCL8 mRNA levels were significantly delayed in the presence of OSM, not approaching those reached in its absence until the 8-hr point. OSM also decreased levels of mRNA corresponding to macrophage inflammatory proteins (MIPs) CCL3, CCL4 and CXCL2. These three genes had similar expression time courses after LPS injection, with maximum levels at t ∇ 1 hr, while OSM significantly reduced their expression at 1-hr and at the 4-hr point in the case of CCL4. The expression of three other chemokines, CCL2, CXCL1 (KC) and CXCL3 (MIP-2β), were not significantly affected by OSM (data not shown).
Fig 5.
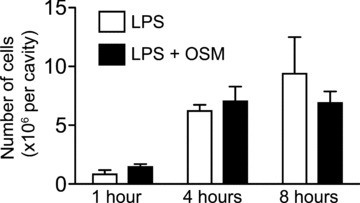
Impact of OSM on the migration of leukocytes to murine dorsal air pouches. Air pouches were injected with LPS (500 ng/ml) alone or in combination with recombinant murine OSM (25 μg/ml). Leukocytes were enumerated in the exudates, at indicated times.
Fig 6.
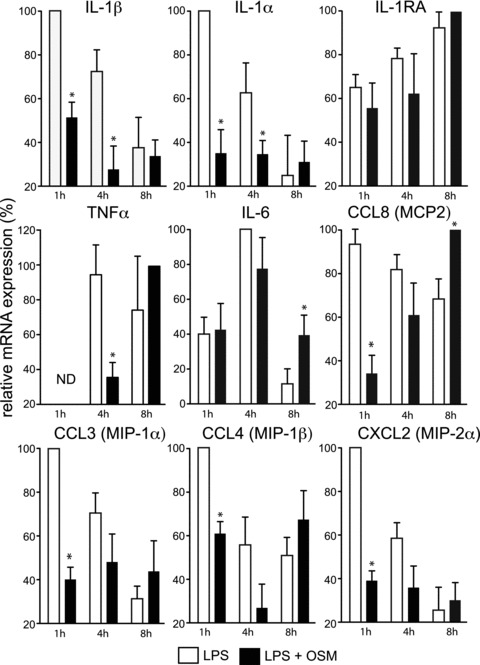
Impact of OSM on mRNA expression in resident cells of murine dorsal air pouches. Murine air pouches were injected with LPS (500 ng/ml) alone or in combination with recombinant murine OSM (25 μg/ml). At the indicated time, cells lining the pouch were collected for RNA extraction and determination of IL-1β, IL-1α, IL-1RA, TNF-α, IL-6, CCL3, CCL4, CCL8 and CXCL2 mRNA levels by real time PCR. Cycle threshold (Ct) values of interest genes are normalized to GAPDH Ct. Results are the mean ± S.E.M. (at least eight mice per group) of the mRNA expression levels expressed in percent, the maximum value for each gene being fixed at 100%. ND: not detected. *Significantly different (P < 0.05) from LPS.
In dorsal pouch exudates, IL-1β concentrations determined by ELISA increased during the 8-hr post-LPS-injection period, but were divided by 4 in the presence of OSM at the 4- and 8-hr points (Fig. 7). In the case of TNF-α, maximum levels were reached 1 hr after injection and dropped below the limit of detection at t ∇ 8 hrs. OSM decreased TNF-α concentrations by approximately 50% at t ∇ 4 hrs. IL-6 levels appeared slightly increased in the presence of OSM, but the increase was not significant (data not shown). Levels of CCL2, CCL3 and CXCL2 in pouch exudate were also investigated but no modulation by OSM was observed (data not shown). OSM neutralization in this model was also tested, but the neutralizing anti-OSM antibody as well as the matched-isotype antibody, elicited non-specific immune responses, thus preventing any interpretation in this experimental setup (data not shown). As a whole, these results support an immunomodulatory role in vivo for OSM, based on regulating the expression of key inflammatory cytokines.
Fig 7.
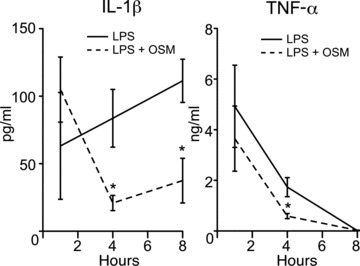
Impact of OSM on cytokine release in murine dorsal air pouches. Murine air pouches were injected with LPS (500 ng/ml) alone or in combination with recombinant murine OSM (25 μg/ml). At indicated times, pouch exudates were collected for determination of the indicated cytokines by specific ELISA. *Significantly different (P < 0.05) from LPS.
Discussion
We observed in this study that recombinant as well as native OSM decreased the release of IL-1β from activated HSFs. IL-1β is a key initiator of inflammatory processes [40-42], stimulating the production of several metalloproteinases and ultimately promoting connective tissue breakdown and inhibiting proteoglycan and type-II collagen synthesis, thereby exerting deletorious pressure on articular cartilage integrity [43, 44]. In bones, IL-1β stimulates the maturation of osteoclasts and participates in the development of bone erosion [45, 46]. Moreover, IL-1β is a major activator of HSFs, which are central effectors in inflammation and joint destruction [26, 47, 48]. In view of the potential impact of OSM as an inhibitor of IL-1β production by activated HSFs, it is conceivable that OSM might contribute to limiting IL-1β-induced tissue damage in inflamed joints.
In addition to their impact on IL-1β expression in activated HSFs, both recombinant and native OSM up-regulated IL-6 and CCL2 expression and down-regulated CXCL8. These results illustrate the anti-inflammatory potential of OSM and consolidate a collection of previous, discrete observations about the effects that recombinant OSM has on different cell types [14–16, 49–53]. Released by activated synoviocytes in the pannus, CXCL8 is a major mediator of inflammation and joint destruction [54, 55]. In contrast, CCL2 is involved in macrophage recruitment typically concurrent with the waning of granulocytes [56] and contributes to tissue reparative processes [57] as well as protecting against viral and bacterial infections [58, 59]. Finally, although the role of IL-6 in inflammation appears complex and multi-faceted [60], it has been shown in vivo to induce the IL-1 receptor antagonist and the soluble TNF receptor p55, both of which attenuate inflammation [61]. The fact that OSM repressed IL-1β and CXCL8 while it increased IL-6 and CCL2 expression suggests the induction of active mechanisms driving inflammation towards its resolution (i.e. receptor engagement and signalling), rather than a passive consequence of IL-1β inhibition. In fact, the multi-pronged impact of OSM on the HSF stimulation profile confirms OSM as an important effector in the attenuation of the inflammatory response [62].
Polymorphonuclear neutrophils predominate in rheumatoid arthritis synovial fluids and have been described as taking part in the aetiology of the disease [24]. Polymorphonuclear neutrophils are also a major source of OSM and our results suggest potential anti-inflammatory functions for PMN-secreted OSM, such as influencing the progression of inflammatory responses and by reducing the activation status of synoviocytes. However, additional studies will be required before a direct link can be made between PMN-derived OSM and the immunomodulatory events reported herein.
Overlapping biological responses between hOSM and hLIF are mediated by the shared type-I receptor (i.e. the LIF receptor), while hOSM manifests its specific responses through OSMRβ [63, 64]. In this study, neither LIF nor IL-6 had the same effect as OSM, on stimulated HSFs. Thus, and also taking into account that fibroblastic cells express the OSMRβ subunit [65, 66], the OSM-induced inhibitions of IL-1β and CXCL8 and the increases in IL-6 and CCL2 observed in the present study specifically suggest engagement and signalling via OSMRβ. Moreover, the similar patterns obtained in vivo strengthen this view because in mice OSM delivers signals only through its specific receptor complex gp130/OSMRβ [67]. A number of studies focusing on OSMRβ signalling, activation of the MAPK and JAK/STAT pathways have been reported with differences according to cell types [68]. In HSFs, OSM mainly activates the transcription factor STAT-1 [15], which also appeared to mediate the OSM-induced decrease in IL-1β and CXCL8 generation, as well as the up-regulation of IL-6 and CCL2. While STAT-1 as an activator of transcription has been well documented, its repression activities are gradually being appreciated. STAT-1 mediates the repression of monocyte IL-10 gene expression in vivo [69], the repression of matrix metalloproteinase 9 in different cell lines [70], the repression of inducible nitric oxide synthase transcription in human fibroblasts [71] and the down-regulation of related neonatal Fc receptor gene expression in human epithelial cells [72]. Mechanisms of the STAT-1-mediated repression are diverse: sequestration of the transcription activator CBP/p300, inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (Nf-κB) functions or interaction with others transcription factors. In our study, OSM differentially affected distinct cytokines, all in a STAT-1-dependent manner, further suggesting a multi-pronged impact of the transcription factor, depending in part on promoter sequences, associated co-factors as well as the transcriptional machinery specific to each target gene. Understanding of the mechanisms of the transcriptional regulation function of STAT-1 in HSFs will require further investigation.
The dorsal air pouch model is regarded as a facsimile of synovial tissues [39]. Presence of OSM in the pouch decreased the inflammatory response to LPS by reducing or delaying mRNA expression of the majority of the important cytokines in this model: IL-1β, TNF-α, CCL8 and MIPs in the cells lining the air pouch, as well as the concentration of IL-1β and TNF-α in the pouch exudate. OSM did not have a significant effect on leucocyte recruitment nor on the ratio of leucocyte subpopulations, indicating that OSM may have a stronger impact on limiting cell activation than on cell movement. Along these lines, OSM induced a reduction in pivotal inflammatory cytokines concentrations while not affecting that of chemokines such as CCL2, CXCL1 or CXCL3. This in vivo model is of course more complex than in vitro experiments, with numerous events intervening, and factors interacting. Nevertheless, testing OSM in the pouch largely corroborated our in vitro observations and showed that this cytokine can decrease the levels of IL-1β and TNF-α, two key cytokines in inflammation. OSM appears as a potential modulator of synovial tissue activation and joint inflammation and could therefore play a major role in directing inflammation toward its resolution.
In conclusion, we showed that OSM decreased the expression of HSF-derived pro-inflammatory factors, in particular IL-1β, while increasing components that can modulate the inflammatory response. These results identify OSM as a modulator of HSF activation and hence as an accessory in the attenuation of inflammation. Because synovial fibroblasts play an important role in the inflammatory phases of rheumatoid arthritis, reduction of their activation by OSM might be a useful therapeutic strategy to prevent inflammatory episodes.
Acknowledgments
This work is supported by an operating grant from the Canadian Institutes of Health Research (CIHR; to M.P.). A.D. is the recipient of a post-doctoral fellowship from the Fonds de la Recherche en Santé du Québec (FRSQ). M.P. is a Senior Investigator Scholar of the FRSQ.
Conflict of interest
Authors confirm that there are no conflicts of interest.
References
- 1.Tanaka M, Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52. doi: 10.1007/s10254-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 2.Zarling JM, Shoyab M, Marquardt H, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci U S A. 1986;83:9739–43. doi: 10.1073/pnas.83.24.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kishimoto T, Akira S, Narazaki M, et al. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54. [PubMed] [Google Scholar]
- 4.Heinrich PC, Behrmann I, Haan S, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosley B, De Imus C, Friend D, et al. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J Biol Chem. 1996;271:32635–43. doi: 10.1074/jbc.271.51.32635. [DOI] [PubMed] [Google Scholar]
- 6.Kuropatwinski KK, De Imus C, Gearing D, et al. Influence of subunit combinations on signaling by receptors for oncostatin M, leukemia inhibitory factor, and interleukin-6. J Biol Chem. 1997;272:15135–44. doi: 10.1074/jbc.272.24.15135. [DOI] [PubMed] [Google Scholar]
- 7.Thoma B, Bird TA, Friend DJ, et al. Oncostatin M and leukemia inhibitory factor trigger overlapping and different signals through partially shared receptor complexes. J Biol Chem. 1994;269:6215–22. [PubMed] [Google Scholar]
- 8.Gadient RA, Patterson PH. Leukemia inhibitory factor, Interleukin 6, and other cytokines using the GP130 transducing receptor: roles in inflammation and injury. Stem Cells. 1999;17:127–37. doi: 10.1002/stem.170127. [DOI] [PubMed] [Google Scholar]
- 9.Modur V, Feldhaus MJ, Weyrich AS, et al. Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J Clin Invest. 1997;100:158–68. doi: 10.1172/JCI119508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boniface K, Diveu C, Morel F, et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J Immunol. 2007;178:4615–22. doi: 10.4049/jimmunol.178.7.4615. [DOI] [PubMed] [Google Scholar]
- 11.Fritz DK, Kerr C, Fattouh R, et al. A mouse model of airway disease: oncostatin M-induced pulmonary eosinophilia, goblet cell hyperplasia, and airway hyperresponsiveness are STAT6 dependent, and interstitial pulmonary fibrosis is STAT6 independent. J Immunol. 2011;186:1107–18. doi: 10.4049/jimmunol.0903476. [DOI] [PubMed] [Google Scholar]
- 12.O’Hara KA, Kedda MA, Thompson PJ, et al. Oncostatin M: an interleukin-6-like cytokine relevant to airway remodelling and the pathogenesis of asthma. Clin Exp Allergy. 2003;33:1026–32. doi: 10.1046/j.1365-2222.2003.01714.x. [DOI] [PubMed] [Google Scholar]
- 13.Wallace PM, Macmaster JF, Rillema JR, et al. In vivo properties of oncostatin M. Ann N Y Acad Sci. 1995;762:42–54. doi: 10.1111/j.1749-6632.1995.tb32313.x. [DOI] [PubMed] [Google Scholar]
- 14.Hurst SM, McLoughlin RM, Monslow J, et al. Secretion of oncostatin M by infiltrating neutrophils: regulation of IL-6 and chemokine expression in human mesothelial cells. J Immunol. 2002;169:5244–51. doi: 10.4049/jimmunol.169.9.5244. [DOI] [PubMed] [Google Scholar]
- 15.Richards CD, Langdon C, Botelho F, et al. Oncostatin M inhibits IL-1-induced expression of IL-8 and granulocyte-macrophage colony-stimulating factor by synovial and lung fibroblasts. J Immunol. 1996;156:343–9. [PubMed] [Google Scholar]
- 16.Brown TJ, Liu J, Brashem-Stein C, et al. Regulation of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor expression by oncostatin M. Blood. 1993;82:33–7. [PubMed] [Google Scholar]
- 17.Loy JK, Davidson TJ, Berry KK, et al. Oncostatin M: development of a pleiotropic cytokine. Toxicol Pathol. 1999;27:151–5. doi: 10.1177/019262339902700201. [DOI] [PubMed] [Google Scholar]
- 18.Wallace PM, MacMaster JF, Rouleau KA, et al. Regulation of inflammatory responses by oncostatin M. J Immunol. 1999;162:5547–55. [PubMed] [Google Scholar]
- 19.Minehata K, Takeuchi M, Hirabayashi Y, et al. Oncostatin m maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int J Hematol. 2006;84:319–27. doi: 10.1532/IJH97.06090. [DOI] [PubMed] [Google Scholar]
- 20.Esashi E, Ito H, Minehata K, et al. Oncostatin M deficiency leads to thymic hypoplasia, accumulation of apoptotic thymocytes and glomerulonephritis. Eur J Immunol. 2009;39:1664–70. doi: 10.1002/eji.200839149. [DOI] [PubMed] [Google Scholar]
- 21.Grenier A, Dehoux M, Boutten A, et al. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood. 1999;93:1413–21. [PubMed] [Google Scholar]
- 22.Okamoto H, Yamamura M, Morita Y, et al. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 1997;40:1096–105. doi: 10.1002/art.1780400614. [DOI] [PubMed] [Google Scholar]
- 23.Cross A, Edwards SW, Bucknall RC, et al. Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis Rheum. 2004;50:1430–6. doi: 10.1002/art.20166. [DOI] [PubMed] [Google Scholar]
- 24.Pillinger MH, Abramson SB. The neutrophil in rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21:691–714. [PubMed] [Google Scholar]
- 25.Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol. 2005;115:118–28. doi: 10.1016/j.clim.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–70. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee DM, Kiener HP, Agarwal SK, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 28.Buckley CD, Filer A, Haworth O, et al. Defining a role for fibroblasts in the persistence of chronic inflammatory joint disease. Ann Rheum Dis. 2004;63:ii92–5. doi: 10.1136/ard.2004.028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HJ, Krenn V, Steinhauser G, et al. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. 1999;162:3053–62. [PubMed] [Google Scholar]
- 30.Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- 31.Pouliot M, Fiset ME, Masse M, et al. Adenosine up-regulates cyclooxygenase-2 in human granulocytes: impact on the balance of eicosanoid generation. J Immunol. 2002;169:5279–86. doi: 10.4049/jimmunol.169.9.5279. [DOI] [PubMed] [Google Scholar]
- 32.Cadieux JS, Leclerc P, St-Onge M, et al. Potentiation of neutrophil cyclooxygenase-2 by adenosine: an early anti-inflammatory signal. J Cell Sci. 2005;118:1437–47. doi: 10.1242/jcs.01737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McColl SR, St-Onge M, Dussault AA, et al. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. Faseb J. 2006;20:187–9. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.St-Onge M, Flamand N, Biarc J, et al. Characterization of prostaglandin E2 generation through the cyclooxygenase (COX)-2 pathway in human neutrophils. Biochim Biophys Acta. 2007;1771:1235–45. doi: 10.1016/j.bbalip.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dussault AA, Pouliot M. Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol Proced Online. 2006;8:1–10. doi: 10.1251/bpo114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korzus E, Nagase H, Rydell R, et al. The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J Biol Chem. 1997;272:1188–96. doi: 10.1074/jbc.272.2.1188. [DOI] [PubMed] [Google Scholar]
- 37.David M, Petricoin E, III, Benjamin C, et al. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269:1721–3. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 38.Winston LA, Hunter T. JAK2, Ras, and Raf are required for activation of extracellular signal-regulated kinase/mitogen-activated protein kinase by growth hormone. J Biol Chem. 1995;270:30837–40. doi: 10.1074/jbc.270.52.30837. [DOI] [PubMed] [Google Scholar]
- 39.Edwards JC, Sedgwick AD, Willoughby DA. The formation of a structure with the features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J Pathol. 1981;134:147–56. doi: 10.1002/path.1711340205. [DOI] [PubMed] [Google Scholar]
- 40.Schiff MH. Role of interleukin 1 and interleukin 1 receptor antagonist in the mediation of rheumatoid arthritis. Ann Rheum Dis. 2000;59:i103–8. doi: 10.1136/ard.59.suppl_1.i103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saijo S, Asano M, Horai R, et al. Suppression of autoimmune arthritis in interleukin-1-deficient mice in which T cell activation is impaired due to low levels of CD40 ligand and OX40 expression on T cells. Arthritis Rheum. 2002;46:533–44. doi: 10.1002/art.10172. [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002;20:S1–13. [PubMed] [Google Scholar]
- 43.Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology. 2003;42:ii11–6. doi: 10.1093/rheumatology/keg327. [DOI] [PubMed] [Google Scholar]
- 44.Pretzel D, Pohlers D, Weinert S, et al. In vitro model for the analysis of synovial fibroblast-mediated degradation of intact cartilage. Arthritis Res Ther. 2009;11:R25. doi: 10.1186/ar2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joosten LA, Helsen MM, Saxne T, et al. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol. 1999;163:5049–55. [PubMed] [Google Scholar]
- 46.Trebec-Reynolds DP, Voronov I, Heersche JN, et al. IL-1alpha and IL-1beta have different effects on formation and activity of large osteoclasts. J Cell Biochem. 2010;109:975–82. doi: 10.1002/jcb.22476. [DOI] [PubMed] [Google Scholar]
- 47.Chen SY, Shiau AL, Shieh GS, et al. Amelioration of experimental arthritis by a telomerase-dependent conditionally replicating adenovirus that targets synovial fibroblasts. Arthritis Rheum. 2009;60:3290–302. doi: 10.1002/art.24940. [DOI] [PubMed] [Google Scholar]
- 48.Ritchlin C. Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000;2:356–60. doi: 10.1186/ar112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown TJ, Rowe JM, Liu JW, et al. Regulation of IL-6 expression by oncostatin M. J Immunol. 1991;147:2175–80. [PubMed] [Google Scholar]
- 50.Langdon C, Leith J, Smith F, et al. Oncostatin M stimulates monocyte chemoattractant protein-1-induced and interleukin-1-induced matrix metalloproteinase-1 production by human synovial fibroblasts in vitro. Arthritis Rheum. 1997;40:2139–46. doi: 10.1002/art.1780401207. [DOI] [PubMed] [Google Scholar]
- 51.Van Wagoner NJ, Choi C, Repovic P, et al. Oncostatin M regulation of interleukin-6 expression in astrocytes: biphasic regulation involving the mitogen-activated protein kinases ERK1/2 and p38. J Neurochem. 2000;75:563–75. doi: 10.1046/j.1471-4159.2000.0750563.x. [DOI] [PubMed] [Google Scholar]
- 52.Bernard C, Merval R, Lebret M, et al. Oncostatin M induces interleukin-6 and cyclooxygenase-2 expression in human vascular smooth muscle cells: synergy with interleukin-1beta. Circ Res. 1999;85:1124–31. doi: 10.1161/01.res.85.12.1124. [DOI] [PubMed] [Google Scholar]
- 53.Repovic P, Mi K, Benveniste EN. Oncostatin M enhances the expression of prostaglandin E2 and cyclooxygenase-2 in astrocytes: synergy with interleukin-1beta, tumour necrosis factor-alpha, and bacterial lipopolysaccharide. Glia. 2003;42:433–46. doi: 10.1002/glia.10182. [DOI] [PubMed] [Google Scholar]
- 54.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 55.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84:1045–9. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–26. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shireman PK, Contreras-Shannon V, Ochoa O, et al. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J Leukoc Biol. 2007;81:775–85. doi: 10.1189/jlb.0506356. [DOI] [PubMed] [Google Scholar]
- 58.Dessing MC, van der Sluijs KF, Florquin S, et al. Monocyte chemoattractant protein 1 contributes to an adequate immune response in influenza pneumonia. Clin Immunol. 2007;125:328–36. doi: 10.1016/j.clim.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Nakano Y, Kasahara T, Mukaida N, et al. Protection against lethal bacterial infection in mice by monocyte-chemotactic and -activating factor. Infect Immun. 1994;62:377–83. doi: 10.1128/iai.62.2.377-383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scholz W. Interleukin 6 in diseases: cause or cure? Immunopharmacol. 1996;31:131–50. doi: 10.1016/0162-3109(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 61.Tilg H, Trehu E, Atkins MB, et al. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumour necrosis factor receptor p55. Blood. 1994;83:113–8. [PubMed] [Google Scholar]
- 62.Wahl AF, Wallace PM. Oncostatin M in the anti-inflammatory response. Ann Rheum Dis. 2001;60:iii75–80. doi: 10.1136/ard.60.90003.iii75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J, Hadjokas N, Mosley B, et al. Oncostatin M-specific receptor expression and function in regulating cell proliferation of normal and malignant mammary epithelial cells. Cytokine. 1998;10:295–302. doi: 10.1006/cyto.1997.0283. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Robledo O, Kinzie E, et al. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J Biol Chem. 2000;275:25273–85. doi: 10.1074/jbc.M002296200. [DOI] [PubMed] [Google Scholar]
- 65.Blanchard F, Wang Y, Kinzie E, et al. Oncostatin M regulates the synthesis and turnover of gp130, leukemia inhibitory factor receptor alpha, and oncostatin M receptor beta by distinct mechanisms. J Biol Chem. 2001;276:47038–45. doi: 10.1074/jbc.M107971200. [DOI] [PubMed] [Google Scholar]
- 66.Hosokawa Y, Hosokawa I, Ozaki K, et al. Catechins inhibit CXCL10 production from oncostatin M-stimulated human gingival fibroblasts. J Nutr Biochem. 2010;21:659–64. doi: 10.1016/j.jnutbio.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 67.Ichihara M, Hara T, Kim H, et al. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood. 1997;90:165–73. [PubMed] [Google Scholar]
- 68.Stahl N, Boulton TG, Farruggella T, et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science. 1994;263:92–5. doi: 10.1126/science.8272873. [DOI] [PubMed] [Google Scholar]
- 69.VanDeusen JB, Shah MH, Becknell B, et al. STAT-1-mediated repression of monocyte interleukin-10 gene expression in vivo. Eur J Immunol. 2006;36:623–30. doi: 10.1002/eji.200535241. [DOI] [PubMed] [Google Scholar]
- 70.Ma Z, Qin H, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-gamma and IFN-beta: critical role of STAT-1alpha. J Immunol. 2001;167:5150–9. doi: 10.4049/jimmunol.167.9.5150. [DOI] [PubMed] [Google Scholar]
- 71.Ganster RW, Taylor BS, Shao L, et al. Complex regulation of human inducible nitric oxide synthase gene transcription by Stat 1 and NF-kappa B. Proc Natl Acad Sci U S A. 2001;98:8638–43. doi: 10.1073/pnas.151239498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu X, Ye L, Bai Y, et al. Activation of the JAK/STAT-1 signaling pathway by IFN-gamma can down-regulate functional expression of the MHC class I-related neonatal Fc receptor for IgG. J Immunol. 2008;181:449–63. doi: 10.4049/jimmunol.181.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]