

Abstract
Alzheimer’s disease (AD) is the major cause of dementia in the elderly, leading to memory loss and cognitive decline. The mechanism underlying onset of the disease has not been fully elucidated. However, characteristic pathological manifestations include extracellular accumulation and aggregation of the amyloid β-peptide (Aβ) into plaques and intracellular accumulation and aggregation of hyperphosphorylated tau, forming neurofibrillary tangles. Despite extensive research worldwide, no disease modifying treatment is yet available. In this review, we focus on gene therapy as a potential treatment for AD, and summarize recent work in the field, ranging from proof-of-concept studies in animal models to clinical trials. The multifactorial causes of AD offer a variety of possible targets for gene therapy, including two neurotrophic growth factors, nerve growth factor and brain-derived neurotrophic factor, Aβ-degrading enzymes, such as neprilysin, endothelin-converting enzyme and cathepsin B, and AD associated apolipoprotein E. This review also discusses advantages and drawbacks of various rapidly developing virus-mediated gene delivery techniques for gene therapy. Finally, approaches aiming at down-regulating amyloid precursor protein (APP) and β-site APP cleaving enzyme 1 levels by means of siRNA-mediated knockdown are briefly summarized. Overall, the prospects appear hopeful that gene therapy has the potential to be a disease modifying treatment for AD.
Keywords: Alzheimer’s disease, gene therapy, gene delivery, neurodegeneration, Aβ metabolism, NGF, BDNF, neprilysin
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease leading to dementia that affects approximately 29 million people around the world [1]. Since life expectancy is increasing in most countries and aging is a major risk factor for AD, the number of patients will continue growing. Unfortunately, no disease-modifying treatment is available today. The disease is clinically characterized by progressive cognitive decline, memory loss and personality change, accompanied with behavioural and psychological symptoms of dementia, such as abnormal behaviour, agitation and mood swings. Pathological characteristics of the AD brain include the amyloid plaques and neurofibrillary tangles (NFTs), which are formed via aggregation of extracellular amyloid β-peptide (Aβ) and intracellular hyperphosphorylated tau, respectively. In addition, pronounced loss of synapses and neurons, accompanied with ventricular enlargements is observed, and the pathology is associated with the clinically observed cognitive decline although the degenerative processes may start substantially before the first clinical manifestations appear.
Aβ is generated from a type I membrane protein, amyloid precursor protein (APP), through sequential proteolytic cleavages performed by β-secretase (β-site APP-cleaving enzyme 1, BACE1) and γ-secretase, which consists of presenilin (PSEN), Aph-1, Pen-2 and nicastrin. β-Secretase mediated cleavage of APP gives rise to a soluble N-terminal fragment (βAPPs) and a membrane bound C-terminal fragment (CTF-β), which is further cleaved by γ-secretase to release Aβ, which consists of 40–43 amino acids. APP, which has been well conserved throughout evolution, is present in almost all cells and is abundantly expressed in neural cells. Its physiological role has not been clarified, although some studies have found APP to have neuroprotective or neurotrophic functions. Genes encoding PSEN1 and PSEN2 and APP have been identified as causal in early-onset familial AD (FAD). FAD-linked mutations in these genes, with an autosomal mode of inheritance, give rise to aggressive pathology and increase either the total Aβ level or the ratio of Aβ42/Aβ40. Aβ42 is more hydrophobic and more prone to aggregate than Aβ40. These observations strongly imply a cause-and-effect relationship between Aβ accumulation and AD pathogenesis. However, amyloid plaques might not necessarily be the major cause underlying neuronal cell death, synaptic loss and eventually brain atrophy in AD. There is evidence that soluble Aβ-oligomers, rather than amyloid plaques, are the major pathological species causing impaired synaptic and neuronal functions. For example, it has been shown that Aβ oligomers are highly toxic, block long-term potentiation (LTP), trigger synaptic retraction and lead to impairment of memory and cognitive functions [2]. On the other hand, amyloid plaques (Aβ fibrils) cause proliferation and activation of glial cells, which secrete cytotoxic factors, and may thus be indirectly involved in neuronal dysfunction. It should be noted, however, that none of the transgenic mouse models that accumulate Aβ oligomers and Aβ plaques have reproduced the neurodegenerative pathologies, so that none of the hypothesis can be considered experimentally proven. Recently it has been shown that also Aβ43 is a component of extracellular plaques in AD brains and may play a role in aggregation ([3] and Saito and Saido [personal communication]). It has been suggested that certain receptors could be involved in Aβ binding, and recent studies have shown that Aβ bound to cellular prion protein or α7 nicotinic acetylcholine receptor (α7nAChR) causes neuropathology, providing further support for a pathophysiological role of Aβ in AD [4–6]. Overall, there is considerable evidence that concentration-dependent Aβ aggregation is one of the major underlying causes of AD, although other factors are also likely to contribute including hyperphosphorylated tau, apolipoprotein E (APOE)-associated lipid metabolism and inflammation [7–9].
Aβ is constantly metabolized in the brain and increased steady-state levels of Aβ may be caused not only by elevated production of Aβ, but also by diminished degradation of Aβ. Several proteases have been implicated in Aβ degradation; neprilysin, insulin-degrading enzyme (IDE), cathepsin B, matrix metalloproteinases, plasmin, endothelin-converting enzyme (ECE) and angiotensin-converting enzyme (ACE) (reviewed in [10, 11]). Among these proteases, neprilysin has been identified as the major Aβ-degrading enzyme [12], while the contributions of ECE and IDE appear to be minor [10]. The Aβ-degrading enzymes could be targets for Aβ-lowering treatments, for example in gene therapeutic approaches aimed at enhancing Aβ-degrading activity.
Given the increasing number of AD patients, the augmenting costs of dementia healthcare, and not least, the personal suffering of patients and their caregivers, all possible treatment strategies should be considered. Acetylcholinesterase (AChE) inhibitors, such as donepezil, rivastigmine and galantamine and an NMDA antagonist, memantine, are currently the only available anti-dementia drugs. They temporarily ameliorate the symptoms but, regrettably, do not stop progression of the disease. However, some promising disease-modifying effects have been achieved with immunotherapy using Aβ-vaccine, and several clinical trials are ongoing [13]. In addition, accumulating data have showed that gene therapy is an efficient way to treat various diseases and could also be a potential treatment for AD.
Gene therapy can directly enhance enzymatic activities and compensate for reduced levels of bioactive substances. This is achieved through the delivery of a transgene mediated by a vector, most commonly a virus that infect host cells, in which the gene is then expressed. Indeed, gene therapy has been successfully applied to several neurodegenerative as well as neurological diseases. To date more than 1340 clinical trials in 28 countries, targeting over 100 genes, have been approved for a variety of diseases, mostly related to cancer [14]. Gene therapy temporarily faced a setback related to the safety of the use of viral vectors (see Table 1 for a summary of viral vectors used in gene therapy and their properties). For example, in a clinical trial of gene therapy for X-linked severe combined immunodeficiency using retroviruses [15, 16], genetic material from the viral vector was inserted into the proximity of an LMO2 oncogene, leading to leukaemia in five patients, of whom one died [17–19]. However, such cases are rare and there have been several successful trials. For instance, retroviral vector-mediated gene delivery has been used for the treatment of metastatic melanoma and chronic granulomatous disease using gene-transferred T cells and blood stem cells, respectively [20, 21]. The development of vectors using oncoviruses, in which genes needed for replication in non-tumour cells are deleted and where expression of the target gene is driven by tumour/tissue-specific promoters to ensure tumour-specific gene expression, was a significant step forward for gene therapy-based cancer treatment [22]. For non-tumour applications, such as neurodegenerative diseases including AD, alternative viral vectors have been developed, e.g. recombinant adeno-associated viral vector (rAAV) and the HIV-derived lentiviral vector. In these vectors, the viral genes for self-replication and, in the case of rAAV, incorporation of genetic material into chromosomal DNA, have been deleted. These vectors are capable of infecting non-dividing neural cells and have been shown to be safe, only weakly immunoreactive, specific and provide long-term expression (up to 6 years [23]). Several AD gene therapy phase I clinical trials are ongoing, and at least one has yielded positive preliminary results. In another neurodegenerative disease, Parkinson’s disease, intra-cranial gene delivery of three targets genes using rAAV vector showed very efficient disease modifying effects without adverse effects (recently reviewed in [24]). This review summarizes reports on experimental gene therapy in AD animal models and clinical trials of AD gene therapy up to date, and discusses the potential of gene therapy as an AD treatment. The molecular targets investigated so far can be divided into four categories according to their physiological function; (i) neurotrophins (NTs) that sustain growth and synaptic activity of neurons (nerve growth factor [NGF] and brain-derived neurotrophic factor [BDNF]), (ii) enzymes directly involved in Aβ degradation (neprilysin, ECE and cathepsin B) (iii) Aβ burden-associated factors (APOE) and (iv) proteins involved in Aβ generation (BACE1 and APP) that are targets for siRNA mediated down-regulation.
Table 1.
Summary of viral vectors used in gene therapy
| Vector type | Virus type | Maximum gene size | Transgene localization |
|---|---|---|---|
| AAV | Parvovirus (ssDNA) | 4.5 kB | Episomes |
| Adenovirus | Adenovirus (dsDNA) | 7 kb | Extrachromosomal |
| HSV | Herpesvirus (dsDNA) | 50 kb | Episomes |
| Lentivirus | Retrovirus (ssRNA) | 10 kB | Integration into chromosomes |
| MLV | Retrovirus (ssRNA) | 7 kb | Integration into chromosomes |
| Sindbis | Alphavirus (ssRNA) | 2–6 kb | Cytoplasm |
Abbreviations: MLV; Moloney leukaemia virus, AAV; Adeno-associated virus, HSV; Herpes simplex virus.
Targets and ongoing research
NGF
Neurotrophic function of NGF
NGF was the first NT to be identified (the NT family also includes BDNF, NT-3 and NT-4/5) and has been intensively studied. NGF was early shown to play an important role during development of the peripheral nervous system where it is secreted by target cells to achieve a balanced production of sensory and sympathetic neurons. In the brain, NGF is synthesized in the hippocampus and the cerebral cortex during development and adult neurogenesis, when continuous generation of new neurons through division of neural stem cells occurs. Although the physiological role of hippocampal neurogenesis is unclear, it may participate in learning and memory functions (reviewed in [25]). NGF is secreted and transported from the hippocampus to axon terminals of basal forebrain cortical neurons (BFCN), where it supports the maintenance and growth of the cholinergic neurons which provide the major innervations to the hippocampus and cerebral cortex. NGF is translated as a pre-pro-protein from two different splice variants of mRNA, giving rise to one long and one short precursor protein, which are further processed in the trans-Golgi network. Thereafter, the signal peptide is removed by furin, a signal peptide peptidase, to generate the mature form of NGF [26]. NGF, together with the other NTs, activates a number of cell signalling pathways such as those involving Ras, extracellular signal-regulated kinases (ERKs) and cAMP response element binding protein, through binding to tropomyosin-related kinase A (TrkA) and p75 receptors present in the BFCN. In addition to its roles in neural growth and cell maintenance, NGF also influences synaptic functions [27, 28]. Furthermore, proNGF, which is more abundant than NGF [29], is believed to be involved in apoptotic cell signalling through binding to the p75 receptor [30]. Gene knockout of NGF in mice led to severe effects on sensory and sympathetic ganglions, but surprisingly no defects of BFCN were observed [31]. On the other hand, infusion of NGF in a mouse model of Down’s syndrome which exhibits failure in retrograde transport and degeneration of the BFCN, could reverse the brain atrophy, indicating a neurotrophic role of NGF [32].
Levels of NGF in AD
In contrast to BDNF (see below), neither the protein nor the mRNA levels of NGF in the AD brain is decreased, but rather the levels are increased in regions affected in AD, such as the hippocampus and cerebral cortex (reviewed in [33]). However, the BFCN exhibit decreases in both NGF and TrkA receptor levels. These phenomena are regarded to be a result of impaired axonal transport of NGF to the BFCN due to NFT formation in the hippocampus. Thus, NGF is accumulated in the hippocampus and TrkA is down-regulated in the BFCN (reviewed in [34]). A decreased level of NGF leads to neuron shrinkage and to lower expression of enzymes involved in acetylcholine metabolism, i.e. choline-acetyl transferase (ChAT) and AChE, in the AD brain. A decrease of these enzymes in the BFCN is already apparent during the mild cognitive impairment phase (MCI), a phase characterized by amnesia without effects on other cognitive functions to such a degree that the daily life is restrained. Approximately half of the patients with MCI develop AD. These findings led to the development of AChE-inhibitors to increase ACh levels. This treatment efficiently but temporarily ameliorates the symptoms of AD patients in the early and middle phases.
Role of NGF in AD
In addition to the neurotrophic properties of NGF, a recent study has indicated that deprivation of NGF in PC12 cells activates the amyloidogenic cascade [35], thereby coupling NGF to Aβ. Worth noting is the increased expression of proNGF in AD; this might be an underlying cause of the neuronal cell death observed in AD, since proNGF induces apoptotic signalling. Furthermore, overexpression of anti-NGF antibody in a transgenic mouse model leads to memory deficits, cognitive impairments and neurodegeneration, accompanied with hyperphosphorylation of tau in the BFCN. The pathological and behavioural effects could be reversed by infusion of NGF (reviewed in [36]). In summary, imbalance between proNGF and NGF and/or between the NGF receptors TrkA and p75, in combination with impaired anterograde transport of NGF, seems to be closely linked to damage of cholinergic neurons and AD pathology, suggesting that NGF may be used in treatments of AD.
NGF as a therapeutic agent
The aim of NGF treatment is to maintain the physiological concentration of NGF to support the cholinergic neurons and enhance their activity to complement the reduced level of ACh in the basal forebrain. NGF administration offers a more persistent effect than AChE-inhibitors, and intervenes in AD pathogenesis to delay clinical deterioration. In fact, the intraventricular infusion of NGF prevented lesion-induced death of cholinergic neurons and improved both learning and memory functions in old rats [32, 37]. Similar results were obtained in primates, where the infusion of NGF rescued cholinergic neurons from dying after lesion [38, 39]. However, NGF treatment would require that sufficient numbers of TrkA and p75 receptors are present on the cell surface of the neurons and therefore co-delivery of these receptor genes with ChAT gene might be preferable to obtain the maximum effect.
After these promising results, the first phase 1 clinical trial with NGF in human beings was initiated [40, 41]. Since NGF is too large to cross the blood brain barrier (BBB), it was administered intraventricularly to AD patients. However, the NGF infusion activated sensory neurons in a nociceptive response, resulting in weight loss and severe back pain, and therefore the trial was halted [42]. Similarly, infusion of NGF in animals caused weight loss, axonal sprouting and migration of Schwann cells into the medulla oblongata and spinal cord [36]. These results revealed a drawback of intraventricular administration i.e. broad and uncontrolled distribution throughout the brain caused unexpected adverse effects. On the other hand, the trial showed some improvement of cognitive function and increased neuronal activity was observed in the patients.
Development of NGF gene therapy
The first clinical trial with direct infusion of NGF demonstrated the need for methods which provide better control of the distribution and the level of NGF in the brain in order to avoid triggering a nociceptive response or other adverse effects. Gene therapy using either ex vivo approaches with engraftment of NGF-expressing cells, or in vivo approaches with direct injection of a vector carrying the NGF gene, offer such possibilities. Ex vivo approaches, for example using fibroblasts transduced with retroviral vectors such as Moloney leukaemia virus (MLV), have been successfully applied in both rodents and monkeys and shown to have long-term expression, at least up to 1 year. Both lesion-induced damage of cholinergic neurons and age-associated brain atrophy were reversed without adverse affects and cognitive functions were improved ([43–50] and Table 2). Therefore, the first phase I clinical trial of gene therapy enrolling eight AD patients, using an MLV vector to express NGF, was started [51]. Skin fibroblasts, transduced with a MLV vector carrying the NGF gene to express NGF (50–75 ng/10E6 cells) were injected into the nucleus basalis of the patients. Cognitive functions were tested and neuronal activity was monitored using positron-emission tomography, and after a 2-year period significant improvements both in glucose metabolism and in cognition were observed with no apparent adverse effects. Regrettably, one patient developed haemorrhages due to movement during the injection and eventually died. Post-mortem analysis revealed a significant growth of cholinergic nerves around the injection site. This was the first direct observation in human showing the neurotrophic effect of NGF. Taken together, the clinical trial showed that local engraftment of fibroblasts expressing NGF to the affected areas of AD brain has neurotrophic effects on the cholinergic system, leading to amelioration of impaired cognitive function without direct adverse effects. Lastly, a clinical trial phase 1b study was initiated at Karolinska Intitutet in 2008 based on encapsulated epithelial cells from retina engineered to express NGF [52]. The study enrolled six patients who received a semipermeable biodelivery implant, which would allow the release of NGF and prevent cell migration, without post-operative complications.
Table 2.
Summary of gene therapy studies in AD
| Target protein | Function in AD | Vector | Site of expression | Effect | References |
|---|---|---|---|---|---|
| NGF | Neurotrophic, synaptic plasticity | MLV (Ex vivo) | B. F. (Fibroblasts) | No acceleration of Aβ deposition | [44] |
| MLV (Ex vivo) | N.B. (Fibroblasts) | Trophic effect on cholinergic neurons and cognitive improvements | [45] | ||
| MLV (Ex vivo) | Rostral-caudal Ch4 (Fibroblasts) | Protection against brain atrophy | [46] | ||
| MLV (Ex vivo) | B.F. | Protection against age-related degeneration of cholinergic innervations | [48] | ||
| MLV (Ex vivo) | Fimbria-fornix (Fibroblasts) | Restorative after lesions | [50] | ||
| MLV (Ex vivo) | N.B.M. (Fibroblasts) | Clinical trial. Trophic effect on cholinergic neurons and cognitive improvements | [51] | ||
| rAAV | Intraseptal/Medial septum | Protection of lesion-induced degeneration | [53] | ||
| rAAV | B.F. | Cholinergic trophic effect and amelioration of memory function | [55] | ||
| rAAV-2 | Septum | Neurotrophic, increased synaptic activity | [57] | ||
| BDNF | Neurotrophic, synaptic plasticity | Lentivirus | Entorhinal cortex | Neurotrophic, cognitive improvements | [98] |
| Neprilysin (membrane-bound form) | Aβ degradation, neuroprotection | rAAV | Hippocampus, dentate gyrus | Reduced soluble Aβ and Aβ burden | [108] |
| (membrane-bound form) | Lentivirus | F. C. and Hippocampus | Reduced Aβ burden | [128] | |
| (membrane-bound form) | HSV | Hippocampus | Reduced Aβ burden | [129] | |
| (membrane-bound form) | Lentivirus (Ex vivo) | Hippocampus (Fibroblasts) | Neuroprotection by cleaved NPY and reduced Aβ burden | [130] | |
| (secreted form) | Lentivirus | Hippocampus | Reduced Aβ burden, and behavioural improvement | [131] | |
| (membrane-bound form) | Lentivirus | F. C. and Hippocampus | Reduced soluble Aβ, reduced Aβ burden, and improved memory function | [132] | |
| (membrane-bound form) | Lentivirus (Ex vivo) | Blood system (bone marrow cells) | Reduced Aβ burden | [133] | |
| (membrane-bound form) | rAAV-8 | Hind limb muscle | Reduced Aβ burden | [134] | |
| ECE | Aβ degradation | rAAV-5 | Hippocampus, Cortex | Reduced Aβ burden | [155] |
| Cathepsin B | Aβ degradation | Lentivirus | Hippocampus | Reduced Aβ burden | [157] |
| APOE2 | Lipoprotein metabolism, Aβ burden | Lentivirus | Hippocampus | Reduced Aβ levels, and reduced Aβ burden | [143] |
| BACE1 | Aβ generation | Lentivirus(siRNA) | Hippocampus | Reduced soluble Aβ, and reduced Aβ burden | [170] |
| APP | Aβ generation | HSV (siRNA) | Hippocampus | Reduced Aβ burden | [129] |
Abbreviations: B.F.; basal forebrain, N.B.; nucleus basalis, N.B.M.; nucleus basalis of Meynert, F.C.; frontal cortex, MLV; Moloney leukaemia virus, rAAV; recombinant Adeno-associated virus, siRNA; short interfering RNA.
In vivo gene delivery of NGF
Since ex vivo gene delivery demands labour-intensive cell-preparative work, less extensive in vivo gene delivery would have significant advantages. Furthermore, first-generation gene delivery studies were based on grafts of cells transduced with retro viruses, e.g. MLV. Although the retroviruses were replication incompetent and non-toxic, the possible oncogenic risk speaks in favour of the use of other viral vectors such as AAV viruses that have been developed for in vivo gene delivery applications (see Table 1 for a summary of viral vectors used in gene therapy and their properties). AAV, which is able to infect both dividing and non-dividing cells, e.g. neurons, is a promising vector for gene therapy in the central nervous system (CNS). Extensive safety studies have shown AAV to be non-toxic, weakly immunoreactive and non-inflammatory, and it can further be engineered to be non-replicative and to have a very low probability of integrating into the chromosome from its episomal stage. Preclinical studies in mice have shown that rAAV-mediated NGF gene delivery can induce stable NGF expression that ameliorates lesion-induced cholinergic degeneration and reverses cognitive deficits in old mice [53–57]. Several serotypes of rAAV exist, and depending on the proteins displayed on the surface of their capsids, they will bind to different cell-surface receptors, leading to varying affinities for different cell types. Hence, the serotypes can be used to determine the specificity of the transduction. Cells in the CNS are efficiently transduced by rAAV serotypes 1, 2 and 4–9 with differential tropism and toxicity [58, 59]. Most gene delivery studies to the CNS have been carried out with AAV-2 and 5, and serotype 5 leads to a more wide-spread infection of neurons and astrocytes compared to the neuron-specific serotype 2. The cytomegalovirus (CMV) promoter is the most extensively used promoter to drive expression of the transgene, although other cell-type-specific promoters have been used, such as the neuron-specific enolase promoter. Further, inclusion of regulatory elements, e.g. Woodchuck hepatitis virus post-transcriptional element increases the transduction frequency. An alternative to AAV is the HIV-derived lentil viral vector; however, although this vector has been engineered to prevent replication and capsid assembly, its HIV origin together with the theoretical oncogenic risk upon chromosomal incorporation has led to some reluctance to use it in clinical trials. On the other hand, chromosomal integration seems not to be a major risk since the large majority of neurons do not divide (neural stem cells being the exception) and development of integration-deficient lentivirus would further increase safety. Ongoing research on the viral infection mechanisms is likely to increase the knowledge and usefulness of rAAV and lentiviruses. The promising preclinical results of NGF gene delivery using rAAV and the data from the clinical trials of ex vivo NGF gene delivery, as well as rAAV-mediated gene therapy in diseases such as PD and cancer (reviewed in [60]), prompted researchers to initiate a clinical study based on rAAV mediated NGF gene therapy [61].
BDNF
Neurotrophic function of BDNF
BDNF is a relatively small (14 kD), homodimeric protein which has been well conserved throughout evolution, and belongs to the NT family. It is well established that BDNF, which shares 50% sequence identity with NGF, participates in development, maintenance and growth of nerve cells both in the CNS and in the peripheral nervous system. BDNF is highly expressed throughout the brain, including regions of importance for cognitive and intellectual functions, such as the hippocampus and the cerebral cortex [62]. BDNF is also expressed in cells outside the brain, e.g. in endothelial and smooth muscle cells [63], where its expression is believed to support peripheral nerve cells. Six different transcripts of the BDNF gene have been identified and multiple promoters ensure tissue-specific expression [64, 65]. BDNF is localized mainly to the cell soma and the dendrites, to which it is transported by secretory and post-Golgi vesicles. BDNF mRNA has been found in dendrites, indicating that local translation occurs. BDNF binds with high affinity to TrkB receptor, which is translated from two different splice variants of mRNA, one of which is a full-length form with kinase activity, while the other is a truncated and kinase-activity-deficient form with low affinity for the p75 receptor [66, 67]. Interestingly, a large amount of BDNF is found in blood platelets, where it may serve as a BDNF reservoir to support peripheral neurons. Importantly, BDNF is involved in synaptic plasticity (reviewed in [27]).
BDNF levels in AD
The BDNF level in brain is decreased not only in AD [68–73], but also in PD [74], Huntington’s disease [75], depression [76, 77] and schizophrenia [78]. Thus, it seems that BDNF-based drugs might find applications to a wide range of brain diseases. Interestingly, BDNF levels are increased by physical exercise and by cognitive stimulation, and decline upon aging in healthy people [79], suggesting that BDNF may be associated with cognitive functions.
RT-PCR analysis revealed that BDNF transcripts 1–3 were decreased two to five times in parietal cortex of AD brains [80] and lowered levels of serum BDNF in AD patients was found in one study (n= 28, MMSE mean of 23.7) [81]. Interestingly, in another study (n= 30) where the AD patients were divided into two groups, one with early stage AD (MMSE > 21, mean of 25.5) and one with late stage AD (MMSE < 21, mean of 13.3) the group of early AD patients showed increased BDNF serum levels while patients with late stage AD showed decreased BDNF serum levels [82]. On the other hand, one extensive study (n= 465) found no significant difference in serum BDNF between patients with dementia and healthy elderly [83]. However, the patients were not divided into subgroups depending on their clinical stage of AD. BDNF has also been identified in cerebrospinal fluid (CSF), where the level is 3000-fold lower than in serum [84].
BDNF function in AD
Much effort has been made to dissect a possible genetic link between either BDNF or TrkB and AD, but the results are not clear. Some studies found a connection between a Val66Met mutation in BDNF and susceptibility to AD, whereas others did not [85–89]. Studies in rodents provide a somehow confusing picture of BDNF function. Administration of BDNF ameliorated impairment of learning and memory functions in a rat dementia model induced by botulinum toxin, whereas a homozygous BDNF knockout mouse model did not present any cholinergic deficits in the basal forebrain [90, 91]. On the other hand, hippocampus-specific deletion of BDNF in mice impaired spatial memory function, and engraftment of neural stem cells into the hippocampus of an AD mouse model rescued cognitive behaviour in a BDNF-dependent manner [92, 93].
In several AD mouse models (APP-NLh, TgCRND8 and App-Swe/PSEN-1) increased levels of neurotoxic Aβ oligomers were found to correlate with decreased BDNF levels [94]. Arancibia and colleagues showed that the neurotoxic effect of Aβ could be counterbalanced by the addition of BDNF to cortical neurons and this effect appeared to be specific, since it was abolished by a TrkB inhibitor [95]. A number of studies have aimed at elucidating the mechanisms behind the decreased BDNF levels in AD. For example it has been shown that BDNF is down-regulated at the mRNA level in neuroblastoma cells treated with Aβ[96]. In addition, Aβ also blocks BDNF-induced expression of activity-regulated cytoskeleton-associated protein (Arc, also termed Arg3.1), which is involved in synaptic plasticity [97].
Towards BDNF gene therapy
An involvement of BDNF in AD pathogenesis seems likely, but it is still unclear whether the decreased expression in the brain either causes AD pathogenesis or is a consequence of the disease. However, supplementing BDNF in AD could be a possible therapeutic treatment. Taking the BBB into account, BDNF should be supplied through intra-cranial administration and a gene therapy approach could be appropriate. Tuszynski and colleagues reported in a first study the effects of BDNF gene delivery in several AD animal models ([98] and Table 2). They found that administration of BDNF has a neuroprotective effect on cells in the entorhinal cortex, which provides the main input to the hippocampus and is a primary affected region in AD, without affecting the Aβ load. Similarly, BDNF administration into the medial entorhinal cortex of aged rats, which showed cognitive decline and aberrant gene expression, improved spatial learning behaviour and increased phosphorylation of ERK – a sign of increased cell signalling. Lastly, the gene expression was restored to at least 30% of normal for approximately 40% of the genes which exhibited altered expression levels.
The APP transgenic mouse J20, expresses APP bearing the Swedish and Indiana mutations. These mutations affect BACE1 and γ-secretase cleavage, respectively, leading to an increase in both total Aβ levels and in Aβ42/Aβ40 ratio, and induce an age-dependent accumulation of Aβ from 3 months of age and a cognitive decline from 6 months of age [99, 100]. Gene delivery of BDNF to J20 mice using a lentiviral vector, injected bilaterally into the entorhinal cortex at the age of 6 months, significantly ameliorated the impairments of learning and memory tasks, as measured by means of Morris water maze and fear conditioning tests, without affecting the Aβ load. Interestingly, BDNF was transported into the hippocampal regions CA1–3 from the injection site. Gene expression profiling revealed that BDNF delivery to some extent restored the aberrant gene expression in the entorhinal cortex and hippocampus. Lastly, lentiviral expression of BDNF in entorhinal cortex in a primate model with lesions of the perforant pathway prevented neuronal cell death and, furthermore, improved visuospatial learning of old primates [98]. Taken together, the BDNF gene delivery studies show promising results and clinical trials are likely to be initiated, when additional in vivo studies have been completed.
Neprilysin
Role of neprilysin in AD
Aggregation of Aβ and subsequent amyloid plaque formation in the brain are linked to an increased concentration of Aβ, especially Aβ42 . The accumulation occurs with aging in normal brains, but is more pronounced in AD brains. Three kinetic factors determine the steady-state concentration of Aβ in the brain: (i) the rate of Aβ generation from APP, (ii) the rate of transport of Aβ across the BBB and (iii) the rate of Aβ degradation in the brain parenchyma [11]. Therefore, even a minor disturbance of the metabolic rates could affect the Aβ concentration and lead to Aβ accumulation and AD pathology. For this reason, the Aβ-generating enzymes have been subjects of intensive research and considerable efforts have been made to design BACE1 and γ-secretase inhibitors. The large active site of BACE1 has been a challenge but recently efficient BACE1 inhibitors have been developed, however the presence of other substrates than APP for BACE1 decreases their usefulness. The multitude of γ-secretase substrates make the development of molecules that specifically inhibit Aβ generation without adverse effects challenging. However, identification of the Aβ-degrading enzymes has both increased our knowledge about Aβ metabolism and opened up a new range of therapeutic targets associated with the catabolic pathways of Aβ.
Aβ degradation can occur through either direct proteolytic degradation, extracellularly or intracellularly, or through receptor-mediated endocytosis (see APOE section) followed by lysosomal degradation. Several proteolytic activities (see above) have been identified in the brain and the main Aβ-degrading enzyme has been shown by in vivo experiments to be the thiorpan and phosphoramidon-sensitive metallo-endopeptidase neprilysin ([12, 101, 102] reviewed in [10, 11, 103]). Gene disruption and further biochemical analysis provided direct evidence for the importance of neprilysin in Aβ metabolism [104]. Although neprilysin knockout mice showed normal morphology, presumably due to the presence of compensating proteases with neprilysin-like activities, a 2-fold increase of Aβ in the brain was observed in homozygous mice and the increase was gene dosage-dependent. Neprilysin is a 90–110 kD membrane-associated glycoprotein with its active site present on the extracellular side and is involved in the degradation of signalling peptides such as enkephalins, neuropeptide Y (NPY), natriuretic peptide and substance P [105]. Neprilysin is mainly localized at the pre-synaptic areas of neurons [106–108], where it participates in Aβ degradation [109]. It is not able to degrade Aβ variants containing the AD-associated Dutch, Flemish, Italian or Arctic mutations. These mutations are associated with severe AD pathology and the inability of neprilysin to degrade Aβ with those mutations further links neprilysin to AD pathology [110]. Genetic analyses of the neprilysin gene have yielded inconclusive results; some studies indicate that polymorphisms, some of which are in the promoter region, are associated with susceptibility to AD [111–114], while others found no genetic association [115–118]. The expression of neprilysin at the cell membrane can be stimulated by somatostatin [119], a neuropeptide that is involved in inhibition of growth hormone secretion and is down-regulated upon aging [120]. At present it is not known what mechanisms underlie somatostatin regulation, which somatostatin receptor subtype is involved or at what level somatostatin affects neprilysin expression.
Neprilysin levels in AD
The connection between AD progression and the expression of neprilysin has been thoroughly investigated both at the mRNA and protein levels, and several studies have shown a decreased neprilysin level in areas affected in AD, such as the hippocampus and the cerebral cortex [121–123]. Although one study found no significant decrease of neprilysin in AD [124], a clear decline of neprilysin and an inverse relationship between levels of neprilysin and insoluble Aβ upon aging has been established, both in AD and normal brains. Further, the neprilysin level in CSF is significantly reduced in MCI and, interestingly, this is followed by an increase associated with progression of the symptoms [125]. In a recent study on a large number of AD and control brain samples, the neprilysin levels were significantly increased in AD when the levels were adjusted for neuronal loss. However, the levels decrease with age [126] possibly reflecting an induction of neprilysin expression in response to the high concentration of Aβ present during the transition from MCI to AD. Lastly, down-regulation of somatostatin (see above) upon aging, and the even more pronounced down-regulation in AD, could be one reason behind the decrease of neprilysin. These data clearly show that (i) neprilysin is the major Aβ-degrading enzyme (ii) neprilysin is down-regulated upon aging, (iii) down-regulation of neprilysin most likely contributes to insufficient catabolism resulting in Aβ accumulation and prodromal AD. Thus, gene delivery of neprilysin could be a useful therapeutic tool to reduce Aβ levels in AD.
Gene delivery of neprilysin in AD animal models
Several studies in animals have indeed shown that gene delivery of neprilysin can efficiently rescue the catabolic pathway and reduce Aβ levels [108] (Fig. 1 and Table 2). Firstly, neprilysin expressed in vitro using a Sindbis viral vector in murine primary cortical neurons decreased the levels of both intra- and extracellular Aβ[127]. Secondly, neprilysin gene transfer by a lentiviral vector, expressed under the control of a CMV promoter and injected into the hippocampus and the frontal cortex of APP transgenic mice, gave rise to cell soma-localized neprilysin and significantly lowered Aβ plaque burden at the ipsilateral side, but not the contralateral side [128]. Further, gene transfer of neprilysin using a rAAV vector, injected into the hippocampal formation of neprilysin KO mice, led to prominent expression of enzymatically active neprilysin. The activity was not confined to cell soma around the injection site, but was further transported along the neuronal projections from the ipsilateral to the contralateral hippocampus, where neprilysin was localized at pre-synaptic sites [108]. Importantly, gene delivery of neprilysin led to efficient degradation of soluble and insoluble Aβ in both neprilysin knockout mice and APP transgenic mice.
Fig 1.
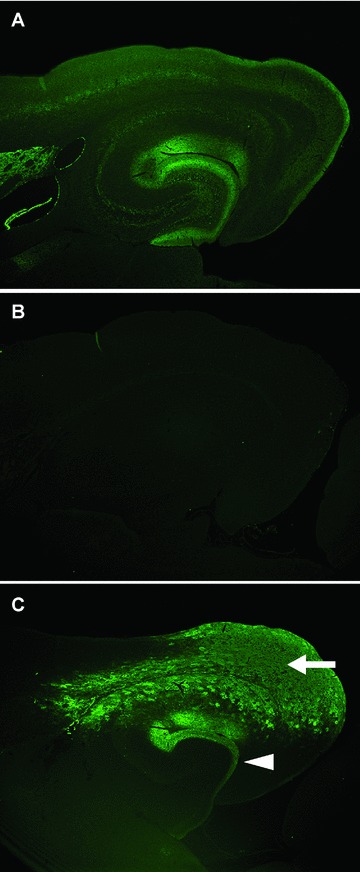
Expression profiles of neprilysin in brain after rAAV5-mediated gene transfer. Expression pattern of neprilysin in the brains was visualized by immunohistochemical staining using an anti-neprilysin antibody. (A) and (B) show typical immunostaining patterns for wild-type and neprilysin-knockout mouse brains, respectively. (C) rAAV5 carrying human neprilysin gene was injected into the lateral entorhinal cortex of neprilysin-knockout mice, and the brain section was immunostained 4 weeks after the injection. The expression of exogenous neprilysin is not only observed in the injection site, lateral entorhinal cortex (arrow), but also occurs in the dentate gyrus (arrowhead), which is the projection site of the lateral entorhinal cortex, and led to increased degradation of Aβ. Thus, neprilysin gene delivery gives a widespread pre-synaptic distribution of exogenous neprilysin and could protect the synaptic function of AD-vulnerable neuronal circuits from Aβ pathology via a reduction of Aβ levels, indicating its potential of gene therapy for AD. Adopted with permission from [108].
Virus-mediated gene delivery to cells or tissues is not only a therapeutic tool, but also offers the possibility of creating new animal models by transduction of mutant genes which induce certain pathologies. In one such AD mouse model, the introduction of mutant APP bearing the Swedish double mutations (K670N/M671L) into mouse brain by a replication-deficient herpes simplex virus (HSV) vector successfully induced high Aβ production [129]. Co-infection of HSV carrying the neprilysin gene in the mouse model lowered the Aβ40 levels by 50% in the hippocampus. In comparison with the rAAV mediated gene delivery of neprilysin, HSV mediated transduction did not lead to widespread expression of neprilysin. Instead, the expression was confined to the ipsilateral hippocampus, being similar to that seen in the case of lentiviral delivery. One possible mechanism explaining the differences in axonal transport of neprilysin is the variation in the 5′ and 3′ UTR in the different viral vectors, which could influence e.g. axonal transport of the mRNA. Another possibility is variation in infection efficiency of the virus, as well as in serotypes. A more widespread distribution of neprilysin following gene delivery would imply a more efficient therapy covering larger areas of the brain. On the other hand, it is possible that neprilysin is also involved in degradation of other physiological peptides, which may lead to adverse effects. In a search for neuropeptides which may undergo processing by neprilysin, it was found that overexpression of neprilysin was associated with increased proteolytic cleavage of NPY [130]. Interestingly, the resulting CTF of NPY exhibited a neuroprotective function in vivo, suggesting that gene delivery of neprilysin would have two disease-modifying effects, i.e. increasing both the degradation of pathogenic Aβ and the production of the neuroprotective NPY fragment.
Ex vivo delivery of a soluble form of neprilysin, in which the transmembrane domain is changed to a signal peptide sequence that induces secretion, was achieved by using grafts of primary fibroblasts transduced with a lentiviral vector and led to a distal reduction of Aβ plaque burden in an AD mouse model [131]. In another study, positive long-term effects, including reduction of Aβ deposition in the cerebral cortex and the hippocampus and improvement of water maze performance, were observed 6 months after injection of a lentiviral vector carrying the neprilysin gene [132]. Two recent studies showed that neprilysin, peripherally expressed on the surface of transduced leukocytes or in skeletal muscle, could lower brain Aβ levels by 30–60% and improve cognitive behaviour without affecting other neuropeptides [133, 134]. These results are in agreement with a sink model, where peripheral degradation of Aβ can sequester brain Aβ. Such a strategy is appealing, taking the simpler injection procedure into account, although more research is needed to verify the feasibility of extracerebral Aβ degradation. The fact that the Km-value of neprilysin for Aβ is in the low micro-molar range is advantageous for a neprilysin-based gene therapy, since neprilysin expression would lead to a more efficient degradation of micro-molar than nano-molar concentrations of Aβ. Hence, a stable and more physiological level of Aβ would be maintained.
Taken together, neprilysin gene delivery in rodents has been extensively studied, with very promising results. Further studies in primates are necessary to evaluate the potential of neprilysin as a possible gene therapy target. For example, it is important to confirm that the levels of other neprilysin substrates, such as the blood pressure-regulating atrial natriuretic peptides and bradykinin, are not affected. If the levels of these peptides are affected by systemic up-regulation of neprilysin, it would have impacts on blood pressure and could lead to hypertension and/or decreased permeability of the BBB [135, 136]. However, local increase in neprilysin activity by gene delivery to the brain would avoid this deleterious effect. Taking into account the ability to reach a large area of the brain with secreted neprilysin, assuming there are no adverse effects, together with the neuroprotective function of the proteolytic fragment of NPY as a second outcome, it appears that neprilysin gene therapy could be a potential treatment for AD.
Potential gene therapy target candidates
APOE
APOE is a 299-amino-acid protein involved in metabolism and transport of lipids including cholesterol and triglycerides. APOE is highly expressed in the liver and in the brain and plays an important role in the delivery of lipoproteins to neurons. In the brain, APOE is mainly expressed in astrocytes and microglia, from which APOE is released to extracellular space. APOE is the genetic risk factor that is most strongly associated with susceptibility to AD (recently reviewed in [7, 137, 138]). The APOE gene exists in 3 isoforms, APOEɛ2, APOEɛ3 and APOEɛ4, among which the ɛ3 allele is the most common (77–78%), followed by ɛ4 (14–16%) and ɛ2 (7–8%). Importantly, this genetic variation of APOE is only seen in human and not in other animals. Carrying one or two ɛ4 alleles significantly increases the incidence of late-onset AD (LOAD) and reduces the average age of onset. Among AD patients, nearly 40% carry one or two ɛ4 alleles.
At the molecular level, the heterogeneity in the APOE gene gives rise to variations of amino acids 112 and 158, where APOE4 has arginine at both positions, APOE3 has a cysteine residue at position 112 and arginine at 158, while APOE2 has cysteine at both positions. The amino acid variations affect the structure of the APOE isoforms, which in turn influence the binding properties of APOE to lipoproteins. The C-terminal part of APOE interacts with lipoproteins and the N-terminal mediates binding to APOE receptors, such as members of the low-density lipoprotein receptor family.
The presence of the two arginines at position 112 and 158 in APOE4 induces an interaction between the N- and C-terminal domains, which is absent in APOE2 or APOE3, and this is believed to be one of the biochemical features underlying the pathological properties of APOE4 related to AD. One consequence of the structural differences is that APOE4 binds preferentially to very low density lipoprotein, whereas APOE2 and 3 binds to high-density lipoprotein. In 1993 it was shown that APOE could bind to Aβ and that APOE was also present in amyloid plaques, with a higher abundance in the brains of AD patients carrying the ɛ4 allele [139]. Accumulating data show the association of APOE4 with in principle all pathological manifestations in AD, i.e. Aβ accumulation, NFTs and neuronal dysfunction and neurodegeneration. However, the mechanism underlying the high incidence of the disease in ɛ4 allele carriers has not yet been fully elucidated. It is also important to clarify whether the effects of APOE4 on AD pathogenesis are attributable to an acquired toxicity or to loss of a protective function. Nevertheless, it is clear that differences in biochemical properties between the APOE isoforms result in a number of physiological changes. For example, all the APOE isoforms promote Aβ42 fibrillization, but APOE4 induces fibrillization to a greater extent than APOE2/3 [140]. On the other hand, the APOE3-lipoprotein complex has higher affinity for Aβ than does the APOE4-lipoprotein complex and therefore, APOE3-associated Aβ is cleared faster through binding to its receptor and subsequent endocytosis and lysosomal degradation [141]. Thus, it is important to separate the effects of APOE itself and the APOE-lipoprotein complex. Furthermore, transgenic mice overexpressing APOE4 exhibit increased phosphorylation of tau. Membrane dynamics play a role in neuronal plasticity, in which cholesterol is one component. Cholesterol level in AD brain is lower compared to normal, and it has been suggested that the secretion of cholesterol from glia is decreased in APOE4 carriers [142]. Therefore, cholesterol imbalance could affect neuronal plasticity. In conclusion, APOE seems to play several roles in AD and the development of gene therapy directed towards APOE will require a careful analysis of the effects on different APOE-related pathologies.
The first gene delivery study in mice, in which different human APOE isoforms were gene-transferred using a lentiviral vector injected into the CA3 region of the hippocampus, provided interesting and promising results. First, in APP transgenic mice (PDAPP) lacking murine APOE, the expression of human APOE4 led to increased Aβ levels and Aβ burden after 5 weeks, while expression of APOE2 and APOE3 did not affect Aβ levels, supporting of the idea that APOE4 is deleterious. Second, in PDAPP mice expressing endogenous APOE, the lentivirus mediated expression of APOE2 significantly decreased both Aβ42 concentration and Aβ burden compared to APOE3 and APOE4 ([143] and Table 2). This proof-of-concept study indicated that gene delivery of APOE2 may have a disease-modifying effect on amyloid pathology, although the study did not provide detailed information concerning other AD-related pathologies, such as tau pathology, synaptic plasticity, neuroinflammation and cognitive effects. It is interesting to note that, since APOE is distributed to a large area of the CNS, APOE gene delivery would be expected to induce widespread effects in the brain and to promote Aβ clearance across the BBB. Although recent data indicate that clearance of Aβ across the BBB is decreased when the APOE complex is formed, clearance occurred more efficiently when Aβ was bound to APOE2/3 as compared to APOE4 [144, 145]. Thus, gene delivery of APOE2 could provide an efficient AD treatment, but could possibly lead to unwanted adverse effects and finely tuned APOE2 expression would probably be required. Finally, it is important to note that in the above study the expression seemed to be confined to neurons. The physiological expression of APOE occurs mainly in microglia and astrocytes, although neuronal expression at a low level has also been observed. However, this study clearly indicates that neuron-derived APOE is able to lower Aβ levels. Alternatively, expression from glial cells may be achieved by using different vectors and/or promoters. A gene therapeutic approach involving APOE could be an alternative to pharmacological modifications of APOE functions, or used in combination with agents directed at other targets. One such target could be the cholesterol-modifying enzyme cholesterol 24-hydroxylase, which converts cholesterol, that cannot be degraded or exported from the brain, to the exportable 24S-hydroxycholesterol. Interestingly, in a recent study AAV-5 was used for gene delivery of cholesterol 24-hydroxylase to the hippocampus and the cerebral cortex of an AD mouse model. The expression of cholesterol 24-hydroxylase in the brain led to decreased levels of Aβ and improved cognitive performance [146]. More investigations are needed to establish in detail the mechanistic roles of APOE in AD pathogenesis, as well as the effects of APOE-based gene therapy.
ECE
ECE is another membrane-integrated protease implicated in Aβ degradation (existing in the two isoforms ECE1 and ECE2) [103, 147]. ECE, which has an extracellular zinc binding active site, is involved in the proteolytic processing of big endothelins, which are peptides considered to regulate a number of physiological functions including vasoconstriction of blood vessels. In the brain, ECE is localized to neurons in hippocampus and layer V of cerebral cortex [148, 149]. ECE can degrade Aβin vitro and in vivo and gene disruption of ECE1+/– or ECE2–/– in mice resulted in a 1.3-fold increase in Aβ levels in the brain [150, 151]. Microarray analysis revealed a significant decrease of ECE2 in the inferior parietal lobe in late-onset AD [152]. When 14 post-mortem temporal neocortics samples from confirmed AD patients were investigated by RT-PCR and Western blot, a 3–9-fold increase in ECE2 mRNA level was found and a 2.5-fold increase in ECE2 protein level. Interestingly, exposing SH-SY5Y neuroblastoma cells to Aβ42 induced a decrease in ECE2 expression after 4 hrs followed by a drastic increase after 24 hrs, though the mechanism behind the regulation needs further investigation [153]. This might indicate that a down-regulation of ECE2 is not the major reason behind Aβ accumulation in AD and, furthermore, that an up-regulation might occur in response to augmented Aβ levels. Moreover, the ECE1 level in CSF was found to be lowered in six AD patients [154]. However, studies including more patients are needed to establish if AD is associated with altered brain ECE levels. Recently, Carty and colleagues reported the potential of ECE gene transfer for AD treatment. Injection of rAAV5-ECE1, where the expression was controlled by the hybrid promoter of chicken β-actin and CMV, unilaterally into the hippocampus and the right anterior cortex of transgenic mice overexpressing APP and PSEN1, leads to expression in the CA3 and CA4 regions in the ipsilateral hippocampus and throughout the cerebral cortex six weeks after injection ([155] and Table 2). Little ECE expression was observed at the contralateral side. Interestingly, six weeks after injection the Aβ burden was analysed immunohistochemically and a 50% reduction was observed. These results show that ECE can participate in Aβ degradation and could be a potential target for a pharmacological or gene therapeutic approach, although more studies are required. No adverse effects, such as weight loss, were observed during the course of the experiments, but it remains to be investigated if ECE gene delivery affects other peptides degraded by ECE.
Cathepsin B
Cathepsin B is suggested to be involved in Aβ degradation, but a rather complex picture of the role of this cysteine protease in AD pathology has been put forward. It was first suggested to be involved in APP processing, having similar properties to those of BACE1. Treatment of mice with cathepsin B inhibitors reduced Aβ40, Aβ42 and CTF-β levels and ameliorated the impaired memory functions in transgenic mice overexpressing wild-type APP, but not APP with a Swedish mutation [156]. On the other hand, cathepsin B was detected immunohistochemically in amyloid plaques, suggesting an association of cathepsin B with Aβ metabolism. Studies of cathepsin B knockout mice have not unambiguously clarified the role of cathepsin B in AD pathogenesis. Cathepsin B deficiency in APP transgenic mice (J20) led to increases in plaque load and in Aβ42/40 ratio in the hippocampus and the cerebral cortex and, furthermore, lentivirus mediated gene delivery of cathepsin B reduced anti-Aβ antibody- and Thioflavin S-positive amyloid plaques, supporting a role for cathepsin B in Aβ degradation ([157] and Table 2). On the other hand, cathepsin B deficiency in wild-type APP transgenic mice significantly decreased Aβ40, Aβ42 and CTF-β levels, supporting a role of cathepsin in APP processing, rather than in Aβ degradation [158]. Further, cathepsin B deficiency in J20 transgenic mice did not alter the levels of Aβ40, Aβ42 and CTF-β, which might be explained by the 50-fold increase in affinity of APP with the Swedish mutation for BACE1 reviewed in [159]. Thus, the role of cathepsin B in Aβ metabolism remains to be established. Variations in amino acid sequence within APP seem to affect the proteolytic cleavage, and if cathepsin B exhibits dual proteolytic properties, i.e. both APP processing and Aβ degradation, gene therapy would have complex implications.
Other Aβ degrading enzymes
In addition to cathepsin B, some attention has been drawn to ACE as an Aβ degrading enzyme. ACE is involved in the processing of angiotensin I to angiotensin II which regulates vasoconstriction and thereby blood pressure. Especially, an indel polymorphism in intron 16 in the ACE gene seems to be associated with AD where the deletion leads to reduced risk of AD [160]. Administration of ACE inhibitors to several AD mouse models have shown some positive effects on cognition but the effects on Aβ levels have not been elucidated, nor has the molecular mechanism behind the improved cognition been shown. Furthermore, knockout of ACE in mouse does not alter the Aβ levels suggesting an indirect role of ACE in improved cognition that does not involve Aβ degradation [11]. Together, the role of ACE in AD needs to be further analysed and the effects of gene delivery of ACE are not obvious and would include effects on blood pressure regulation which needs to be considered.
Another metallo-endopeptidase is IDE which has been shown to contribute to Aβ degradation. For example, gene knockout of IDE in mice leads to slightly increased Aβ levels [161, 162]. IDE can degrade Aβ as well as the type 2 diabetes-associated insulin and amylin peptides. It is interesting to note that both genetic variations in IDE and hyperinsulinaemia are associated with increased risk of AD and the common link might be IDE [163]. Gene delivery of IDE in a gene therapeutic approach could possibly enhance Aβ degradation and influence AD pathology, but the effects on insulin levels have to be carefully examined in such approach.
Down-regulation of AD-associated proteins by siRNA
BACE1
The initial step in Aβ generation is β-secretase cleavage of APP at the extracellular N-terminal site of Aβ, and is performed by the aspartic protease BACE1 (reviewed in [164]). This cleavage is a prerequisite for the subsequent γ-cleavage to generate the Aβ peptide (see above). BACE1 is a type 1 transmembrane protein which has two essential catalytic aspartic acids in the active site and mutation of either of them inactivates the enzyme. BACE1 is expressed at high levels in the brain, especially in neurons. Since Aβ production strongly depends on BACE1 activity, great effort has been made to find potential inhibitors of this enzyme. One of the challenging aspects of this effort is the open structure of the active site. Many inhibitors have been discovered and lately the main focus has been on non-peptidic inhibitors that more easily pass the BBB. One such inhibitor has shown Aβ lowering effects in clinical trials [165]. However, the presence of several substrates for BACE1 interfere with its usefulness. For example, myelination of neurons in adults could be affected by BACE1 inhibitor treatment. Furthermore, BACE1-knockout mice are viable and without any severe deficits, possibly due to redundancy of BACE1 activity, though some emotional changes such as hyperactivity are observed [166]. Therefore, if BACE1 is to be targeted, it will be crucial to obtain a well controlled down-regulation.
Increased levels of both BACE1 mRNA and protein have been observed in the brain at late stages of AD development [167–169].
Therefore, an alternative to pharmacological treatment would be genetic down-regulation of BACE1. Singer and colleagues first reported the effects of BACE1-targeted siRNA delivery into the brain using an AD mouse model ([170] and Table 2). Lentivirus mediated gene transfer of BACE1-targeting siRNA into the hippocampus of APP transgenic mice caused a significant drop in BACE1 expression and decreased both APP-CTF-β and Aβ42 generation. In addition, the siRNA-mediated knockdown of BACE1 ameliorated the impaired neuronal integrity, as measured in terms of the levels of the dendritic marker MAP2 and the synaptic marker synaptophysin, and resulted in improved performance of the mice in the Morris water maze. To summarize, these are the first in vivo data showing a direct effect on amyloid pathology as a result of altering gene expression of an Aβ-anabolizing enzyme. The results seem very promising but therapeutic applications involving BACE1 will require more research in both rodents and primates.
An interesting alternative to intracranial distribution of the viral vector is extra cerebral administration that allows delivery to the brain through receptor-mediated transport across the BBB. An example is the development of the so-called Trojan horse liposomes, which are liposomes containing a super-coiled plasmid carrying a transgene or shRNA. In the liposomes, the DNA or RNA is stabilized with polyethylene glycol linked to a BBB receptor-specific ligand which can mediate transport across the BBB [171].
Also the composition of the vehicle is of importance for efficient delivery of the gene or siRNA and an improvement of in vivo siRNA delivery was recently published by Love and colleagues [172]. They performed a screen of a combinatorial epoxide-derived lipidoid library, composed of non-degradable amino alcohols. In the screen, lipidoids with drastically improved delivery efficacy and increased serum stability were identified and shown to silence genes in nonhuman primates at a dose of 0.03 mg siRNA/kg bodyweight, orders of magnitude lower doses than today’s available siRNA silencing techniques. Combined with brain-specific receptor ligands that deliver the lipidoids over the BBB, these lipidoids could be used for highly efficient RNAi-based therapeutics for AD.
APP
A possible way to reduce the generation of Aβ could be the direct down-regulation of APP by using siRNA. Both in vitro and in vivo studies in mice have shown that siRNA mediated knockdown of APP can reduce Aβ levels. HSV delivery of shRNA directed against APP in a mouse model, where APP is expressed using a lentiviral vector, efficiently reduced the amount of APP mRNA ([129] and Table 2). The shRNA expression reduced the Aβ40 levels by more than 50%. However, accumulating data indicate that APP itself, or proteolytic fragments thereof (see above), have important physiological functions which would be affected by down-regulating APP. For example, APP-knockout mice, although viable, exhibit several adverse phenotypes including reduced weight and locomotor activity [173, 174]. Additionally, siRNA mediated knockdown of APP in aged mice impairs spontaneous alternation in the Y-maze test, a short-term and spatial working memory test [175]. Therefore, gene therapy targeting APP using siRNA should be examined with care.
Concluding remarks
Today no disease modifying treatment for AD exists and research in many areas of AD is undertaken. So far, focus has been on the development of inhibitors for the Aβ-generating enzymes, BACE1 and γ-secretase, immunotherapy directed to Aβ, as well as anti Aβ aggregation agents. No substance has yet been approved for AD treatment, though several are tested in clinical trials where the results so far are not entirely convincing. A lot of effort has been spent on developing an Aβ vaccine. The results have not been successful, with adverse effects such as meningoencephalitis, until recently when active immunization, using either the 6 or 17 first amino acids of Aβ as antigen (CAD106 and ACC-001), has given promising results and the studies are now in phase 2 clinical trial [176]. Difficulties have involved low immunogenicity in elderly and control of serum antibody levels. An alternative is passive immunization using anti-Aβ antibodies, which allows for adjustable serum antibody concentrations but requires repeated costly injections. At present, there are several studies in phase 3. In contrast, the durability of a single injection of a viral vector in a gene therapeutic approach for PD has been reported to last up till 6 years [23].
In the majority of AD cases the accumulation of Aβ is due to an imbalance between Aβ generation and clearance. Thus, a gene therapy involving Aβ degrading enzymes would be a possible alternative to the strategies above. Gene therapy has recently been successfully used to treat a wide range of diseases. In particular, the development of new viral vectors offers advantages such as increased safety and specificity of gene expression. Especially the oncogenic risk of several viral vectors used today has been eliminated (discussed above). In addition, up-regulation of an enzyme might be more feasible using gene therapy than by using small molecule therapeutics.
Gene therapy has been studied extensively in several AD animal models with promising results, and the first clinical trials using ex vivo gene delivery have been accomplished resulting in amelioration of AD pathogenesis. Given the lengthy procedures needed for ex vivo gene delivery, clinical trials of in vivo approaches are likely to be favoured in the future. Other important aspects of gene therapy, besides safety, include correct gene dosage and appropriate targeting of gene expression. Increased knowledge of viral vectors should allow us to further modify the vectors to achieve both highly site-specific expression and adjustable expression level. Vector systems with promoters that can be switched on and off, such as the tetracycline system, should be further considered.
Another promising inducible system is based on administration of tamoxifen, an oestrogen receptor binding ligand. In this system, tamoxifen binds to a mutated oestrogen ligand binding domain fused to Cre recombinase, leading to its activation, which in turn catalyses recombination of a target construct, which is dependent on a flox recombination event for activity [177]. Such systems offer the possibility of both correct timing and dosage. Intra-cranial delivery includes brain surgery which is a moment of risk. The development of extra-cranial methods of gene delivery across the BBB may solve this issue and simplify the procedure. In summary, gene therapy does appear to have the potential to become a disease-modifying treatment for AD.
Acknowledgments
Work in the authors’ laboratories was supported by a research grant from RIKEN Brain Science Institute, Grants-in-aid for Scientific Research on Priority Areas (12210019, 13035055, 17025046 and 18023037) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and Grants-in-aid for Scientific Research (12672167, 14370762, 17500244) from the Japan Society for the Promotion of Science, Special Coordination Funds for promoting Science and Technology of STA (13073–2125-14), a research grant from the Ministry of Health and Welfare of Japan. P.N. is financially supported by the Swedish Research Council. L.O.T. and B.W. are partly financed by Dainippon Sumitomo Pharma, Japan.
References
- 1.Wimo A, Winblad B, Jönsson L. An estimate of the total worldwide societal costs of dementia in 2005. Alzheimers Dementia. 2007;3:81–91. doi: 10.1016/j.jalz.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Welander H, FrÂnberg J, Graff C, et al. Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J Neurochem. 2009;110:697–706. doi: 10.1111/j.1471-4159.2009.06170.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang H-Y, Lee DHS, D’Andrea MR, et al. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. J Biol Chem. 2000;275:5626–32. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 5.Wang H-Y, Lee DHS, Davis CB, et al. Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem. 2000;75:1155–61. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- 6.Lauren J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–32. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–44. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 9.Salminen A, Ojala J, Kauppinen A, et al. Inflammation in Alzheimer’s disease: amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87:181–94. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-β peptide and Alzheimer’s disease. Pharmacol Ther. 2005;108:129–48. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Saido TC, Iwata N. Metabolism of amyloid β peptide and pathogenesis of Alzheimer’s disease: towards presymptomatic diagnosis, prevention and therapy. Neurosci Res. 2006;54:235–53. doi: 10.1016/j.neures.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 12.Iwata N, Tsubuki S, Takaki Y, et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 13.Hawkes CA, McLaurin J. Clinical immunotherapy trials in Alzheimer’s disease. Drug Discov Today Ther Strateg. 2008;5:177–83. [Google Scholar]
- 14.Gillet J, Macadangdang B, Fathke R, et al. The development of gene therapy: from monogenic recessive disorders to complex diseases such as cancer. Methods Mol Biol. 2009;542:5–54. doi: 10.1007/978-1-59745-561-9_1. [DOI] [PubMed] [Google Scholar]
- 15.Cavazzana-Calvo M, Hacein-Bey S, De Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–72. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 16.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-Associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 17.Davé UP, Akagi K, Tripathi R, et al. Murine leukemias with retroviral insertions at Lmo2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genet. 2009;5:e1000491. doi: 10.1371/journal.pgen.1000491. DOI: 10.1371/journal.pgen.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–50. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deakin CT, Alexander IE, Kerridge I. Accepting risk in clinical research: is the gene therapy field becoming too risk-averse? Mol Ther. 2009;17:1842–8. doi: 10.1038/mt.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 22.Liu TC, Kirn D. Gene therapy progress and prospects cancer: oncolytic viruses. Gene Ther. 2008;15:877–84. doi: 10.1038/gt.2008.72. [DOI] [PubMed] [Google Scholar]
- 23.Bankiewicz KS, Forsayeth J, Eberling JL, et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther. 2006;14:564–70. doi: 10.1016/j.ymthe.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Björklund T, Kirik D. Scientific rationale for the development of gene therapy strategies for Parkinson’s disease. Biochim Biophys Acta. 2009;1792:703–13. doi: 10.1016/j.bbadis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Lee E, Son H. Adult hippocampal neurogenesis and related neurotrophic factors. BMB reports. 2009;42:239–44. doi: 10.5483/bmbrep.2009.42.5.239. [DOI] [PubMed] [Google Scholar]
- 26.Mouri A, Nomoto H, Furukawa S. Processing of nerve growth factor: the role of basic amino acid clusters in the pro-region. Biochem Biophys Res Commun. 2007;353:1056–62. doi: 10.1016/j.bbrc.2006.12.136. [DOI] [PubMed] [Google Scholar]
- 27.Poo M-m. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 28.Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–9. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- 29.Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci. 2001;18:210–20. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- 30.Nykjaer A, Lee R, Teng KK, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–8. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- 31.Crowley C, Spencer SD, Nishimura MC, et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–11. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 32.Cooper JD, Salehi A, Delcroix J-D, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci USA. 2001;98:10439–44. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schindowski K, Belarbi K, Buée L. Neurotrophic factors in Alzheimer’s disease: role of axonal transport. Genes Brain Behav. 2008;7:43–56. doi: 10.1111/j.1601-183X.2007.00378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heese K, Low JW, Inoue N. Nerve growth factor, neural stem cells and Alzheimer’s disease. Neurosignals. 2006;15:1–12. doi: 10.1159/000094383. [DOI] [PubMed] [Google Scholar]
- 35.Matrone C, Di Luzio A, Meli S, et al. Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J Alzheimers Dis. 2008;13:81–96. doi: 10.3233/jad-2008-13109. [DOI] [PubMed] [Google Scholar]
- 36.Cattaneo A, Capsoni S, Paoletti F. Towards Non Invasive Nerve Growth Factor Therapies for Alzheimer’s Disease. J Alzheimers Dis. 2008;15:255–83. doi: 10.3233/jad-2008-15210. [DOI] [PubMed] [Google Scholar]
- 37.Fischer W, Wictorin K, Björklund A, et al. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–8. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- 38.Tuszynski M, U H, Amaral D, Gage F. Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J Neurosci. 1990;10:3604–14. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koliatsos V, Nauta H, Clatterbuck R, et al. Mouse nerve growth factor prevents degeneration of axotomized basal forebrain cholinergic neurons in the monkey. J Neurosci. 1990;10:3801–13. doi: 10.1523/JNEUROSCI.10-12-03801.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seiger A, Nordberg A, Von Holst H, et al. Intracranial infusion of purified nerve growth factor to an Alzheimer patient: the first attempt of a possible future treatment strategy. Behav Brain Res. 1993;57:255–61. doi: 10.1016/0166-4328(93)90141-c. [DOI] [PubMed] [Google Scholar]
- 41.Eriksdotter Jönhagen M, Nordberg A, Amberla K, et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9:246–57. doi: 10.1159/000017069. [DOI] [PubMed] [Google Scholar]
- 42.Nauta HJW, Wehman JC, Koliatsos VE, et al. Intraventricular infusion of nerve growth factor as the cause of sympathetic fiber sprouting in sensory ganglia. J Neurosurg. 1999;91:447–53. doi: 10.3171/jns.1999.91.3.0447. [DOI] [PubMed] [Google Scholar]
- 43.Blesch A, Grill RJ, Tuszynski MH, et al. Neurotrophin gene therapy in CNS models of trauma and degeneration. Prog Brain Res. 1998;117:473–84. doi: 10.1016/s0079-6123(08)64033-9. [DOI] [PubMed] [Google Scholar]
- 44.Tuszynski MH, Smith DE, Roberts J, et al. Targeted intraparenchymal delivery of human NGF by gene transfer to the primate basal forebrain for 3 months does not accelerate β-amyloid plaque deposition. Exp Neurol. 1998;154:573–82. doi: 10.1006/exnr.1998.6956. [DOI] [PubMed] [Google Scholar]
- 45.Chen K, Gage F. Somatic gene transfer of NGF to the aged brain: behavioral and morphological amelioration. J Neurosci. 1995;15:2819–25. doi: 10.1523/JNEUROSCI.15-04-02819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DE, Roberts J, Gage F, et al. Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc Natl Acad Sci USA. 1999;96:10893. doi: 10.1073/pnas.96.19.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emerich D, Winn S, Harper J, et al. Implants of polymer-encapsulated human NGF-secreting cells in the nonhuman primate: rescue and sprouting of degenerating cholinergic basal forebrain neurons. J Comp Neurol. 1994:349. doi: 10.1002/cne.903490110. [DOI] [PubMed] [Google Scholar]
- 48.Conner JM, Darracq MA, Roberts J, et al. Nontropic actions of neurotrophins: subcortical nerve growth factor gene delivery reverses age-related degeneration of primate cortical cholinergic innervation. Proc Natl Acad Sci USA. 2001;98:1941–6. doi: 10.1073/pnas.98.4.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuszynski MH, Gabriel K, Gage FH, et al. Nerve growth factor delivery by gene transfer induces differential outgrowth of sensory, motor, and noradrenergic neurites after adult spinal cord Injury. Exp Neurol. 1996;137:157–73. doi: 10.1006/exnr.1996.0016. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg M, Friedmann T, Robertson R, et al. Grafting genetically modified cells to the damaged brain: restorative effects of NGF expression. Science. 1988;242:1575–8. doi: 10.1126/science.3201248. [DOI] [PubMed] [Google Scholar]
- 51.Tuszynski MH, Thal L, Pay M, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–5. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- 52.Eriksdotter-Jonhagen M, Almqvist PM, Lind GSÅ, et al. Encapsulated cell biodelivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients. Abstract P01172, Neurol Am Acad Neurol. 2009:A49. [Google Scholar]
- 53.Mandel RJ, Gage FH, Clevenger DG, et al. Nerve growth factor expressed in the medial septum following in vivo gene delivery using a recombinant adeno-associated viral vector protects cholinergic neurons from fimbria-fornix lesion-induced degeneration. Exp Neurol. 1999;155:59–64. doi: 10.1006/exnr.1998.6961. [DOI] [PubMed] [Google Scholar]
- 54.Andsberg G, Kokaia Z, Klein RL, et al. Neuropathological and behavioral consequences of adeno-associated viral vector-mediated continuous intrastriatal neurotrophin delivery in a focal ischemia model in rats. Neurobiol Dis. 2002;9:187–204. doi: 10.1006/nbdi.2001.0456. [DOI] [PubMed] [Google Scholar]
- 55.Klein RL, Hirko AC, Meyers CA, et al. NGF gene transfer to intrinsic basal forebrain neurons increases cholinergic cell size and protects from age-related, spatial memory deficits in middle-aged rats. Brain Res. 2000;875:144–51. doi: 10.1016/s0006-8993(00)02634-2. [DOI] [PubMed] [Google Scholar]
- 56.Ramirez JJ, Caldwell JL, Majure M, et al. Adeno-associated virus vector expressing nerve growth factor enhances cholinergic axonal sprouting after cortical injury in rats. J Neurosci. 2003;23:2797–803. doi: 10.1523/JNEUROSCI.23-07-02797.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu K, Meyers CA, Guerra NK, et al. The effects of rAAV2-mediated NGF gene delivery in adult and aged rats. Mol Ther. 2004;9:262–9. doi: 10.1016/j.ymthe.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 58.Howard DB, Powers K, Wang Y, et al. Tropism and toxicity of adeno-associated viral vector serotypes 1, 2, 5, 6, 7, 8, and 9 in rat neurons and glia in vitro. Virology. 2008;372:24–34. doi: 10.1016/j.virol.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tenenbaum L, Chtarto A, Lehtonen E, et al. Recombinant AAV-mediated gene delivery to the central nervous system. J Gene Med. 2004;6:S212–22. doi: 10.1002/jgm.506. [DOI] [PubMed] [Google Scholar]
- 60.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–93. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. http://www.clinicaltrials.gov/ct2/show/ NCT00087789?term=NGF&rank=4. Last accessed March 7, 2010.
- 62.Castren E, Thoenen H, Lindholm D. Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience. 1995;64:71–80. doi: 10.1016/0306-4522(94)00386-j. [DOI] [PubMed] [Google Scholar]
- 63.Lommatzsch M, Braun A, Mannsfeldt A, et al. Abundant production of brain-derived neurotrophic factor by adult visceral epithelia. Am J Pathol. 1999;155:1183–93. doi: 10.1016/S0002-9440(10)65221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Timmusk T, Palm K, Metsis M, et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–89. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- 65.Aoyama M, Asai K, Shishikura T, et al. Human neuroblastomas with unfavorable biologies express high levels of brain-derived neurotrophic factor mRNA and a variety of its variants. Cancer Lett. 2001;164:51–60. doi: 10.1016/s0304-3835(00)00715-1. [DOI] [PubMed] [Google Scholar]
- 66.Klein R, Nanduri V, Jing S, et al. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Middlemas DS, Lindberg RA, Hunter T. TrkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–53. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Connor B, Young D, Yan Q, et al. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Mol Brain Res. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- 69.Fahnestock M, Garzon D, Holsinger R, et al. Neurotrophic factors and Alzheimer’s disease: are we focusing on the wrong molecule? J Neural Transm Suppl. 2002;62:241–52. doi: 10.1007/978-3-7091-6139-5_22. [DOI] [PubMed] [Google Scholar]
- 70.Tapia-Arancibia L, Aliaga E, Silhol M, et al. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–20. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 71.Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–22. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- 72.Narisawa-Saito M, Wakabayashi K, Tsuji S, et al. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport. 1996;7:2925–8. doi: 10.1097/00001756-199611250-00024. [DOI] [PubMed] [Google Scholar]
- 73.Hock C, Heese K, Hulette C, et al. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived nneurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000;57:846–51. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- 74.Howells DW, Porritt MJ, Wong JYF, et al. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000;166:127–35. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 75.Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 76.Karege F, Perret G, Bondolfi G, et al. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002;109:143–8. doi: 10.1016/s0165-1781(02)00005-7. [DOI] [PubMed] [Google Scholar]
- 77.Strauss J, Barr CL, George CJ, et al. Brain-derived neurotrophic factor variants are associated with childhood-onset mood disorder: confirmation in a Hungarian sample. Mol Psychiatry. 2005;10:861–7. doi: 10.1038/sj.mp.4001685. [DOI] [PubMed] [Google Scholar]
- 78.Pillai A. Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. Neurosignals. 2008;16:183–93. doi: 10.1159/000111562. [DOI] [PubMed] [Google Scholar]
- 79.Lommatzsch M, Zingler D, Schuhbaeck K, et al. The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol Aging. 2005;26:115–23. doi: 10.1016/j.neurobiolaging.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 80.Garzon D, Yu G, Fahnestock M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J Neurochem. 2002;82:1058–64. doi: 10.1046/j.1471-4159.2002.01030.x. [DOI] [PubMed] [Google Scholar]
- 81.Laske C, Stransky E, Leyhe T, et al. Decreased brain-derived neurotrophic factor (BDNF)- and β -thromboglobulin (β -TG)- blood levels in Alzheimer’s disease. Thromb Haemost. 2006;96:102–3. doi: 10.1160/TH06-03-0173. [DOI] [PubMed] [Google Scholar]
- 82.Laske C, Stransky E, Leyhe T, et al. Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J Neural Transm. 2006;113:1217–24. doi: 10.1007/s00702-005-0397-y. [DOI] [PubMed] [Google Scholar]
- 83.Ziegenhorn AA, Schulte-Herbrüggen O, Danker-Hopfe H, et al. Serum neurotrophins-A study on the time course and influencing factors in a large old age sample. Neurobiol Aging. 2007;28:1436–45. doi: 10.1016/j.neurobiolaging.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 84.Laske C, Stransky E, Leyhe T, et al. BDNF serum and CSF concentrations in Alzheimer’s disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res. 2007;41:387–94. doi: 10.1016/j.jpsychires.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 85.Akatsu H, Yamagata HD, Kawamata J, et al. Variations in the BDNF gene in autopsy-confirmed Alzheimer’s disease and dementia with Lewy bodies in Japan. Dement Geriatr Cogn Disord. 2006;22:216–22. doi: 10.1159/000094933. [DOI] [PubMed] [Google Scholar]
- 86.Li Y, Rowland C, Tacey K, et al. The BDNF val66met polymorphism is not associated with late onset Alzheimer’s disease in three case-control samples. Mol Psychiatry. 2005;10:809–10. doi: 10.1038/sj.mp.4001702. [DOI] [PubMed] [Google Scholar]
- 87.Nacmias B, Piccini C, Bagnoli S, et al. Brain-derived neurotrophic factor, apolipoprotein E genetic variants and cognitive performance in Alzheimer’s disease. Neurosci Lett. 2004;367:379–83. doi: 10.1016/j.neulet.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 88.Vepsäläinen S, Castren E, Helisalmi S, et al. Genetic analysis of BDNF and TrkB gene polymorphisms in Alzheimer’s disease. J Neurol. 2005;252:423–8. doi: 10.1007/s00415-005-0667-5. [DOI] [PubMed] [Google Scholar]
- 89.Huang R, Huang J, Cathcart H, et al. Genetic variants in brain-derived neurotrophic factor associated with Alzheimer’s disease. J Med Genet. 2007;44:e66. doi: 10.1136/jmg.2006.044883. DOI: 10.1136/jmg.2006.044883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones KR, Fariñas I, Backus C, et al. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–99. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ando S, Kobayashi S, Waki H, et al. Animal model of dementia induced by entorhinal synaptic damage and partial restoration of cognitive deficits by BDNF and carnitine. J Neurosci Res. 2002;70:519–27. doi: 10.1002/jnr.10443. [DOI] [PubMed] [Google Scholar]
- 92.Heldt SA, Stanek L, Chhatwal JP, et al. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–70. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blurton-Jones M, Kitazawa M, Martinez-Coria H, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA. 2009;106:13594–9. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peng S, Garzon DJ, Marchese M, et al. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2009;29:9321–9. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arancibia S, Silhol M, Mouliüre F, et al. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–26. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 96.Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–35. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Echeverria V, Berman DE, Arancio O. Oligomers of β-amyloid peptide inhibit BDNF-induced arc expression in cultured cortical neurons. Curr Alzheimer Res. 2007;4:518–21. doi: 10.2174/156720507783018190. [DOI] [PubMed] [Google Scholar]
- 98.Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–7. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shankar GM, Leissring MA, Adame A, et al. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Aβ assembly forms throughout life. Neurobiol Dis. doi: 10.1016/j.nbd.2009.07.021. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mucke L, Masliah E, Yu G-Q, et al. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Takaki Y, Iwata N, Tsubuki S, et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid β peptide in the brain. J Biochem. 2000;128:897–902. doi: 10.1093/oxfordjournals.jbchem.a022839. [DOI] [PubMed] [Google Scholar]
- 102.Shirotani K, Tsubuki S, Iwata N, et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem. 2001;276:21895–901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- 103.Miners JS, Baig S, Palmer J, et al. Aβ-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008;18:240–52. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 105.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23:261–9. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 106.Fukami S, Watanabe K, Iwata N, et al. Aβ-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Aβ pathology. Neurosci Res. 2002;43:39–56. doi: 10.1016/s0168-0102(02)00015-9. [DOI] [PubMed] [Google Scholar]
- 107.Barnes K, Turner A, Kenny A. Membrane localization of endopeptidase-24.11 and peptidyl dipeptidase A (angiotensin converting enzyme) in the pig brain: a study using subcellular fractionation and electron microscopic immunocytochemistry. J Neurochem. 1992;58:2088–96. doi: 10.1111/j.1471-4159.1992.tb10950.x. [DOI] [PubMed] [Google Scholar]
- 108.Iwata N, Mizukami H, Shirotani K, et al. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J Neurosci. 2004;24:991–8. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang S-M, Mouri A, Kokubo H, et al. Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–51. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- 110.Tsubuki S, Takai Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Aβ to physiologically relevant proteolytic degradation. Lancet. 2003;361:1957–8. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]
- 111.Helisalmi S, Hiltunen M, Vepsäläinen S, et al. Polymorphisms in neprilysin gene affect the risk of Alzheimer’s disease in Finnish patients. J Neurosurg Psychiatry. 2004;75:1746–8. doi: 10.1136/jnnp.2004.036574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sakai A, Ujike H, Nakata K, et al. Association of the neprilysin gene with susceptibility to late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord. 2004;17:164–9. doi: 10.1159/000076351. [DOI] [PubMed] [Google Scholar]
- 113.Wood LS, Pickering EH, McHale D, et al. Association between neprilysin polymorphisms and sporadic Alzheimer’s disease. Neurosci Lett. 2007;427:103–6. doi: 10.1016/j.neulet.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 114.Shi J, Zhang S, Tang M, et al. Mutation screening and association study of the neprilysin gene in sporadic Alzheimer’s disease in Chinese persons. J Gerontol A Biol Sci Med Sci. 2005;60:301–6. doi: 10.1093/gerona/60.3.301. [DOI] [PubMed] [Google Scholar]
- 115.Lilius L, Forsell C, Axelman K, et al. No association between polymorphisms in the neprilysin promoter region and Swedish Alzheimer’s disease patients. Neurosci Lett. 2003;337:111–3. doi: 10.1016/s0304-3940(02)01300-9. [DOI] [PubMed] [Google Scholar]
- 116.Oda M, Morino H, Maruyama H, et al. Dinucleotide repeat polymorphisms in the Neprilysin gene are not associated with sporadic Alzheimer’s disease. Neurosci Lett. 2002;320:105–7. doi: 10.1016/s0304-3940(02)00057-5. [DOI] [PubMed] [Google Scholar]
- 117.Sodeyama N, Mizusawa H, Yamada M, et al. Lack of association of neprilysin polymorphism with Alzheimer’s disease and Alzheimer’s disease-type neuropathological changes. J Neurol Neurosurg Psychiatry. 2001;71:817–8. doi: 10.1136/jnnp.71.6.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blomqvist ME, McCarthy S, Blennow K, et al. Evaluation of neprilysin sequence variation in relation to CSF β-Amyloid levels and Alzheimer disease risk. Int J Mol Epidemiol Genet. 2010;1:47–52. [PMC free article] [PubMed] [Google Scholar]
- 119.Saito T, Iwata N, Tsubuki S, et al. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat Med. 2005;11:434–9. doi: 10.1038/nm1206. [DOI] [PubMed] [Google Scholar]
- 120.Lu T, Pan Y, Kao S-Y, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 121.Akiyama H, Kondo H, Ikeda K, et al. Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid β-protein (Aβ) deposition. Brain Res. 2001;902:277–81. doi: 10.1016/s0006-8993(01)02390-3. [DOI] [PubMed] [Google Scholar]
- 122.Yasojima K, Akiyama H, McGeer EG, et al. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- 123.Carpentier MB, Robitaille YM, Desgroseillers LP, et al. Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:849–56. doi: 10.1093/jnen/61.10.849. [DOI] [PubMed] [Google Scholar]
- 124.Hellström-Lindahl E, Ravid R, Nordberg A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: inverse correlation with Aβ levels. Neurobiol Aging. 2008;29:210–21. doi: 10.1016/j.neurobiolaging.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 125.Maruyama M, Higuchi M, Takaki Y, et al. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer’s disease. Ann Neurol. 2005;57:832–42. doi: 10.1002/ana.20494. [DOI] [PubMed] [Google Scholar]
- 126.Miners JS, Baig S, Tayler H, et al. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J Neuropathol Exp Neurol. 2009;68:902. doi: 10.1097/NEN.0b013e3181afe475. [DOI] [PubMed] [Google Scholar]
- 127.Hama E, Shirotani K, Masumoto H, et al. Clearance of extracellular and cell-associated amyloid β peptide through viral expression of neprilysin in primary neurons. J Biochem. 2001;130:721–6. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- 128.Marr RA, Rockenstein E, Mukherjee A, et al. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–6. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hong CS, Goins WF, Goss JR, et al. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-β peptide in vivo. Gene Ther. 2006;13:1068–79. doi: 10.1038/sj.gt.3302719. [DOI] [PubMed] [Google Scholar]
- 130.Rose JB, Crews L, Rockenstein E, et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J Neurosci. 2009;29:1115–25. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hemming ML, Patterson M, Reske-Nielsen C, et al. Reducing amyloid plaque burden via ex vivo gene delivery of an Aβ-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 2007;4:e262. doi: 10.1371/journal.pmed.0040262. DOI: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Spencer B, Marr R, Rockenstein E, et al. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice. BMC Neuroscience. 2008;9:109. doi: 10.1186/1471-2202-9-109. DOI: 10.1186/1471-2202-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guan H, Liu Y, Daily A, et al. Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer’s disease. J Neurosci Res. 2009;87:1462–73. doi: 10.1002/jnr.21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu Y, Studzinski C, Beckett T, et al. Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol Ther. 2009;17:1381–6. doi: 10.1038/mt.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Su J, Cui M, Tang Y, et al. Blockade of bradykinin B2 receptor more effectively reduces postischemic blood-brain barrier disruption and cytokines release than B1 receptor inhibition. Biochem Biophys Res Commun. 2009;388:205–11. doi: 10.1016/j.bbrc.2009.07.135. [DOI] [PubMed] [Google Scholar]
- 136.Borlongan CV, Emerich DF. Facilitation of drug entry into the CNS via transient permeation of blood brain barrier: laboratory and preliminary clinical evidence from bradykinin receptor agonist, Cereport. Brain Res Bull. 2003;60:297–306. doi: 10.1016/s0361-9230(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 137.Cedazo-MÌnguez A. Apolipoprotein E and Alzheimer’s disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11:1227–38. doi: 10.1111/j.1582-4934.2007.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmechel D, Saunders A, Strittmatter W, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ma J, Yee A, Brewer HB, et al. Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 141.LaDu MJ, Falduto MT, Manelli AM, et al. Isoform-specific binding of apolipoprotein E to β-amyloid. J Biol Chem. 1994;269:23403–6. [PubMed] [Google Scholar]
- 142.Ledesma MD, Dotti CG. Amyloid excess in Alzheimer’s disease: what is cholesterol to be blamed for? FEBS Lett. 2006;580:5525–32. doi: 10.1016/j.febslet.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 143.Dodart J-C, Marr RA, Koistinaho M, et al. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2005;102:1211–6. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2006;27:909–18. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Deane R, Sagare A, Hamm K, et al. ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–13. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hudry E, Van Dam D, Kulik W, et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther. 2009 doi: 10.1038/mt.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Macours N, Poels J, Hens K, et al. Structure, evolutionary conservation, and functions of angiotensin- and endothelin-converting enzymes. Int Rev Cytol. 2004;239:47–97. doi: 10.1016/S0074-7696(04)39002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sluck JM, Lin RCS, Katolik LI, et al. Endothelin converting enzyme-1-, endothelin-1-, and endothelin-3-like immunoreactivity in the rat brain. Neuroscience. 1999;91:1483–97. doi: 10.1016/s0306-4522(98)00692-7. [DOI] [PubMed] [Google Scholar]
- 149.Walkden BJ, Wilkinson TC, Turner AJ. Expression of endothelin-converting enzyme in both neuroblastoma and glial cell lines and its localization in rat hippocampus. J Neurochem. 1997;68:570–7. doi: 10.1046/j.1471-4159.1997.68020570.x. [DOI] [PubMed] [Google Scholar]
- 150.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer’s amyloid β peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–8. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 151.Eckman EA, Watson M, Marlow L, et al. Alzheimer’s sisease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–4. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 152.Weeraratna AT, Kalehua A, DeLeon I, et al. Alterations in immunological and neurological gene expression patterns in Alzheimer’s disease tissues. Exp Cell Res. 2007;313:450–61. doi: 10.1016/j.yexcr.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Palmer JC, Baig S, Kehoe PG, et al. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by A{beta} Am J Pathol. 2009;175:262–70. doi: 10.2353/ajpath.2009.081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yoshizawa T, Iwamoto H, Mizusawa H, et al. Cerebrospinal fluid endothelin-1 in Alzheimer’s disease and senile dementia of Alzheimer type. Neuropeptides. 1992;22:85–8. doi: 10.1016/0143-4179(92)90059-6. [DOI] [PubMed] [Google Scholar]
- 155.Carty NC, Nash K, Lee D, et al. Adeno-associated viral (AAV) serotype 5 vector mediated gene delivery of endothelin-converting enzyme reduces Aβ deposits in APP + PS1 transgenic mice. Mol Ther. 2008;16:1580–6. doi: 10.1038/mt.2008.148. [DOI] [PubMed] [Google Scholar]
- 156.Hook VYH, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce β-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, β-secretase site of the amyloid precursor protein. J Biol Chem. 2008;283:7745–53. doi: 10.1074/jbc.M708362200. [DOI] [PubMed] [Google Scholar]
- 157.Mueller-Steiner S, Zhou Y, Arai H, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51:703–14. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 158.Hook VYH, Kindy M, Reinheckel T, et al. Genetic cathepsin B deficiency reduces β-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res Commun. 2009;386:284–8. doi: 10.1016/j.bbrc.2009.05.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stockley J, O’Neill C. Understanding BACE1: essential protease for amyloid-β production in Alzheimer’s disease. Cell Mol Life Sci. 2008;65:3265–89. doi: 10.1007/s00018-008-8271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lehmann DJ, Cortina-Borja M, Warden DR, et al. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am J Epidemiol. 2005;162:305–17. doi: 10.1093/aje/kwi202. [DOI] [PubMed] [Google Scholar]
- 161.Miller BC, Eckman EA, Sambamurti K, et al. Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA. 2003;100:6221–6. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-[beta] peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–8. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 164.Cole SL, Vassar R. The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. J Biol Chem. 2008;283:29621–5. doi: 10.1074/jbc.R800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Silvestri R. Boom in the development of non-peptidic beta-secretase (BACE1) inhibitors for the treatment of Alzheimer’s disease. Med Res Rev. 2009;29:295–338. doi: 10.1002/med.20132. [DOI] [PubMed] [Google Scholar]
- 166.Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–2. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 167.Fukumoto H, Cheung BS, Hyman BT, et al. β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–9. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 168.Holsinger RMD, McLean CA, Beyreuther K, et al. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol. 2002;51:783–6. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 169.Yang L-B, Lindholm K, Yan R, et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 170.Singer O, Marr RA, Rockenstein E, et al. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–9. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- 171.Boado R. Blood–brain barrier transport of non-viral gene and RNAi therapeutics. Pharm Res. 2007;24:1772–87. doi: 10.1007/s11095-007-9321-5. [DOI] [PubMed] [Google Scholar]
- 172.Love KT, Mahon KP, Levins CG, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. 2010;107:1864–69. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zheng H, Jiang M, Trumbauer ME, et al. β-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–31. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 174.Müller U, Cristina N, Li Z-W, et al. Behavioral and anatomical deficits in mice homozygous for a modified β-amyloid precursor protein gene. Cell. 1994;79:755–65. doi: 10.1016/0092-8674(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 175.Senechal Y, Kelly PH, Cryan JF, et al. Amyloid precursor protein knockdown by siRNA impairs spontaneous alternation in adult mice. J Neurochem. 2007;102:1928–40. doi: 10.1111/j.1471-4159.2007.04672.x. [DOI] [PubMed] [Google Scholar]
- 176. http://www.clinicaltrials.gov.
- 177.Li X-g, Okada T, Kodera M, et al. Viral-mediated temporally controlled dopamine production in a rat model of Parkinson disease. Mol Ther. 2006;13:160–6. doi: 10.1016/j.ymthe.2005.08.009. [DOI] [PubMed] [Google Scholar]