

Abstract
Tumour hypoxia is a well-known microenvironmental factor that causes cancer progression and resistance to cancer treatment. This involves multiple mechanisms of which the best-understood ones are mediated through transcriptional gene activation by the hypoxia-inducible factors (HIFs). HIFs in turn are regulated in response to oxygen availability by a family of iron- and 2-oxoglutarate-dependent dioxygenases, the HIF prolyl hydroxylases (PHDs). PHDs inactivate HIFs in normoxia by activating degradation of the HIF-α subunit but release HIF activation in poorly oxygenated conditions. The function of HIF in tumours is fairly well characterized but our understanding on the outcome of PHDs in tumours is much more limited. Here we review the function of PHDs on the HIF system, the expression of PHDs in human tumours as well as their putative function in cancer. The PHDs may have either tumour promoting or suppressing activity. Their outcome in cancer depends on the cell and cancer type-specific expression and on the availability of diverse natural PHD inhibitors in tumours. Moreover, besides the action of PHDs on HIF, recent data suggest PHD function in non-HIF signalling. Together the data illustrate a complex operation of the oxygen sensors in cancer.
Keywords: cancer, hypoxia, oxygen-sensing, VHL
Hypoxia in tumours
Most if not all solid tumours are at least partially hypoxic. This is due to limited amount of vasculature, leaky or otherwise poorly functional tumour vessels with chaotic architecture. These vessels may have good perfusion but at the same time flow-through where the blood does not reach many parts of the cancer tissue and cannot supply the intensely proliferating tumour tissue with sufficient oxygen [1]. Up to 50–60% of locally advanced tumours exhibit hypoxic or anoxic tissue areas that are heterogeneously distributed within the tumour mass. These areas are not static either, but the oxygenation varies temporarily. At a given time-point tumour cells are exposed to an oxygen gradient decreasing gradually from efficient oxygenation to near anoxia, e.g. at perinecrotic areas (for a review see [2]).
Hypoxia is associated with restrained proliferation and differentiation, it impairs energy production and cell viability, arrests cell growth and induces cell death in normal cells (reviewed in [2, 3]). However, in a subset of carcinoma cells hypoxia can also function as a selection pressure for more malignant phenotype as a number of survival responses are activated in the hypoxic cancer cells. These include the activation of anaerobic metabolism, growth factor signalling and neovascularization as well as cell cycle regulation, cell proliferation and protein catabolism that enable a subset of tumour cells to survive in poorly oxygenated conditions (reviewed in [4, 5]). Moreover, hypoxia affects cell-matrix anchorage, activates breakdown of tight cell–cell junctions, activates cell migration and invasion [6, 7]. These increase the potential of cell invasion making the cells more prone to initiate metastatic programs. Finally, hypoxia may enhance the expansion of tumour cells with diminished apoptotic potential [8]. Cells that activate these responses and generate a more aggressive phenotype may be further selected from the tumour cell population by the hypoxia-created genetic instability [9].
It is a long-known fact that hypoxia causes resistance to cancer therapy, namely radiation therapy and also chemotherapy (reviewed in [10]). Already in the early 1900s it was demonstrated that cells irradiated under hypoxic or anoxic conditions are less sensitive to radiation as compared to cells irradiated in oxygenated conditions (for a review see [11]). Accordingly, over 50 years, hypoxia has been understood to negatively impact radiation therapy [12, 13]. At least partially this has been explained by the reduced formation of reactive oxygen species in hypoxia but other apoptosis resistance mechanisms are likely to be involved. Hypoxic cells are also more resistant to many chemotherapeutic agents. This may be due to diminished availability of the agents in poorly vascularized region, to induction of multidrug resistance in hypoxia [14] but also because some cytostatic prodrugs require oxygen for activation [10]. In keeping with these, hypoxia correlates with poor prognosis in several cancer types [15]. These include at least breast cancers, head and neck squamous cell carcinoma (HNSCC), lung cancer and colorectal cancers [16–18].
Overview of the oxygen-sensing mechanisms
To adequately respond to hypoxia, a rapidly responding machinery that can be tightly controlled over a wide range of lowered oxygen tension has evolved. The best-characterized molecular responses to hypoxia are mediated through the activation of gene transcription (reviewed in [15, 19]). Hypoxia-inducible factors (HIFs) have been recognized as transcription factors mainly responsible for the hypoxic gene activation [20] that has been demonstrated to occur in a wide range of human cell types [21]. The active HIF complex is composed of one α-subunit (HIF-α) that is regulated by oxygen tension and one constitutively expressed β-subunit. Three HIF-α family members are known (HIF-1α, -2α and -3α). Out of these HIF-1α and-2α seem to be the main gene activators.
Under well-oxygenated conditions HIF-α is post-translationally hydroxylated within the central oxygen-dependent degradation domain at one or two prolyl residues (Pro402 and 564 in human HIF-1α) [22–25]. The hydroxyprolines serve as recognition sites for von Hippel-Lindau tumour suppressor protein (pVHL), an E3 ubiquitin ligase complex. The formation of pVHL-HIF complex leads to polyubiquitylation and subsequently to the destruction of HIF-α subunits in proteasomes [26–30]. Three human HIF prolyl hydroxylases that can hydroxylate the prolyl residues of HIF-α have been characterized [31, 32]. The HIF hydroxylases have been termed prolyl hydroxylase domain proteins (PHDs, used hitherto), HIF prolyl hydroxylases or Egl-9 homologues. PHDs belong to a larger family of dioxygenases that use O2 and 2-oxoglutarate as co-substrates (reviewed in [19, 33]). Under diminished tissue oxygen tension the PHD activity decreases, binding of pVHL to HIF-α and the degradation of HIF-α is attenuated. This causes ‘automatic’ stabilization and accumulation of the HIF-α subunit, which forms an active transcription factor complex with HIF-β and other transcriptional co-factors such as p300 [20, 34]. The HIF target genes encode proteins that enable cells to survive the oxygen depletion (Fig. 1). Tumour hypoxia and the HIF-system are intensely involved in the process of tumour progression, conferring growth advantage to tumour cells and to the development of a more malignant phenotype. The understanding as to how hypoxia operates at the molecular level has generated great optimism that specific therapeutics for the PHD-HIF system could be designed to overcome the hypoxia-induced chemo- and radiation resistance.
Fig 1.
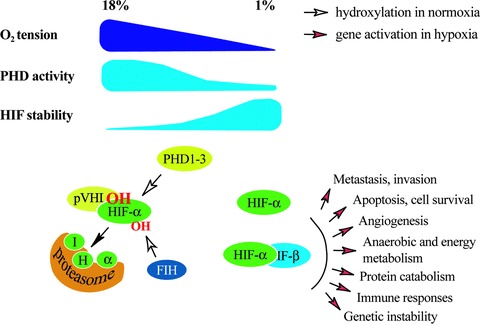
Overview of the known molecular responses to varying tissue oxygen tension. Cellular oxygen levels are sensed by a family of HIF PHD1–3 that use O2 as a co-substrate. Upon oxygenated conditions the PHDs hydroxylate the α subunits of HIF at two prolyl residues. These are recognized by pVHL that ubiquitylates HIF causing its proteasomal degradation. When O2 availability is lowered the hydroxylation and degradation of HIF gradually decreases making HIF stable and transcriptionaly active. Another member of the dioxygenase superfamily, FIH, hydroxylates one asparaginyl residue at the transcriptional domain of HIF causing suppression of the transcriptional activity of HIF.
The family of HIF hydroxylases
Currently the HIF hydroxylases are known to consist of three prolyl hydroxylases, the PHDs and one asparaginyl hydroxylase, FIH (factor inhibiting HIF). PHDs belong to an evolutionarily conserved superfamily of non-haem iron containing dioxygenases [31, 32]. The family comprises at least three prolyl hydroxylases, PHD-1, -2 and -3. PHD isoenzymes 1, 2 and 3 consist of 407, 426 and 239 amino acids, respectively [35] (Fig. 2). All PHDs demonstrate expression of several mRNA species due to alternative splicing or translation initiation with poorly understood function [36, 37]. Similar to other dioxygenases, PHDs require Fe2+, 2-oxoglutarate and molecular oxygen (O2) for their enzymatic function. Ascorbate is a relative requirement for PHDs by keeping the iron in the reduced Fe2+ form. The dioxygenases catalyse the incorporation of one oxygen atom into the substrate forming a hydroxyl group. In the hydroxylation reaction CO2 and succinate are formed as by-products in the decarboxylation of the 2-oxoglutrate (reviewed in [33]).
Fig 2.

Schematic model of the structure of the HIF hydroxylases and naturally occurring activators and inhibitors of their activity. All hydroxylases have a conserved dioxygenase domain at the C-terminal ends. PHD2 contains a zf-MYND (zinc-finger domain) that may interact with regulatory proteins. Both PHD1 and PHD2 contain potential subcellular localization signal sequences, nuclear localization signal or nuclear export signal. The known inhibitors, such as TCA cycle intermediates (TCA IMs) affect the enzymatic activity of the hydroxylases. The expression or activity of the hydroxylases can also be modulated in a post-translational manner (PT). The activators mainly increase the hydroxylase expression by activating transcription (T).
Overall the PHDs show a 42–59% sequence similarity. PHD2 is most closely related ancestrally to the HIF PHDs in Drosophila melanogaster and Caenorhabditis elegans[35]. The hydroxylase domains at the C-termini are well conserved while the N-terminal parts are more divergent [32]. Given the high Km values of PHDs [36], they respond fairly linearly to the wide range of physiological oxygen tension and thereby seem to be well suited as cellular oxygen sensors. Interestingly though, PHD2 and PHD3 have been shown to retain significant activity in hypoxic conditions [38]. All PHDs can hydroxylate HIF-αin vitro[31, 32, 36, 39]. However, PHD2 has been proposed to be the main regulator of HIF-1α, as the RNA interference directed against it is sufficient to induce HIF-α subunits in normoxia, whereas silencing of PHD1 and PHD3 had no effect on the stability of HIF-1α in normoxia or upon re-oxygenation of cells briefly exposed to hypoxia [40]. This might have to do with the relative abundance of PHD2, as it is the most abundant isoform in normoxic conditions in most cell types. While PHD2 is the main regulator of HIF-1α in normoxia and mild hypoxia, PHD3 might regulate HIF in more severe and prolonged hypoxia [38, 40].
The reason for the evolution of three PHD isoforms in mammals is poorly understood. However, in vitro studies have suggested that different PHDs have different specificities towards the different HIF hydroxylation sites [32, 36, 41]. PHD3 mainly hydroxylates the C-terminal hydroxylation site Pro564 (Pro531 in HIF-2α) and has essentially no activity on the N-terminal hydroxylation site Pro402 (Pro405 in HIF-2α). No marked difference was found in the efficiencies of PHD1 and -2 in this respect [32, 36, 38]. PHD2 on the other hand has relatively more influence on HIF-1α than HIF-2α, whereas PHD3 more efficiently regulates HIF-2α[38]. Noticeably, PHD2 has been reported to repress HIF-1α transcriptional activity also in hypoxia. PHD2 was seen to bind HIF-1α in hypoxia without affecting the proteolysis of HIF-1α and thereby inhibiting the HIF-1α N-terminal transcriptional activity [42]. This implies that the PHDs may operate as HIF repressors also in hypoxia and fine tune HIF activity to avoid excessive gene activation.
Shortly after the demonstration of the oxygen-dependent proline hydroxylation, hydroxylation of an asparaginyl residue of HIF-α by another dioxygenase was revealed [43]. The asparagine hydroxylation at the C-terminal transactivation domain of HIF-1α (N803) and HIF-2α (N851) was shown to occur by a formerly described inhibitor of HIF that was termed FIH. FIH prevents the HIF transcriptional activation by hydroxyasparaginyl-dependent inhibition of the interaction between HIF and coactivators, such as p300 and its paralogue CREB-binding protein [43, 44].
Expression of PHDs in normal tissues
The expression of the three PHD isoforms has been studied at protein and mRNA levels in some normal human and murine tissues. PHDs have unique but also partially overlapping expression patterns. Most of the initial expression studies were performed at the mRNA level before the availability of PHD-specific antibodies. A summary of the PHD expression in tissues and some cell lines is presented in Table 1. PHD2 demonstrates most abundant mRNA expression across tissues while the tissue distribution of PHD1 and -3 is more restricted [36, 45, 46]. PHD1 mRNA is particularly abundant in the testis and placenta and is slightly less expressed in the brain, liver, heart and adipose tissue [45–48]. PHD2 mRNA is widely expressed in most tissues and has a more uniform expression pattern. The expression seems particularly abundant in adipose tissue and heart. PHD3 mRNA is also expressed at low level in many tissues but is most abundant in the heart and placenta [45, 46, 48] and to a lesser extent in skeletal muscle and adipose tissue [45, 46, 48]. In cell culture conditions the PHD expression has mainly been studied using cancer-derived cell lines. In normoxic conditions PHD3 mRNA expression is below detection levels in most cell lines studied, while PHD2 is rather uniformly expressed. In cultured cells the protein levels of PHD2 and 3 seem to correlate well with the mRNA levels but the PHD1 protein levels were found to be lower than would be expected from the mRNA levels [38].
Table 1.
Expression of PHD1–3 isoforms in normal tissues and cultured cancer cells at mRNA and protein level
| mRNA expression | Protein expression | |||||
|---|---|---|---|---|---|---|
| Cell/tissue | PHD1 | PHD2 | PHD3 | PHD1 | PHD2 | PHD3 |
| Brain | + | ++ | + | ++ | ++ | + |
| Heart | + | +++ | +++ | N/A | ++ | ++ |
| Skeletal muscle | + | +++ | ++ | N/A | + | N/A |
| Liver | ++ | ++/+++ | ++ | +/− | +/− | +/− |
| Kidney | + | ++ | + | ++ | ++ | ++ |
| Lung | + | + | + | + | − | + |
| Pancreas | N/A | ++ | N/A | + | +/− | +/++ |
| Testis | +++ | ++ | + | ++ | ++ | ++ |
| Placenta | +++ | N/A | +++ | ++/− | ++ | +++ |
| Adipose tissue | ++ | +++ | N/A | − | − | − |
| Endothelium | N/A | N/A | N/A | − | −/++/+++ | −/++ |
| Epidermis, basal layer | N/A | ++ | N/A | |||
| Epidermis, upper layers | N/A | − | N/A | |||
| Carcinoma cells | + | + | + | |||
| Breast cancer | + | ++ | +/− | |||
| Testicular cancer | ++ | + | +/− | |||
| Osteosarcoma cells | + | ++ | + | |||
There are little studies comparing head-to-head the mRNA and protein expression and for many normal tissues only mRNA expression seems to be available. Furthermore, perhaps depending on the antibodies used, the expression of PHDs is not completely overlapping between different investigations. However, the immunohistochemical studies show partially different distribution of PHDs compared to their mRNA expression. PHD1 shows staining in the epithelial cells of human pancreatic and salivary gland ducts, gall bladder and renal tubules [49]. The expression is also detected in myoepithelial and luminal cells of breast ducts and in testicular Leydig and Sertoli cells. In this study the distribution of PHD2 was found mainly similar to PHD1, except that PHD2 showed strong staining in tracheal respiratory epithelial cells and the staining was weak in gallbladder epithelium [49]. In another study very strong PHD2 expression was seen in endothelial cells of skin and in the basal proliferating layer of epidermis. Possibly related to its proliferation-inducing function seen in other cell types, PHD2 expression was detected in the basal proliferating layer of the epidermis and was lost in the upper differentiated layers [50]. PHD3 shows epithelial staining in several intestinal organs such as oesophagus, gastric mucosa, intestine, pancreas and renal tubules. It has been detected also in endometrium, breast myoepithelial and luminal cells, respiratory epithelium, testicular cells, as well as lymphocytes [49]. For FIH prominent staining is seen at least in testicular cells [49]. Suggesting an important function in the central nervous system, all PHDs have been reported to be expressed in the brain. Here the expression level increases in an age-dependent manner [51]. Such age-dependent increase in expression has been previously described for PHD3 in the heart as well [52].
Subcellular localization patterns of PHDs have been studied in cultured cells and at least for PHD2 in human tumours. The subcellular localization of PHD isoforms and FIH-1 were first determined with forced expression of EGFP-fusion proteins in cultured cells. PHD1 was found to be exclusively nuclear. PHD2 and FIH-1 were mainly cytoplasmic while PHD3 was distributed evenly between the nucleus and the cytoplasm [53]. Explaining its primarily nuclear localization, PHD1 has been reported to contain a functional nuclear localization signal [54]. Also PHD2 contains putative subcellular localization signals in the N-terminus. Since EGFP might influence the intracellular distribution of tagged proteins it has been important to verify these results with smaller tags as well as with endogenous proteins. In line with the previous studies, overexpressed FLAG-tagged PHD1 showed predominantly nuclear localization and PHD2 and PHD3 appeared mostly cytoplasmic in COS-1 cells [39]. Immunohistochemical studies of normal human tissues showed similar subcellular localization for PHD2 and 3, but PHD1 was shown to be mostly cytoplasmic with only weak nuclear staining in a proportion of cells [49].
Besides mainly cytoplasmic localization, more recent studies demonstrate also nuclear localization for endogenous PHD2 at least in a subpopulation of cells. This has been detected for example in cancer samples of squamous carcinoma cells and pancreatic endocrine tumours (PET) but also in matching cultured cells [50, 55]. Noticeably, in contrast to the overall expression, hypoxia does not influence the subcellular localization of PHDs [53, 125]. In line with the cell culture studies, FIH staining is reported to be mostly cytoplasmic in a wide range of epithelial cells.
Regulation of PHDs
Besides the regulation of PHD activity by oxygen and other co-substrate availability, their expression is regulated at transcriptional level, by proteasomal destruction and by protein interactions. Moreover, the activity of PHDs is influenced by diverse enzyme inactivating molecules. The latter include PHD enzymatic inhibition by several TCA cycle intermediates and naturally occurring molecules found in tumour tissues. Synthetic pharmacological PHD inhibitors are reviewed elsewhere, for example, recently by Fraisl and colleagues [56].
PHD2 and PHD3 expression levels have been shown by several laboratories to be up-regulated in hypoxia. In contrast, PHD1 levels remain stable regardless of the oxygen tension [32, 40, 45, 57, 58], or may even be reduced under hypoxia [38, 59]. The hypoxic induction of PHD2 and -3 expressions is thought to generate a negative feedback loop that can restrict excessive HIF activity in lowered oxygen tension. In line with this, HIF is commonly found to be down-regulated in continuously hypoxic conditions. Supporting the negative feedback loop, attenuation of the expression of all PHD isoforms extends the time that HIF-1α remains expressed in hypoxia [60]. PHD2 gene contains a functional HRE within the promoter 0.5 kb upstream of the translation start site and is a direct HIF target [61]. PHD3 gene has a functional HRE as well, located within an enhancer region of the first intron 12 kb downstream of the transcription initiation site [62]. PHD1 promoter on the other hand has been reported to contain binding site for ARNT/HIF-1β by which HIF-1β might regulate the hypoxic down-regulation of PHD1 [63]. PHD2 mRNA and protein levels have been proposed to be up-regulated also in an aryl hydrocarbon receptor dependent manner under normoxia [64].
Besides hypoxia, several other microenvironmental factors, such as growth factors and hormones regulate PHD expression. PHD1 mRNA levels are known to be oestrogen inducible [38]. In fact, PHD1 was first cloned as an oestrogen-induced gene in a breast cancer cell line [65]. Out of the growth factors at least TGF-β1 has been shown to markedly down-regulate PHD2 mRNA and protein expression levels, thereby increasing the levels of HIF-1α[66]. Given the up-regulation of some TGF-β superfamily ligands by HIF and the fact that both hypoxia and TGF-β are potent inducers of angiogenesis and invasion of carcinoma cells, a potential positive feedback loop between these factors may operate to further enhance tumour progression.
The fact that PHDs have an absolute requirement for a TCA cycle intermediate 2-oxoglutarate and that several glycolysis and TCA cycle metabolites can inhibit the enzyme activity of PHDs adds to the complexity of the oxygen sensing mechanism. These findings have recently shed light to pathological mechanisms of particular cancer types. The glucose metabolites pyruvate, oxaloacetate, citrate, isocitrate, malate and fumarate all have been reported to inhibit PHDs by binding to the 2-oxoglutarate sites of the enzymes [67–72]. However, there are differences as to which intermediates are the most potent ones as well as to which dioxygenases are preferentially inactivated (Fig. 2). Fumarate and succinate have the most consistent effects. They inhibit all three PHDs competitively with respect to 2-oxoglutarate but neither of them inhibits FIH. In contrast, FIH seems to be more sensitive to citrate and oxaloacetate [68, 70]. In which cancers these inhibitors relate to the activity of different hydroxylase isoforms depend on the TCA cycle enzyme mutations in diverse neoplasias. For example, RCC and leiomyomas are known to bear inactivating mutations leading to the accumulation of the TCA intermediates. It has been proposed that the inhibition of hydroxylase activity is a crucial mediator in the initiation and progression of these cancers [69, 72].
Nitric oxide has been known for some time to modify the hypoxic response. However, the outcome of increased nitric oxide in normoxia and hypoxia is opposite. In normoxia nitric oxide causes stabilization of HIF but in hypoxia increased degradation, both of which may operate through the PHDs. The normoxic stabilization of HIF is likely to occur by direct inhibition of the PHD2 enzymatic activity perhaps by affecting the oxidation of iron [73, 74]. In contrast, the hypoxic reduction in HIF accumulation that is observed with nitric oxide treatment, has generated more controversy [75–77]. It has been proposed that nitric oxide, by inhibiting the mitochondrial respiration chain, allows more oxygen to be available for the PHDs in the proximity of the mitochondria. This would result in enhanced PHD activity and HIF degradation [77]. The proposed mechanism however, has not been fully supported by other studies [78].
Finally, the PHD activity can be controlled by post-translational mechanisms including proteasomal degradation and modulation of the PHD activity by interacting proteins. For example, PHD3 and to a lesser extent PHD1 are targeted to proteasomal degradation by Siah ubiquitin ligases [79]. Interestingly, in this study the HIF-α expression was reported to be completely inhibited in a Siah2-deficient cell line in hypoxia, while PHD2 expression was not affected. Since Siah2 did interact with PHD2, although much more weakly compared to PHD3, it is tempting to speculate that the ubiquitin ligase still could introduce some inhibitory activity also on PHD2 without influencing its expression. Such protein interaction dependent regulation of PHD2 has been described for example for OS9, which promotes PHD2 activity towards HIF in a hydroxylation-independent manner [80]. Also ING4, a tumour suppressor protein, interacts with PHD2 and enhances HIF activity without modulating PHD2 expression or its hydroxylase activity [55]. Another known protein that modulates PHD2 activity is FKBP38. FKBP38 binds PHD2, but not PHD1 or -3, and suppresses PHD2 hydroxylase activity, partially by regulating the PHD2 protein stability though [81].
PHDs in cell growth and differentiation
Due to their function as inhibitors of HIF-1α stability, the PHDs have been proposed to function as tumour suppressors in different cancer types. However, recent studies on PHD expression and their outcome in cancer cells draw a much more complicated picture (Fig. 3). A line of data implicates that PHDs have an essential function in normal cell growth and proliferation. Drosophila PHD is essential for cell growth in fat bodies and wing imaginal discs [82]. PHD1 was cloned as an oestrogen inducible gene in a breast cancer cell line [65] and in in vitro studies PHD1 stimulated cell proliferation of breast cancer cells [65]. Likewise, the mouse PHD1 functioned as a growth stimulator in mouse embryo fibroblasts in response to DNA damage [59]. However, in contrast to the growth promoting functions, ectopic expression of PHD1 suppressed tumour growth in a xenograft tumour mouse model using HCT116 colon carcinoma cells [59].
Fig 3.
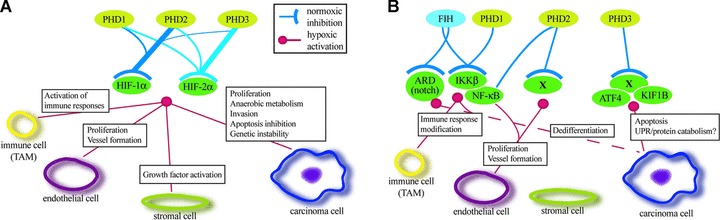
Schematic model of the HIF hydroxylase function that occurs in HIF-dependent (A) or HIF-independent manner (B). (A) The activities of different PHD isoforms towards the two HIF-α isoforms are illustrated by the line thickness. Main HIF-dependent functions of PHDs on different tumour cell types are indicated. (B) Some of the activities of the hydroxylases on potential non-HIF hydroxylation targets and their function on different tumour cell types. X indicates non-characterized hydroxylase targets.
Similar to PHD1, PHD2 has been implicated both as an activator and inhibitor of cell proliferation depending on the cell type. As expected, PHD2 has a critical role in suppressing excessive erythropoiesis that is demonstrated by erythrocytosis caused by a hydroxylase inactivating mutation in the PHD2 gene [83]. PHD2 has also been shown to suppress hypoxia-induced endothelial precursor cell proliferation [84]. Accordingly, PHD2 suppresses the recruitment of endothelial precursor cells and thereby vasculogenesis [85]. Much of the functions of the PHDs are not easily explained solely by their function as inducers of HIF degradation. Also the above two studies implied hydroxylase-independent action either by PHD2’s ability to bind and recruit binding partners to HIF-1α, such as OS-9 and ING4, or through PHD2 activity on the NF-κB pathway. Perhaps related to its function in vascular formation, PHD2 is reported to be required for normal growth and FGF-2 and PDGF-induced proliferation of human pulmonary artery smooth muscle cells as well [86]. Besides the reported cell growth activating functions of PHD2, other evidence implies PHD2 as a potential tumour suppressor. A recent animal model suggests that partial inactivation of PHD2 can activate endothelial cells to form better functioning tumour vessels [87]. Also in line with its possible tumour suppressor function, PHD2 has been reported to induce senescence in carcinoma cells of endometrial cancer [88].
The rat SM-20, a PHD3 homologue, was identified as a growth-factor responsive gene in smooth muscle cells [89] and as a mitochondrial apoptotic factor in neuronal cells in normoxic conditions [90]. Accordingly, PHD3 was shown to be both necessary and sufficient for apoptosis of neuronal cells after withdrawal of neural growth factor [91]. The PHD3 mediated apoptosis required hydroxylation activity, which however, did not seem to be HIF-dependent [91]. In line with this, KIF1Bβ was demonstrated to be the downstream effector of PHD3 [92]. Knockout studies underline the importance of the PHD3-induced apoptosis in the proper development of several neural cell-derived organs [93]. Also in a subset of normoxic carcinoma cells the forced PHD3 expression has been shown to lead to caspase-dependent apoptosis. Surprisingly, this associated with oxygen-dependent protein aggregation. Since the aggregates contain diverse proteasomal components this might be partially due to PHD3-induced malfunction of the proteasomal system in a condition where PHD3 expression is retained in normoxic or reoxygenated conditions in some cell types [94].
PHDs have also been implicated to have functions in cellular differentiation. For example, PHD3 regulates skeletal muscle differentiation [95] as well as differentiation of sympathoadrenal organs apparently by activating appropriate apoptosis [93]. PHD2 is needed for myocardial development in the mouse [96]. Whether the effects on differentiation of normal cells have implications in cancer cell aggressiveness, is yet unclear. In summary, the three PHD isoforms have clearly separate functions on cell survival, proliferation and death. Their function further varies depending on the cell type and the conditions they are expressed in. Moreover, while many of the PHD functions are clearly conveyed through the HIF system, more data are accumulating that the functions at least partially depend on non-HIF signalling.
Expression of PHDs in cancer
Immunohistochemical studies in human cancers have revealed mainly increased but variable staining of PHDs (Table 2). Simplistically one might expect PHD expression to correlate with HIF-α inversely in normoxic tumour regions or positively in hypoxic areas. This seems however, not to be true in many tumours studied so far. For example, prominent PHD2 expression has been detected in large tumour regions simultaneously with HIF-1α. Also PHD3, whose expression is very low in most tissues and cultured cancer cells, shows strong expression in seemingly non-hypoxic tumours [97] (Jokilehto et al., unpublished). In prostate cancer all PHDs are expressed at least moderately. Here the PHD2 expression inversely correlated with HIF-2α, but no correlation was seen between the other PHDs or HIF-1α[97]. In PET all PHDs and FIH are expressed at weak or moderate levels [98]. Similarly, in breast cancers all PHDs and FIH are moderately or strongly expressed [49, 50, 99]. These include the two major forms of breast cancer, ductal and lobular carcinomas. Whether the hydroxylase expression associates either with HER2 amplification or with oestrogen or progesterone receptor expression is unclear. Given that PHD1 is known to be oestrogen-inducible [65], it would be sensible to investigate any association between the hormone status and PHD1 expression. While not addressing any association between molecular markers and PHD expression, for PHD2 an increased expression has been reported to associate with the aggressiveness of breast cancer and HNSCC [50]. In contrast to the above cancers, decreased expression of the hydroxylases has also been reported. In broncogenic carcinomas, renal cell carcinoma and follicular lymphomas the overall staining suggested a slight decrease in the amount of PHD-1, -2 and -3, while an increased FIH was detected in these neoplasia compared to corresponding normal tissues [49].
Table 2.
Expression of PHDs in human cancers
| Cancer type | Hydroxylase isoform | References | |||
|---|---|---|---|---|---|
| PHD1 | PHD2 | PHD3 | FIH | ||
| Protein | |||||
| HNSCC | N/A | +++ | N/A | N/A | [50] |
| Breast cancer | N/A | ++ | N/A | N/A | [50] |
| Breast cancer | ++ | ++ | ++ | ++ | [99] |
| Breast cancer | N/A | N/A | N/A | +++ | [99] |
| Prostate cancer | ++/+++ | ++/+++ | ++/+++ | N/A | [97] |
| Pancreatic endocrine Tumours | ++ | ++ | + | +/++ | [98] |
| Broncogenic carcinoma | +/++ | +/++ | +/++ | +++ | [49] |
| Breast carcinoma | +/++ | +/++ | +/++ | +++ | [49] |
| RCC | −/+ | −/+ | −/+ | +++ | [49] |
| Follicular lymphomas | −/+ | −/+/++/+++ | −/+/++/+++ | ++/+++ | [49] |
| mRNA | |||||
| RCC | N/A | N/A | +++ | N/A | Amatschek et al. 2008 |
| LSCC | N/A | N/A | +++ | N/A | Amatschek et al. 2008 |
Besides the overall expression, a varying intracellular distribution of the hydroxylases in human cancers has been described by several studies. PHD2 expression is not only increased in HNSCC and breast carcinomas but in these carcinomas PHD2 is translocated into the cell nuclei in a subset of cells. Here the nuclear expression associates with tumour aggressiveness using any clinical measurements. These include pathological grade, higher tumour size, TNM classification and stage, higher rate of recurrence as well as poor overall survival [50, 98]. A recent study further suggests that the PHD2 nuclear translocation may associate with cancer therapy response. In line with the association between nuclear PHD2 expression and tumour aggressiveness, in HNSCC a low nuclear staining of PHD2 (<10% of PHD2 positive nuclei) in the primary tumour was found to associate with good radiation response [100]. These imply that the nuclear localization of PHD2 would be beneficial for carcinoma cell growth and somewhat contradicts to the supposed functions of PHD2 as a down-regulator of HIF expression. However, as mentioned, no clear association between HIF-1α and PHD2 was detected neither in HNSCC nor in prostate cancer [50, 97].
Also the subcellular localizations of PHD1, -3 and FIH have shown association with tumour behaviour. Cytoplasmic FIH-1 has been shown to associate with tumour grade in invasive breast cancer [99]. Patients with tumours expressing only cytoplasmic FIH-1 had a significantly shorter survival compared with those with exclusive nuclear expression. In this study the cytoplasmic FIH-1 expression was also an independent prognostic factor for poor disease-free survival in breast cancer. In a set of rarer cancers, the PET, high nuclear PHD1 or PHD3 expression was associated with a poorer survival. In line with the breast cancer study, also cytoplasmic FIH was significantly higher in malignant PETs and in PETs with lymph node metastases [98]. Interestingly, in addition to the cancer cells, the stromal expression of FIH correlated not only with its higher nuclear expression but also with poorer disease-free survival [98].
In summary, all hydroxylases seem to be overexpressed at least to some extent in the common cancers of the breast, prostate and head and neck region as well as in a rarer cancer type of the pancreas. This seemingly contradicts their known function as inhibitors of HIF, a well-characterized tumour progression factor. In a subpopulation of tumours this discrepancy might be explainable by the induction of PHD2 and -3 by hypoxia. However, e.g. for PHD3, which has been described as a pro-apoptotic protein by several laboratories, the up-regulation in cancer tissue is not easy to explain. One needs to bear in mind, however, that none of the studies has demonstrated that the hydroxylases are functional or how much diverse PHD inhibitors, such as the TCA cycle intermediates, might influence their function in cancer tissue.
Function of the PHDs in cancer
While all PHDs can down-regulate HIF-α in in vitro, the specific roles for each PHD in cancer still remain inconclusive. Their contribution is clearly dependent on several conditions, such as their relative abundance in a particular cell or relative influence on the HIF isoforms as well as the two prolyl residues in each isoform. For example, PHD2 is the most abundantly expressed isoform in normoxia and has the highest specific activity towards HIF-1α while PHD3 has more influence on HIF-2α than on HIF-1α. Moreover, as PHD1 and -2 hydroxylates both the N-terminal and C-terminal prolyl hydroxylation sites, PHD3 has activity only towards the N-terminal proline [38–40]. The outcome of this complexity on hypoxic gene expression and cell fate under normoxia and hypoxia is incompletely understood. Also the involvement of diverse inhibitors together with other hydroxylation targets will further complicate the picture. Importantly, information on the functions of PHD that would have been obtained from gene inactivation experiments in animal cancer models is still scarce. Therefore, given the uncertainties in specific PHD function, their role in cancer is inconclusive at the best. It is likely that in the forthcoming years a number of genetics-based information from animal models and possibly from characterizing gene mutations in human tumours will clarify the picture. However, the role of PHDs in cell proliferation and survival, together with the expression patterns in cancer and some tumour specific data have shed light on their possible influence on tumour progression and will be discussed here.
PHD2 is the only isoform reported to display inactivating mutations in human beings. Germline mutations in the PHD2 gene have been reported in patients with familial erythrocytosis [83]. A germline PHD2 mutation (H374R) was also found in a patient with erythrocytosis and recurrent paraganglioma [101]. In line with this, a partial interruption of the PHD2-HIF pathway has been experimentally observed to cause erythrocytosis by activating a set of genes such as erythropoietin. These data point to a possible tumour suppressor function for PHD2 at least in the erythroid cell lineage [102, 103]. PHD2 has been also suggested to work as a tumour suppressor in carcinoma cells. In one study PHD2 gene was found to be mutated in 60% of resected endometrial cancers. In line with this, another study showed that introduction of wild-type PHD2 into endometrial cancer cell lines induced cellular senescence [88].
A role for PHD2 as a key modulator of endothelial proliferation and vessel formation has been independently described in animal models by three laboratories. First, disruption of the Phd2 gene in mouse led to increased vascularization of multiple normal organs, while the knockdown of either PHD1 or PHD3 had no apparent vascular phenotype. Here, PHD2 seemed to operate at least partially in an HIF-independent manner [84, 102]. Another more recent study showed that PHD2 suppresses the recruitment of bone marrow-derived cells to restrict vasculogenesis. In this study PHD2 also seemed to operate in an HIF-independent manner and the vascular suppressing function of PHD2 involved the regulation of NF-κB and subsequently the expression of IL-8 and angiogenin [85]. The third study demonstrated that heterozygous deficiency of Phd2 in endothelial cells partially normalizes the endothelial lining and the stability and barrier function of tumour vessels. These changes were reported to improve tumour perfusion and oxygenation as well as reduce metastasis formation [87]. Taken together, these studies imply PHD2 as either a suppressor of angiogenesis or as an activator by ‘normalization’ of the tumour vasculature. However, given the strong overexpression of PHD2 in common human carcinomas together with its reported function as a growth suppressor in other cell types [86, 88, 104], it is feasible that PHD2 has a dual role in tumour progression. Indeed, a biphasic role for PHD2 in modulating tumour-forming potential has been proposed. In transformed, but not cancerous cells, PHD2 levels were inversely correlated with the tumour-forming potential, whereas the loss of PHD2 activity in malignant cells leads to loss of the tumorigenic phenotype [104].
Although PHD3 is overexpressed in several cancers, including carcinomas, the role of it has mainly been studied in neuronal cells. Before understanding the role of PHD3 in the oxygen sensing pathway, the murine homolog (SM-20) was described to activate neuronal apoptosis in stressed conditions [90]. Later studies have shown that the loss of PHD3 is associated with development of pheochromocytomas by failure in apoptosis [91] that is mediated by a kinesin KIF1Bβ[92]. What the role of PHD3 is in carcinomas remains elusive. On the other hand a clearly enhanced expression is seen in carcinomas, while the normoxic apoptosis-inducing function has been reported not only in neuronal cells but also under normoxic conditions in a subset of HeLa carcinoma cells [94].
At least one experimental animal study, together with the previous cell culture and cancer expression data, implies PHD1 as a suppressor of tumour growth. In this study the ectopic expression of PHD1 in mouse xenografts inhibited tumour growth of colon carcinoma cells and suppressed the accumulation of HIF-1α. The inhibition of tumour growth was correlated with increased necrosis and a decrease in microvessel density [59]. Another study with genetic inactivation of PHD1 demonstrated lowered oxygen consumption in skeletal muscle [105]. This occurred by switch of the glucose metabolism to switch from oxidative to more anaerobic ATP production. If this function of PHD1 was operational also in cancer cells, one might expect PHD1 mutations to be common in cancers.
Besides the alterations in their cancer expression or inactivating mutations, the outcome of PHDs in cancer is strongly influenced by several microenvironmental and cellular inhibitors or activators (Fig. 2). Some genetic mutations on the TCA cycle enzymes that influence the activity of all hydroxylases have been described. Heterozytotic germline mutations in fumarate hydratase or succinate dehydrogenase subunits B, C or D predispose to tumours. Germline mutations have been implicated in the development of the hereditary leiomyomatosis and renal-cell cancer syndrome and the hereditary paraganglioma–pheochromocytoma syndrome [106]. In tumours deficient for fumarate hydratase and succinate dehydrogenase, accumulation of the substrates (fumarate and succinate) inhibits PHD function and thereby causes overexpression of HIF [69, 72]. The forthcoming years are likely to reveal more cancers that display PHD inhibition by the TCA intermediates. Interestingly, it was described already in the 1930s that even normoxic tumours have a strong glycolytic activity, accumulation of lactate and thereby insufficient progression of pyruvate to the TCA cycle (the Warburg effect)[107]. Whether this is linked to the accumulation of the TCA intermediates or the inhibition of PHDs, remains to be investigated.
Finally, a group of recent investigations imply that the cellular protein and organelle catabolism through autophagy, which now is linked to the PHD-HIF system, may be a prominent feature of tumour cell survival [108–113]. Together these studies imply that the PHD-HIF system is an important regulator of protein catabolism, which is understandable since by autophagy the catabolism of organelle components provides nutrient-depleted cells with a source of carbohydrates, lipids and amino acids. For example, the HIF-driven proapoptotic proteins BNIP3 and BNIP3L have been proposed to trigger autophagy in cells and thereby rather to promote survival than induce cell death [108, 111]. Accordingly, HIF activates mitophagy (mitochondrial autophagy) through BNIP [113]. Moreover, p62, a protein that regulates autophagosomal protein transport, is down-regulated in hypoxia adding another layer to the hypoxic regulation of autophagosomal protein catabolism [109, 114].
Other dioxygenase targets in cancer and future directions
In the past years several laboratories have engaged in an attempt to reveal non-HIF hydroxylation targets. Several putative targets have been reported but in many cases they lack a demonstration of an actual hydroxylated target residue. Hydroxylation of asparagine residues in ankyrin-repeat domain containing proteins by FIH have been characterized [115–118]. The ankyrin-repeat domain proteins include an inhibitor of the NF-κB signalling, IKKβ, which has been implicated as a PDH1 target as well [119]. Hydroxylation of IKKβ leads to inactivation of NF-κB signalling that may have several functions not only modulating the tumour-associated immune cell function but also to the response of carcinoma cells under immunological attack. Interestingly, a recent study links PHD2 to NF-κB regulation, suggesting that similar to HIF, several hydroxylases may regulate this pathway [85]. Also the Notch-signalling pathway, a crucial factor in regulating cellular differentiation, is modulated by the hypoxia-sensing machinery. HIF-1α directly interacts with Notch, stabilizes Notch and thereby increases the Notch activity that in turn inhibits differentiation [120]. Moreover, FIH directly hydroxylates the Notch intracellular domain causing negative regulation on the Notch signalling. Under hypoxia and diminished FIH activity the Notch signalling is released. It is feasible that this could promote dedifferentiation of carcinoma cells [115, 117].
Although PHD2 has been linked to non-HIF signalling, to our knowledge PHD2 hydroxylation targets other than HIF-α have not been described. Since PHD2 is evolutionarily the most conserved isoform and the main regulator of HIF-α, it is tempting to speculate that PHD2 is the most HIF-specific isoform, while the other hydroxylases have evolved stronger capacity to target non-HIF proteins as well. PHD3 has been reported to target ATF-4 transcription factor [121] and myogenin [95] in a hydroxylation-dependent manner but actual hydroxylation has not been demonstrated. However, the data suggest that through PHD3-dependent hydroxylation ATF-4 could regulate genes, such as the unfolded protein response genes that HIF is not able to control. PHD3 has also been implicated to signal downstream to KIF1B kinesin and thereby to activate neuronal apoptosis but the kinesin does not seem to be the hydroxylation target in this pathway [92]. Finally, degradation of RNAPolII has been reported to occur in a hydroxylation-dependent manner, while the dioxygenase responsible for this has not been characterized [122]. This raises, together with several other data on the hydroxylases, a possibility that hydroxylation could shut down S-phase progression. Importantly, the halt of cells in the G1 phase and an inability to continue to S-phase is a characteristic feature of cells encountering hypoxia. In order for tumours to progress a subset of cancer cells needs to escape this regulation.
The characterization of the HIF hydroxylases as cellular oxygen sensors has led to intense development of small molecule inhibitors. These are expected to find use, e.g. in mimicking hypoxia in order to activate erythropoiesis and to replace the erythropoietin analogues as a drug of choice in anaemia, or to induce artificial hypoxic preconditioning and cytoprotection in ischemic diseases (reviewed in [56]). Most of the pharmaceuticals are small molecule inhibitors of the hydroxylase enzymatic activity, such as 2-oxoglutarate analogues, that bind the dioxygenase enzyme pocket [123]. As these are highly conserved among the PHD isoforms (although less in FIH), it will be a challenge to design isoform-specific SM inhibitors. For example in anaemia, non-specific inhibitors are likely to activate cellular programs beyond erythropoiesis. The most advanced PHD inhibitors are currently in phase II clinical trials and at least one of them might have been associated with a patient death, raising concerns on poor toxicity profile. Importantly, most of the data so far have implied the PHDs and HIF suppression as inhibitors of tumour progression. Although data that contradict this exist and our knowledge on the effects of PHD inhibition in carcinogenesis and tumour progression is clearly incomplete, one should be cautious with a wide PHD inhibition. Long-term non-specific PHD inhibition could potentially predispose patients to activation of dormant neoplasias. Even short-term inhibition in, e.g. cancer-associated anaemia, could be detrimental by enhancing cancer progression, as has been suspected to occur with the use of erythropoietin analogues.
Acknowledgments
The study was supported by The Academy of Finland (grant 200462), Emil Aaltonen Foundation, Sigrid Juselius Foundation, Turku University Hospital (EVO13031) and the Turku University Foundation. P.M.J. is a senior fellow of the Finnish Cancer Institute.
References
- 1.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 2.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–39. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 3.Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nature Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 4.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–84. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 5.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Esteban MA, Tran MG, Harten SK, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–75. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 7.Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–6. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 8.Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 9.Koshiji M, To KK, Hammer S, et al. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 11.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–75. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams GE, Dische S, Fowler JF, et al. Hypoxic cell sensitisers in radiotherapy. Lancet. 1976;1:186–8. doi: 10.1016/s0140-6736(76)91285-x. [DOI] [PubMed] [Google Scholar]
- 13.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9:539–49. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comerford KM, Wallace TJ, Karhausen J, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–94. [PubMed] [Google Scholar]
- 15.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 16.Kaanders JH, Wijffels KI, Marres HA, et al. Pimonidazole binding and tumor vascularity predict for treatment outcome in head and neck cancer. Cancer Res. 2002;62:7066–74. [PubMed] [Google Scholar]
- 17.Giatromanolaki A, Koukourakis MI, Georgoulias V, et al. Angiogenesis vs. response after combined chemoradiotherapy of squamous cell head and neck cancer. Int J Cancer. 1999;80:810–7. doi: 10.1002/(sici)1097-0215(19990315)80:6<810::aid-ijc3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 18.Nordsmark M, Bentzen SM, Rudat V, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol. 2005;77:18–24. doi: 10.1016/j.radonc.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–28. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 20.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 21.Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3’ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci USA. 1993;90:2423–7. doi: 10.1073/pnas.90.6.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 23.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 24.Yu F, White SB, Zhao Q, et al. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001;98:9630–5. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masson N, Appelhoff RJ, Tuckerman JR, et al. The HIF prolyl hydroxylase PHD3 is a potential substrate of the TRiC chaperonin. FEBS Lett. 2004;570:166–70. doi: 10.1016/j.febslet.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 26.Cockman ME, Masson N, Mole DR, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–41. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 27.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis [see comments] Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 28.Tanimoto K, Makino Y, Pereira T, et al. Mechanism of regulation of the hypoxia-inducible factor-1alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–7. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 30.Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–7. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 31.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 32.Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 33.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 34.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–8. [PubMed] [Google Scholar]
- 35.Taylor MS. Characterization and comparative analysis of the EGLN gene family. Gene. 2001;275:125–32. doi: 10.1016/s0378-1119(01)00633-3. [DOI] [PubMed] [Google Scholar]
- 36.Hirsila M, Koivunen P, Gunzler V, et al. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 37.Tian YM, Mole DR, Ratcliffe PJ, et al. Characterization of different isoforms of the HIF prolyl hydroxylase PHD1 generated by alternative initiation. Biochem J. 2006;397:179–86. doi: 10.1042/BJ20051996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–65. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 39.Huang J, Zhao Q, Mooney SM, et al. Sequence determinants in hypoxia inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277:39792–800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berra E, Benizri E, Ginouves A, et al. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–90. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masson N, Willam C, Maxwell PH, et al. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.To KK, Huang LE. Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol Chem. 2005;280:38102–7. doi: 10.1074/jbc.M504342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lando D, Peet DJ, Gorman JJ, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cioffi CL, Liu XQ, Kosinski PA, et al. Differential regulation of HIF-1 alpha prolyl-4-hydroxylase genes by hypoxia in human cardiovascular cells. Biochem Biophys Res Commun. 2003;303:947–53. doi: 10.1016/s0006-291x(03)00453-4. [DOI] [PubMed] [Google Scholar]
- 46.Lieb ME, Menzies K, Moschella MC, et al. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–6. doi: 10.1139/o02-115. [DOI] [PubMed] [Google Scholar]
- 47.Oehme F, Ellinghaus P, Kolkhof P, et al. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochem Biophys Res Commun. 2002;296:343. doi: 10.1016/s0006-291x(02)00862-8. [DOI] [PubMed] [Google Scholar]
- 48.Willam C, Maxwell PH, Nichols L, et al. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol. 2006;41:68–77. doi: 10.1016/j.yjmcc.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Soilleux EJ, Turley H, Tian YM, et al. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology. 2005;47:602–10. doi: 10.1111/j.1365-2559.2005.02280.x. [DOI] [PubMed] [Google Scholar]
- 50.Jokilehto T, Rantanen K, Luukkaa M, et al. Overexpression and nuclear translocation of hypoxia-inducible factor prolyl hydroxylase PHD2 in head and neck squamous cell carcinoma is associated with tumor aggressiveness. Clinical cancer research. 2006;12:1080–7. doi: 10.1158/1078-0432.CCR-05-2022. [DOI] [PubMed] [Google Scholar]
- 51.Ndubuizu OI, Chavez JC, Lamanna JC. Increased prolyl 4-hydroxylase expression and differential regulation of hypoxia-inducible factors in the aged rat brain. Am J Physiol Regul Integr Comp Physiol. 2009;297:R158–65. doi: 10.1152/ajpregu.90829.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohrbach S, Simm A, Pregla R, et al. Age-dependent increase of prolyl-4-hydroxylase domain (PHD) 3 expression in human and mouse heart. Biogerontology. 2005;6:165–71. doi: 10.1007/s10522-005-7950-9. [DOI] [PubMed] [Google Scholar]
- 53.Metzen E, Berchner-Pfannschmidt U, Stengel P, et al. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003;116(Pt 7):1319–26. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 54.Yasumoto KI, Kowata Y, Yoshida A, et al. Role of the intracellular localization of HIF-prolyl hydroxylases. Biochim Biophys Acta. 2009;1793:792–7. doi: 10.1016/j.bbamcr.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 55.Ozer A, Wu LC, Bruick RK. The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF) Proc Natl Acad Sci USA. 2005;102:7481–6. doi: 10.1073/pnas.0502716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–52. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- 57.D’Angelo G, Duplan E, Boyer N, et al. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem. 2003;278:38183–7. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- 58.Marxsen JH, Stengel P, Doege K, et al. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381:761–7. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erez N, Milyavsky M, Eilam R, et al. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res. 2003;63:8777–83. [PubMed] [Google Scholar]
- 60.Ginouves A, Ilc K, Macias N, et al. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci USA. 2008;105:4745–50. doi: 10.1073/pnas.0705680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metzen E, Stiehl DP, Doege K, et al. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: identification of a functional hypoxia-responsive element. Biochem J. 2005;387:711–7. doi: 10.1042/BJ20041736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pescador N, Cuevas Y, Naranjo S, et al. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005;390:189–97. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Erez N, Stambolsky P, Shats I, et al. Hypoxia-dependent regulation of PHD1: cloning and characterization of the human PHD1/EGLN2 gene promoter. FEBS Lett. 2004;567:311–5. doi: 10.1016/j.febslet.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 64.Seifert A, Katschinski DM, Tonack S, et al. Significance of prolyl hydroxylase 2 in the interference of aryl hydrocarbon receptor and hypoxia-inducible factor-1 alpha signaling. Chem Res Toxicol. 2008;21:341–8. doi: 10.1021/tx7001838. [DOI] [PubMed] [Google Scholar]
- 65.Seth P, Krop I, Porter D, et al. Novel estrogen and tamoxifen induced genes identified by SAGE (serial analysis of gene expression) Oncogene. 2002;21:836–43. doi: 10.1038/sj.onc.1205113. [DOI] [PubMed] [Google Scholar]
- 66.McMahon S, Charbonneau M, Grandmont S, et al. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–81. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- 67.Dalgard CL, Lu H, Mohyeldin A, et al. Endogenous 2-oxoacids differentially regulate expression of oxygen sensors. Biochem J. 2004;380:419–24. doi: 10.1042/BJ20031647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hewitson KS, Lienard BM, McDonough MA, et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007;282:3293–301. doi: 10.1074/jbc.M608337200. [DOI] [PubMed] [Google Scholar]
- 69.Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 70.Koivunen P, Hirsila M, Gunzler V, et al. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 71.Lu H, Dalgard CL, Mohyeldin A, et al. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–39. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 72.Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 73.Berchner-Pfannschmidt U, Yamac H, Trinidad B, et al. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem. 2007;282:1788–96. doi: 10.1074/jbc.M607065200. [DOI] [PubMed] [Google Scholar]
- 74.Metzen E, Zhou J, Jelkmann W, et al. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–81. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berchner-Pfannschmidt U, Tug S, Trinidad B, et al. Nuclear oxygen sensing: induction of endogenous prolyl-hydroxylase 2 activity by hypoxia and nitric oxide. J Biol Chem. 2008;283:31745–53. doi: 10.1074/jbc.M804390200. [DOI] [PubMed] [Google Scholar]
- 76.Sandau KB, Fandrey J, Brune B. Accumulation of HIF-1alpha under the influence of nitric oxide. Blood. 2001;97:1009–15. doi: 10.1182/blood.v97.4.1009. [DOI] [PubMed] [Google Scholar]
- 77.Hagen T, Taylor CT, Lam F, et al. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 78.Doege K, Heine S, Jensen I, et al. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106:2311–7. doi: 10.1182/blood-2005-03-1138. [DOI] [PubMed] [Google Scholar]
- 79.Nakayama K, Frew IJ, Hagensen M, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–52. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Baek JH, Mahon PC, Oh J, et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell. 2005;17:503–12. doi: 10.1016/j.molcel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 81.Barth S, Nesper J, Hasgall PA, et al. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27:3758–68. doi: 10.1128/MCB.01324-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frei C, Edgar BA. Drosophila cyclin D/Cdk4 requires Hif-1 prolyl hydroxylase to drive cell growth. Dev Cell. 2004;6:241–51. doi: 10.1016/s1534-5807(03)00409-x. [DOI] [PubMed] [Google Scholar]
- 83.Percy MJ, Zhao Q, Flores A, et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA. 2006;103:654–9. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takeda K, Fong GH. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension. 2007;49:178–84. doi: 10.1161/01.HYP.0000251360.40838.0f. [DOI] [PubMed] [Google Scholar]
- 85.Chan DA, Kawahara TL, Sutphin PD, et al. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15:527–38. doi: 10.1016/j.ccr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schultz K, Murthy V, Tatro JB, et al. Prolyl hydroxylase 2 deficiency limits proliferation of vascular smooth muscle cells by hypoxia-inducible factor-1{alpha}-dependent mechanisms. Am J Physiol Lung Cell Mol Physiol. 2009;296:L921–7. doi: 10.1152/ajplung.90393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mazzone M, Dettori D, Leite de Oliveira R, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–51. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kato H, Inoue T, Asanoma K, et al. Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. International journal of cancer. 2006;118:1144–53. doi: 10.1002/ijc.21488. [DOI] [PubMed] [Google Scholar]
- 89.Wax SD, Tsao L, Lieb ME, et al. SM-20 is a novel 40-kd protein whose expression in the arterial wall is restricted to smooth muscle. Lab Invest. 1996;74:797–808. [PubMed] [Google Scholar]
- 90.Lipscomb EA, Sarmiere PD, Crowder RJ, et al. Expression of the SM-20 gene promotes death in nerve growth factor- dependent sympathetic neurons. J Neurochem. 1999;73:429–32. doi: 10.1046/j.1471-4159.1999.0730429.x. [DOI] [PubMed] [Google Scholar]
- 91.Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer cell. 2005;8:155–67. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 92.Schlisio S, Kenchappa RS, Vredeveld LC, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884–93. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bishop T, Gallagher D, Pascual A, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3-/- mice. Mol Cell Biol. 2008;28:3386–400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rantanen K, Pursiheimo J, Hogel H, et al. Prolyl hydroxylase PHD3 activates oxygen-dependent protein aggregation. Mol Biol Cell. 2008;19:2231–40. doi: 10.1091/mbc.E07-11-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fu J, Menzies K, Freeman RS, et al. EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J Biol Chem. 2007;282:12410–8. doi: 10.1074/jbc.M608748200. [DOI] [PubMed] [Google Scholar]
- 96.Takeda K, Ho VC, Takeda H, et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–46. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boddy JL, Fox SB, Han C, et al. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin Cancer Res. 2005;11:7658–63. doi: 10.1158/1078-0432.CCR-05-0460. [DOI] [PubMed] [Google Scholar]
- 98.Couvelard A, Deschamps L, Rebours V, et al. Overexpression of the oxygen sensors PHD-1, PHD-2, PHD-3, and FIH is associated with tumor aggressiveness in pancreatic endocrine tumors. Clin Cancer Res. 2008;14:6634–9. doi: 10.1158/1078-0432.CCR-07-5258. [DOI] [PubMed] [Google Scholar]
- 99.Tan EY, Yan M, Campo L, et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br J Cancer. 2009;100:405–11. doi: 10.1038/sj.bjc.6604844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luukkaa M, Jokilehto T, Kronqvist P, et al. Expression of the cellular oxygen sensor PHD2 (EGLN-1) predicts radiation sensitivity in squamous cell cancer of the head and neck. Int J Radiat Biol. 2009;85:900–8. doi: 10.1080/09553000903074104. [DOI] [PubMed] [Google Scholar]
- 101.Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–92. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 102.Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–81. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- 103.Minamishima YA, Moslehi J, Bardeesy N, et al. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–44. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee K, Lynd JD, O’Reilly S, et al. The biphasic role of the hypoxia-inducible factor prolyl-4-hydroxylase, PHD2, in modulating tumor-forming potential. Mol Cancer Res. 2008;6:829–42. doi: 10.1158/1541-7786.MCR-07-2113. [DOI] [PubMed] [Google Scholar]
- 105.Aragones J, Schneider M, Van Geyte K, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–80. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 106.Eng C, Kiuru M, Fernandez MJ, et al. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- 107.Warburg O. The metabolism of tumours. London: Arnold Constable; 1930. [Google Scholar]
- 108.Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pursiheimo JP, Rantanen K, Heikkinen PT, et al. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28:334–44. doi: 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- 110.Samokhvalov V, Scott BA, Crowder CM. Autophagy protects against hypoxic injury in C. elegans. Autophagy. 2008;4:1034–41. doi: 10.4161/auto.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tracy K, Macleod KF. Regulation of mitochondrial integrity, autophagy and cell survival by BNIP3. Autophagy. 2007;3:616–9. doi: 10.4161/auto.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is a HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 114.Jaakkola PM, Pursiheimo JP. p62 degradation by autophagy: another way for cancer cells to survive under hypoxia. Autophagy. 2009;5:410–2. doi: 10.4161/auto.5.3.7823. [DOI] [PubMed] [Google Scholar]
- 115.Coleman ML, McDonough MA, Hewitson KS, et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282:24027–38. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 116.Cockman ME, Lancaster DE, Stolze IP, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc Natl Acad Sci USA. 2006;103:14767–72. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zheng X, Linke S, Dias JM, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA. 2008;105:3368–73. doi: 10.1073/pnas.0711591105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ferguson JE, 3rd, Wu Y, Smith K, et al. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol. 2007;27:6407–19. doi: 10.1128/MCB.00511-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Developmental cell. 2005;9:617–28. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 121.Koditz J, Nesper J, Wottawa M, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;15:3610–7. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 122.Kuznetsova AV, Meller J, Schnell PO, et al. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci USA. 2003;100:2706–11. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ivan M, Haberberger T, Gervasi DC, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:13459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Amatschek S, Koenig U, Auer H, et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res. 2008;64:844–56. doi: 10.1158/0008-5472.can-03-2361. [DOI] [PubMed] [Google Scholar]
- 125.Jokilehto T, Högel H, Heikkinen P, et al. Retention of prolyl hydroxylase PHD2 in the cytoplasm prevents PHD2-induced anchorage-independent carcinoma cell growth. Exp Cell Res. 2010 doi: 10.1016/j.yexcr.2010.02.012. . doi: 10.1016/j.yexcr.2010.02.012. [DOI] [PubMed] [Google Scholar]