

Abstract
The impact of angiotensin II receptor blockers (ARBs) on electrical remodelling after myocardial infarction (MI) remains unclear. The purpose of the present study was to evaluate the effect of valsartan on incidence of ventricular arrhythmia induced by programmed electrical stimulation (PES) and potential link to changes of myocardial connexins (Cx) 43 expression and distribution in MI rats. Fifty-nine rats were randomly divided into three groups: Sham (n = 20), MI (n = 20) and MI + Val (20 mg/kg/day per gavage, n = 19). After eight weeks, the incidence of PES-induced ventricular tachycardia (VT) and fibrillation (VF) was compared among groups. mRNA and protein expressions of Cx43, angiotensin II type 1 receptor (AT1R) in the LV border zone (BZ) and non-infarct zone (NIZ) were determined by real-time PCR and Western blot, respectively. Connexins 43 protein and collagen distribution were examined by immunohistochemistry in BZ and NIZ sections from MI hearts. Valsartan effectively improved the cardiac function, reduced the prolonged QTc (163.7 ± 3.7 msec. versus 177.8 ± 4.5 msec., P < 0.05) after MI and the incidence of VT or VF evoked by PES (21.1% versus 55%, P < 0.05). Angiotensin II type 1 receptor expression was significantly increased in BZ and NIZ sections after MI, which was down-regulated by valsartan. The mRNA and protein expressions of Cx43 in BZ were significantly reduced after MI and up-regulated by valsartan. Increased collagen deposition and reduced Cx43 expression in BZ after MI could be partly attenuated by Valsartan. Valsartan reduced the incidence of PES-induced ventricular arrhythmia, this effect was possibly through modulating the myocardial AT1R and Cx43 expression.
Keywords: ventricular arrhythmia, Angiotensin II receptor blocker, valsartan, myocardial infarction, connexin
Introduction
Myocardial infarction (MI) is a common clinical disease associated with high morbidity and mortality. Fibrotic scar tissue after MI could result in haemodynamic instability and electrical conduction disorders which is responsible for the occurrence of ventricular tachyarrhythmias [1, 2]. Ventricular tachyarrhythmia is the main cause of sudden death in patients after MI. Modern medications and approaches for MI treatment should thus be aimed not only to improve the cardiac function, but also to reduce the incidence of arrhythmia.
The renin-angiotensin system (RAS) plays an important role in the regulation of cardiovascular function. Up-regulation of the RAS has been implicated during MI, which is linked with an increased risk of arrhythmia [3]. Previous studies showed that angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II (ANG II) receptor blockers (ARBs) could attenuate myocardial ischaemia and improve heart function [4-6] and these beneficial effects are associated with inhibiting myocardial angiotensin II type 1 (AT1R) receptor and reducing myocardial fibrosis [7]. Recently, some studies demonstrated that ARBs use was also linked to reduced arrhythmia post-ischaemia [8]. In The Losartan Intervention For Endpoint reduction in Hypertension (LIFE) study, Losartan reduced the incidences of new-onset atrial fibrillation (AF) and complications associated with new-onset or pre-existing AF [9, 10]. In chronic post-infarction, the role of gap junctions is less clear, but a general down-regulation of Cx43 and inhomogeneity in the post-infarction area has been described [11]. Loss of Cx43 expression greatly increased the risk of ventricular tachyarrhythmias and sudden death in adult mice model [12]. Danik et al. demonstrated that a marked slowing of conduction velocity and abrupt onset of spontaneous ventricular tachyarrhythmias were caused by Cx43 deficiency [13]. It remains unknown if chronic ARB therapy after MI could affect myocardial Cx43 expression and linked to their beneficial effects on reducing arrhythmias. We, therefore, evaluated the effects of valsartan on the incidence of ventricular arrhythmia induced by PES in rats with experimental MI and myocardial expression of Cx43.
Materials and methods
Animal preparation
The animal experiments were approved and performed according to the regulation of the Animal Ethics Committee of Shanghai Xinhua Hospital and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (NIH). Female Sprague–Dawley (SD) rats (SPF class, 220–250 g) were used. They were housed under standard laboratory conditions at 25 ± 2°C, relative humidity of 50 ± 15% and normal photoperiod (12 hrs dark and 12 hrs light) with regular food and tap water.
Myocardial infarction
Myocardial infarction was induced in rats by ligation of the left coronary artery [14]. Briefly, 90 rats were anaesthetized with intraperitoneal pentobarbital (40 mg/kg) and then intubated. The left coronary arteries were ligated at approximately 2–3 mm from the origin with 5.0 silk sutures in 70 rats. Twenty rats treated similarly without coronary artery ligation served as Sham group. After operation, all rats were housed and fed in separate cages. Three days after surgery, 40 rats with LV ejection fraction (LVEF) measured by echocardiography less than 45% were randomly divided into two groups: MI group (n = 20, 2 ml saline per gavage per day for 8 weeks) and MI + Val group (n = 20, valsartan 20 mg/kg per gavage per day for 8 weeks). Valsartan (Novartis Pharma, Basel, Switzerland) was dissolved in saline by adding 0.1 mol/l NaOH followed by 0.1 mol/l HCl to titrate pH to 7.4 [15].
Doppler echocardiography study
Transthoracic echocardiography equipped with M-mode transducer (Sequoia 512 and 15-MHz probe; Siemens Medical, CA, USA) was performed on 3 days and 8 weeks after MI [16] in anaesthetized rats [intraperitoneal administration of pentobarbital: 40 mg/kg bodyweight (bw)]. Two-dimensional short-axis views of the left ventricle and M-mode tracings were recorded through the anterior and posterior LV walls at the papillary muscle level to measure LV end-diastolic dimension (LVDd) and LV end-systolic dimension (LVDs). LV ejection fraction was obtained using Simpson approach. All measurements were performed by an experienced technician who was blinded to study groups.
Programmed electrical stimulation
Next day after final echocardiography examinations, electrical vulnerability in vivo by burst and extrastimulus pacing protocols was performed. For the programmed electrical stimulation (PES), animals were anaesthetized again with pentobarbital (40 mg/kg bw). Surface six-lead ECGs were recorded using 25 gauge subcutaneous electrodes. ECG channels were filtered out below 10 Hz and above 100 Hz. Standard criteria were used for interval measurements: RR, PR, QRS and QTc (QT interval corrected for the heart rate using Bazett’s formula QTc = QT/√RR). The right ventricle was catheterized from the right external jugular vein to the right atrium and through the tricuspid valve with a 1.9-F eight-polar catheter for small-animal electrophysiology (Scisence, Ontario, Canada). The threshold potential for stable pacing was assessed at a cycle length of 200 msec. The stimulation study was then initiated twice as much as the threshold. Pacing was performed by applying a 1-msec. pulse pacing of width which was two times higher than capture threshold. Standard clinical PES protocols were used, including burst, single, double and triple extra stimuli applied under spontaneous rhythm. The coupling interval of the last extra stimulus was decreased by 2-msec. steps from 80 msec. down to the ventricular effective refractory period (VERP). The PES protocol was invoked until sustained ventricular tachycardia (SVT) or ventricular fibrillation (VF) was induced or until the protocol was exhausted. Ventricular fibrillation was defined as a run of three or more consecutive, rapid ventricular premature beats (VPBs) which were defined as discrete and identifiable premature QRS complexes. Sustained ventricular tachycardia was defined as fast ventricular rhythm of 15 or more beats. Pacing protocols were interrupted if SVT or VF was induced.
All PES were performed in 10 min. intervals. All the arrhythmic events were recorded during PES. The severity of arrhythmias was quantified by a scoring system [17, 18]: hearts with no VPBs, VT or VF were given a score of 0; occurrences of premature ventricular contractions were given a score of 1; less than five occurrences of VT was given a score of 2; more than five occurrences of VT, or one occurrence of VF or both was given a score of 3; two to five occurrences of VF was given a score of 4; more than five occurrences of VF was given a score of 5.
ELISA for serum B-type natriuretic peptide and ANG II
After EPS, plasma was collected and stored at −80°C. Serum B-type natriuretic peptide (BNP) and ANG II were measured using commercially available rat ELISA kit (R&D, Minneapolis, MN, USA).
Preparation of tissue samples
Thereafter, rats were weighed and killed in deep anaesthesia with overdose pentobarbital (80 mg/kg bw), the hearts were immediately excised, rinsed and weighed (4°C). The atriums, great vessels and valves were trimmed away before the left ventricle was dissected free and weighed. A middle ring was cut and paraffin-embedded for the measurement of infarct size. Left ventricle was divided into remote non-infarct zone (NIZ) and border zone (BZ); BZ was taken at 2 mm away from infarct edge [19, 20]. Parts of myocardial samples were quick frozen in liquid nitrogen and stored at −80°C for the molecular analysis, the rest myocardial samples were fixed in 4% paraformaldehyde and embedded in paraffin for histological analysis.
Western blot analysis
For Western blot analysis, the proteins of each example were extracted from the NIZ and the BZ of left ventricles, 10 μg protein was taken and 1/5 volume of 5χ loading buffer was added and mixed. Then the mixture denatured by boiling at 100°C for 5 min., then loaded to a 10% SDS-PAGE and transferred to the polyvinylidene fluoride membrane. The membrane was blocked in 5% (wt/vol) non-fat dry milk in 0.1% phosphate buffered saline with Tween-20 (PBST) and probed overnight at 4°C with primary antibodies (Cx43 and Tublin; Santa Cruz, CA, USA; AT1R; Abcam, Cambridge, UK) followed by the horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000 dilution; Jackson, PA, USA) for 2 hrs at room temperature. Protein concentrations were measured by chemiluminescence and Bio-Rad imaging system. Images were analysed by Quantity One software (Bio-Rad, CA, USA).
Extraction of total RNA and real-time reverse transcriptase–PCR
For mRNA analysis of AT1R and Cx43, frozen tissue samples were homogenized and total RNA was extracted from NIZ and BZ of left ventricles using Trizol (Invitrogen, CA, USA). Chloroform was added and total RNA was precipitated from the aqueous phase with isopropyl alcohol after centrifugation. The quality and integrity were controlled by running samples on a 1% denaturing agarose gel to ensure the presence of the 28S and 18S ribosomal bands and measured at the 260–280 nm UV absorbance ratio. The first-strand cDNA was synthesized by the Toyobo First-Strand Synthesis kit (TOYOBO, Osaka, Japan) according to the manufacturer’s instructions. The relative quantification of gene expression by real-time reverse transcriptase–PCR was determined by comparing the target amplified product against actin (internal standard) within the same sample. PCR amplification was carried on Gene Rotor 3000 using real-time PCR kit (TOYOBO). Primer concentration was 200 pM. The reaction cycles were: denaturation at 95°C for 20 sec. followed by 40 cycles of denaturation at 95°C for 15 sec., annealing at 60°C for 20 sec. and extension at 72°C for 20 sec. The primer sequences were shown in Table 1.
Table 1.
The primer sequences of AT1R and Cx43
| Gene | Primer sequence |
|---|---|
| AT1R | |
| Foreword | GCTGGCAGGCACAGTTACATATT |
| Reverse | TCACCACCAAGCTGTTTCCA |
| Cx43 | |
| Foreword | ACAGCGCAGAGCAAAATCG |
| Reverse | ATGGCTGGAGTTCATGTCCAG |
| Actin | |
| Foreword | GCTCCTCCTG AGCGCAAGTA |
| Reverse | CCTGCTTGCTGATCCACATCT |
Infarct size measurement
Paraffin-embedded LV middle ring was sectioned into 5-μm-thick slices and applied Mallory’s trichrome staining. The inner and outer perimeters of the left ventricle were traced with a digital image processing system (Leica Qwin Plus 3.0, Leica, Solms, Germany). Infarct size (%) was expressed as the mean of the percentage of infarcted LV versus total LV inner and outer circumferences.
Immunohistochemical staining
Paraffin-embedded LV tissue slides were dewaxed by xylol twice, each time 10 min., then gradient hydration (100%, 85%, 75% ethanol) to water and air-dried. The endogenous peroxidase was removed by incubating with 3% methanol–hydrogen peroxide for 10 min. at room temperature (RT), washed in PBS (three times for 5 min.). Then specimens were retrieved antigen for 20 min. in sodium citrate buffer (pH 6.0) at 98°C, cooled to RT, and blocked for 1 hr using PBS containing 10% normal goat serum (Sigma-Aldrich, St. Louis, MO, USA) at RT. Next, the slides were incubated with anti-Cx43 antibody (Abcam) 1:1000 dilution at 4°C overnight. After being washed three times by PBS, the slices were re-incubated with HRP enzyme–labelled goat anti-rabbit IgG (Boehringer Mannheim Corp., Inc., Mannheim, Germany) 1:1000 dilution at 37°C for 1 hr. The Cx43 staining was visualized with a 3,3′-diaminobenzidine substrate system, slides were mounted in neutral gum medium and observed with a (10χ ocular lens) light microscope (Leica DM LB2, Leica, Germany).
Statistical analysis
All statistical analyses were performed with SPSS 13.0 software. Data presented as mean ± standard deviation and compared by one-way ANOVA followed by Tukey’s test for individual significant difference. Frequency of PES-induced arrhythmia was compared using chi-square test. P < 0.05 was considered as statistically significant.
Results
Effects of valsartan on plasma BNP, ANG II, LV weight and the LV weight/bw ratio
In our study, the total survival rate among rats subjected to the MI operation was 64% (45/70) and 40 rats with EF < 45% were included in this study. One rat in MI + Val group died on the 7th day after operation. As shown in Table 2, bw at 8 weeks after operation was significantly reduced in both MI group and Sham group whereas LV weights were similar among groups. LV/bw ratio was significantly higher in MI groups than in Sham group (P < 0.01) and MI + Val group (P < 0.01). Plasma BNP and ANG II levels were significantly increased in MI group compared to those in Sham group which could be significantly attenuated by valsartan.
Table 2.
Plasma BNP, ANG II, bw and LV weight, the LV weight/bw ratio at 8 weeks after operation
| Sham group (n = 20) | MI MI group (n = 20) | MI + Val group (n = 19) | |
|---|---|---|---|
| bw (g) | 332.1 ± 8.9 | 311.0 ± 10.3* | 310.7 ± 11.4* |
| LV (mg) | 690.8 ± 23.0 | 699. 9 ± 31.0 | 679.1 ± 26.1 |
| LV/bw (mg/g) | 2.08 ± 0.02 | 2.27 ± 0.07* | 2.16 ± 0.02*,† |
| BNP (pg/ml) | 4.16 ± 0.16 | 11.92 ± 1.06* | 7.31 ± 0.56*,† |
| ANG II (ng/ml) | 64.10 ± 6.46 | 111.10 ± 20.98* | 80.96 ± 11.67*,† |
Sham: Sham-operated rats; MI: myocardial infarction; bw: bodyweight; LV: left ventricle weight. *P < 0.01 versus Sham group. †P < 0.01 versus MI group.
Effects of valsartan on LV function
Echocardiographic assessment of rats was shown in Figure 1. LV end-diastolic dimension was similar among groups at 3 days after operation and significantly increased at 8 weeks after operation in MI group (versus Sham group, P < 0.01) which could be partly attenuated by valsartan in MI + Val group (versus MI group, P = 0.004 < 0.01) (Fig. 1A). LV end-diastolic dimension was significantly increased in MI groups and MI + Val group than in Sham group at 3 days after operation. LV end-diastolic dimension was further increased in MI group, but remained unchanged in MI + Val group 8 weeks after operation compared with that at 3 days after operation (P > 0.05) (Fig. 1B). LV ejection fraction was significantly lower in MI and MI + Val groups than in Sham group at 3 days after operation and LVEF was significantly higher in MI + Val group compared to MI group at 8 weeks after operation (P < 0.01) (Fig. 1C). Figure 1D–F shows echocardiography examples of individual group at 8 weeks after operation.
Fig 1.
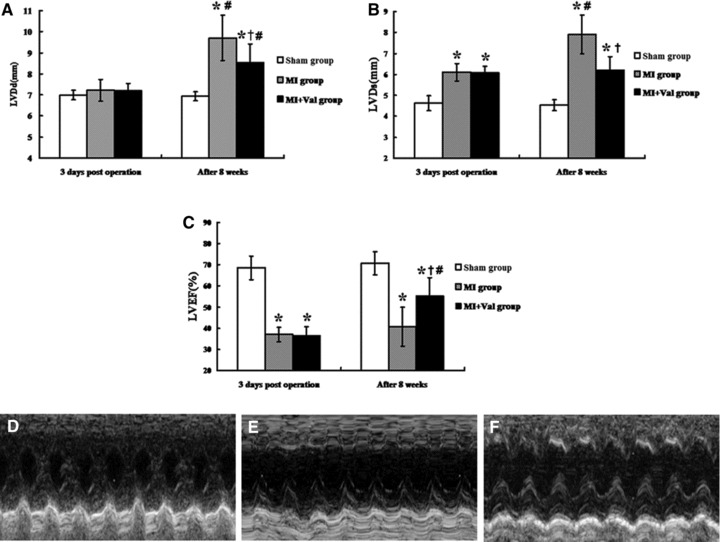
Echocardiography results and images of three groups. M-mode images were taken from the level of the LV outflow tract and the mitral valve was studied during the activity of ventricular wall, especial anterior wall. (A–C) The changes of LVDd, LVDs and LVEF 3 days after operation and after 8 weeks. The LV wall motion in Sham group (D) was better than the other groups. The condition of MI + Val group (F) was improved better compared with the MI group (E). LVDd: LV dimension end diastole; LVDs: LV dimension end systole; LVEF: LV ejection fraction; *P < 0.01 versus Sham group; †P < 0.01 versus MI group; #P < 0.05 versus 3 days after operation.
Effects of valsartan on incidence of PES-induced ventricular tachyarrhythmia
ECG parameters were summarized in Table 3, the QTc in MI group was significantly prolonged than that in Sham group (P = 0.001) and which could be reversed by valsartan treatment (P = 0.001 versus MI + Val group) (Fig. 2B), the remaining parameters including duration of P-wave and PR interval were similar among three groups (P > 0.05).
Table 3.
Results of ECG parameters among three groups
| RR (msec.) | P (msec.) | PR (msec.) | QRS (msec.) | QTc (msec.) | |
|---|---|---|---|---|---|
| Sham group (n = 20) | 156.5 ± 10.1 | 17.0 ± 2.2 | 52.3 ± 3.9 | 22.2 ± 1.5 | 159.3 ± 4.9 |
| MI group (n = 20) | 142.7 ± 10.8* | 16.1 ± 1.5 | 50.5 ± 2.5 | 23.8 ± 1.6* | 177.8 ± 4.5* |
| MI + Val group (n = 19) | 153.8 ± 10.5† | 16.3 ± 1.9 | 50.8 ± 3.2 | 22.7 ± 1.2† | 163.7 ± 3.7*,† |
Sham: Sham-operated rats; MI: myocardial infarction. *P < 0.01 versus Sham group. †P < 0.05 versus MI group.
Fig 2.
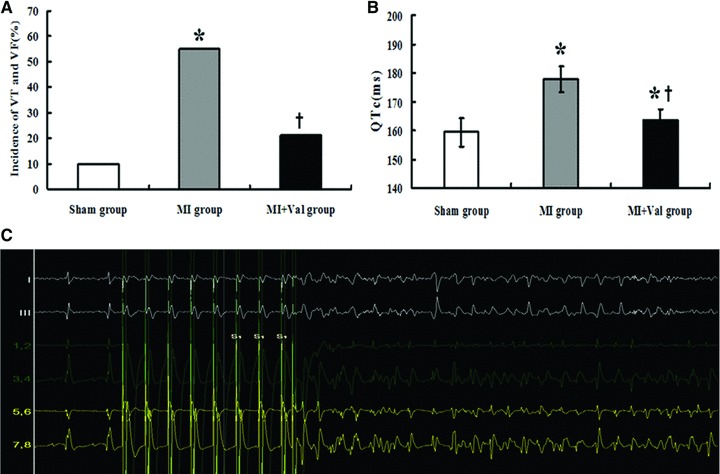
Results displayed the incidence of inducible VT or VF, and QTc in ECG parameters among three groups. (A) Incidence of VT and VF induced by PES. The incidence of ventricle arrhythmias was significantly increased in MI group (*P < 0.05 versus Sham group); and declined in MI + Val group (†P < 0.05 versus MI group). (B) Mean QTc in ECG before PES was significantly longer in MI group (*P < 0.05 versus Sham group); and shorter in MI + Val group (†P < 0.05 versus MI group). (C) VT induced by PES. Sham group (n = 20), MI group (n = 20) and MI + Val group (n = 19).
Incidence of PES-induced VT or VF was significantly increased in MI group than in Sham group (55% versus 10%, P = 0.003), which could be significantly reduced in MI + Val group (21.1%, P = 0.031 versus MI group) (Fig. 2A). As shown in Table 4, the arrhythmic scoring points were significantly higher in MI group than in Sham group, which could be significantly reduced in MI + Val group. Figure 2C shows an example of PES-induced VT.
Table 4.
The incidence of PES-induced VT or VF among three groups
| 0 | 1 | 2 | 3 | 4 | 5 | Total | |
|---|---|---|---|---|---|---|---|
| Sham group (n = 20) | 13 | 5 | 2 | 0 | 0 | 0 | 9 |
| MI group (n = 20) | 0 | 9 | 5 | 4 | 1 | 1 | 40* |
| MI + Val group (n = 19) | 5 | 11 | 2 | 0 | 1 | 0 | 19*,† |
Sham: Sham-operated rats; MI: myocardial infarction. *P < 0.05 versus Sham group. †P < 0.05 versus MI group.
Valsartan reduced expressions of AT1R and up-regulated Cx43 expression in MI rats
As shown in Figure 3A, AT1R mRNA level in BZ and NIZ of MI group was significantly higher than that of Sham group (P < 0.05) and AT1R mRNA was significantly down-regulated in NIZ and BZ in MI + Val group (all P < 0.05). Angiotensin II type 1 receptor protein levels are shown in Figure 3C, which were significantly up-regulated in MI group than in Sham group and were significantly down-regulated in MI + Val group (all P < 0.05).
The mRNA level of Cx43 in BZ and NIZ of MI group was significantly lower than in Sham group (P < 0.05) and the reduction was more significant in BZ than in NIZ; the mRNA level of Cx43 in BZ and NIZ was significantly higher in MI + Val group than in MI group. The protein expression of Cx43 in BZ was significantly reduced in MI group than in Sham group which could be reversed by valsartan treatment (Fig. 3D).
Fig 3.
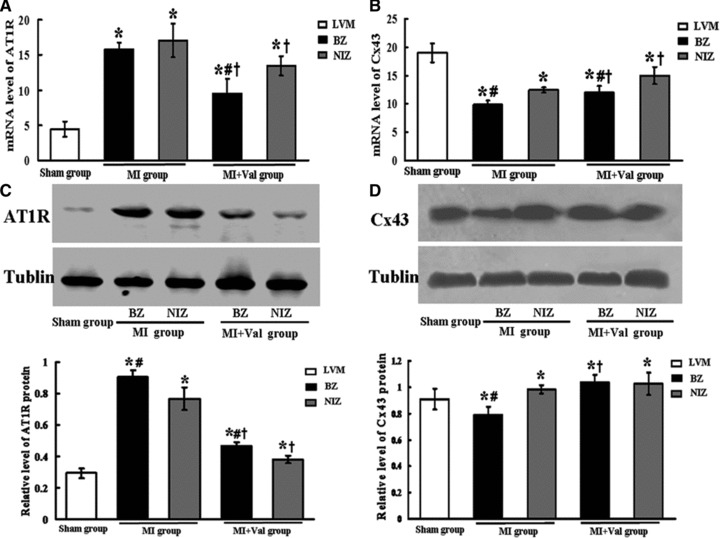
Effects of valsartan on expressions of AT1R and Cx43. The mRNA levels of (A) AT1R and (B) Cx43 were evaluated by the real-time PCR (n = 6). The protein levels of (C) AT1R and (D) Cx43 were analysed by Western blot (n = 7). Proteins loading of each sample in different groups were normalized by Tublin, the house keeping gene. BZ: border zone; NIZ: non-infarct zone. *P < 0.05 versus Sham group; #P < 0.05 versus NIZ; †P < 0.05 versus MI group.
Effect of Valsartan on Cx43 distribution and fibrosis in MI rats
Changes in Cx43 distribution were examined by immunohistochemistry in both fractions of BZ and NIZ from MI heats with or without valsartan treatment. Connexins 43 distribution was significantly reduced in BZ than in the NIZ in MI group, and the Cx43 distribution in BZ tended to be higher in MI + Val group than in MI group (data not shown). Valsartan treatment also effectively attenuated collagen hyperplasia in BZ after MI (Fig. 4A and B).
Fig 4.
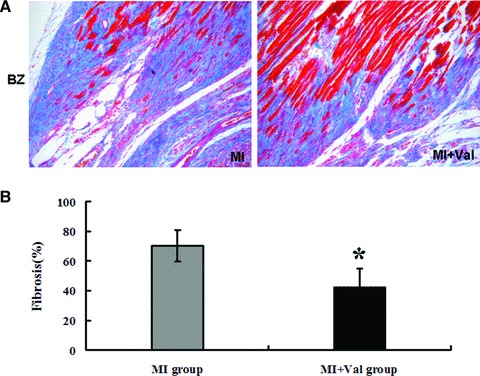
Effect of valsartan on the collagen staining in the LV border zone section after MI. (A) Collagen staining of myocardial tissues with Masson’s trichrome (stained in blue) in rat hearts 8 weeks after MI. (B) Quantitative data evaluated the levels of collagen deposition in both BZ and NIZ sections (n = 7 in MI group, n = 6 in MI + Val group. *P < 0.05 versus MI group).
Infarct size after 8 weeks
There was no significant difference in infarct size between MI group and MI + Val group at eight weeks after operation (Fig. 5).
Fig 5.
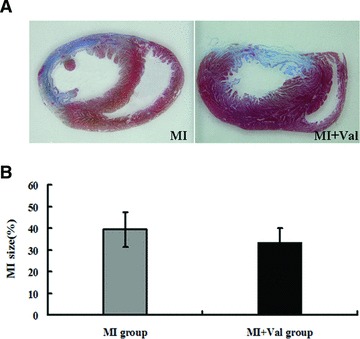
Heart infarct sizes in rats of MI and MI + Val groups were detected after 8 weeks by (A) Mallory’s trichrome staining, and quantified by (B) Qwin Plus3.0 (Leica, Germany). MI group (n = 7) and MI + Val group (n = 6).
Discussion
In the present study, we examined the effects of the ARB, valsartan, on cardiac function and occurrence of PES-induced ventricular arrhythmia in rats with experimental MI. As expected, our results indicated that valsartan could attenuate ventricular remodelling and efficiently improved the cardiac function after MI. Moreover, a novel finding from this study was that valsartan effectively reduced the incidence of PES-induced ventricular arrhythmia in MI rats, which might be linked on the modulating effects of valsartan on myocardial AT1R and Cx43 expressions.
Myocardial infarction is associated with activation of RAS [21], in line with this concept, our results showed up-regulated myocardial AT1R mRNA and protein expressions after MI. Increased RAS might affect the stability of electrical conduction and prone to arrhythmia [22]. Previous studies [23-31] showed beneficial effects on arrhythmia of ACEI and ARB. Use of these agents increased the threshold of VF and monophasic action potential and reduced duration of VF [29]. In hypertrophied rat hearts, severe ventricular arrhythmias could be reduced by short-term treatment with ACEI [27]. In this study, we tested the hypothesis that ARB,
an important agent class of RAAS inhibition, may have similar effect on arrhythmias as ACEI. Losartan can prevent the stretch-induced electrical re-modelling process in atrium [32] and had the clinically proved protection against AF. Zhang et al. [31] reported that ARB, candesartan, had anti-arrhythmic effect on mouse model of LV hypertrophy. Our results suggested down-regulation of AT1R mRNA in NIZ and protein expression in both BZ and NIZ might be one of the mechanisms for the observed beneficial effects on EPS-induced arrythmias of valsartan. Clinically, valsartan-based treatment reduced the development of new-onset AF in hypertensive patients in the VALUE trial [26] and Val-HeFT study showed that therapy with valsartan for heart failure significantly reduced the incidence of AF [25]. It is to note that there was a discrepancy between protein and mRNA expression in BZ on MI + Val rats, the underlying reason remains to be explored.
Besides RAS activation, Cxs, especially Cx43, remodelling is of importance for increased arrhythmic episodes after MI [33]. Cxs are intercellular low-resistance pathways and allow intercellular communication by electrical and metabolic coupling between neighbouring cells [34], which benefit coordination of electrical and mechanical activity. As expression and distribution of Cxs form the basis of the biophysics of heart and could influence the networking, changes in the Cxs pattern may lead to cardiac arrhythmia, which is characteristic of chronic heart diseases. It was shown that Cx43 was the predominant Cx in ventricular cardiomyocytes. Connexins 43 may negatively affect cardiac function after MI. Remodelling of cardiomyocyte interconnections may be an important determinant of VT in regions bordering healed infarcts. Loss of Cx43 expression greatly increased the risk of ventricular tachyarrhythmias and sudden death in adult mice model [12].
Previous studies [35, 36] also showed that the heterogeneous gap junction uncoupling, alteration in contraction would induce arrhythmia in heart failure, which was highly associated with expression and distribution of Cx43. Connexins 43 could enhance cell connection and intercellular communication, ischaemia caused translocation of Cx43 and altered the normal distribution of Cx43 in cardiomyocytes. Unuma et al. [37] reported that coronary occlusion induced transient Cx43 translocation, redistribution, dephosphorylation and reduction.
In line with this finding, our results showed reduced Cx43 expression and increased collagen deposition in BZ after MI, which might be responsible for the abnormal electrical rhythms after MI. Up-regulating Cx43 expression might contribute to the beneficial effects of valsartan on PES-induced arrhythmias. Previous study showed that loss of the regulatory domain of Cx43 led to an increase in infarct size and increased susceptibility to arrhythmia following MI [12, 38]. The incidence of hearts exhibiting pacing-induced VT was clearly increased after mutations in the Cx43 gene, which all suggested a crucial role of gap junction remodelling in the arrhythmogenesis [39]. Interestingly, valsartan significantly up-regulated the Cx43 mRNA expression in both BZ and NIZ and protein expression in BZ, up-regulation of Cx43 by valsartan might therefore serve as another important mechanism for the observed improved electro-stability by PES in valsartan-treated MI rats. In our previous study [40], we found that skeletal myoblast (SKM) transplantation in MI rats could reduce the arrhythmogenic effect by increasing the expression of Cx43 in SKMs. To prove the causal relationship between up-regulation of myocardial Cx43 and the arrhythmogenic effect of valsartan, in vivo studies exploring the valsartan effect on PES-induced VT or VF after knocking-down Cx43 are essential and future studies are therefore warranted.
In conclusions, the present study documented that valsartan not only improved cardiac function by attenuating ventricular remodelling, but also reduced the incidence of PES-induced ventricular arrhythmia after MI. These effects were possibly linked with the modulating effects of valsartan on myocardial AT1R and Cx43 expression.
Study limitations
A major limitation of the present study is the lack of impact in terms of translational medicine, as ACEI and ARB are already widely used in patients with ischaemic diseases.
Acknowledgments
This work is supported by National Natural Science Foundation of China (No. 30670831, No. 30871082 and No. 81070154) and Shanghai Excellent Academic Leaders Project (No. 10xd1402800).
We thank Dr. Kai Hu from University Wrzburg (Germany) and Dr. Yunzeng Zou from Zhongshan Hospital, Fudan University (China) for their help in the preparation of this manuscript.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Brezins M, Elyassov S, Elimelech I, et al. Comparison of patients with acute myocardial infarction with and without ventricular fibrillation. Am J Cardiol. 1996;78:948–50. doi: 10.1016/s0002-9149(96)00474-2. [DOI] [PubMed] [Google Scholar]
- 2.Pascale P, Schlaepfer J, Oddo M, et al. Ventricular arrhythmia in coronary artery disease: limits of a risk stratification strategy based on the ejection fraction alone and impact of infarct localization. Europace. 2009;11:1639–46. doi: 10.1093/europace/eup314. [DOI] [PubMed] [Google Scholar]
- 3.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–15. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura Y, Yoshiyama M, Omura T, et al. Beneficial effects of combination of ACE inhibitor and angiotensin II type 1 receptor blocker on cardiac remodeling in rat myocardial infarction. Cardiovasc Res. 2003;57:48–54. doi: 10.1016/s0008-6363(02)00644-2. [DOI] [PubMed] [Google Scholar]
- 5.Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–906. doi: 10.1056/NEJMoa032292. [DOI] [PubMed] [Google Scholar]
- 6.Tsutsui H, Matsushima S, Kinugawa S, et al. Angiotensin II type 1 receptor blocker attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Hypertens Res. 2007;30:439–49. doi: 10.1291/hypres.30.439. [DOI] [PubMed] [Google Scholar]
- 7.Stein M, Boulaksil M, Jansen JA, et al. Reduction of fibrosis-related arrhythmias by chronic renin-angiotensin-aldosterone system inhibitors in an aged mouse model. Am J Physiol Heart Circ Physiol. 2010;299:H310–21. doi: 10.1152/ajpheart.01137.2009. [DOI] [PubMed] [Google Scholar]
- 8.Yahiro E, Ideishi M, Wang LX, et al. Reperfusion-induced arrhythmias are suppressed by inhibition of the angiotensin II type 1 receptor. Cardiology. 2003;99:61–7. doi: 10.1159/000069722. [DOI] [PubMed] [Google Scholar]
- 9.Wachtell K, Devereux RB, Lyle PA, et al. The left atrium, atrial fibrillation, and the risk of stroke in hypertensive patients with left ventricular hypertrophy. Ther Adv Cardiovasc Dis. 2008;2:507–13. doi: 10.1177/1753944708093846. [DOI] [PubMed] [Google Scholar]
- 10.Wachtell K, Hornestam B, Lehto M, et al. Cardiovascular morbidity and mortality in hypertensive patients with a history of atrial fibrillation: the losartan intervention for end point reduction in hypertension (LIFE) study. J Am Coll Cardiol. 2005;45:705–11. doi: 10.1016/j.jacc.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 11.Severs NJ, Coppen SR, Dupont E, et al. Gap junction alterations in human cardiac disease. Cardiovasc Res. 2004;62:368–77. doi: 10.1016/j.cardiores.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 12.van Rijen HV, Eckardt D, Degen J, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–55. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- 13.Danik SB, Liu F, Zhang J, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–41. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim SY, Davidson SM, Paramanathan AJ, et al. The novel adipocytokine visfatin exerts direct cardioprotective effects. J Cell Mol Med. 2008;12:1395–403. doi: 10.1111/j.1582-4934.2008.00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jugdutt BI, Sawicki G. AT1 receptor blockade alters metabolic, functional and structural proteins after reperfused myocardial infarction: detection using proteomics. Mol Cell Biochem. 2004;263:179–88. [PubMed] [Google Scholar]
- 16.Cury AF, Bonilha A, Saraiva R, et al. Myocardial performance index in female rats with myocardial infarction: relationship with ventricular function parameters by Doppler echocardiography. J Am Soc Echocardiogr. 2005;18:454–60. doi: 10.1016/j.echo.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 17.Curtis MJ, Walker MJ. Quantification of arrhythmias using scoring systems: an examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc Res. 1988;22:656–65. doi: 10.1093/cvr/22.9.656. [DOI] [PubMed] [Google Scholar]
- 18.Walker MJ, Curtis MJ, Hearse DJ, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447–55. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 19.Chen YF, Kobayashi S, Chen J, et al. Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol. 2008;44:180–7. doi: 10.1016/j.yjmcc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohno T, Anzai T, Naito K, et al. Angiotensin-receptor blockade reduces border zone myocardial monocyte chemoattractant protein-1 expression and macrophage infiltration in post-infarction ventricular remodeling. Circ J. 2008;72:1685–92. doi: 10.1253/circj.cj-08-0115. [DOI] [PubMed] [Google Scholar]
- 21.Sun Y. Intracardiac renin-angiotensin system and myocardial repair/remodeling following infarction. J Mol Cell Cardiol. 2010;48:483–9. doi: 10.1016/j.yjmcc.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer R, Dechend R, Gapelyuk A, et al. Angiotensin II-induced sudden arrhythmic death and electrical remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H1242–53. doi: 10.1152/ajpheart.01400.2006. [DOI] [PubMed] [Google Scholar]
- 23.Fogari R, Derosa G, Ferrari I, et al. Effect of valsartan and ramipril on atrial fibrillation recurrence and P-wave dispersion in hypertensive patients with recurrent symptomatic lone atrial fibrillation. Am J Hypertens. 2008;21:1034–9. doi: 10.1038/ajh.2008.217. [DOI] [PubMed] [Google Scholar]
- 24.Goette A, Staack T, Rocken C, et al. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation. J Am Coll Cardiol. 2000;35:1669–77. doi: 10.1016/s0735-1097(00)00611-2. [DOI] [PubMed] [Google Scholar]
- 25.Maggioni AP, Latini R, Carson PE, et al. Valsartan reduces the incidence of atrial fibrillation in patients with heart failure: results from the Valsartan Heart Failure Trial (Val-HeFT) Am Heart J. 2005;149:548–57. doi: 10.1016/j.ahj.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 26.Schmieder RE, Kjeldsen SE, Julius S, et al. Reduced incidence of new-onset atrial fibrillation with angiotensin II receptor blockade: the VALUE trial. J Hypertens. 2008;26:403–11. doi: 10.1097/HJH.0b013e3282f35c67. [DOI] [PubMed] [Google Scholar]
- 27.Shimada Y, Gunasegaram S, Yokoyama H, et al. Inhibition of angiotensin-converting enzyme reduces susceptibility of hypertrophied rat myocardium to ventricular fibrillation. Circ J. 2002;66:1045–53. doi: 10.1253/circj.66.1045. [DOI] [PubMed] [Google Scholar]
- 28.Shimoni Y. Inhibition of the formation or action of angiotensin II reverses attenuated K+ currents in type 1 and type 2 diabetes. J Physiol. 2001;537:83–92. doi: 10.1111/j.1469-7793.2001.0083k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Tang W, Ristagno G, et al. The potential mechanisms of reduced incidence of ventricular fibrillation as the presenting rhythm in sudden cardiac arrest. Crit Care Med. 2009;37:26–31. doi: 10.1097/CCM.0b013e3181928914. [DOI] [PubMed] [Google Scholar]
- 30.Zankov DP, Omatsu-Kanbe M, Isono T, et al. Angiotensin II potentiates the slow component of delayed rectifier K+ current via the AT1 receptor in guinea pig atrial myocytes. Circulation. 2006;113:1278–86. doi: 10.1161/CIRCULATIONAHA.104.530592. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, Yasuno S, Kuwahara K, et al. Blockade of angiotensin II type 1 receptor improves the arrhythmia morbidity in mice with left ventricular hypertrophy. Circ J. 2006;70:335–41. doi: 10.1253/circj.70.335. [DOI] [PubMed] [Google Scholar]
- 32.Wachtell K, Gerdts E, Aurigemma GP, et al. In-treatment reduced left atrial diameter during antihypertensive treatment is associated with reduced new-onset atrial fibrillation in hypertensive patients with left ventricular hypertrophy: The LIFE Study. Blood Press. 2010;19:169–75. doi: 10.3109/08037051.2010.481811. [DOI] [PubMed] [Google Scholar]
- 33.Chen CC, Lin CC, Lee TM. 17beta-Estradiol decreases vulnerability to ventricular arrhythmias by preserving connexin43 protein in infarcted rats. Eur J Pharmacol. 2010;629:73–81. doi: 10.1016/j.ejphar.2009.11.050. [DOI] [PubMed] [Google Scholar]
- 34.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 35.Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:1762–70. doi: 10.1152/ajpheart.00346.2004. [DOI] [PubMed] [Google Scholar]
- 36.Shintani-Ishida K, Unuma K, Yoshida K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ J. 2009;73:1661–8. doi: 10.1253/circj.cj-09-0079. [DOI] [PubMed] [Google Scholar]
- 37.Unuma K, Shintani-Ishida K, Tsushima K, et al. Connexin-43 redistribution and gap junction activation during forced restraint protects against sudden arrhythmic death in rats. Circ J. 2010;74:1087–95. doi: 10.1253/circj.cj-09-1019. [DOI] [PubMed] [Google Scholar]
- 38.Maass K, Chase SE, Lin X, et al. Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc Res. 2009;84:361–7. doi: 10.1093/cvr/cvp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalcheva N, Qu J, Sandeep N, et al. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci U S A. 2007;104:20512–6. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li YG, Zhang PP, Jiao KL, et al. Knockdown of microRNA-181 by lentivirus mediated siRNA expression vector decreases the arrhythmogenic effect of skeletal myoblast transplantation in rat with myocardial infarction. Microvasc Res. 2009;78:393–404. doi: 10.1016/j.mvr.2009.06.011. [DOI] [PubMed] [Google Scholar]