

Abstract
Tenascins are large glycoproteins found in embryonic and adult extracellular matrices. Of the four family members, two have been shown to be overexpressed in the microenvironment of solid tumours: tenascin-C and tenascin-W. The regular presence of these proteins in tumours suggests a role in tumourigenesis, which has been investigated intensively for tenascin-C and recently for tenascin-W as well. In this review, we follow a malignant cell starting from its birth through its potential metastatic journey and describe how tenascin-C and tenascin-W contribute to these successive steps of tumourigenesis. We consider the importance of the mechanical aspect in tenascin signalling. Furthermore, we discuss studies describing tenascin-C as an important component of stem cell niches and present examples reporting its role in cancer therapy resistance.
Keywords: tenascin, genomic instability, stem cells, primary tumour, EMT, angiogenesis, extracellular matrix, metastasis, mechanical forces, cancer therapy, microenvironment, stroma
Introduction
Tumourigenesis has traditionally been described as a cell-autonomous process when a cell, after accumulation of genetic alterations, loses its control over growth, undergoes aberrant proliferation and thus gives rise to a tumour. It is now accepted that the microenvironment surrounding a potential tumour cell plays a crucial role influencing its fate [1]. For instance, experiments exposing pre-neoplastic or tumour cells to various mesenchymal environments have underlined the critical role of the stroma in this context [2–4]. Among the proteins known to be overexpressed in tumour-associated stroma are tenascin-C and tenascin-W. Tenascins are large glycoproteins found in the extracellular compartment of various tissues. The tenascin family has four members: tenascin-C, tenascin-R, tenascin-X and tenascin-W. They all share a characteristic modular structure with an oligomerization domain followed by EGF-like repeats, fibronectin (FN) type III repeats and a fibrinogen globe (see Ref. [5] and Fig. 1). In the case of tenascin-C and tenascin-R, alternative splicing can lead to the generation of multiple isoforms that contain additional FN type III repeats. Only two of the tenascin members, the original tenascin-C [6, 7] and the more recently discovered tenascin-W [8], have been shown to be overexpressed in tumours compared to healthy tissues (for review, see Ref. [9]). In most cases, healthy tissues are not completely deprived of tenascin-C [10]. However, corresponding tumour tissues express much higher levels of tenascin-C or harbour different, larger isoforms [11]. Tenascin-W expression is much more restricted in adult healthy tissues [10], which makes it an excellent tumour marker: it is absent in healthy tissue but present in most breast [12], colon [13] and brain [14] tumours. In contrast, tenascin-R and tenascin-X expression is rather constant and is not modulated by tissues malignancy (for review, see Ref. [15]). In this review, we will follow the major steps in the life of a tumour cell and descendants and describe how tenascin-C and tenascin-W influence their fate.
Fig 1.
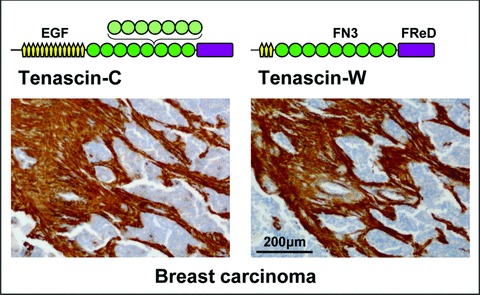
Tenascin structure and tumour stroma staining. A schematic model representing one subunit of the hexameric tenascin-C and tenascin-W proteins is shown above a breast carcinoma section stained with antibodies against the two proteins. Both tenascins are built from the following domains: a central, N-terminal oligomerization domain (black line), EGF-like repeats (EGF, yellow), FN type III repeats (FN3, green; FN3 repeats subject to alternative splicing in light green) and a C-terminal fibrinogen related domain (FReD, violet). Antibodies against tenascin-C and tenascin-W strongly stain the stroma of the breast carcinoma, although the epithelial tumour nests (nuclei in blue) are negative.
Birth of a malignant cell
The birth of a malignant cell is a multi-step process, which starts with the acquisition of alterations in genes encoding proteins implicated in cellular growth control. For this reason, the first defense of an organism against cancer relies on the prevention of genomic alterations. At the cellular level, many responses to mutagenic challenges have been developed. For instance, cell cycle arrest is triggered to enable proofreading of DNA and ensure genomic stability. Interestingly, the state of cell adhesion is suspected to alter pathways controlling genomic stability (for review, see Ref. [16]). Because tenascins modulate the adhesion status of cells [17], it is tempting to speculate that they may also influence genomic stability [9]. Consistent with this hypothesis, BRCA1-associated RING domain 1 (Brad1), H2A histone family member X (H2AX), as well as other molecules with assigned functions in genome stability were found to be down-regulated in the presence of tenascin-C in glioblastoma cells [18]. The hypothesis that tenascin-C could favour the birth of malignant cells implies its expression before the development of the tumour. Remarkably, transgenic mice expressing an autoactivating mutant of the matrix metalloproteinase stromelysin-1 in mammary epithelia show inappropriate expression of tenascin-C, starting by day 6 of pregnancy and followed by the development of mammary pre-neoplastic and neoplastic lesions [19]. Furthermore, fibroblasts expanded from Gorlin syndrome patients, which are prone to develop basal cell carcinomas, show phenotypic traits reminiscent of cancer-associated fibroblasts. In particular, tenascin-C is highly expressed by these cells [20]. It is believed that the dermis contributes to the predisposition of these patients to develop basal cell carcinomas, which is compatible with the hypothesis that a tenascin-C rich environment favours the birth of malignant cells.
Niches for self-renewable cells
The capacity to proliferate indefinitely and to self-renew is a shared hallmark of stem cells and tumour cells. Hence, major signalling pathways initially aimed at regulating normal stem cells, such as Wnt, Shh and Notch signalling are re-utilized by the tumour cell machinery (for review, see Ref. [21]). It is interesting to note that tenascin-C was shown to be associated with stem cell niches in the neural, haematopoietic and epidermal system (for review, see Ref. [22]). Tenascin-C in brain is highly expressed in specialized germinal zones such as the subventricular zone where it may play an important role in the regulation of stem cells because transgenic mice lacking tenascin-C show altered numbers of central nervous system stem cells [23]. Specific carbohydrate side chains of tenascin-C could play a key role in the regulation of embryonic neural stem cell proliferation. For instance, human natural killer 1 (HNK1) carbohydrate epitopes carried by large splice variants of tenascin-C are involved in the proliferation of neural stem cells via modulation of the expression level of epidermal growth factor (EGF) receptor [24]. In parallel, tenascin-C was identified as a cell surface glycoprotein marker for glioblastoma-derived stem-like cells [25]. Tenascin-C is also present in the haematopoietic stem cell microenvironment [26], where it may regulate stem cell recruitment because mice lacking tenascin-C show reduced haematopoiesis [27]. In addition, tenascin-C could be part of the specialized extracellular matrix characterizing the niche of epidermal stem cells because it is strongly up-regulated in the hair follicle bulge, which is known to be a stem cell reservoir [28].
Expression patterns in tumours
In tumours arising from epithelial organs (i.e. in most carcinomas), the source of tenascins is restricted to the structural, mesenchymal compartment: very strong tenascin immunostaining is found in the tumour stroma surrounding tenascin-free tumour nests (see Fig. 1, for a typical staining pattern). The stromal tenascin-C expression can be influenced by cancer cells because co-culture of fibroblasts with tumour cells has been shown to stimulate tenascin-C expression in the fibroblasts [29, 30]. In other tumours such as melanoma and brain tumours, the tumour cells themselves are the source of tenascin-C [31, 32]. Interestingly, brain tumours arise from structural cells (oligodendrocytes for oligodendrogliomas, astrocytes for astrocytomas and glioblastomas), which may explain that these cells have common features with mesenchymal cells rather than with carcinoma cells. Melanomas develop from neural crest-derived melanocytes. Neural crest cells express high levels of tenascin-C [33], which is required for their migration in embryogenesis [34]. In contrast to tenascin-C, tenascin-W expression has yet to be described in tumour cells themselves. However, tenascin-W is also expressed in the stroma of breast and colon carcinomas. In brain tumours, both tenascin-C and tenascin-W can be observed around blood vessels. However, their pattern of expression is distinct because tenascin-C surrounds both endothelial and pericyte cell layers, whereas tenascin-W is restricted to the endothelial cell layer [14].
Promotion of tumour cell proliferation
An important feature of tumour cells is their ability to overproliferate. Interestingly, tenascin-C has been shown to promote tumour cell proliferation in standard cell culture conditions [7, 35]. More specifically, culturing glioblastoma and breast carcinoma cells on mixed fibronectin/tenascin-C substrata not only attenuated their adhesion but also increased their proliferation rate compared to a pure fibronectin substratum [35]. Similar observations were recently made in three-dimensional mammary epithelial tissues reconstructed in vitro [36]. In this model, human mammary epithelial cells form polarized multicellular acini characterized by the presence of a hollow lumen. In the presence of tenascin-C, acini fail to generate a continuous basement membrane and cell proliferation is increased. Similarly, growth of melanoma spheres (known to be enriched for stem-like cells) is significantly reduced in absence of tenascin-C [37]. In vivo, expression of tenascin-C at the invasion border of early breast cancers was associated with a higher proliferation rate assessed by Ki-67 staining and the analysis of the fraction of cells in S-phase [38].
Promotion of tumour cell migration
Tenascin-C was initially described as an anti-adhesive molecule that antagonizes the capacity of tumour cells to adhere and spread on fibronectin [39]. Experiments performed in three-dimensional fibrin matrices containing fibronectin have revealed that tenascin-C suppresses actin stress fibre formation via inhibition of RhoA activation, and instead induces actin-rich filopodia [40]. A very specific adhesion state is required for the cell to be able to migrate (for review, see Ref. [41]). When a cell adheres too strongly, the links between the cytoskeleton and the extracellular matrix are difficult to break and the cell remains fixed to the substratum. Conversely, when a cell adheres weakly, contractile forces required for motility are missing [42]. Adhesion to a tenascin-C–rich matrix can be defined as intermediate, and thus favours both cell motility [43, 44] and invasion [45]. Interestingly, the site of tenascin-C responsible for focal adhesion disassembly has been located in the alternatively spliced FN type III repeats [46]. In agreement, large tenascin-C variants containing FN type III extra-repeats expressed by co-cultured fibroblasts elicit increased cell migration and invasion in breast cancer models [47]. In contrast to tenascin-C, tenascin-W does not interfere with the cell adhesive function of fibronectin [12]. However, it also stimulates migration of mammary cancer cells [12, 48].
Contribution to epithelial–mesenchymal transition (EMT)
Epithelial-mesenchymal transition is a developmental process re-utilized by cancer cells and characterized by loss of cell adhesion, repression of E-cadherin expression as well as increased cell mobility. Tenascin-C has been shown to be associated with EMT because tumour cell lines undergoing transforming growth factor-β (TGF-β) induced EMT secrete tenascin-C [49]. Furthermore, vimentin (an EMT marker) and tenascin-C expressions are associated in cancer cells [50], and cancer cell lines with clear epithelial morphology secrete far less tenascin-C than cancer cell lines characterized by a fibroblastic morphology (our observations). Finally, a recent study reports that tenascin-C is required for injury-induced EMT of lens epithelium [51]. However, except for the last example, it remains to be determined whether tenascin-C expression is only a consequence of EMT or may also be required for this process.
Promotion of angiogenesis
When a tumour reaches a critical size, it has to attract the formation of new vessels to supply nutrients and oxygen to the proliferating cells. Both tenascin-C and tenascin-W can been classified as pro-angiogenic factors because they trigger endothelial cells to acquire a sprouting phenotype and increase their migration capacities [14, 52, 53]. Tenascin-C is known to play a crucial role in postnatal cardiac angiogenesis because tenascin-C–deficient mice fail to vascularize cardiac allografts in an established cardiac transplantation model [54]. Furthermore, in a xenograft tumour model it was shown that tumours were much more vascularized when they were grown in wild-type mice than in tenascin-C–deficient mice, suggesting that the presence of tenascin-C in the microenvironment is very important for tumour angiogenesis [55]. Tenascin-C was also associated with lymphangiogenesis [56], particularly in the context of podoplanin induction [57]. The proximity of tenascin-C and tenascin-W around blood vessels prompts the question whether or not these proteins can extravasate from the tumour site and circulate in the bloodstream. Indeed, tenascin-C was found in the serum of cancer patients, but because its presence in serum was also associated with conditions other than cancer (e.g. as a consequence of infection and inflammation, see Ref. [10]), tenascin-C turned out to be a questionable serum tumour marker [58]. In contrast, tenascin-W function in adults seems to be restricted to tumourigenesis and osteogenesis (for review, see Ref. [59]), so it may become a better tumour biomarker. Indeed, tenascin-W levels in serum from breast and colorectal cancer patients were found to be elevated compared to serum collected from healthy, control individuals [13]. However, individual tenascin-W levels were heterogeneous, with some patients exhibiting highly elevated values and others with values within the range of controls. Interestingly, most patients that developed early tumour recurrence were initially characterized by high serum tenascin-W levels. A follow-up study including a large cohort of patients should therefore be performed to confirm this tendency. Together, these results suggest that tenascin-W is a promising tumour biomarker, but should be used in combination with other markers to avoid generation of false negative distortions.
Promotion of metastasis
After escape from the primary tumour and circulation in the blood or lymphatic vessels cancer cells will, as an ultimate step, colonize other organs of the body. The nature of the ‘soil’ influences greatly the choice of host tissues in which the tumour cells will ‘seed’[60]. Interestingly, extracellular matrix proteins produced by tumour cells themselves also determine their metastatic capacity. For instance, knocking-down tenascin-C in melanoma cells significantly decreased their capacity to form pulmonary metastases [37]. This holds true as well for certain breast tumour xenograft models [61, 62]. In support for a human relevance of these observations, high tenascin-C expression was associated with an 8-fold increased risk of metastasis in classic giant cell tumours of bone [63]. A recent study suggests that the fibrillar organization of tenascin-C, requiring stromal fibroblasts and active MMP2, is associated with metastatic pancreatic cancers [64]. However, activation of tenascin-C–dependent metastatic pathways may depend on the initial oncogenic alterations because tenascin-C is not found in all metastatic transcript profiles [65]. To our knowledge, tenascin-W expression has not been associated with metastasis in human beings. However, this possibility is difficult to investigate in experimental models because cancer cell lines expanded from any type of tumours were negative for tenascin-W expression (our own observation).
How do tenascins signal to cells?
We described above the effects triggered by tenascins on tumour and endothelial cells, but a main question remains: how do the signals activated by tenascins reach the nucleus where regulation of gene expression takes place? There are many possibilities how this can occur. For example, the signal can be transmitted by an indirect, ‘classical’ pathway, that is through activation of transmembrane receptor proteins, cascades of kinases, and activation of transcription factors, which finally act in the nucleus to modulate gene expression. Alternatively, the signal could also be transduced mechanically in a more direct manner through the cell cytoskeleton, which physically connects the outside of a cell with the chromatin [66]. Concerning indirect ways of activation, there is no consensus for a given signalling pathway, suggesting that tenascin-C can activate many parallel context- and tissue-specific signalling cascades. On the level of cell surface receptors, a large set of integrins have been identified as receptors for tenascin-C (α2β1, αvβ3, α7β1, α8β1, α9β1, α5β3, α5β6 [67–71]) and for tenascin-W (α8β1, αvβ1, α4β1 [12, 48]). The fact that tenascins contain epidermal growth factor motifs suggests that they could bind to EGF receptors. Tenascin-C was classified as an ‘insoluble growth factor ligand’[72] because it can bind to EGFR with a low affinity insufficient to trigger ligand internalization. This binding favours activation of motility rather than proliferation responses [73]. Interestingly, tenascin-C–enriched medium can stimulate invasion of colon cancer cells only in presence of a functional EGFR [45]. The receptor of hepatocyte growth factor, c-Met, was shown to work in close association with tenascin-C both in colon and mammary culture models [36, 45]. Tenascin-C triggers in vitro the formation of filled (instead of hollow) acini surrounded by an abnormal basement membrane. This effect is abrogated in the absence of functional c-Met receptors [36]. Another transmembrane protein, TLR4, a member of the toll-like receptor family which is known to contribute to infection and inflammation responses, is activated by tenascin-C [74]. Whether tenascin-C binds as a ligand to TLR4 remains to be investigated.
In addition to EGFR, HGF/c-Met and TLR4, two other well-described pathways are suspected to be modulated by tenascin-C: these are TGF-β and Wnt signalling pathways. Gene expression profile analysis of T98G glioblastoma cells cultured in presence of tenascin-C revealed, for instance, down-regulation of follistatin, a known inhibitor of activins [18]. This study also led to the identification of Dkk1, a known inhibitor of the Wnt pathway, as a gene down-regulated by tenascin-C. Interestingly, a potential mechanism of cell cycle regulation by tenascin-C could also be through induction of 14-3-3τ[75] followed by p21 down-regulation [76].
Another more direct way for tenascin-C–triggered signals to reach the nucleus has been recently investigated in our laboratory [77]. These studies show that disruption of the physical link connecting the cell membrane to the nucleus perturbs the mechanical control of cell differentiation in the context of myogenesis. This suggests that tenascin-C–induced cytoskeletal rearrangements could also affect mechanotransduction in the frame of tumourigenesis.
It is important to note that transduction of tenascin signals to cells could also be influenced by other extracellular matrix molecules. For instance, through their binding to tenascin-C, fibronectin [78], perlecan [79], lectican [80], heparin [81], contactin [82, 83] and SMOC1 [84] have been shown to modify its effects.
Importance of the mechanical aspect
An important parameter for the progression of a tumour is the stiffness of the tissue in which it develops. Reconstruction of mammary epithelial acini in vitro has shown that the rigidity of the matrix strongly influences epithelial morphogenesis [85]. Consistently, in vivo reduction of lysyl oxidase-mediated collagen crosslinking to weaken the collagen fibres and to reduce their tensile strength decreases mammary tumour incidence [86]. Interestingly, mechanical strain not only influences features of the epithelial tumour cells themselves but may also stimulate neovascularization through activation of angiogenic genes [87]. It is therefore interesting to note that tenascin-C is induced by mechanical strain, requiring integrin-linked kinase [88] followed by RhoA/ROCK-dependent actin contractility [89]. Interestingly, fibroblasts cultured on attached, restrained collagen gels express much more tenascin-C than those cultured on floating, unrestrained collagen gels [90]. This may be an explanation why cancer-associated fibroblasts, which are exposed to high mechanical strain, overexpress tenascin-C compared to ‘normal’ fibroblasts populating healthy tissues. Stiffness of the tumour environment may also favour transdifferentiation of cancer-associated fibroblasts into myofibroblasts as myodifferentiation of primary lung fibroblasts has been recently reported to be influenced by cyclic mechanical stress [91].
Evasion of tumour cells from conventional therapy
Interestingly, tenascin-C has been shown to mediate chemotherapy-resistance in various contexts. Knocking-down tenascin-C sensitizes melanoma cells to doxorubicin [37]. This happens most likely via down-regulation of ATP-binding cassette B5, a transporter known to provide the cells high efflux capacity for anti-mitotic drugs. In the frame of breast cancer, tenascin-C could be associated with resistance to tamoxifen therapy [92] and doxorubicin therapy [76]. Interestingly, the doxorubicin-induced G1/S arrest could be abrogated through p21 regulation, which in turn is known to be affected by the presence of tenascin-C. It was also shown that large isoforms of tenascin-C and cell surface protein annexin A2 can interact and together decrease gemcitabine-induced cytotoxicity in pancreatic cancer cells [93]. Tenascin-C might also be linked to resistance to anti-angiogenesis treatment because hypoxic tumour cells adapt by inducing new sets of genes, among which tenascin-C is found [94].
Conclusions
We have summarized how tenascins have been shown to influence the successive steps of tumourigenesis (summarized in Fig. 2). Based on our knowledge of the properties of tenascin-C, two types of anti-cancer therapies have been established. The first type aims at the neutralization of its pro-tumourigenic effects by down-regulating its expression using anti-sense oligonucleotides or aptamers (for review, see Refs. [95, 96], respectively). The second type takes advantage of the specific overexpression of tenascin-C isoforms at tumour sites to selectively target tumour cells, using anti–tenascin-C antibody fragments coupled to a variety of anti-cancer molecules (for review, see Ref. [97]). Further studies will be necessary to extend our knowledge to tenascin-W, but studies reported so far suggest a promising future for tenascin-W as a tumour marker and potential drug target.
Fig 2.
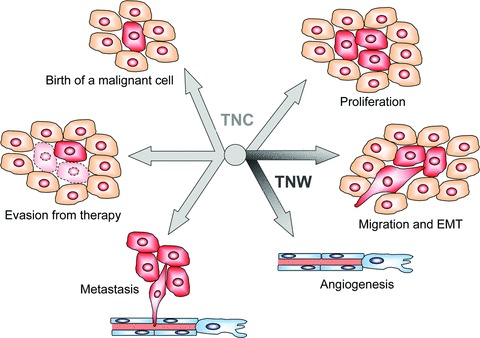
Summary of the actions of tenascins in cancer. The centre of the figure depicts the six-armed tenascin-C (TNC) and tenascin-W (TNW) protein pointing with their arms towards processes that are influenced by their presence. Although TNC is involved in all processes depicted, including the birth of a malignant cell, proliferation, migration and EMT, angiogen-esis, metastasis and evasion from therapy, TNW was shown to affect cell migration and angiogenesis (indicated by the darker arms).
Acknowledgments
We thank Richard P. Tucker for critical reading of the paper and helpful suggestions. This work is supported by the grant 31003A-120235 from the Swiss National Science Foundation SNF to R.C.-E.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Allen M, Louise Jones J. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol. 2011;223:162–76. doi: 10.1002/path.2803. [DOI] [PubMed] [Google Scholar]
- 2.Milas L, Hirata H, Hunter N, et al. Effect of radiation-induced injury of tumour bed stroma on metastatic spread of murine sarcomas and carcinomas. Cancer Res. 1988;48:2116–20. [PubMed] [Google Scholar]
- 3.Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma-associated fibroblasts direct tumour progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shekhar MP, Werdell J, Santner SJ, et al. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumour development and progression. Cancer Res. 2001;61:1320–6. [PubMed] [Google Scholar]
- 5.Chiquet-Ehrismann R. Tenascins. Int J Biochem Cell Biol. 2004;36:986–90. doi: 10.1016/j.biocel.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Bourdon MA, Wikstrand CJ, Furthmayr H, et al. Human glioma-mesenchymal extracellular matrix antigen defined by monoclonal antibody. Cancer Res. 1983;43:2796–805. [PubMed] [Google Scholar]
- 7.Chiquet-Ehrismann R, Mackie EJ, Pearson CA, et al. Tenascin: an extracellular matrix protein involved in tissue interactions during foetal development and oncogenesis. Cell. 1986;47:131–9. doi: 10.1016/0092-8674(86)90374-0. [DOI] [PubMed] [Google Scholar]
- 8.Scherberich A, Tucker RP, Samandari E, et al. Murine tenascin-W: a novel mammalian tenascin expressed in kidney and at sites of bone and smooth muscle development. J Cell Sci. 2004;117:571–81. doi: 10.1242/jcs.00867. [DOI] [PubMed] [Google Scholar]
- 9.Orend G. Potential oncogenic action of tenascin-C in tumourigenesis. Int J Biochem Cell Biol. 2005;37:1066–83. doi: 10.1016/j.biocel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Sports. 2009;19:511–9. doi: 10.1111/j.1600-0838.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 11.Jones PL, Jones FS. Tenascin-C in development and disease: gene regulation and cell function. Matrix Biol. 2000;19:581–96. doi: 10.1016/s0945-053x(00)00106-2. [DOI] [PubMed] [Google Scholar]
- 12.Degen M, Brellier F, Kain R, et al. Tenascin-W is a novel marker for activated tumour stroma in low-grade human breast cancer and influences cell behavior. Cancer Res. 2007;67:9169–79. doi: 10.1158/0008-5472.CAN-07-0666. [DOI] [PubMed] [Google Scholar]
- 13.Degen M, Brellier F, Schenk S, et al. Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int J Cancer. 2008;122:2454–61. doi: 10.1002/ijc.23417. [DOI] [PubMed] [Google Scholar]
- 14.Martina E, Degen M, Ruegg C, et al. Tenascin-W is a specific marker of glioma-associated blood vessels and stimulates angiogenesis in vitro. Faseb J. 2010;24:778–87. doi: 10.1096/fj.09-140491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucker RP, Chiquet-Ehrismann R. The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta. 2009;1793:888–92. doi: 10.1016/j.bbamcr.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Tlsty TD. Cell-adhesion-dependent influences on genomic instability and carcinogenesis. Curr Opin Cell Biol. 1998;10:647–53. doi: 10.1016/s0955-0674(98)80041-0. [DOI] [PubMed] [Google Scholar]
- 17.Chiquet-Ehrismann R, Tucker R. Tenascins and the importance of adhesion modulation. Cold Spring Harb Perspect Biol. 2011;3:a004960. doi: 10.1101/cshperspect.a004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz C, Huang W, Hegi ME, et al. Growth promoting signaling by tenascin-C [corrected] Cancer Res. 2004;64:7377–85. doi: 10.1158/0008-5472.CAN-04-1234. [DOI] [PubMed] [Google Scholar]
- 19.Thomasset N, Lochter A, Sympson CJ, et al. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am J Pathol. 1998;153:457–67. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valin A, Barnay-Verdier S, Robert T, et al. PTCH1 +/− dermal fibroblasts isolated from healthy skin of Gorlin syndrome patients exhibit features of carcinoma associated fibroblasts. PLOS One. 2009;4:e4818. doi: 10.1371/journal.pone.0004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 22.von Holst A. Tenascin C in stem cell niches: redundant, permissive or instructive? Cells Tissues Organs. 2008;188:170–7. doi: 10.1159/000112848. [DOI] [PubMed] [Google Scholar]
- 23.Garcion E, Halilagic A, Faissner A, et al. Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development. 2004;131:3423–32. doi: 10.1242/dev.01202. [DOI] [PubMed] [Google Scholar]
- 24.Yagi H, Yanagisawa M, Suzuki Y, et al. HNK-1 epitope-carrying Tenascin-C spliced variant regulates the proliferation of mouse embryonic neural stem cells. J Biol Chem. 2010;285:37293–301. doi: 10.1074/jbc.M110.157081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He J, Liu Y, Xie X, et al. Identification of cell surface glycoprotein markers for glioblastoma-derived stem-like cells using a lectin microarray and LC-MS/MS approach. J Proteome Res. 2010;9:2565–72. doi: 10.1021/pr100012p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein G, Beck S, Muller CA. Tenascin is a cytoadhesive extracellular matrix component of the human hematopoietic microenvironment. J Cell Biol. 1993;123:1027–35. doi: 10.1083/jcb.123.4.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta M, Sakai T, Saga Y, et al. Suppression of hematopoietic activity in tenascin-C-deficient mice. Blood. 1998;91:4074–83. [PubMed] [Google Scholar]
- 28.Kloepper JE, Tiede S, Brinckmann J, et al. Immunophenotyping of the human bulge region: the quest to define useful in situ markers for human epithelial hair follicle stem cells and their niche. Exp Dermatol. 2008;17:592–609. doi: 10.1111/j.1600-0625.2008.00720.x. [DOI] [PubMed] [Google Scholar]
- 29.Chiquet-Ehrismann R, Kalla P, Pearson CA. Participation of tenascin and transforming growth factor-beta in reciprocal epithelial-mesenchymal interactions of MCF7 cells and fibroblasts. Cancer Res. 1989;49:4322–5. [PubMed] [Google Scholar]
- 30.Jotzu C, Alt E, Welte G, et al. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumour-derived factors. Anal Cell Pathol (Amst) 2010;33:61–79. doi: 10.3233/ACP-CLO-2010-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukunaga-Kalabis M, Santiago-Walker A, Herlyn M. Matricellular proteins produced by melanocytes and melanomas: in search for functions. Cancer Microenviron. 2008;1:93–102. doi: 10.1007/s12307-008-0009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirata E, Arakawa Y, Shirahata M, et al. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009;100:1451–9. doi: 10.1111/j.1349-7006.2009.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tucker RP, McKay SE. The expression of tenascin by neural crest cells and glia. Development. 1991;112:1031–9. doi: 10.1242/dev.112.4.1031. [DOI] [PubMed] [Google Scholar]
- 34.Tucker RP. Abnormal neural crest cell migration after the in vivo knockdown of tenascin-C expression with morpholino antisense oligonucleotides. Dev Dyn. 2001;222:115–9. doi: 10.1002/dvdy.1171. [DOI] [PubMed] [Google Scholar]
- 35.Huang W, Chiquet-Ehrismann R, Moyano JV, et al. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumour cell proliferation. Cancer Res. 2001;61:8586–94. [PubMed] [Google Scholar]
- 36.Taraseviciute A, Vincent BT, Schedin P, et al. Quantitative analysis of three-dimensional human mammary epithelial tissue architecture reveals a role for tenascin-C in regulating c-met function. Am J Pathol. 2010;176:827–38. doi: 10.2353/ajpath.2010.090006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukunaga-Kalabis M, Martinez G, Nguyen TK, et al. Tenascin-C promotes melanoma progression by maintaining the ABCB5-positive side population. Oncogene. 2010;29:6115–24. doi: 10.1038/onc.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jahkola T, Toivonen T, Virtanen I, et al. Tenascin-C expression in invasion border of early breast cancer: a predictor of local and distant recurrence. Br J Cancer. 1998;78:1507–13. doi: 10.1038/bjc.1998.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiquet-Ehrismann R, Kalla P, Pearson CA, et al. Tenascin interferes with fibronectin action. Cell. 1988;53:383–90. doi: 10.1016/0092-8674(88)90158-4. [DOI] [PubMed] [Google Scholar]
- 40.Wenk MB, Midwood KS, Schwarzbauer JE. Tenascin-C suppresses Rho activation. J Cell Biol. 2000;150:913–20. doi: 10.1083/jcb.150.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001;107:785–90. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palecek SP, Loftus JC, Ginsberg MH, et al. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–40. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 43.Deryugina EI, Bourdon MA. Tenascin mediates human glioma cell migration and modulates cell migration on fibronectin. J Cell Sci. 1996;109:643–52. doi: 10.1242/jcs.109.3.643. [DOI] [PubMed] [Google Scholar]
- 44.Nishio T, Kawaguchi S, Yamamoto M, et al. Tenascin-C regulates proliferation and migration of cultured astrocytes in a scratch wound assay. Neuroscience. 2005;132:87–102. doi: 10.1016/j.neuroscience.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 45.De Wever O, Nguyen QD, Van Hoorde L, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. Faseb J. 2004;18:1016–8. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- 46.Murphy-Ullrich JE, Lightner VA, Aukhil I, et al. Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol. 1991;115:1127–36. doi: 10.1083/jcb.115.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hancox RA, Allen MD, Holliday DL, et al. Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009;11:R24. doi: 10.1186/bcr2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scherberich A, Tucker RP, Degen M, et al. Tenascin-W is found in malignant mammary tumours, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene. 2005;24:1525–32. doi: 10.1038/sj.onc.1208342. [DOI] [PubMed] [Google Scholar]
- 49.Maschler S, Grunert S, Danielopol A, et al. Enhanced tenascin-C expression and matrix deposition during Ras/TGF-beta-induced progression of mammary tumour cells. Oncogene. 2004;23:3622–33. doi: 10.1038/sj.onc.1207403. [DOI] [PubMed] [Google Scholar]
- 50.Dandachi N, Hauser-Kronberger C, More E, et al. Co-expression of tenascin-C and vimentin in human breast cancer cells indicates phenotypic transdifferentiation during tumour progression: correlation with histopathological parameters, hormone receptors, and oncoproteins. J Pathol. 2001;193:181–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH752>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka S, Sumioka T, Fujita N, et al. Suppression of injury-induced epithelial-mesenchymal transition in a mouse lens epithelium lacking tenascin-C. Mol Vis. 2010;16:1194–205. [PMC free article] [PubMed] [Google Scholar]
- 52.Canfield AE, Schor AM. Evidence that tenascin and thrombospondin-1 modulate sprouting of endothelial cells. J Cell Sci. 1995;108:797–809. doi: 10.1242/jcs.108.2.797. [DOI] [PubMed] [Google Scholar]
- 53.Chung CY, Murphy-Ullrich JE, Erickson HP. Mitogenesis, cell migration, and loss of focal adhesions induced by tenascin-C interacting with its cell surface receptor, annexin II. Mol Biol Cell. 1996;7:883–92. doi: 10.1091/mbc.7.6.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ballard VL, Sharma A, Duignan I, et al. Vascular tenascin-C regulates cardiac endothelial phenotype and neovascularization. Faseb J. 2006;20:717–9. doi: 10.1096/fj.05-5131fje. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka K, Hiraiawa N, Hashimoto H, et al. Tenascin-C regulates angiogenesis in tumour through the regulation of vascular endothelial growth factor expression. Int J Cancer. 2004;108:31–40. doi: 10.1002/ijc.11509. [DOI] [PubMed] [Google Scholar]
- 56.Clasper S, Royston D, Baban D, et al. A novel gene expression profile in lymphatics associated with tumour growth and nodal metastasis. Cancer Res. 2008;68:7293–303. doi: 10.1158/0008-5472.CAN-07-6506. [DOI] [PubMed] [Google Scholar]
- 57.Cueni LN, Hegyi I, Shin JW, et al. Tumour lymphangiogenesis and metastasis to lymph nodes induced by cancer cell expression of podoplanin. Am J Pathol. 2010;177:1004–16. doi: 10.2353/ajpath.2010.090703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schenk S, Muser J, Vollmer G, et al. Tenascin-C in serum: a questionable tumour marker. Int J Cancer. 1995;61:443–9. doi: 10.1002/ijc.2910610402. [DOI] [PubMed] [Google Scholar]
- 59.Martina E, Chiquet-Ehrismann R, Brellier F. Tenascin-W: an extracellular matrix protein associated with osteogenesis and cancer. Int J Biochem Cell Biol. 2010;42:1412–5. doi: 10.1016/j.biocel.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 60.Mendoza M, Khanna C. Revisiting the seed and soil in cancer metastasis. Int J Biochem Cell Biol. 2009;41:1452–62. doi: 10.1016/j.biocel.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calvo A, Catena R, Noble MS, et al. Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumour growth and lung metastasis. Oncogene. 2008;27:5373–84. doi: 10.1038/onc.2008.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pazzaglia L, Conti A, Chiechi A, et al. Differential gene expression in classic giant cell tumours of bone: Tenascin C as biological risk factor for local relapses and metastases. Histopathology. 2010;57:59–72. doi: 10.1111/j.1365-2559.2010.03597.x. [DOI] [PubMed] [Google Scholar]
- 64.Chen J, Chen Z, Chen M, et al. Role of fibrillar Tenascin-C in metastatic pancreatic cancer. Int J Oncol. 2009;34:1029–36. [PubMed] [Google Scholar]
- 65.Qiu TH, Chandramouli GV, Hunter KW, et al. Global expression profiling identifies signatures of tumour virulence in MMTV-PyMT-transgenic mice: correlation to human disease. Cancer Res. 2004;64:5973–81. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 66.Crisp M, Liu Q, Roux K, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramos DM, Chen BL, Boylen K, et al. Stromal fibroblasts influence oral squamous-cell carcinoma cell interactions with tenascin-C. Int J Cancer. 1997;72:369–76. doi: 10.1002/(sici)1097-0215(19970717)72:2<369::aid-ijc28>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 68.Sriramarao P, Mendler M, Bourdon MA. Endothelial cell attachment and spreading on human tenascin is mediated by alpha 2 beta 1 and alpha v beta 3 integrins. J Cell Sci. 1993;105:1001–12. doi: 10.1242/jcs.105.4.1001. [DOI] [PubMed] [Google Scholar]
- 69.Yokosaki Y, Palmer EL, Prieto AL, et al. The integrin alpha 9 beta 1 mediates cell attachment to a non-RGD site in the third fibronectin type III repeat of tenascin. J Biol Chem. 1994;269:26691–6. [PubMed] [Google Scholar]
- 70.Varnum-Finney B, Venstrom K, Muller U, et al. The integrin receptor alpha 8 beta 1 mediates interactions of embryonic chick motor and sensory neurons with tenascin-C. Neuron. 1995;14:1213–22. doi: 10.1016/0896-6273(95)90268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yokosaki Y, Monis H, Chen J, et al. Differential effects of the integrins alpha9beta1, alphavbeta3, and alphavbeta6 on cell proliferative responses to tenascin. Roles of the beta subunit extracellular and cytoplasmic domains. J Biol Chem. 1996;271:24144–50. doi: 10.1074/jbc.271.39.24144. [DOI] [PubMed] [Google Scholar]
- 72.Swindle CS, Tran KT, Johnson TD, et al. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol. 2001;154:459–68. doi: 10.1083/jcb.200103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iyer AK, Tran KT, Griffith L, et al. Cell surface restriction of EGFR by a tenascin cytotactin-encoded EGF-like repeat is preferential for motility-related signaling. J Cell Physiol. 2008;214:504–12. doi: 10.1002/jcp.21232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–80. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 75.Martin D, Brown-Luedi M, Chiquet-Ehrismann R. Tenascin-C signaling through induction of 14-3-3 tau. J Cell Biol. 2003;160:171–5. doi: 10.1083/jcb.200206109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang B, Liu K, Lin HY, et al. 14-3-3 Tau regulates ubiquitin-independent proteasomal degradation of p21, a novel mechanism of p21 downregulation in breast cancer. Mol Cell Biol. 2010;30:1508–27. doi: 10.1128/MCB.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brosig M, Ferralli J, Gelman L, et al. Interfering with the connection between the nucleus and the cytoskeleton affects nuclear rotation, mechanotransduction and myogenesis. Int J Biochem Cell Biol. 2010;42:1717–28. doi: 10.1016/j.biocel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Chung CY, Zardi L, Erickson HP. Binding of tenascin-C to soluble fibronectin and matrix fibrils. J Biol Chem. 1995;270:29012–7. doi: 10.1074/jbc.270.48.29012. [DOI] [PubMed] [Google Scholar]
- 79.Chung CY, Erickson HP. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci. 1997;110:1413–9. doi: 10.1242/jcs.110.12.1413. [DOI] [PubMed] [Google Scholar]
- 80.Day JM, Olin AI, Murdoch AD, et al. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J Biol Chem. 2004;279:12511–8. doi: 10.1074/jbc.M400242200. [DOI] [PubMed] [Google Scholar]
- 81.Fischer D, Chiquet-Ehrismann R, Bernasconi C, et al. A single heparin binding region within the fibrinogen-like domain is functional in chick tenascin-C. J Biol Chem. 1995;270:3378–84. doi: 10.1074/jbc.270.7.3378. [DOI] [PubMed] [Google Scholar]
- 82.Zacharias U, Norenberg U, Rathjen FG. Functional interactions of the immunoglobulin superfamily member F11 are differentially regulated by the extracellular matrix proteins tenascin-R and tenascin-C. J Biol Chem. 1999;274:24357–65. doi: 10.1074/jbc.274.34.24357. [DOI] [PubMed] [Google Scholar]
- 83.Czopka T, von Holst A, ffrench-Constant C, et al. Regulatory mechanisms that mediate tenascin C-dependent inhibition of oligodendrocyte precursor differentiation. J Neurosci. 2010;30:12310–22. doi: 10.1523/JNEUROSCI.4957-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brellier F, Ruggiero S, Zwolanek D, et al. SMOC1 is a tenascin-C interacting protein overexpressed in brain tumours. Matrix Bio. 2011:225–33. doi: 10.1016/j.matbio.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 85.Paszek MJ, Zahir N, Johnson KR, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 86.Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumour progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang R, Amir J, Liu H, et al. Mechanical strain activates a program of genes functionally involved in paracrine signaling of angiogenesis. Physiol Genom. 2008;36:1–14. doi: 10.1152/physiolgenomics.90291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maier S, Lutz R, Gelman L, et al. Tenascin-C induction by cyclic strain requires integrin-linked kinase. Biochim Biophys Acta. 2008;1783:1150–62. doi: 10.1016/j.bbamcr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 89.Sarasa-Renedo A, Tunc-Civelek V, Chiquet M. Role of RhoA/ROCK-dependent actin contractility in the induction of tenascin-C by cyclic tensile strain. Exp Cell Res. 2006;312:1361–70. doi: 10.1016/j.yexcr.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 90.Chiquet-Ehrismann R, Tannheimer M, Koch M, et al. Tenascin-C expression by fibroblasts is elevated in stressed collagen gels. J Cell Biol. 1994;127:2093–101. doi: 10.1083/jcb.127.6.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blaauboer ME, Smit TH, Hanemaaijer R, et al. Cyclic mechanical stretch reduces myofibroblast differentiation of primary lung fibroblasts. Biochem Biophys Res Commun. 2011;404:23–7. doi: 10.1016/j.bbrc.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 92.Helleman J, Jansen MP, Ruigrok-Ritstier K, et al. Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin Cancer Res. 2008;14:5555–64. doi: 10.1158/1078-0432.CCR-08-0555. [DOI] [PubMed] [Google Scholar]
- 93.Gong XG, Lv YF, Li XQ, et al. Gemcitabine resistance induced by interaction between alternatively spliced segment of tenascin-C and annexin A2 in pancreatic cancer cells. Biol Pharm Bull. 2010;33:1261–7. doi: 10.1248/bpb.33.1261. [DOI] [PubMed] [Google Scholar]
- 94.Lal A, Peters H, St Croix B, et al. Transcriptional response to hypoxia in human tumours. J Natl Cancer Inst. 2001;93:1337–43. doi: 10.1093/jnci/93.17.1337. [DOI] [PubMed] [Google Scholar]
- 95.Caruso G, Caffo M, Raudino G, et al. Antisense oligonucleotides as innovative therapeutic strategy in the treatment of high-grade gliomas. Recent Pat CNS Drug Discov. 2010;5:53–69. doi: 10.2174/157488910789753503. [DOI] [PubMed] [Google Scholar]
- 96.Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol Cancer Ther. 2006;5:2957–62. doi: 10.1158/1535-7163.MCT-06-0172. [DOI] [PubMed] [Google Scholar]
- 97.Schliemann C, Neri D. Antibody-based vascular tumour targeting. Recent Results Cancer. 2010;180:201–16. doi: 10.1007/978-3-540-78281-0_12. [DOI] [PubMed] [Google Scholar]