

Abstract
Compound K (20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol, CK), an intestinal bacterial metabolite of panaxoside, has been shown to inhibit tumour growth in a variety of tumours. However, the mechanisms involved are largely unknown. We use human gastric carcinoma cell lines BGC823, SGC7901 and human gastric carcinoma xenograft in nude mice as models to study the mechanisms of CK in gastric cancers. We found that CK significantly inhibits the viabilities of BGC823 and SGC7901 cells in dose- and time-dependent manners. CK-induced BGC823 and SGC7901 cells apoptosis and cell cycle arrest in G2 phase by up-regulation of p21 and down-regulation of cdc2 and cyclin B1. Further studies show that CK induces apoptosis in BGC823 and SGC7901 cells mainly through mitochondria-mediated internal pathway, and that CK induces the translocation of nuclear Bid to mitochondria. Finally, we found that CK effectively inhibited the tumour formation of SGC7901 cells in nude mice. Our studies show that CK can inhibit the viabilities and induce apoptosis of human gastric carcinoma cells via Bid-mediated mitochondrial pathway.
Keywords: gastric carcinoma, compound K, Bid, apoptosis
Introduction
Gastric carcinoma is one of common malignant tumours from human digestive system, and a serious threat to human health. Gastric carcinoma currently ranks second in global tumour mortality [1]. Particularly in Asia and parts of South America, it is the most common epithelial malignancy and leading cause of cancer-related deaths [1, 2]. The aetiology of gastric carcinoma is very complex, including mutations of oncogenes and tumour suppressor genes and abnormalities in growth factors and their receptors [3–6]. However, it has not been effectively treated so far because of its biological characteristics. Most patients are either diagnosed at advanced stages, or developed a relapse after surgery with curative intent [1, 2]. So a clear understanding of the mechanisms of the development and progression of gastric carcinoma is crucial for developing new effective therapeutic drugs.
Compound K (20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol, CK; Fig. 1A), also known as IH-901 or M1, is an intestinal bacterial metabolite of panaxoside Rb1, Rb2 and Rc [7]. It has a wide range of pharmacological effects. Some CK-related studies have been performed in various medical fields, such as neuropathy [8], dermatonosis [9], endocrinopathy [10] and digestive disease [11]. Studies confirmed that CK can inhibit the growth of a variety of tumour cells, for example, hepatoma [12, 13], lung carcinoma [14], colorectal carcinoma [15], glioma [16], etc. Further studies show that it can inhibit proliferation and induce apoptosis in tumour cells [12–20]. However, the mechanisms involved are not clearly understood.
Fig 1.
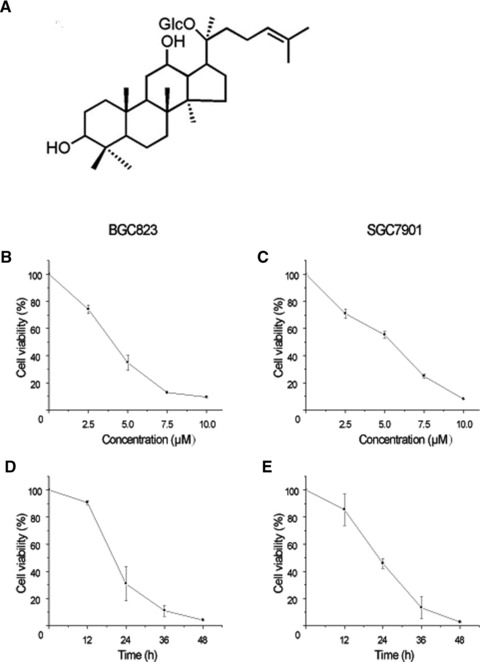
Effect of CK on the viabilities of human gastric carcinoma cells. (A) The chemical structure of Compound K (CK). (B) BGC823 cells were treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs. (C) SGC7901 cells were treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs. (D) BGC823 cells were treated with 5.0 μmol/l CK for different time periods (0, 12, 24, 36 and 48 hrs). (E) SGC7901 cells were treated with 5.0 μmol/l CK for different time periods (0, 12, 24, 36 and 48 hrs). The cell viabilities were determined by MTT assay. Data are reported as means ± S.D. of three separate experiments.
Bid, a BCL2 homology (BH)3 domain-only agonist, is a BH3-only Bcl-2 family member with multiple functions [21, 22]. In most cases it functions in a truncated form. The cleaved form of Bid (tBid) is myristoylated, and translocated to the mitochondria where it activates oligomerization of Bak or Bax and induces cytochrome c release. Cytochrome c in turn activates the apoptotic cascade of caspases-9 and -3, leading to cell death [23, 24]. However, the cleavage of Bid may not be an absolute requirement for Bid to be pro-apoptotic. Full-length Bid can also translocate to mitochondria and get activated without cleavage [25–27]. Studies suggest that Bid also has functions in DNA damage repair [28, 29]. Bid has emerged as a central player linking death signals through surface death receptors to the core apoptotic mitochondrial pathway [30]. Bid can also induce the activation of different signalling pathways to promote or inhibit tumour development, and it may have different biological functions in different systems [31, 32]. Studies from our group indicate that Bid might be a potential target for tumour therapy [27, 30, 33].
In this study, we use human gastric carcinoma cell lines BGC823, SGC7901 and the model of human gastric carcinoma xenograft in nude mice to investigate the mechanisms of CK-induced apoptosis, especially CK's possible roles on Bid in human gastric carcinoma cells.
Materials and methods
Cell culture and cell viability assay
The human gastric carcinoma cell lines BGC823 (poorly differentiated) and SGC7901 (moderately differentiated) were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences, Shanghai, China, and were maintained in RPMI1640 (Roswell Park Memorial Institute) supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin, at 37°C in a water-saturated atmosphere of 5% CO2. CK was purchased from Sigma Chemical Co. (St. Louis, MO, USA). The structure of CK is shown in Figure 1A. Cell viability was assessed by methyl thiazolyl tetrazolium (MTT) assay as previously described [27].
Cell morphological analysis
Cells were treated with 5.0 μM CK or 0.1% (v/v) dimethyl sulfoxide (DMSO) (control) for 24 hrs. Then they were incubated with 10 μg/ml Hoechst 33342 (Sigma) and observed under fluorescence microscope (Leica DMIRB; Leica, Wetzler, Germany).
Annexin V assays
Cells were cultured and treated with different concentrations of CK. At various time-points, cells were trypsinized and harvested. After centrifugation, the cells were washed, resuspended and stained for annexin V and propidium iodide (PI) as described in the manufacturer's instructions (Pharmingen, San Diego, CA, USA). The samples were analysed by Flow cytometry (FACScan; Becton Dickinson, Franklin Lakes, NJ, USA).
Cell cycle analysis
Cell cycle was measured by propidium iodide labelling cellular nuclear DNA. The cells were incubated overnight to allow them to attach to the plate, and then treated with CK. Cell cycle was analysed by Becton Dickinson FACScan. The ratio of cells in the G0/G1, S and M phases of the cell cycle was determined by their DNA content.
Immunofluorescence double staining
Immunofluorescence double staining was performed as previously described [27] with some modifications. Briefly, After CK treatment, cells were fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Then, cells were incubated with mouse monoclonal antibody against Bid (1:200), and washed and subsequently incubated with a rabbit polyclonal antibody against Hsp60 (1:200) for another 1 hr at room temperature. The cells were then washed and subsequently incubated with both fluoresceine isothiocyanate (FITC)-conjugated goat antimouse and tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit secondary antibodies at a dilution of 1:200 for 1 hr at room temperature. After rinsing, cells were mounted in ProLong Antifade solution onto glass slides and observed under fluorescence microscope (Leica DMIRB).
Cytosolic, nuclear and mitochondrial protein isolation
The cytosolic, nuclear and mitochondrial protein fractions were isolated according to our previously reported procedure [27]. Briefly, after treatment as indicated, cells were collected and resuspended in five volumes of ice-cold extract buffer A and were homogenized. The homogenates were centrifuged at 750 × g for 10 min. The supernatant was collected and centrifuged at 10,000 × g for 15 min. to obtain the mitochondria pellets. The supernatants were further centrifuged at 100,000 × g for 1 hr to collect the cytosolic fraction. For the isolation of the nuclear proteins, ice-cold extract buffer B was added to the pellet after 750 × g centrifugation. The pellet and buffer B were mixed by gently pipetting and kept on ice for 15 min. The mixture was then treated with 10% NP-40, and centrifuged at 10,000 × g for 30 sec. The supernatant was discarded and the pellet was resuspended and homogenized in extract buffer C, and then centrifuged at 10,000 × g for 20 min. The supernatant was collected as the nuclear fraction.
Western blot analysis
Western blot was performed as previously described [34]. Protein Assay Kit for protein quantity analysis was purchased from Bio-Rad (Hercules, CA, USA). The enhanced chemiluminescence detection system was purchased from GE/Amersham (Piscataway, NJ, USA). The mouse monoclonal antibody against cytochrome c was obtained from ZYMED Laboratories, Inc. (South San Francisco, CA, USA). All other antibodies were provided by Santa Cruz (Santa Cruz, CA, USA).
Xenograft assays in nude mice
In vivo animal experiments with female Balb/c nude mice were carried out in the Cancer Research Center, Medical College of Xiamen University. SGC7901 cells (2 × 106/mouse) were implanted by subcutaneous injection into the right hind leg of the mouse. Then different doses of CK were administered to the mice by subcutaneous injection every other day for 4 weeks. Mice in untreated control groups were given normal saline alone. Shortly after treatment of CK, all mice were killed and examined for the growth of subcutaneous tumours and micrometastasis formation. Six nude mice were used in each set of experiment.
Immunohistochemical staining
The subcutaneous tumours and metastases were fixed in 10% formaldehyde and embedded in paraffin. Sections were then cut and immunohistochemical staining was performed as previously described [34]. For negative control, the primary antibody was substituted with normal mouse IgG.
Statistic analysis
Data are presented as the means ± S.D. for at least three separate determinations for each group. The differences between the groups were examined for statistical significance using the Student's t-test with SPSS software.
Results
CK inhibits human gastric carcinoma cells survival
We determined the viabilities of BGC823 and SGC7901 cells treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs and 5.0 μmol/l CK for different time periods (0, 12, 24, 36 and 48 hrs) using MTT assay. As shown in Figure 1, the cell viabilities were decreased in dose- and time-dependent manners, and BGC823 cells were more sensitive to CK treatment than SGC7901 cells.
CK induces apoptosis in human gastric cells
Hoechst-33342 and annexin-V/PI staining assays were combined to investigate whether CK can induce human gastric carcinoma cells apoptosis. First, we observed the apoptotic morphology of BGC823 and SGC7901 cells treated with 5.0 μmol/l CK for 24 hrs, and found that the CK-treated cells had manifest brighter granular blue fluorescence and more apoptotic bodies compared with control group (Fig. 2A). Furthermore, we assessed the apoptotic rate of the cells treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs and 5.0 μmol/l CK for different time periods (0, 12, 24, 36 and 48 hrs) using annexin V/PI staining assay. As shown in Figure 2B and C, CK-induced apoptosis in BGC823 and SGC7901 cells in a dose- and time-dependent manner and BGC823 cells were more sensitive to CK treatment than SGC7901 cells.
Fig 2.
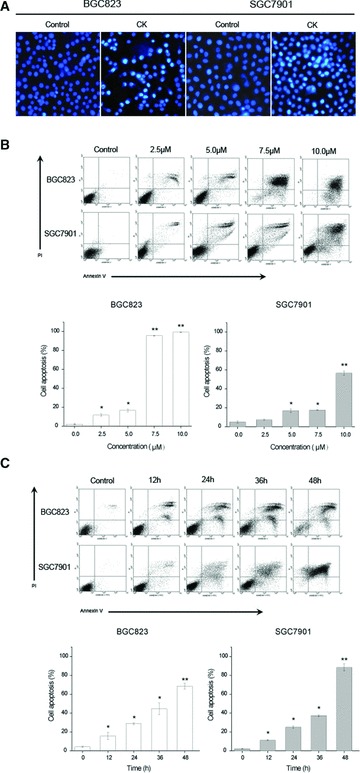
CK induces apoptosis in human gastric carcinoma cells. (A) Effect of CK on the apoptotic morphology of human gastric carcinoma cells. Cells were stained with Hoechst-33342, and images were captured by fluorescence microscopy. Magnification, 100×. (B) Cells were treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs. (C) Cells were treated with 5 μmol/l CK for different time periods (0, 12, 24, 36 and 48 hrs), and the apop-tosis was determined by annexin V/PI staining assay. Data are reported as means ± S.D. of three separate experiments. * and ** indicate P < 0.05 and P < 0.01 compared with control group, respectively.
CK induces cell cycle arrest in G2 phase in human gastric carcinoma cells
To identify the effect of CK on cell cycle of human gastric carcinoma cells, we determined the cell cycle distribution of BGC823 and SGC7901 cells treated with different concentrations of CK (0, 2.5 and 5.0 μmol/l) for 24 hrs using PI staining assay. As shown in Figure 3A, we found that CK-induced cell cycle arrest in G2 phase in BGC823 and SGC7901 cells. Further investigation was performed to locate the molecular mechanisms on how CK regulates cell cycle arrest in G2 phase in human gastric carcinoma cells. The Western blot analysis showed that CK up-related expression of p21 and down-related expression of cdc2 and cyclin B1. The statistic tests showed the significant differences between control and CK treatment groups (Fig. 3B).
Fig 3.
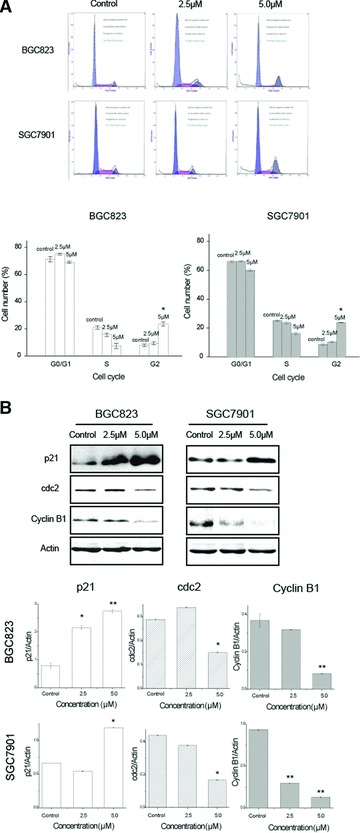
Effect of CK on the cell cycle distribution of human gastric carcinoma cells. (A) Cells were treated with different concentrations of CK (0, 2.5 and 5.0 μmol/l) for 24 hrs. The cell cycle distribution was determined by PI staining assay. Data are reported as means ± S.D. of three separate experiments. * and ** indicate P < 0.05 and P < 0.01 compared with control group, respectively. (B) Cells were treated with different concentrations of CK (0, 2.5 and 5.0 μmol/l) for 24 hrs. The total protein was extracted and the expression of cell cycle-related proteins was analysed by Western blot assay. Data are reported as means ± S.D. of three separate experiments. * and ** indicate P < 0.05 and P < 0.01 compared with control group, respectively.
Expression of apoptosis-related proteins in human gastric carcinoma cells treated with CK
To further clarify the apoptotic mechanisms of human gastric carcinoma cells induced by CK treatment, we analysed total proteins of BGC823 and SGC7901 cells treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs using Western blot assay. We found that expression levels of Fas, Fas-L and Bcl-XL were not changed significantly, but Bid and Bcl-2 were down-regulated, and poly ADP-ribose polymerase (PARP) was shown cleavage activation. Bcl-2 and Bid expression was analysed by statistic tests and significant differences between control and CK treatment groups were shown (Fig. 4). The results indicate that CK induces apoptosis in BGC823 and SGC7901 cells mainly through mitochondria-mediated internal pathway.
Fig 4.
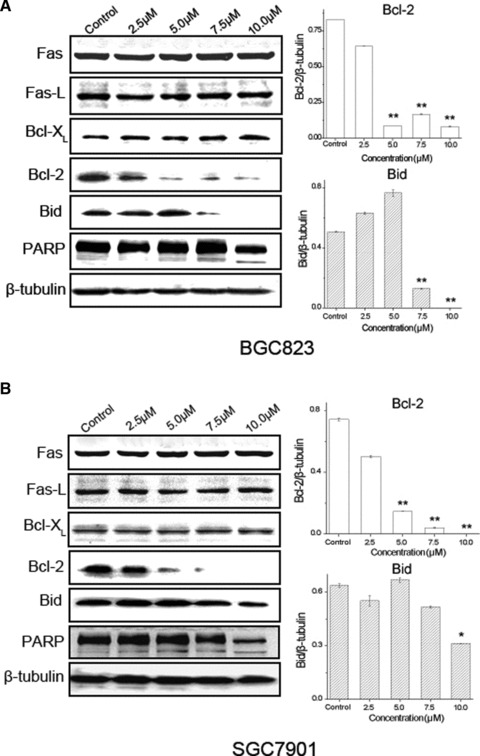
Expression of apoptosis-related proteins in human gastric carcinoma cells treated with CK. (A) BGC823 cells were treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs. (B) SGC7901 cells were treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs. The total protein was extracted and the expression of apoptosis-related proteins was analysed by Western blot assay. Data are reported as means ± S.D. of three separate experiments, * and ** indicate P < 0.05 and P < 0.01 compared with control group, respectively.
CK induces Bid translocation in human gastric carcinoma cells
To detect the clear site-specific effect of Bid in BGC823 and SGC7901 induced by 5.0 μmol/l CK treatment for 24 hrs, we combined immunofluorescence and cell fractionation methods. 4,6-diamino-2-phenylindole (DAPI) and Hsp60 were used to stain nucleus and mitochondria, respectively. As shown in Figure 5A, Bid located in nucleus and cytoplasm in control groups. In CK-treated groups, we observed merged fluorescence between Bid and Hsp60, indicating that upon CK treatment, nuclear Bid translocated to mitochondria in BGC823 and SGC7901 cells. To further confirm this result, we separated nuclear, cytoplasmic and mitochondrial proteins of BGC823 and SGC7901 cells treated with 5.0 μmol/l CK for 24 hrs and analysed them using Western blot assay. Lamin B, β-tubulin and Hsp60 were chosen as specific markers to indicate nuclear, cytoplasmic and mitochondrial proteins, respectively. As shown in Figure 5B, we found that, upon CK treatment, expression level of nuclear Bid manifestly decreased. However, we did not observe obvious increase of Bid in mitochondria fraction, which might be due to further cleavage of Bid to t-Bid in mitochondria. We also measured the release of cytochrome c, an important indicator of apoptosis, and found that cytochrome c released to cytosol upon CK treatment.
Fig 5.
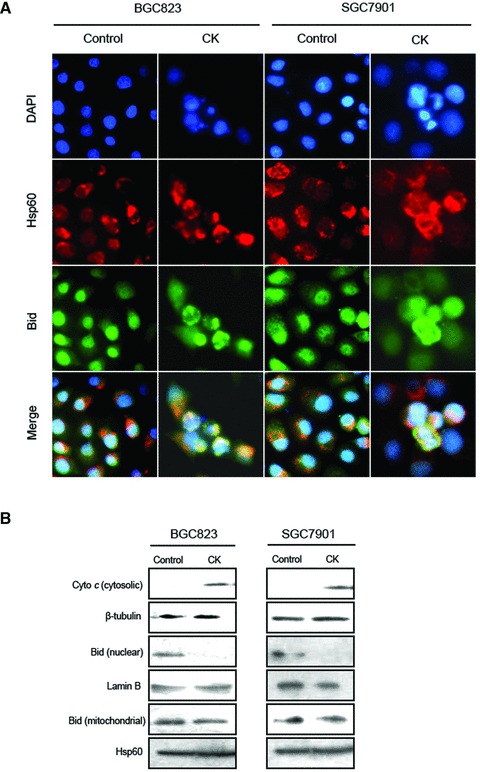
CK induces Bid translocation in human gastric carcinoma cells. (A) Effect of the site-specific morphology of Bid in human gastric carcinoma cells induced by CK treatment. Cells were treated with 5.0 μmol/l of CK for 24 hrs. The cellular locations of Bid were determined by immunofluorescence assay. DAPI and Hsp60 represent nucleus and mitochondria staining, respectively. Images were captured by fluorescence microscopy. Magnification, 400×. (B) Expression of cytosolic, nuclear and mitochondrial Bid and cytochrome c in human gastric carcinoma cells with CK treatment. Cells were treated with 5.0 μmol/l of CK for 24 hrs. The nuclear, cytoplasmic and mitochondrial proteins were segregated and analysed by Western blot assay.
CK inhibits the proliferation of SGC7901 cells in the nude mice
We established the model of human gastric carcinoma xenograft in nude mice by injecting SGC7901 cells. Volume and weight of the transplanted tumours from the nude mice treated with different of doses of CK (0, 2.5, 5.0 and 10.0 mg/kg) were assessed. As shown in Figure 6, CK inhibited the proliferation of SGC7901 cells in nude mice. Meanwhile, the subcutaneous tumour tissues were examined by haematoxylin and eosin staining (Fig. 6E), and Bid, MMP-9 were analysed by immunohistochemical staining. Tumours from 10.0 mg/kg CK treatment group showed obviously decreased expression of Bid and MMP-9, together with the cellular debris and morphological changes associated with apoptosis. The results of Bid expression in immunohistochemical staining are in accordance with the results of the Western blot analysis in human gastric cancer cells treated with CK in vitro (Fig. 4).
Fig 6.
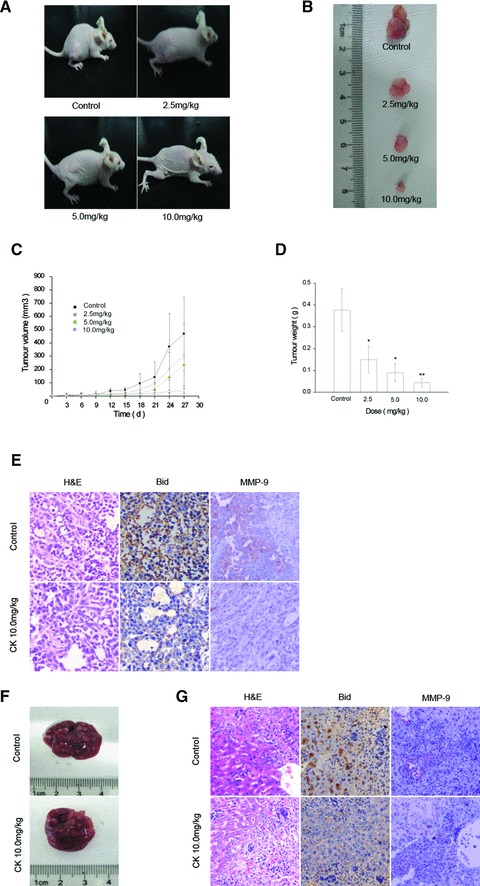
CK inhibits the proliferation of SGC7901 cells in nude mice. (A) and (B) Typical photographs of tumour samples from the nude mice treated with different doses of CK. SGC7901 cells were implanted by subcutaneous injection into the right hind leg of the mouse. The different doses of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) were administered to the mice by subcutaneous injection every other day. Mice in untreated control group were given normal saline alone. After 4 weeks, the mice were sacrificed and analysed. (C) Volume of tumour samples from nude mice treated with different doses of CK. (D) Weight of tumour samples from nude mice treated with different doses of CK. Data are reported as means ± S.D. of three separate experiments. * and ** indicate P < 0.05 and P < 0.01 compared with control group, respectively. (E) The tumour samples of untreated and 10.0 mg/kg CK-treated were immunostained with anti-Bid and anti-MMP-9 antibody and analysed by haema-toxylin and eosin staining. (F) and (G) Liver samples of untreated and 10.0 mg/kg CK-treated were photographed. The samples were also analysed by immunohistochemical staining of Bid and MMP-9 and haematoxylin and eosin staining.
On the other side, the reduction effect of CK on spontaneous gastric carcinoma metastasis cannot be clearly quantified in the liver samples of nude mice due to overcrowded matasis on the liver. However, in immunohistochemical staining, CK treatment group showed similarly depressed expression of Bid and MMP-9 (Fig. 6G).
Discussion
Ginseng saponins have been widely reported to exert anticancer activity. CK is the main metabolite of protopanaxadiol type ginseng saponin by intestinal bacteria after oral administration of ginseng extract, and is speculated to be the major form of protopanaxadiol saponins absorbed from the intestine [35–37]. Previous studies showed that CK possesses various chemopreventive and chemotherapeutic activities including anti-tumour activity [38, 39]. Researchers also found that CK can effectively kill HepG2, a hepatocellular carcinoma cell line, whereas it has much less cytotoxic effect in normal human liver cell line [40], suggesting that CK might be a very good candidate drug for cancer targeting therapy. The effect of CK on the suppression of hepatocellular carcinoma cells survival has been studied in our previous report [13]. However, the roles of CK on gastric carcinoma are not clearly understood, and the molecular mechanisms of CK on inducing apoptosis in human gastric carcinoma cells have never been reported. Therefore, here, we focus on the cellular location and distribution of Bid, a key molecule in the process of CK-induced apoptosis in human gastric carcinoma cell lines BGC823 and SGC7901. In this report, we observed that CK significantly inhibits the viabilities of human gastric carcinoma cells in a dose- and time-dependent manner (Fig. 1). Using multiple in vitro and in vivo models, we reported that CK potently induced BGC823 and SGC7901 cells apoptosis (Fig. 2) and attenuated subcutaneous tumour growth (Fig. 6). Although the effect of CK on spontaneous gastric carcinoma metastasis has not been clearly quantified in the nude mice due to overcrowded metastasis sites on the liver, the immunohistochemical staining results showed that CK effectively reduced the expression of MMP-9, a metastasis related protein in the nude mice (Fig. 6), suggesting CK's inhibitory effect on tumour metastasis.
Cell cycle redistribution usually occurs in CK-induced apoptosis. Some studies reported that CK-induced tumour cell arrest in G0/G1 phase [12, 13, 41]. However, we found that CK-induced cell cycle arrest in G2 phase in BGC823 and SGC7901 cells, which indicates that CK might induce different types of cell cycle arrest in different tumour cell lines. There are different check points during cell cycle and cells can be arrested at different check points for repair or apoptosis. Our studies indicate that 5 μM CK treatment for 24 hrs can induce BGC823 and SGC7901 not only apoptosis but also cell cycle arrest in G2 phase. The reason might be that cells at different phases in cell cycle have different sensitivities to CK treatment. Some cells may arrest in G2 phase to perform repair, but some just enter apoptosis directly.
Entry of eukaryotic cells into mitosis is regulated by activation of cdc2 kinase. The physiological process is controlled at several steps including cyclin B1 binding. As one of cell cycle suppressor molecules, the important function of p21 is to induce cell cycle arrest by inhibiting CDKs. Our study indicates that CK induces cell cycle arrest in G2 phase in BGC823 and SGC7901 cells not only by inhibiting expression level of cyclin B1, but also by up-regulating p21 and down-regulating cdc2 indirectly. Cells at different cell cycle phases have different sensitivities to radiation in mammals. It is known that the cells at G2 and M phases are much more sensitive to radiation. Chae's group [14] found that pre-treatment of NCI-H460 human lung cancer cells with CK enhanced γ-ray radiation-induced cell death, indicating that CK possesses radiosensitizing effect. Our studies provided further evidence that CK might increase radiosensitivity of tumour cells by blockade of cells at G2 phase. However, cell cycle regulation is complicated and cell specific. Further study is essential to disclose the mechanism of CK on cell cycle arrest.
There are two classic apoptotic pathways in mammalian cells, namely internal pathway and external pathway, also known as mitochondria-mediated apoptotic pathway and death receptor-mediated apoptotic pathway, and they cross-talk. Some studies show that both pathways are involved in CK-induced tumour cell apoptosis [12–20]. To further clarify the apoptotic molecular mechanisms of human gastric carcinoma cells induced by CK, we analysed total proteins of BGC823 and SGC7901 cells treated with different concentrations of CK (0, 2.5, 5.0, 7.5 and 10.0 μmol/l) for 24 hrs using Western blot assay. We found that expression levels of Fas, Fas-L and Bcl-XL were not changed significantly, but Bid and Bcl-2 were down-regulated by CK, and PARP showed cleavage activation. Cytochrome c release to cytoplasm induced by CK was also detected. The results show that CK induces apoptosis in BGC823 and SGC7901 cells mainly through mitochondria-mediated internal pathway (Fig. 7).
Fig 7.
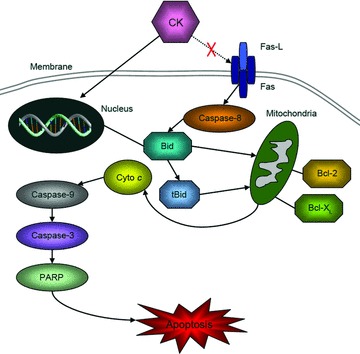
The tentative mechanism of CK-induced apoptosis in human gastric carcinoma cells.
Bid, a BH3 domain-only agonist, is a BH3-only Bcl-2 family member with multiple functions [21, 22]. Bid has emerged as a central player linking death signals through surface death receptors to the core apoptotic mitochondrial pathway [31]. The tBid is initially found to be myristoylated and translocated into mitochondria in response to death receptor-mediated apoptotic signalling. tBid is very unstable and has a half-life of less than 1.5 hrs [42]. However, the recent findings from our group and others have shown that the cleavage of Bid may not be an absolute requirement for Bid to be pro-apoptotic [25–27, 30, 43]. For example, the mutant Bid that lacks the caspase-8 cleavage site can still kill cells [26], and Bid has been shown to translocate to the mitochondria without cleavage [27]. Our results showed that Bid located in nucleus and cytoplasm and nuclear Bid translocated to mitochondria in BGC823 and SGC7901 cells by CK treatment (Fig. 5). Although we observed partial relocation of nuclear Bid to mitochondria in BGC823 and SGC7901 cells, mitochondrial Bid expression did not change significantly. In the mitochondria-mediated apoptosis pathway, Bid is mainly cleaved to tBid by activated Caspase-8, then tBid translates into mitochondria where it interact with some Bcl-2 family proteins and induces cytochrome c release into cytoplasm [23, 24]. tBid is a 15 KD protein and is easily degraded via ubiquitination pathway. So this might be a possible reason that we could not detect the change of mitochondrial Bid expression in BGC823 and SGC7901 cells with CK treatment.
In conclusion, the present studies demonstrated that CK, a novel ginseng saponin metabolite, could significantly inhibit the proliferation and induce apoptosis in human gastric carcinoma cells via Bid-mediated mitochondrial pathway. The elucidation of the mechanism of CK-induced tumour cell apoptosis suggests a possible therapeutic role of CK and its potential of combination with other chemotherapies targeting Bid to treat gastric carcinoma.
Acknowledgments
This work was supported by National Natural Science Foundation of China grants (30971524, 31071187 and 81072015), Program for New Century Excellent Talents in University, the Fundamental Research Funds for the Central Universities (2010121102 and 2010121103), Ministry of Science and Technology grant 2009CB522200, the 985 Project grant of Xiamen University and the research grant GCP200906 from Key Laboratory for Green Chemical Process of Ministry of Education and School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer. 1999;80:827–41. doi: 10.1002/(sici)1097-0215(19990315)80:6<827::aid-ijc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.Siewert JR, Bottcher K, Stein HJ, et al. Relevant prognostic factors in gastric cancer: ten-year results of the German Gastric Cancer Study. Ann Surg. 1998;228:449–61. doi: 10.1097/00000658-199810000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan AO, Luk JM, Hui WM, et al. Molecular biology of gastric carcinoma: from laboratory to bedside. J Gastroenterol Hepatol. 1999;14:1150–60. doi: 10.1046/j.1440-1746.1999.02000.x. [DOI] [PubMed] [Google Scholar]
- 5.Zheng L, Wang L, Ajani J, et al. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004;7:61–77. doi: 10.1007/s10120-004-0277-4. [DOI] [PubMed] [Google Scholar]
- 6.Xie K, Wei D, Huang S. Transcriptional anti-angiogenesis therapy of human pancreatic cancer. Cytokine Growth Factor Rev. 2006;17:147–56. doi: 10.1016/j.cytogfr.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Akao T, Kida H, Kanaoka M, et al. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J Pharm Pharmacol. 1998;50:1155–60. doi: 10.1111/j.2042-7158.1998.tb03327.x. [DOI] [PubMed] [Google Scholar]
- 8.Choi K, Kim M, Ryu J, et al. Ginsenosides compound K and Rh(2) inhibit tumor necrosis factor-alpha-induced activation of the NF-kappaB and JNK pathways in human astroglial cells. Neurosci Lett. 2007;421:37–41. doi: 10.1016/j.neulet.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 9.Shin YW, Bae EA, Kim SS, et al. Effect of ginsenoside Rb1 and compound K in chronic oxazolone-induced mouse dermatitis. Int Immunopharmacol. 2005;5:1183–91. doi: 10.1016/j.intimp.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Yoon SH, Han EJ, Sung JH, et al. Anti-diabetic effects of compound K versus metformin versus compound K-metformin combination therapy in diabetic db/db mice. Biol Pharm Bull. 2007;30:2196–200. doi: 10.1248/bpb.30.2196. [DOI] [PubMed] [Google Scholar]
- 11.Kim do Y, Yuan HD, Chung IK, et al. Compound K, intestinal metabolite of ginsenoside, attenuates hepatic lipid accumulation via AMPK activation in human hepatoma cells. J Agric Food Chem. 2009;57:1532–7. doi: 10.1021/jf802867b. [DOI] [PubMed] [Google Scholar]
- 12.Oh SH, Lee BH. A ginseng saponin metabolite-induced apoptosis in HepG2 cells involves a mitochondria-mediated pathway and its downstream caspase-8 activation and Bid cleavage. Toxicol Appl Pharmacol. 2004;194:221–9. doi: 10.1016/j.taap.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Ming YL, Song G, Chen LH, et al. Anti-proliferation and apoptosis induced by a novel intestinal metabolite of ginseng saponin in human hepatocellular carcinoma cells. Cell Biol Int. 2007;31:1265–73. doi: 10.1016/j.cellbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Chae S, Kang KA, Chang WY, et al. Effect of compound K, a metabolite of ginseng saponin, combined with gamma-ray radiation in human lung cancer cells in vitro and in vivo. J Agric Food Chem. 2009;57:5777–82. doi: 10.1021/jf900331g. [DOI] [PubMed] [Google Scholar]
- 15.Kim do Y, Park MW, Yuan HD, et al. Compound K induces apoptosis via CAMK-IV/AMPK pathways in HT-29 colon cancer cells. J Agric Food Chem. 2009;57:10573–8. doi: 10.1021/jf902700h. [DOI] [PubMed] [Google Scholar]
- 16.Jung SH, Woo MS, Kim SY, et al. Ginseng saponin metabolite suppresses phorbol ester-induced matrix metalloproteinase-9 expression through inhibition of activator protein-1 and mitogen-activated protein kinase signaling pathways in human astroglioma cells. Int J Cancer. 2006;118:490–7. doi: 10.1002/ijc.21356. [DOI] [PubMed] [Google Scholar]
- 17.Wakabayashi C, Murakami K, Hasegawa H, et al. An intestinal bacterial metabolite of ginseng protopanaxadiol saponins has the ability to induce apoptosis in tumor cells. Biochem Biophys Res Commun. 1998;246:725–30. doi: 10.1006/bbrc.1998.8690. [DOI] [PubMed] [Google Scholar]
- 18.Lee SJ, Ko WG, Kim JH, et al. Induction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-mediated activation of caspase-3 protease. Biochem Pharmacol. 2000;60:677–85. doi: 10.1016/s0006-2952(00)00362-2. [DOI] [PubMed] [Google Scholar]
- 19.Choi HH, Jong HS, Park JH, et al. A novel ginseng saponin metabolite induces apoptosis and down-regulates fibroblast growth factor receptor 3 in myeloma cells. Int J Oncol. 2003;23:1087–93. [PubMed] [Google Scholar]
- 20.Park EJ, Zhao YZ, Kim J, et al. A ginsenoside metabolite, 20-O-beta-D-glucopyranosyl-20(S)-protopanaxadiol, triggers apoptosis in activated rat hepatic stellate cells via caspase-3 activation. Planta Med. 2006;72:1250–3. doi: 10.1055/s-2006-947223. [DOI] [PubMed] [Google Scholar]
- 21.Wang K, Yin XM, Chao DT, et al. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–69. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 22.Yin XM. Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene. 2006;369:7–19. doi: 10.1016/j.gene.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Zhu H, Xu CJ, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 24.Luo X, Budihardjo I, Zou H, et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 25.Sarig R, Zaltsman Y, Marcellus RC, et al. BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem. 2003;278:10707–15. doi: 10.1074/jbc.M210296200. [DOI] [PubMed] [Google Scholar]
- 26.Valentijn AJ, Gilmore AP. Translocation of full-length Bid to mitochondria during anoikis. J Biol Chem. 2004;279:32848–57. doi: 10.1074/jbc.M313375200. [DOI] [PubMed] [Google Scholar]
- 27.Song G, Chen GG, Chau DK, et al. Bid exhibits S phase checkpoint activation and plays a pro-apoptotic role in response to etoposide-induced DNA damage in hepatocellular carcinoma cells. Apoptosis. 2008;13:693–701. doi: 10.1007/s10495-008-0195-8. [DOI] [PubMed] [Google Scholar]
- 28.Kamer I, Sarig R, Zaltsman Y, et al. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell. 2005;122:593–603. doi: 10.1016/j.cell.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 29.Zinkel SS, Hurov KE, Ong C, et al. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–91. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 30.Song G, Chen GG, Hu T, et al. Bid stands at the crossroad of stress-response pathways. Curr Cancer Drug Target. 2010;10:584–92. doi: 10.2174/156800910791859515. [DOI] [PubMed] [Google Scholar]
- 31.Miao J, Chen GG, Chun SY, et al. Bid sensitizes apoptosis induced by chemotherapeutic drugs in hepatocellular carcinoma. Int J Oncol. 2004;25:651–9. [PubMed] [Google Scholar]
- 32.Miao J, Chen GG, Chun SY, et al. Adenovirus-mediated tBid overexpression results in therapeutic effects on p53-resistant hepatocellular carcinoma. Int J Cancer. 2006;119:1985–93. doi: 10.1002/ijc.22040. [DOI] [PubMed] [Google Scholar]
- 33.Song G, Chen GG, Yun JP, et al. Association of p53 with Bid induces cell death in response to etoposide treatment in hepatocellular carcinoma. Curr Cancer Drug Target. 2009;9:871–80. doi: 10.2174/156800909789760302. [DOI] [PubMed] [Google Scholar]
- 34.Song G, Mao YB, Cai QF, et al. Curcumin induces human HT-29 colon adenocarcinoma cell apoptosis by activating p53 and regulating apoptosis-related protein expression. Braz J Med Biol Res. 2005;38:1791–8. doi: 10.1590/s0100-879x2005001200007. [DOI] [PubMed] [Google Scholar]
- 35.Karikura M, Miyase T, Tanizawa H, et al. Studies on absorption, distribution, excretion and metabolism of ginseng saponins. VII. Comparison of the decomposition modes of ginsenoside-Rb1 and -Rb2 in the digestive tract of rats. Chem Pharm Bull. 1991;39:2357–61. doi: 10.1248/cpb.39.2357. [DOI] [PubMed] [Google Scholar]
- 36.Hasegawa H, Sung JH, Matsumiya S, et al. Main ginseng saponin metabolites formed by intestinal bacteria. Planta Med. 1996;62:453–7. doi: 10.1055/s-2006-957938. [DOI] [PubMed] [Google Scholar]
- 37.Akao T, Kanaoka M, Kobashi K. Appearance of compound K, a major metabolite of ginsenoside Rb1 by intestinal bacteria, in rat plasma after oral administration–measurement of compound K by enzyme immunoassay. Biol Pharm Bull. 1998;21:245–9. doi: 10.1248/bpb.21.245. [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa H, Sung JH, Huh JH. Ginseng intestinal bacterial metabolite IH901 as a new anti-metastatic agent. Arch Pharm Res. 1997;20:539–44. doi: 10.1007/BF02975208. [DOI] [PubMed] [Google Scholar]
- 39.Choo MK, Sakurai H, Kim DH, et al. A ginseng saponin metabolite suppresses tumor necrosis factor-alpha-promoted metastasis by suppressing nuclear factor-kappaB signaling in murine colon cancer cells. Oncol Rep. 2008;19:595–600. [PubMed] [Google Scholar]
- 40.Oh SH, Yin HQ, Lee BH. Role of the Fas/Fas ligand death receptor pathway in ginseng saponin metabolite-induced apoptosis in HepG2 cells. Arch Pharm Res. 2004;27:402–6. doi: 10.1007/BF02980081. [DOI] [PubMed] [Google Scholar]
- 41.Kang KA, Kim YW, Kim SU, et al. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch Pharm Res. 2005;28:685–90. doi: 10.1007/BF02969359. [DOI] [PubMed] [Google Scholar]
- 42.Breitschopf K, Zeiher AM, Dimmeler S. Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J Biol Chem. 2000;275:21648–52. doi: 10.1074/jbc.M001083200. [DOI] [PubMed] [Google Scholar]
- 43.Konig HG, Rehm M, Gudorf D, et al. Full length Bid is sufficient to induce apoptosis of cultured rat hippocampal neurons. BMC Cell Biol. 2007;8:7. doi: 10.1186/1471-2121-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]