

Abstract
Maintenance of cellular pH homeostasis is fundamental to life. A number of key intracellular pH (pHi) regulating systems including the Na+/H+ exchangers, the proton pump, the monocarboxylate transporters, the HCO3− transporters and exchangers and the membrane-associated and cytosolic carbonic anhydrases cooperate in maintaining a pHi that is permissive for cell survival. A common feature of tumours is acidosis caused by hypoxia (low oxygen tension). In addition to oncogene activation and transformation, hypoxia is responsible for inducing acidosis through a shift in cellular metabolism that generates a high acid load in the tumour microenvironment. However, hypoxia and oncogene activation also allow cells to adapt to the potentially toxic effects of an excess in acidosis. Hypoxia does so by inducing the activity of a transcription factor the hypoxia-inducible factor (HIF), and particularly HIF-1, that in turn enhances the expression of a number of pHi-regulating systems that cope with acidosis. In this review, we will focus on the characterization and function of some of the hypoxia-inducible pH-regulating systems and their induction by hypoxic stress. It is essential to understand the fundamentals of pH regulation to meet the challenge consisting in targeting tumour metabolism and acidosis as an anti-tumour approach. We will summarize strategies that take advantage of intracellular and extracellular pH regulation to target the primary tumour and metastatic growth, and to turn around resistance to chemotherapy and radiotherapy.
Keywords: acidosis, carbonic anhydrases, energy production, glycolysis, hypoxia-inducible factor, monocarboxylate transporters, Na+/H+ exchanger, oncogenes activation, pH homeostasis, tumour microenvironment
Introduction
The regulation of the cellular hydrogen potential (pH) is a key step that controls many vital cellular processes such as energy balance, cell proliferation and migration of all mammalian cells. The low extracellular pH or pHout (pHo), in the range of 5.6 to 6.8, and the rather neutral/alkaline intracellular pH (pHi), in the range of 7.2 to 7.5, are hallmarks of malignant cells [1]. Recent techniques using nuclear magnetic resonance 31P spectroscopy for investigation into the pHi and pHo of tumours have confirmed the capacity of tumour cells to acidify the extracellular environment and to maintain a rather neutral/alkaline pHi [2–4]. The extracellular acidic stress in which tumour cells evolve and adapt is a consequence of poor blood perfusion, low oxygen availability, increased metabolism of glucose and production of metabolic acids, such as lactic acid [1, 4–9]. As the tumour develops and oxygen and nutrients become limiting, a gradient of protons extending from blood vessels to the necrotic area of the tumour is established. This so called pH gradient is the inverse of the oxygen gradient. The lower the oxygen level, the more protons (H+) accumulate. The amount of H+ within solid tumours is the consequence of oncogene activation, loss of tumour suppressor activity, adaptation to hypoxia and the ability of tumour cells to extrude acids such as lactic acid and carbonic acid that are generated by a shift in glucose metabolism from oxidative phosphorylation (OXPHOS) to glycolysis. The master transcription factor hypoxia-inducible factor HIF, and particularly HIF-1, induces the expression of glycolytic enzymes to elevate the glycolytic metabolic flux for ATP generation [10, 11]. This metabolic shift is responsible for the increase in production of metabolic acid that is regulated by ubiquitously expressed pHi-regulating proteins in cooperation with the HIF-1-induced pHi-regulating systems. Continual re-adjustment of the pHi is essential since a variation of 0.1 units can disrupt multiple biological functions including ATP production, protein synthesis, cell proliferation, migration and apoptosis through caspase activation [10, 12–15]. Among the constitutively expressed plasma membrane-bound transporters and exchangers, the Na+/H+ exchanger NHE-1 [16] and its isoforms [17], the vacuolar-type H+-ATPase (V-ATPase) [18], the monocarboxylate transporters (MCTs) [19] and HCO3− transporters and exchangers (NBCs and AEs) [20, 21] cooperate to protect the cytosol from acidification. Moreover, in a low oxygen environment, hypoxic tumour cells develop supplemental key pHi-regulating systems such as MCT4 [22] and the membrane-associated carbonic anhydrases (CAs, EC: 4.2.1.1) CAIX and CAXII, which restore a permissive pHi that favours cell survival [23, 24]. It is essential to not only regulate production and export of intracellular acid but also the acidic tumour microenvironment. Extracellular tumour acidosis is an indicator of bad prognosis and has the property to facilitate tumour invasion by the degradation of the extracellular matrix [25, 26] and to reduce immunosurveillance by inhibiting the activity of natural killer (NK) cells [27] and the cytolytic activity of cytotoxic T lymphocytes [28]. Thus, acidosis promotes tumour cell migration and exerts immunosuppressive effects that encourage tumour progression. Tumour acidosis has also been linked to multi-drug resistance (MDR) due to the neutralization of weak base chemotherapeutic drugs, which makes the drugs less efficient [29], and is also associated with increased chromosomal instability in vitro[30]. The difference between the pHo of normal and tumour tissue offers a selective and specific strategy for developing new treatments for targeting solid tumours [31, 32]. The identification of the most pertinent pHi-regulating systems, the function of which is essential to maintain continuous and efficient production of ATP, represents a potential new anticancer approach that exploits both the addiction of cells to glycolysis and the regulation of pH (pHi and pHo) homeostasis. Such a strategy would block the export of metabolic acids and thus would poison tumour cells with their own acid. Consequently, ATP production would be compromised and would drive the cells of the primary tumour to necrotic cell death. Moreover, this approach would also reduce extracellular acidification of the tumour environment that would decrease metastatic formation and sensitize the cells to chemotherapeutic agents. This strategy is mostly dedicated to hypoxic tumour cells, but since a large variety of tumours are addicted to glycolysis due to oncogenes and tumour suppressor gene mutations they may be highly sensitive to inhibition of pHi regulation.
Intracellular pH regulation controls energy balance and cell proliferation: chemical and biological proof of principle
Chemical proof of principle
Why should we be concerned about pHi levels? Human physiology is constantly controlled by the pH, which can be either acid (pH 1 to 6.9), neutral (pH 7.0 to 7.4) or alkaline (pH above 7.4). Since most of the human body is water based, the pH level has profound effects on body chemistry, health and disease. This is illustrated in the case of mountain sickness, which is treated with a drug that inhibits CAs. This drug modifies the blood pH but also changes the perception of carbonated beverages in the mouth [33]. One of the major pH-dependent reactions within prokaryotic and eukaryotic cells concerns the cytosolic glycolytic pathway that generates two highly energetic molecules of ATP for one molecule of glucose consumed. The uptake of glucose and its sequestration by hexokinase (HK, EC: 2.7.1.1) are key steps in initiating glycolysis, which once complete satisfies the cell’s energy demands. HK1/2 catalyse the phosphorylation of glucose to glucose-6-phosphate (G-6-P) in the presence of ATP and Mg2+. This first chemical step of the glycolytic pathway is very important for several reasons: (i) because of its negative charges, G-6-P cannot diffuse across the plasma-membrane and the phosphoryl group destabilizes glucose, which facilitates glucose metabolism and then continuous energy production, (ii) this reaction is pH dependent, which means that whatever the extracellular environment, strict control of the pHi is required to access the subsequent reactions of glycolysis that lead to ATP production. Several studies have demonstrated that a pH threshold controls the activity of enzymes such as HK and phosphofructokinase (PFK) [34–36]. It should be pointed out that the transfer of the phosphoryl group from ATP to glucose is a nucleophilic substitution reaction that cannot occur in a low pHi. Thus, pHi maintenance is necessary in initiating glycolysis and promoting ATP formation.
Biological proof of principle: the role of the Na+/H+ exchanger-1
Key intracellular processes such as glycolysis-dependent ATP production and protein synthesis require close regulation of the pHi, affecting therefore proliferation, migration, and cell survival [12–14]. The main source of acid in the context of biological processes concerns, so far, the direct generation of H+, or that coming from weak acids such as lactic acid and carbonic acid (H2CO3). A variety of constitutively expressed membrane-associated systems are involved in the regulation of the pHi of all mammalian cells: (i) the family of growth factor-activated NHEs [16], and the vacuolar-type H+-ATPase (V-ATPase), that requires ATP to actively extrude two H+[18, 37, 38], (ii) the MCTs (MCT1, MCT2, MCT4) that transport lactic acid bidirectionally [19, 39–41], (iii) Na+-HCO3− co-transporters (NBCs) and Cl−/HCO3− exchangers (AEs) that, in the opposite direction to H+ extrusion, favour HCO3− influx and thus contribute to cytoplasmic alkalinization [42, 43], (iv) the family of carbonic anhydrases (CAs) and particularly, the cytosolic CAII, have been shown to play a key role in the regulation of the physiological pH by catalysing the hydration of carbon dioxide to protons and bicarbonate ions [44]. Among this spectra of coexisting pHi-regulating systems, the first evidence that pH regulation is a key process in mammalian growth control was demonstrated through the study of NHE-1, the first identified molecularly member of a large family [16]. Nine isoforms of NHEs (NHE-1 to NHE-9) have been identified but only NHE-1 to NHE-3 have been well characterized. NHE-1 is ubiquitously expressed, while NHE-2 and NHE-3 are found in the kidney and the gastrointestinal epithelium [45–48]. NHE-4 and NHE-5 are restricted to the gastrointestinal tract and the brain, however little is known about their function [49]. Isoforms NHE-6 to NHE-9 are localized to subcellular organelles [50–52]. The amiloride sensitive and growth factor-activated Na+/H+ antiport NHE-1 has been actively studied in our laboratory [53]. It is activated by all mitogens, integrins and oncogenic transformation [53]. NHE-1 is activated by intracellular H+ at an allosteric modifier site and extrudes H+ against entry of Na+. Growth factors increase the affinity of this H+ modifier site leading to a persistent increase in the pHi (0.2 to 0.3 pHi units more alkaline) [53]. In non-neoplastic Chinese hamster lung fibroblasts expressing endogenous NHE-1, NHE-2 and NHE-3, the H+-suicide technique [13], which consists in delivering a massive acid load into cells through a NH4+ pre-pulse, demonstrated the high potential of NHE-1 in regulating the pHi in acidic conditions, based on the rapid recuperation on the pHi values permissive with cell survival (Fig. 1). These observations, on the potential of NHE-1 to maintain the pHi do not only concern normal cells but also tumour cells, as we found for colon carcinoma LS174Tr cells and cervix carcinoma HeLa cells (data not shown). In contrast, mutant cells lacking NHE-1 do not recover their pHi after an acid-load and die within 1 hr as a result of persistent acidification (Fig. 1). This tight regulation of the pHi is a mitogenic signal for the control of cell growth. Previous studies have reported that the regulation of the pHi via the NHE-1 system constitutes a limiting step for growth factor stimulated ribosomal protein S6 phosphorylation [54], which points to protein synthesis as a pH-sensitive process. These data support the idea that Na+-dependent extrusion of H+via the activation of NHE-1 is a key event in the mechanism of action of growth factors. Although a rise in the pHi is not mitogenic per se, it has been demonstrated that its increase, above a critical threshold value of pHi 7.2, is a prerequisite for growth factor-stimulated quiescent cells to progress into the cell cycle [55]. However, in response to growth factors, quiescent mutant fibroblasts lacking NHE-1 activity fail to increase their pHi and to reinitiate DNA synthesis at an acidic pHo [13]. These data clearly demonstrate that reinitiation of DNA synthesis is a highly pHi-dependent process. H-ras-transformed stable mutants of originally non-neoplastic Chinese hamster lung fibroblasts impaired either in respiration (CCL39-res−) or in glycolysis (CCL39-gly−) or in the NHE-1 activity (CCL39-nhe-1−) were obtained from the parental cell line CCL39-wt to study the control of the pHi on tumour incidence [13, 56]. Mutant cells lacking NHE-1 (CCL39-nhe-1−) formed tumours less frequently than the wt cells 10 days after transplantation in nude mice [57]. Once they make tumours, 80% of these tumours lacking NHE-1 activity regress [58] (Fig. 2). Human bladder carcinoma MGH-U1 cells deficient in NHE-1 activity also failed to form spheroids in culture, and the few tumours that were obtained after in vivo implantation were revertants that recovered NHE-1 activity [59]. CCL39-res− (respiration negative) mutants consume almost no oxygen and secrete around three times more lactic acid than wt cells. Thus, this mutant relies only on glycolysis to survive and has a low frequency of tumour take (75%) with apparition after 8 to 9 weeks after injection to mice and a low in vivo proliferation rate. In addition, some of these tumours when formed showed a high frequency of regression (80% of regressed tumours) (Fig. 2). These observations were attributed to the high amount of lactic acid they produce in addition to the high demand of glucose that could be unsatisfied when they reached a threshold size [60]. The CCL39-gly− (glycolysis negative) mutant deficient in phosphoglucose-isomerase activity, do not produce lactic acid but consume more oxygen than wt cells and 100% of the mice slowly developed tumours 4 to 5 weeks after inoculation. This suggested that lactic acid production via glycolysis is not the only cause of extracellular acidosis within solid tumours [60–62] and that carbonic acid may be highly involved in acidosis of the tumour microenvironment. Moreover, to study the contribution of NHE-1 activity in the context of tumour growth of this ‘metabolically’-mutated cell, double mutants gly− nhe-1− or res− nhe-1− were generated from the individual gly− or res− mutants, respectively [58]. As early as in the 1980s, the authors suggested that the lactic acid produced by the cells was the main cause of cell death when NHE-1 was mutated [58]. To validate this hypothesis, the double mutant res− nhe-1− never gave rise to tumours since the large amount of lactic acid produced by this mutant could not be regulated by NHE-1 (Fig. 2). As a control for no lactic acid production during tumour development, the double mutation gly− nhe-1− gave 100% tumour-bearing mice meaning that the function of NHE-1 is not necessary for cells producing almost no lactic acid (Fig. 2). These striking and interesting results proved that the pHi-regulating system NHE-1 plays a key role in in vivo growth and validate the hypothesis that targeting the pHi-regulating system of highly glycolytic cells may be a novel and efficacious anti-tumour strategy since tumour cells maintain their energy demand by shifting their glucose metabolism from OXPHOS to aerobic glycolysis or anaerobic glycolysis (due to oncogene activation, loss of tumour suppressor genes, or hypoxia itself).
Fig 1.
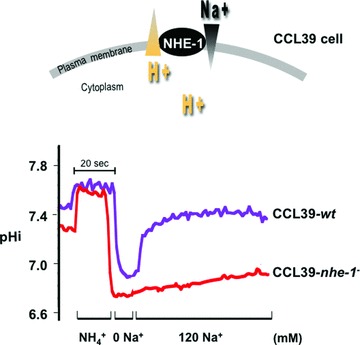
pHi recovery after an acid-load (ammonium pre-pulse) in CCL39 wild-type (CCL39-wt) or mutant cells lacking NHE-1 (CCL39-nhe-1−). Exponentially growing non-neoplastic Chinese hamster lung fibroblast CCL39 cells seeded on glass cover slips were incubated for 30 min. in glucose/saline solution bicarbonate-free, HEPES-buffered solution adjusted to pHo 7.4. The pH-sensitive fluorescent dye 2′,7′ bis (carboxyethyl)-5-(6)-carboxyfluorescein (BCECF-AM) was then added to the cells for 5 min. before transfer of cells to a laminar flow cell chamber perfused with the same solution. Cells were maintained for 10 sec. and then a NH4Cl solution (NH4+) that causes alkalinization (ammonium pre-pulse) was added. The cells were shifted to a pHo 7.4 Na+/NH4Cl-free solution (0 mM Na+) (containing choline chloride instead of NaCl), which causes intracellular acidification due to extrusion of NH3 while preventing exchanger activity. Re-introduction of Na+ (120 mM Na+) allowed for exchanger activity. Ratiometric measurement of the fluorescence of 50 randomly selected individual cells per cover slip was performed in a workstation (Acquacosmos). The pHi was estimated by in situ two-point calibration (pHo 6.6 to 7.6) with perfusion of glucose/saline KCl/nigericin containing solutions to measure the pHi as a function of the fluorescence ratio.
Fig 2.
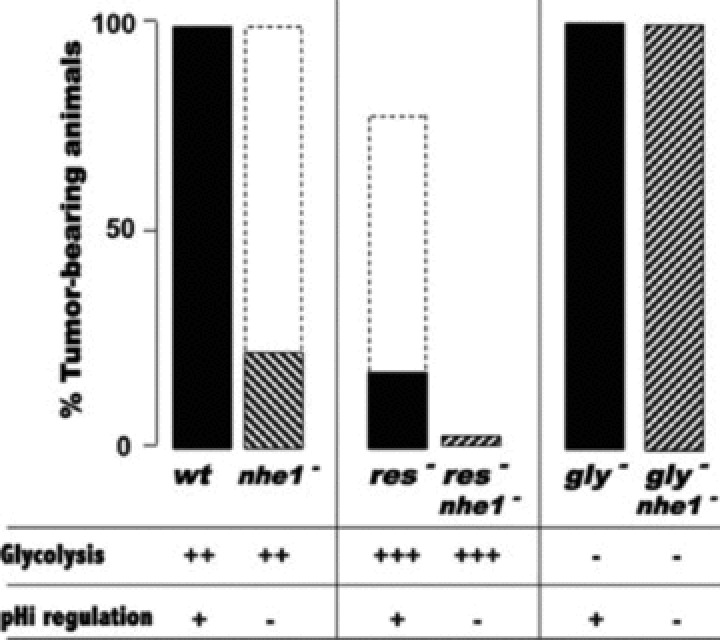
Role of NHE-1 in the control of energy metabolism, pH regulation and nude mice tumorigenicity. Tumour incidence in nude mice of the hamster lung cell lines derived from CCL39 cells. Parental hamster lung fibroblasts (wt), mutant cells impaired in respiration (res−), mutant cells impaired in glycolysis due to a lack of phosphoglucose isomerase activity (gly−), mutant cells lacking NHE-1 activity (nhe-1−) and mutant cells lacking both functions res− nhe-1− and gly−nhe-1−, were transformed with the H-ras oncogene and inoculated (1 × 106 cells/mice) by subcutaneous injection into athymic nude mice. The combination of a defect in pHi regulation through mutation of nhe-1 (nhe-1−) or of high glycolysis (res−) resulted in transient tumour take (percentage of tumour bearing mice represented by dotted lines) but which regressed substantially. The full line shows the final tumour take. Combination of exacerbated glycolysis and a defect in pHi regulation through nhe-1 mutation (res− nhe-1−) resulted rarely in tumour take.
In previous studies, all the in vitro experiments performed to highlight the role of NHE-1 in pHi regulation were monitored in the absence of extracellular bicarbonate to limit the involvement of anion exchangers. The existence of additional pHi-regulating mechanisms, such as the Na+-dependent HCO3−/Cl− exchanger have been identified [63]. In a bicarbonate-buffered medium, the growth and pHi of fibroblasts was not affected by the inhibition of NHE-1 (methyl, ethyl-amiloride analogues), suggesting the implication of HCO3− transporters. In a HCO3− containing medium, the Na+-dependent HCO3−/Cl− exchanger was shown to have two essential physiological functions in cells lacking NHE-1: (i) it protects the cells against excessive cytoplasmic acidification and (ii) establishes a steady-state pHi that is permissive for growth, in a neutral and slightly acidic pHo environment.
These studies have shown that: (i) unchecked regulation of the pHi interrupts many cellular enzyme activities and functions such as energy production, protein synthesis, cell cycle entry, and DNA damage repair, which will ultimately impair tumour growth, (ii) NHE-1 is a key pHi-regulating system in mammalian cells since no other system was able to recover in the absence of NHE-1 in CCL39-nhe-1− cells after an acid-load (Fig. 1) and its mutation has a notable effect on tumour growth evolution and tumour regression. Thus, the challenge is to target pHi-regulating systems, particularly in the context of tumour growth. This requires investigation of the function of the different plasma membrane systems related to pH homeostatsis in tumour cell lines. Once the main tumour pHi regulators are identified, their inhibition would lead to a decline in ATP formation as a consequence of lactic acid accumulation, thus causing metabolic deficiency and cell death. Many studies have been performed in the last 20 years with the aim of understanding tumour pH regulation, metabolic reprogramming and aggressiveness, which results from oncogene activation and hypoxia, and of identifying the key targets involved in metabolic adaptation.
Oncogene activation and transformation cause acidosis
Hyper activation of oncogenes and inhibition of tumour-suppressor genes are considered to be essential in the initiation of tumorigenesis [64]. A strong correlation has now been established between such genetic transformation, glucose addiction, ATP maintenance and lactic acid production in an aerobic environment.
Warburg effect (aerobic glycolysis)
In the 1920s, the pioneering investigations of the Nobel Prize winner Otto Warburg revealed that in contrast to normal differentiated cells, which rely primarily on mitochondrial OXPHOS to generate ATP, tumour cells have a high rate of glycolysis in conditions of high oxygen tension (aerobic glycolysis) [65]. This phenomenon also called the ‘Warburg effect’, gives rise to increased lactic acid production and glucose uptake, which are now known indicators of tumour aggressiveness and poor patient prognosis [66]. Based on the observation of ‘glucose addiction’ of tumour cells, positron emission tomography (PET) has been developed to measure the in vivo tumour glucose capture with a non-metabolizable analogue of glucose, the fluorine-18 deoxyglucose (FDG) and to clinically characterize patients’ tumours and metastases. An example of an image obtained with this clinical procedure is illustrated in a patient with lymphoma in a review by Gatenby and Gillies [6]. However, the ‘Warburg effect’ is not applicable to all cancers [67]. The contradiction resides in the fact that the catabolism of glucose to lactate generates only two ATP molecules per molecule of glucose consumed, whereas complete OXPHOS leads to 36 ATP molecules per glucose molecule. The challenge was to understand why proliferating cells metabolize glucose to lactate through aerobic glycolysis that causes acidosis, rather than more efficient ATP production through mitochondrial OXPHOS. Although aerobic glycolysis is an inefficient way of synthesizing ATP it is the quickest way of producing ATP. It is suggested that cancer cells have increased metabolic autonomy leading to extracellular acidification of the tumour microenvironment, and the acquisition of a more invasive phenotype [68]. More than 60 years later, Warburg’s observations have been resurrected. Although complex, key aspects and related signalling pathways have been elucidated with the aim of linking the Warburg theory to mechanisms of metabolic reprogramming of cancer cells. Warburg thought that a defect in mitochondrial respiration was the cause of the shift to glycolytic metabolism [65]. However, more recently, Thompson and colleagues, and other groups have not linked the Warburg effect directly to a mitochondrial defect, but to oncogenic activation and mutation in the signalling pathways that regulate glucose uptake [69]. Together, the activity of the phosphatidylinositol 3-kinase (PI3K), the serine/threonine Akt and the mammalian target of rapamycin pathway (mTOR) (PI3K/Akt/mTOR pathway) and the effects of the transcriptional activation of c-Myc or of HIF-1 participate in the glycolytic metabolic shift.
Inhibition of tumour suppressor genes and oncogene activation drive the ‘Warburg effect’ and cause acidosis
The tumour suppressor p53 has been shown to decrease the activity of glycolytic enzymes such as phosphoglycerate mutase [70] and to inhibit glycolysis through the induction of TIGAR, a regulatory protein for p53-induced glycolysis and apoptosis that modulates the levels of fructose-2, 6-bisphosphate [71]. p53 also directly regulates the expression and activity of synthesis of cytochrome c oxidase 2 (SCO2) that is required for the formation of the mitochondrial cytochrome c oxidase complex (COX), which is involved in the electron transport chain of mitochondrial respiration [72]. However, a variety of tumours often mutate or lose p53 to ensure their replicative potential. Thus, p53-deficient tumour cells can directly contribute to the ‘Warburg effect’ through aerobic glycolytic compensation accompanied by increased lactic acid production.
The binding of growth factors to their receptors leads to activation of the PI3K/Akt/mTOR pathway. In some cancers, this pathway is constitutively active due to PI3K constitutively active mutations or inactivation of the tumour suppressors phosphatase and tensin homologue deleted on chromosome ten (PTEN) and tuberous sclerosis complex (TSC1 or TSC2). Activation of the PI3K/AKT/mTOR pathway increases the expression of nutrient transporters (glucose, amino acids) at the cell surface [73]. Akt activation increases glycolysis through increased expression of HIF-1α, as discussed later, leading to increased expression and activity of glycolytic enzymes [10]. Thus, Akt activation is sufficient to explain in part the ‘Warburg effect’[69, 74, 75] that leads to acidosis of the tumour environment. Other oncogenes such as active Ras can increase glycolysis by activating PI3K and promoting accumulation of the α subunit of the transcription factor HIF-1 and lactic acid production, as discussed below [76]. The oncogenic transcription factor Myc has been proposed to regulate the metabolic activity required for G1/S transition. Indeed, Myc increases expression of many metabolic and glycolytic enzymes, such as LDH-A as well as several enzymes required for nucleotides synthesis and ATP production [77, 78]. Thus, oncogenic transformed cells become addicted to glycolysis for ATP production. Myc not only regulates glucose metabolism but also activates glutaminolysis, as demonstrated recently [79]. Glutamine plays a key role in cell viability and proliferation [80] and some tumours of patients have been reported to be ‘glutamine addicted’[81, 82]. Thus, Thompson and colleagues have shown that Myc is also required in order to ensure glutamine uptake even in the presence of glucose [79]. Myc expression leads to an increase in the expression of glutamine transporters, glutaminase and LDH-A. This intermediate metabolism regulated by Myc is required to maintain mitochondrial tricarboxylic acid cycle (TCA) cycle integrity. Glutaminolysis is a source of the electron donors NADPH, an important molecule required to satisfy the energy demands of cell proliferation [83]. Glutaminolysis confers on Myc the ability to be glucose-independent for NADPH production. Finally, the production of a high amount of intracellular lactic acid that is the consequence of aerobic glycolysis, causes acidification of the extracellular milieu due to the action of multiple proton-extruding enzymes and transporters that are regulated by oncogenic transformation, as seen in the next section.
Neoplastic transformation drives intracellular alkalinization and extracellular acidification through the activation and up-regulation of pHi-regulating systems
There are two biochemical phenomena common to all tumour cells: the shift to glycolytic metabolism and cellular alkalinization in spite of the large amount of intracellular produced lactic acid. Intracellular alkalinization results from the activity of membrane-bound transporters. Expression of oncogenic c-H-Ras leads to cytoplasmic alkalinization by activating NHE-1 and Na+-dependent HCO3−/Cl− exchangers even when in the presence of extracellular bicarbonate [84]. The importance of pHi regulation in cellular transformation has been shown by ectopic expression of the plasma membrane V-ATPase in fibroblasts. V-ATPase expression led to tumorigenic transformation and intracellular alkalinization [85, 86]. To better understand the link between pHi control and the induction of cellular events that lead to malignant transformation, recent studies have focused on NIH3T3 cells and human HPKIA keratinocytes, which express the HPV16 E7 oncoprotein [87]. For both cell lines, E7 transformation induced cytoplasmic alkalinization driven by stimulation of NHE-1 expression and activity via an increase in the affinity of the intracellular NHE-1 proton regulatory site [87]. These authors concluded that this step is an early and essential physiological event that is necessary for the development of other transformed phenotypes and for in vivo tumour formation in nude mice. The mechanism, by which HPV16 E7-dependent transformation activates NHE-1 in pHi regulation and thus transformation, has been elucidated recently. Activation of NHE-1 by the E7 oncogene is driven by PKA-dependent phosphorylation of RhoA, a small G-protein, and leads to inhibition of p38 MAPK and a cellular rise in cAMP [88].
Hypoxia promotes acidosis by shifting from oxidative phosphorylation to glycolytic metabolism
As tumour cells develop, the supporting vasculature is often limiting, disorganized and not functional, which leads to insufficient irrigation of tissues and thus to a deficient supply in oxygen (hypoxia) and nutrients. There are two factors that are always strongly associated with malignancy, no matter what the oncogene mutation present: acidic pH and lack of oxygen. One of the principal mechanisms driving the tumour glycolytic shift in hypoxia (low oxygen availability) and the resulting acidosis is increased transcription. Stabilization of HIF-1α and subsequent dimerization with HIF-1β leads to transcriptional activation of an arsenal of genes involved in oxygen sensing, decreased respiration, erythropoiesis, angiogenesis, glycolytic metabolism, pHi and pHo regulation, autophagy (cell survival), migration and invasion [11] (Fig. 3). Thus dynamic cycles of hypoxia/reoxygenation, i.e. of respectively survival/proliferation states make tumour cells more aggressive [89]. Moreover, in the context of solid tumour progression, hypoxia is associated with bad prognosis and resistance to radio- and chemo-therapy [11].
Fig 3.
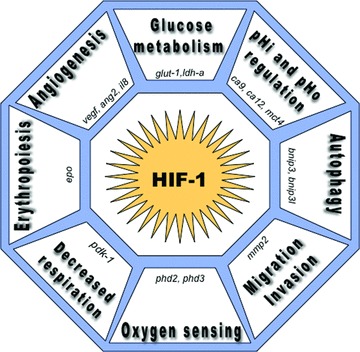
HIF-1 mediates microenvironmental changes and cellular adaptation to tumour hypoxia. After translocation to the nucleus, dimerization of HIF-1α with the constitutive HIF-1β, forms an active transcription factor that only exists in a low oxygen tension (hypoxia). Heterodimerization and DNA binding involve interaction between the N-terminal basic-helix-loop-helix (bHLH) and PerArntSim domains. HIF-1 binds hypoxia responsive elements (HRE) in target genes and activates the transcription of genes enhancing adaptation and cell survival in a hypoxic environment. These target genes play key roles in oxygen sensing via the prolyl hydroxylases 2 and 3 (phd2, phd3), in decreased mitochondrial respiration through the induction of pdk1, in erythropoiesis (epo), in angiogeneisis via the expression of vascular endothelial growth factor (vegf), angiopoietin 2 (ang2), interleukin 8 (il8), in increased glycolytic enzymes as for example glucose transporters (glut-1), lactate dehydrogenase-a (ldh-a), in pHi and pHo regulation with HIF-1 induction of the carbonic anhydrase 9 and 12 (ca9, ca12) and the mct4, in autophagy triggered by Bcl-2/adenovirus EIB 19-kD interacting protein 3 (bnip3) and bnip3l, and in migration/invasion mediated by HIF induction of matrix metalloproteinase 2 (mmp2).
HIF mediates cellular adaptation to low oxygen availability
Hypoxia is not only a pathological event (ischemia, atherosclerosis, psoriasis, diabetes, Alzheimer’s disease, inflammatory disorders and cancer) [90] but also occurs in physiological situations during embryonic development and is mediated by the activation of HIF. HIF is a heterodimeric complex that is composed of an oxygen-dependent α-subunit and a constitutively expressed non oxygen-dependent β-subunit. Three isoforms of the α-subunit (HIF-1α, HIF-2α, HIF-3α) and two isoforms of the β-subunit have been described to contribute to the in vivo response to hypoxia [91, 92]. The transcriptional activation of HIF requires the post-translational stabilization of the α-subunit. This means that the oxygen concentration does not impact on hif-α mRNA levels, but on HIF-α protein post-translational stability. Under normoxic conditions (>5% O2), the half-life of HIF-α is less than five minutes [93]. The rapid post-translational degradation of HIF-α involves two key molecules, oxygen and a Krebs cycle intermediate 2-oxoglutarate (2-OG), which are the substrates of the three major oxygen sensors of the Fe(II)-dependent dioxygenases family also called the prolyl hydroxylase domain (PHD) proteins [94]. Hydroxylation by PHDs on two proline residues Pro402 and Pro564 in the oxygen-dependent degradation domain (ODDD) of human HIF-1α leads to interaction with the ubiquitin E3-containing ligase von Hippel-Lindau (VHL), which attaches polyubiquitin chains to HIF-1α that initiates its recognition and degradation by the proteasome [95]. In case a pool of HIF-α escapes PHD hydroxylation and proteasomal degradation, another oxygen sensor termed factor inhibiting HIF (FIH) maintains HIF-α in an inactive state. FIH is a dioxygenase, which catalyses asparaginyl hydroxylation (Asn803 in HIF-1α) in the C-terminal transcriptional activation domain (C-TAD) of HIF-1α and HIF-2α[96]. This post-translational modification impairs the interaction between the C-TAD and the coactivator p300/CREB binding protein (CBP) [97, 98]. Other post-translational modifications of HIF-1α can occur and might act to modify the stability and the activity of HIF. These include phosphorylation, SUMOylation and S-nitrosylation, as extensively reviewed [99, 100]. As the oxygen level decreases, which establishes a hypoxic gradient, the half-life of HIF-α increases (60 min.), since the oxygen-dependent enzymes PHDs and FIH progressively become inactive [101]. The HIF-α protein is consequently stabilized and translocates into the nucleus as a result of the presence of a nuclear localization signal in its C-terminal region [102]. Once in the nucleus, HIF-α heterodimerizes with HIF-β and HIF binds to hypoxia response elements (HRE) in target genes (Fig. 3). HIF transcriptional activity is not only enhanced in response to hypoxic stress but also as a result of oncogene activation, including that of Ras, Raf, PI3K, Akt, mTOR, Myc or the loss of tumour suppressor genes such as p53, ING4, PTEN and VHL [11, 103–105]. Via its two TAD located in the N-terminal (N-TAD) and C-terminal (C-TAD) regions, HIF controls even as much as 5% of the human genome [106]. Through induction or repression of genes and corresponding proteins, HIF assists hypoxic cells in surviving and adapting to a low oxygen environment [11, 107]. HIF-induced genes are involved in oxygen sensing (phd2, phd3), reduced respiration (pdk1), erythropoiesis (epo), angiogenesis (e.g. vegf, ang2, il8), glycolytic metabolism (e.g. glut1, hk2, pgk1, ldh-a), pH homeostasis (e.g. ca9, ca12, mct4), autophagy (e.g. bnip3, bnip3l), migration (e.g. mmp2), among others [11] (Fig. 3). Thereby HIF reinforces tumour growth and promotes metastasis in a drastic acidic environment deprived of oxygen and nutrients. HIF-1α and HIF-2α may have either distinct target genes or overlap in the activation of some genes [108, 109]. Little is known about HIF-3α, which can act as a dominant negative of HIF transcriptional activity [110].
HIF-induced metabolic reprogramming in response to tumour hypoxia causes acidosis
Since mitochondria consume the majority of the cell’s oxygen during OXPHOS, which leads to production of high levels of ATP, they have been proposed as candidates in cellular oxygen sensing [111]. Mitochondria also produce damaging reactive oxygen species (ROS), such as H2O2 and superoxide anions, and some studies have shown an increase in ROS production in hypoxia at the electron transport chain with subsequent stabilization of HIF-α[111]. However, the mechanism of stabilization remains controversial [112] and may implicate inactivation of the PHD activity by ROS [113] or TCA intermediates [114]. Little information is available concerning the impact of ROS on pH homeostasis. ROS production by articular chondrocytes, which are normally exposed to relatively low levels of oxygen due to the lack of contact with the vasculature, was reported to be decreased in hypoxia possibly by depolarization of the mitochondrial membrane potential. In addi tion, less ROS correlated with reduced activity of NHE and the inability of the cells to regulate their pHi [115]. However, studies into pulmonary arterial myocytes showed that the expression and activity of NHE-1 was enhanced in hypoxia [116], but no implication of ROS was investigated.
In most cases, in hypoxia, HIF-1 compromises oxygen consuming OXPHOS by inducing the expression of pyruvate dehydrogenase kinase-1 (PDK1), which inhibits mitochondrial pyruvate dehydrogenase (PDH), the enzyme that converts pyruvate into acetyl-CoA. In non-hypoxic tissues, acetyl-CoA enters the TCA cycle to produce NADPH and 5,10-methylenetetrahydrofolate reductase (FADH2), which are essential for mitochondrial respiration [117, 118]. HIF-1 also impacts on OXPHOS by transcriptionally activating the cytochrome c oxidase (COX) isoform COX4–2 and the mitochondrial protease LON that degrades another isoform COX4–1, which optimizes mitochondrial respiration in a low oxygen environment [119]. Thus in hypoxia, COX4–2, which is more efficient than COX4–1 in oxygen utilization, allows hypoxic cells to exploit more efficiently low available oxygen levels and avoid production of ROS. However, as mentioned above, since global OXPHOS is reduced, hypoxic cells are forced to enhance anaerobic glycolysis in order to maintain the production of ATP for cell survival. HIF-1 increases the expression of glucose transporters (GLUT-1 and GLUT-3) to increase glucose capture and induces the expression of down-stream glycolytic enzymes such as HK2, phosphoglycerate kinase (PGK1), LDH-A [11, 120] (Fig. 4). In addition, oncogenes such as Myc and the activation of the PI3K pathway also contribute to HIF-1α accumulation and thus promote aerobic glycolysis by induction of HK2, LDH-A and PDK1 [121]. HIF-1-mediated metabolic reprogramming redirects pyruvate to the cytosolic enzyme LDH-A, a HIF-1-targeted gene that catalyses the conversion of pyruvate into lactate [122]. The knockdown of LDH-A suppresses lactic acid production under both normoxic and hypoxic conditions and stimulates mitochondrial respiration, which strongly compromises in vitro cell proliferation in hypoxia. The reduction of pyruvate into lactate by LDH-A, in the presence of NADH, is the key reaction that regenerates oxidized NAD+ required to ensure continuous glycolysis and ATP production (Fig. 4). Since only a low level of NAD+ exists in cells it has to be regenerated. Thus, the effect observed in LDH-A-silenced cells is attributed to the inability of cells to regenerate NAD+ and to produce ATP. Mouse breast cancer LDH-A deficient cells orthotopically transplanted into the mammary gland of mice show a substantial delay in tumour take and a reduction in the tumour growth rate [122]. This study reinforced the idea that manipulating hypoxic tumour metabolism by targeting specific HIF-induced glycolytic enzymes could participate in bringing about a metabolic deficiency through an indirect block in ATP production [10, 123]. When HIF-1-induced LDH-A is fully active, pyruvate is converted to lactic acid that tumour cells must dispose of in order to maintain pHi levels and associated biological functions. However, hypoxic acidosis is not only due to increased lactic acid but also to diminished vascular dispersion of CO2[124]. In addition to the ubiquitously expressed pHi-regulating systems such as NHE-1 and MCT1 that export acid, and AEs, and NBCs, that trap intracellular H+ by transporting HCO3−, HIF-1 induces specific genes dedicated to pH homeostasis to ensure a pHi value that is permissive with cell survival and energy maintenance. Hypoxic tumour cells also express the HIF-inducible membrane-associated CAIX and CAXII that catalyse the hydration of extracellular CO2 into H+ and HCO3− ions. Through this simple reaction CAIX and CAXII regulate tumour pH in contributing to acidification of the tumour environment [125, 126] and alkalinization of the cytoplasm [24, 127]. The former action favours invasion and metastasis while the latter maintains cell proliferation and survival. MCT4 is up-regulated in hypoxia and cooperates with the constitutive MCT1 in extruding the huge amount of lactic acid that is generated [22].
Fig 4.
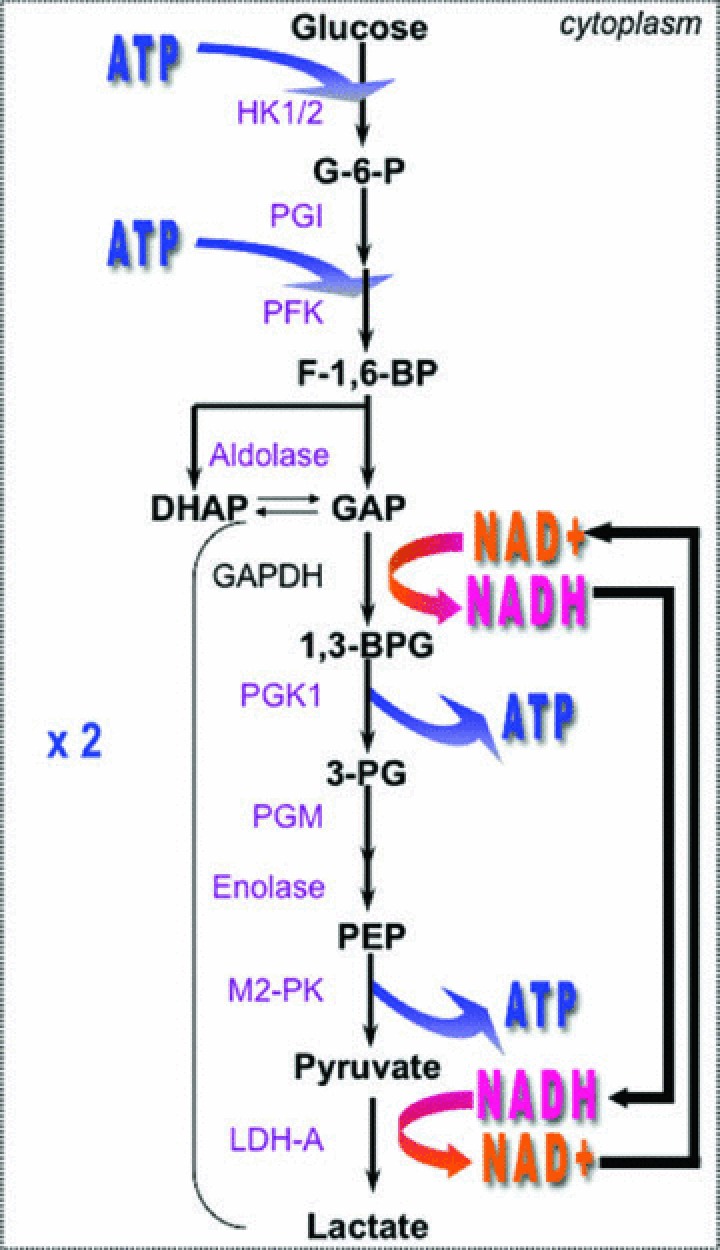
Diagram of the glycolytic pathway. Glycolysis produces two ATP molecules from one glucose molecule consumed. The expression of many of the genes coding for enzymes of the pathway are HIF-1-induced (and oncogenes-induced); including HK1/2, phosphoglucose isomerase, PFK, aldolase, PGK1, phosphoglycerate mutase, enolase, pyruvate kinase (M2-PK) and the lactate dehydrogenase-a (LDH-A). NAD+ recycling is of capital importance for ATP generation. Glyceraldehydes-3-phosphate dehydrogenase (GAPDH).
Acidosis may affect HIF-α stabilization and HIF-induced gene regulation
It has been proposed that extracellular acidosis may also modulate HIF-1α stabilization by protecting HIF-1α from proteasomal degradation [128]. In this interesting study, extracellular acidification led to the nuclear relocalization of VHL, a component of the HIF-1α ubiquitin ligase complex. However, the mechanism is still not clear and results from our laboratory do not concord with these findings, as also published recently by another group [129]. Moreover, little is known about how hypoxia-induced acidosis is related to the in vivo adaptive phenotype of cancer cells. An in vitro acidic pHo gave an increase in HIF-1-induced expression of angiogenic interleukin 8 (IL-8) and vascular endothelial growth factor (VEGF) [130–132]. More recently, the stimulation of the production of angiogenic IL-8 by a low pHo has been associated with activation of NF-κB and the p38 MAPK pathways [133], which are known, respectively, to enhance transcription and translation of HIF-1α[134, 135]. SNAIL, a HIF-1 target gene that is implicated in metastasis and epithelial–mesenchymal transition (EMT), was overexpressed in tumours of mice injected with melanoma cells first exposed to acidosis and then transferred to a neutral environment before injection [136, 137]. This suggests that many of the changes linked to hypoxia are also triggered by acidosis.
Hypoxia enhances the expression and activity of pHi-regulating systems to promote cell survival and invasion
Despite a low pHo, the pHi of tumour cells is maintained around neutral values or even slightly more alkaline. This has been reported to result from HIF-mediated up-regulation of plasma membrane transporters, exchangers, pumps and enzymes that are involved in pH homeostasis.
Hypoxia increases NHE-1 expression and activity
Exposure of pulmonary arterial smooth muscle cells to chronic hypoxia was shown to increase the pHi compared to that in normoxia [138]. This result was obtained in the presence of extracellular bicarbonate, but the effect was more obvious in HEPES-buffered bicarbonate-free media. Chronic hypoxia-induced intracellular alkalinization correlated with an increase in the H+ efflux, as a result of nhe-1 up-regulation at both the mRNA and protein levels, and with enhanced NHE-1 activity, while expression of nhe-2 and nhe-3 was not modified in chronic hypoxia. A more recent study, suggested that the promoter region of the nhe-1 gene might have a candidate binding site for HIF-1α[116], but full characterization of the HRE is still missing. Based on the previous study, the authors tested, in pulmonary arterial smooth muscle cells, the hypothesis that hypoxic induction of nhe-1 and a consequential pHi increase, are mediated by HIF-1. hif-1α+/+ and hif-1α+/− mice were exposed to a normoxic or chronic hypoxic environment. The chronic hypoxia-induced increase in the pHi was reduced in pulmonary arterial smooth muscle cells isolated from hif-1α+/− mice, which are partially deficient in HIF-1α, compared to hif-1α+/+ cells. This result correlated with inhibition of chronic hypoxia-induced nhe1 expression (mRNA and protein) and thus with a reduction in the NHE-1 activity in cells of hif-1α+/− mice. Moreover, overexpression of HIF-1α in pulmonary arterial smooth muscle cells in normoxia resulted in an increase in NHE-1 protein expression and activity, which was sufficient to increase the basal pHi. However, a direct correlation between HIF-1α and NHE-1 transcription is still missing and requires further investigation. Results from our laboratory did not detect hypoxic induction of nhe-1, neither at the mRNA nor at the protein level in a variety of human tumour cell lines (data not published). However, Gatenby and colleagues showed higher NHE-1 expression in central hypoxic regions of ductal carcinoma in situ[139]. Yet the NHE-1 activity was enhanced in hypoxia, since hypoxia generates a high amount of acid, which is known to activate NHE-1. Many studies have also clearly reported the impact of the NHE-1 activity on tumour cell migration and invasion in human breast carcinoma [140], and human melanoma cells [141, 142]. However, a strict demonstration of an increase in expression of NHE-1 in vitro in human tumour cell lines has to be clarified. In addition, NHE-6, a protein localized in subcellular organelles was reported to be regulated by hypoxia in renal cell carcinoma (RCC) [118]. However, using the same cell line and the same experimental conditions we were unable to reproduce this data (data not shown).
The hypoxia-induced membrane-associated carbonic anhydrases are key enzymes involved in pH homeostasis, cell survival and migration in a hypoxic/acidic microenvironment
The CAs are a family of zinc metalloenzymes that reversibly catalyse the hydration of cell-generated CO2 to H+ and HCO3− ions [143] (Fig. 5). Thirteen active and three inactive isoforms of CAs are expressed in mammalian cells. The active isoforms contain a conserved active site and variable levels of expression and activity. They differ in their tissue distribution and cellular localization. CA isoforms such as CAIV, CAIX, CAXII, CAXIV are plasma-membrane localized and have been identified to possess an extracellular catalytic site (Fig. 5). The mature CAIX protein is a unique member among the CAs since it contains a proteoglycan-like (PG) domain, a CA domain (CA), a small transmembrane domain (TM) and a short intracellular C-terminal tail (IC) [144]. In contrast CAXII and the other CAs as the cytosolic CAII lack the PG-like domain (Fig. 5). In multiple epithelial tumour types, only the expression of the membrane-associated CAIX and CAXII is controlled by oxygen levels through a HIF-1-mediated mechanism and only the two hypoxia-induced CAs, CAIX and CAXII, have been proposed to contribute to tumour pH homeostasis based on the catalytic reaction [23, 145].
Fig 5.
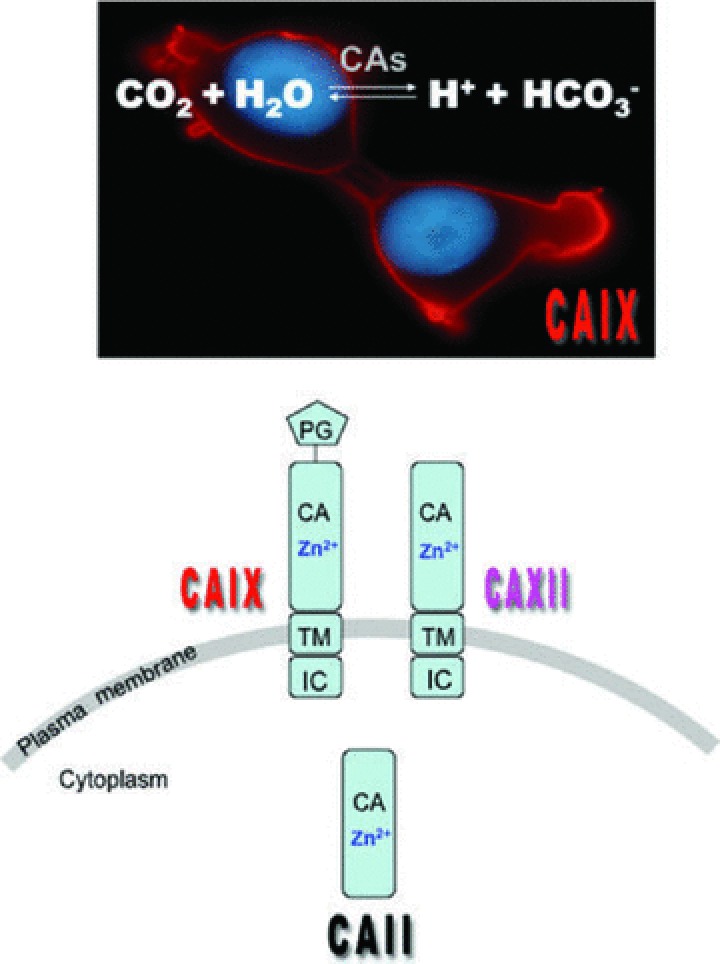
The carbonic anhydrase catalytic reaction, localization and domain structure CAs catalyse the reversible hydration of CO2 to H+ and HCO3−. CAIX and CAXII are cell membrane located, as demonstrated by immunofluorescence of CAIX-expressing cells, in contrast to the cytosolic distribution of CAII. Domain organization of the membrane-bound hypoxia inducible CAIX and CAXII, and the constitutive cytosolic CAII: PG = proteoglycan-like domain, CA = catalytic domain, TM = transmembrane domain, IC = intracellular C-terminal tail.
CAIX regulation and expression
The predominant full-length carbonic anhydrase 9 (ca9) was cloned in HeLa cells, in the 1990s [144, 146], before the discovery that it was one of the most highly hypoxia-induced genes [11]. In addition, recent RT-PCR detection has revealed an alternative spliced variant, lacking exons 8/9 among 11 exons, which is constitutively expressed (that does not depend on hypoxia) and has no enzymatic activity [147]. Chromatin immunoprecipitation assays have shown that both HIF-1α and HIF-2α can bind to the HRE of the full-length ca9[148]. However, ca9 has been identified as a specific HIF-1-induced gene [107, 145], while it fails to be expressed in cells in which hypoxia is not mediated by HIF-1 [107] or when the ca9 promoter is methylated [149]. The mechanism of such a level of selectivity remains unclear, but it was suggested that the ca9 promoter is the most sensitive sensor of HIF-1 activity [150]. In addition to the HRE that allows regulation of ca9 by HIF-1 the ca9 promoter contains five important regions PR1-PR5 that play a role in transcription of ca9 via the binding of SP1/3 transcription factors [151]. Moreover, the C-TAD of HIF-1 regulates preferentially the transcription of ca9[107]. The inhibition of tumour suppressors and the activation of oncogenes that participate in HIF-1α accumulation in non-hypoxic environments, may also contribute to ca9 up-regulation. Most RCCs have a mutation in the tumour suppressor vhl. Non-functional VHL is unable to polyubiquitylate HIF-1α and thus, HIF-1α is no longer recognized by the proteasomal degradation system. This leads to constitutive activation of HIF-1 even in an oxygenated environment [152]. The consequence of vhl inactivation is high and uniform expression of CAIX, and of other HIF-1-induced genes [145, 153, 154]. Mutation in vhl concerns, so far, RCCs and tumours of the retina. While mutation in the tumour suppressor p53 had no effect on ca9 expression, activation of p53 by genotoxic stress (DNA damage) increases the degradation of HIF-1α and thus leads to down-regulation of hypoxia-inducible ca9 expression [155]. Oncogenic pathways also contribute to ca9 regulation. Inhibitors of the ERK pathway down-regulate CAIX expression; however, in HeLa cells, CAIX expression and ERK activity were inversely correlated [156, 157]. The MAPK activity may influence ca9 transcription by an increase in HIF-1 activity indirectly through p300/CBP [158] or SP1/SP3 [157]. Cell density-dependent transcriptional activation of ca9 requires separate but interdependent pathways requiring cooperation of SP1 and involving activation of the PI3K pathway, and minimal stabilization of HIF-1α[159, 160]. However, constitutive activation of the PI3K pathway increases HIF-1α but decreases ca9 expression at the mRNA and protein level [105]. These data underline the existence of other potential pathways involved in HIF-independent regulation of ca9. More recently, a convincing study revealed that activation of the unfolded protein response (UPR) by hypoxia cooperates with HIF for hypoxic activation of ca9 transcription [161]. In severe hypoxia, UPR activation leads to a reduction in overall protein synthesis. However, under this condition, the endoplasmic reticulum kinase PERK, a component of the UPR, phosphorylates the eukaryotic translation initiation factor eiF2α that activates mRNA translation of the transcription factor ATF4 [161]. Thus, ATF4 was found to bind directly to the ca9 promoter and to be necessary for ca9 induction in hypoxia. Mutation in eiF2α decreased CAIX expression at the mRNA and protein level without affecting the HIF-1 activity and ca9 mRNA stability. The regulation of ca9 by others environmental factors such as acidosis, glucose deprivation or bicarbonate depletion has also been reported. Controversial data have shown that acidification of the culture media increases the expression of CAIX (mRNA and protein) in glioblastoma cells, due to the stabilization of HIF-1α and activation of the ERK pathway under acidic conditions [162]. However, acidosis or glucose deprivation did not induce ca9 expression in normoxic HeLa cells [163], while the combination of hypoxia and low glucose or low bicarbonate in the media, moderately enhanced hypoxia-induced CAIX expression [163]. The clear demonstration that microenvironmental factors may have an effect on CAIX expression via a HIF-independent mechanism has not been demonstrated since the activation of ca9 occurs with the concurrent activation of HIF-1.
Under physiological conditions, the expression of CAIX is essentially restricted to a few tissues including the epithelium of the stomach (gastric mucosa), biliary tree, crypt cells of the duodenum, and the epithelium of the small intestine [23, 164]. A strong signal for ca9 mRNA was also detected by RT-PCR in the kidney and skeletal muscle of mice, while immunoblotting and immunohistochemical analysis showed no signal for the protein, which suggests tissue-specific post-translational control of CAIX expression, but is yet to be elucidated [165]. CAIX is expressed during embryonic development as shown in the mouse [166] but is more highly expressed in many cancers such as oligodendroglial brain tumours [167], colorectal cancer [168], ovarian tumours [169], gastric cancer [170], pancreatic cancer [171] and breast cancer associated with c-erbB2 overexpression [172]. In most cases, CAIX expression correlates with the pattern of HIF-1α expression in cancers in hypoxic/perinecrotic regions of the tumour, distant from the blood vessel (Fig. 6) and also colocalizes with the hypoxic indicator (hypoxyprobe) stain [24]. However, exceptions have been observed where expression of CAIX is detected in the absence of HIF-1α staining [173]. These observations could be interpreted as the existence of dynamic cycles of acute hypoxia/ rapid reoxygenation within the tumour, and the short half-life of HIF-1α compared to CAIX where the latter is highly stable once induced [163]. The recent demonstration showing that ca9 expression is regulated by the UPR pathway may also explain the absence of HIF-1α and CAIX colocalization [161]. Some tumours also contain hypoxic regions without CAIX expression [174], which could be explained by the lag in HIF target transcriptional activation and protein expression. The requirement of UPR signalling could also explain why HIF-1α+ tumours are negative for CAIX staining [161]. As for HIF-1α, CAIX has been proposed as a marker of an aggressive malignant phenotype of solid tumours, since a strong link between its expression and poor patient survival has been established by many groups [175–182]. Only one case reported that low expression of CAIX in RCC tumours is a factor of bad prognosis [183]. However, the status of HIF within these RCC tumours was not indicated and the presence of HIF-2α instead of HIF-1α may explain this phenomenon [184].
Fig 6.
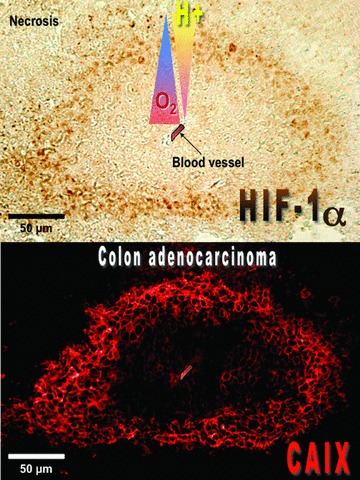
Immunohistochemical analysis of HIF-1α and CAIX expression in mice xenograft tumours. Tumour sections obtained from subcutaneous injection of colon carcinoma LS174Tr cells were stained with an anti-CAIX monocalonal antibody M75 and showed correlation between CAIX expression and regions of HIF-1α+ staining. A relationship between CAIX localization and regions of hypoxia and necrosis, was also noted. Staining of CAIX is predominantly membranous while HIF-1α is localized in the nucleus. Magnification 20×.
CAXII regulation and expression
The cDNA of ca12 was cloned in 1998 from RCC tumours [185]. Its chromosomal localization mapped differently to that of ca9. The ca12 gene is also induced in hypoxia [23, 145], via a HIF-1-dependent mechanism [24]. However, the HRE of ca12 has not been identified. The fold induction of ca12 is lower than that for ca9 because the basal level of ca12 mRNA and protein is higher than for ca9 in tumour cells in normoxia. ca12 mRNA is up-regulated in VHL-defective renal tumours due to constitutive stabilization and activation of HIF-1 [153]. In breast cancer, oestrogen receptor (ER-α) activation leads to transcriptional up-regulation of ca12 via a hormone-responsive enhancer [186]. An alternatively spliced variant of ca12, lacking an exon, was detected in infiltrating astrocytomas [187]. The shorter form that was found to predominate in brain tumours was lacking residues that are located in the transmembrane domain. Thus, this spliced isoform may affect the quaternary structure of dimeric CAXII and signalling pathways involving protein kinase C and A, due to disruption in a potential phosphorylation site of these kinases in the C-terminal tail of CAXII. CAXII is a 39 kD protein with a conserved Zn-binding catalytic site, a transmembrane domain and a short intracellular C-terminal extension containing potential phosphorylation sites for casein II kinase, protein kinase C, and c-AMP-dependent kinase [185]. Under physiological conditions, CAXII expression was detected in human and mouse endometrial epithelia, suggesting a role in reproductive physiology [188, 189]. Unlike CAIX expression, the endogenous expression of CAXII is less tissue specific and is not only restricted to normal epithelia. CAXII is expressed in a variety of normal tissues but its expression becomes stronger in tumours. CAXII expression was found in normal human pancreas, kidney, gut and the gastrointestinal tract [190–193]. Prominent expression of CAXII was observed during embryonic eye development but was significantly decreased in the adult eye [194]. However, in glaucoma (high intraocular pressure), ca12 mRNA was strongly increased in the adult eye, while ca9 mRNA was not detectable. CAXII is highly expressed in gastric tumours compared to weak expression in normal gastric mucosa [195] and its expression is also high in various tumours [190–193]. While tumour expression of HIF-1α and CAIX correlate with poor patient survival, the significance of CAXII is less obvious and the distribution of CAXII is not always associated with CAIX expression [24, 153]. However, CAXII distribution was associated with high malignancy grade and poor prognosis for astrocytic gliomas [187].
The activity and functions of CAIX and CAXII
The two hypoxia-inducible CAIX and CAXII are not highly homologous in their protein sequence (30% sequence homology only), but they share the same catalytic site with the three conserved zinc-binding histidine residues. To date, the cytosolic CAII was reported to have the highest catalytic efficiency with a kcat/Km = 1.5 × 108/M/s [44]. However, recent biochemical characterization of the hypoxia-induced CAIX showed it was the most active carbonic anhydrase isozyme with a surprisingly high CA activity of kcat/Km = 3.5 × 109/M/sec., which was associated with the presence of the PG-like domain of CAIX [196]. However, the mechanism by which the PG-like domain contributes to an increase in the CO2 hydration activity of the CA is still unknown. It was suggested that metal ions, such as Zn2+ might bind to the catalytic site and probably relieve the electrostatic repulsion of the PG-like domain. CAIX was also shown to have a higher activity for CO2 hydration in an acidic environment (around pHo 6.5) compared to in a neutral environment (pHo 7.4) [197]. The activity of CAXII was reported to be moderate compared to that of CAIX [143, 198, 199]. One of the major challenges in the study of HIF-induced CAs is to understand the advantage tumour cells derive from overexpression of enzymes that catalyse an already spontaneous reaction that hydrates CO2 to generate H+ and HCO3−. It is well known that H+, the main acidifier and the couple of CO2/HCO3− (pKa = 6.37), the main pH buffering system, governs body pH homeostasis and the pH of intracellular and extracellular compartments. Thus, since the 1990s the tumour-associated hypoxia-inducible CAs were suggested to be functionally linked to the regulation of the tumour pH, in particular for CAIX. Firstly, CAIX expression was clearly shown to contribute to extracellular acidification (decreased pHo) of tumour cells in hypoxia, in a way that does not depend on lactic acid production [125]. Moreover, this study revealed that the expressed CAIX was functionally active only when cells were exposed to a hypoxic environment [125, 200]. Investigations into the implication of CAIX in the regulation of the pHi have shown to take place in cell monolayers and in three-dimensional cell culture [24, 127, 201]. These studies showed that: (i) CAIX expression in human bladder carcinoma RT112 cells was not able to regulate the pHi of isolated cells in neutral and bicarbonate-buffered medium but contribute to pHi regulation at the core of growing spheroids [127]. In agreement with this study, our laboratory showed that an excess in extracellular bicarbonate (that would saturate bicarbonate transporters at the cell surface) would not allow the study of the contribution of CAIX to pHi regulation in a cell monolayer [24], (ii) when cells were exposed to a nominally bicarbonate-free and acidic milieu (pHo 6.2 to 6.8), CAIX-expressing cells display a more alkaline resting pHi promoting cell viability and proliferation. This demonstrates the role of CAIX on a pHi-threshold value that is permissive with survival and growth of non-tumour (fibroblasts) and tumour cell lines such as adecomarcinoma LS174Tr cells [24]. Moreover, pHi regulation was reversed in the presence of diisothiocyanatostilbene-2′, 2-disulfonic acid, an inhibitor of HCO3− transporters, suggesting that the local production of the weak base HCO3−, which was rapidly generated in the presence of CAIX, was recaptured and capable of counteracting intracellular acidification [24], (iii) in growing spheroids of RT112 cells and HCT116 (colon carcinoma) cells the pHi of cells evolving within the spheroid core was higher when cells overexpressed CAIX compared to control non-expressing cells, while the extracellular environment was more acidic. These effects on pHi and pHo were reversed when the CAIX activity was reduced with non-permeable inhibitors of CA activity [127, 201]. Thus, the authors concluded that tumour cells express CAIX to facilitate CO2 extrusion, thereby reducing the pHo and increasing the pHi and promoting cell survival. The contribution of CAIX expression to pHi was also detected in fibroblasts impaired in NHE-1 expression (nhe-1− mutant), which ensured the absence of interference by this major player in pHi homeostasis. This finding indicates that CAIX is a key pHi-regulating system [24]. The mechanism by which membrane-bound CAIX regulate the pHi occurs through the efficient uptake of HCO3− locally formed in the ‘mouth’ of CAs through Cl−/HCO3− exchangers (AE) and/or Na+/HCO3− co-transporters [202]. Interaction of the catalytic domain of CAIX with bicarbonate transporters has been reported and was shown to increase the AE exchanger activity [202] (Fig. 7). However, further investigations are required to evaluate the key bicarbonate transporters coupled to CAIX (and CAXII) in pHi regulation in hypoxic tumour cells. Both CAIV and CAXIV were shown to play a key role in pHi regulation of neurons by enhancing the expression of AE3-mediated Cl−/HCO3− exchanger [203]. While the role of CAIX in tumour pH homeostasis has been investigated, the contribution of CAXII was not examined until recently. Evidence of a conjugated role of CAIX and XII in tumour pHi regulation and tumour growth has been suggested by investigations in our laboratory. Knockdown of ca9 did not change the rate of catalysis of acidification and pHi regulation in an acidic environment due to compensation by CAXII expression (at the mRNA and protein level) and activity in hypoxia [24]. Even in absence of changes in pH (pHo and pHi), silencing of ca9 reduced spheroid and tumour growth in nude mice [24] as shown in in vitro cell proliferation assays in bicarbonate-containing media [126]. Combined silencing of both membrane-associated hypoxia-inducible CAIX and CAXII led to a reduction in extracellular acidification and pHi regulation, which correlated with a dramatic decrease in the rate of xenograft tumours growth [24]. Both CAIX and XII play a key role in pHi regulation in hypoxia by contributing to removal of metabolically generated CO2, Thus, such regulation is a key event controlling cell viability, and in vivo tumour growth in a hostile acidic and hypoxic microenvironment. Accordingly, simultaneous expression of the hypoxia-induced CAIX and CAXII predicted extremely poor prognosis for patients with astrocytomas tumours, compared to the survival observed in astrocytomas lacking CAIX and CAXII expression [187]. While the knockout of ca12 in mice has not been investigated, the knockout of ca9 was not lethal and resulted in limited phenotypic changes, prominently giving gastric hyperplasia, but no change was observed in gastric secretion or in other tissues known to express CAIX. This indicated that other CA isoforms might compensate for the lack of CAIX [204, 205]. Knockout of ca9 in mice led to up-regulation of ca2 expression (mRNA and protein) in the stomach and inversely, knockout of ca2 was accompanied by an increase in ca9 (mRNA and protein) levels, in the same tissue [206]. Interestingly, we also observed that knockdown of ca9 in colon carcinomas LS174Tr cells led to partial compensation by up-regulation of ca12 (Fig. 8) in vitro and in vivo with a reproducible 1.5-fold induction compared to the level of hypoxic induction of ca12[24] and ca2 (data not shown) at mRNA and protein levels, suggesting that cross talk between the CAs is needed to maintain a threshold level of activity for cell pH homeostasis. The mechanism of such specific cross talk between the CAs remains unclear and is presently in progress. Accordingly, study of normal and malignant lesions of the large intestinal mucosa revealed high expression of the cytosolic CAI and CAII in normal tissue compared to weak expression in colorectal tumours while the opposite pattern of expression was detected for the membrane-associated CAIX and CAXII that exert strong expression in tumours and are barely detectable in normal intestinal mucosa [207]. Thus, not only high levels of CAIX and CAXII in tumours but also decreased expression of CAI and CAII may contribute to progression during malignant transformation. The possible implication of these CAs as signalling molecules, independent of their function as pH-regulating enzymes, is another important point that has not been extensively investigated and may explain the partial decrease in xenograft growth of ca9 silenced cells despite maintenance of the CA catalytic activity due to up-regulation of CAXII. Signalling through the EGF pathway by phosphorylation of a cytoplasmic tyrosine residue of CAIX may either activate CAIX or enhance its expression by increasing translation of HIF-1α[208]. In addition, phosphorylation activates PI3-K resulting in phosphorylation of Akt and cell survival. CAIX expression was shown to interact with β-catenin and thus was suggested to play an important role in the modulation of E-cadherin mediated cell adhesion [209]. When renal carcinoma cells were treated with acetazolamide, an inhibitor of carbonic anhydrases, their capacity to invade was diminished [210]. In addition, CAIX expression was strongly associated with the development of metastasis in patients with cervical tumours, while CAXII expression was associated with less metastasis [211], but the invasion of breast carcinoma cells in vitro was not influenced by CAIX in another study [126].
Fig 7.
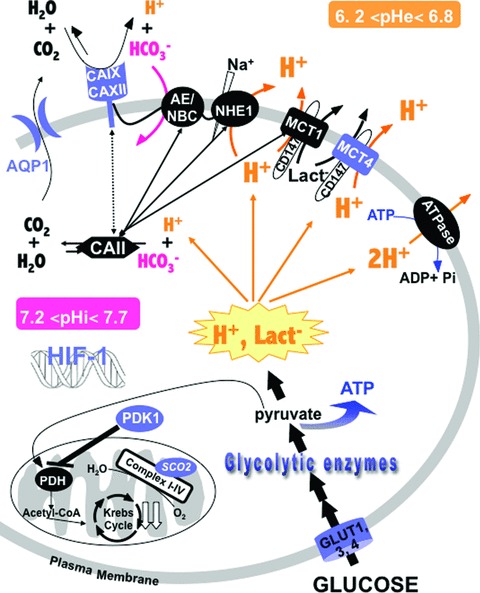
Up-regulation of glycolysis and pHi regulation: hallmarks of invasive cancers. HIF-1 stimulates glycolysis by activating the expression of the glucose transporters GLUT and glycolytic enzymes such as HK2, lactate dehydrogenase A, and PDK1, an inhibitor of PDH, that inhibits mitochondrial uptake of pyruvate. Mutation of p53 blocks the expression of SCO2 that is critical for regulating the COX complex, the major site of mitochondrial oxygen utilization. Despite the huge production of lactate−/H+ (represented in orange), tumour cells maintain the pHi compatible with cell viability by activating their constitutively expressed pHi-regulating system (black symbols) such as the Na+/H+ exchanger-1 (NHE-1), the bicarbonate transporters such as Cl−/HCO3− exchangers (AE), Na+-dependent HCO3− transporter (NBC), the MCT1, the H+-pump (V-ATPase), the cytoplasmic carbonic anhydrase II (CAII) and the HIF-1-induced pHi-regulating systems (blue symbols) such as the MCT4, carbonic anhydrases IX and XII (CAIX and CAXII) and aquaporin 1 (AQP1). Many of these components, which regulate the pHi, may interact to form a ‘metabolon’ that enhances metabolic flux of H+, lactate− and HCO3− (represented by double arrows).
Fig 8.
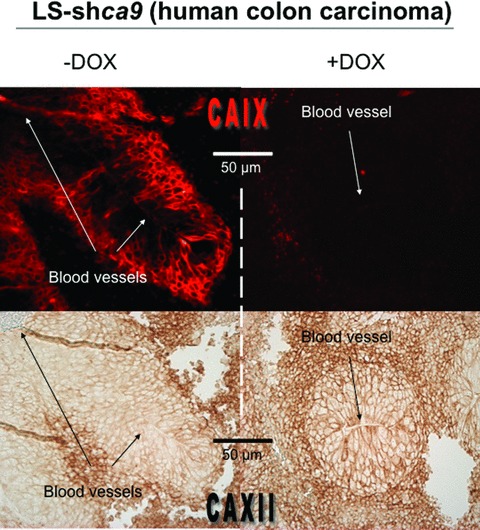
Cross talk between CAIX and CAXII expression. Reduced expression of CAIX by doxycycline (DOX)-induced shRNA interference in LS174Tr human colon carcinoma cells (LS-shca9) leads to up-regulation of CAXII. Co-staining for CAIX (Immunofluorescence) (upper panel) and CAXII (Immunohistochemistry) (lower panel) was observed in mice xenograft tumours of inducible colon carcinoma LS-shca9 cells with (+DOX) or without (–DOX) doxycycline in the drinking water of mice. Magnification 20×.
The hypoxia-induced monocarboxylate transporter MCT4, the constitutively expressed MCT1 and their chaperone CD147 are key plasma-membrane proteins involved in pH regulation, energy balance, tumour progression and metastasis
Among the 14 MCT isoforms, only MCT1, MCT2, MCT3 and MCT4 have been demonstrated to enhance H+-linked transport of metabolic carboxylates such as lactate [212]. In particular, the cell-surface expression of MCT1 and MCT4 which is mediated by the presence of a chaperone CD147 [213], has been associated with tumour aggressiveness [214]. It has been proposed that they are powerful regulators of the tumour pHi by extruding lactic acid produced through the highly glycolytic nature of tumour cells.
MCT regulation, expression, structure and implication of their chaperone CD147
The MCTs have 12 transmembrane domains, intracellular N- and C-termini residues and a large intracellular loop between the transmembrane domains 6 and 7 [212, 215]. These plasma membrane located isoforms differ in their tissue distribution and affinity for lactate [19, 212]. MCT1 is ubiquitously expressed in all tissues whereas MCT2 is essentially located in the kidney and the brain, while MCT3 is found in the retinal pigmented epithelium. Surprisingly, MCT4 is strongly expressed in glycolytic tissues such as skeletal muscles and is induced in hypoxia in most human tumour cell lines through a HIF-1-dependent mechanism via a HRE [22, 216]. Interestingly MCT1 and MCT4 require the expression of the CD147 chaperone for functional expression and lactate transport at the cell surface [213, 217] but CD147 is not a HIF-induced gene. CD147, also named basigin or EMMPRIN is a multifunctional, highly conserved glycoprotein that is associated with poor patient prognosis in renal carcinomas [218], gastric tumours [219] and head and neck cancers [220].
The MCT1, MCT4 and CD147 activity and functions
Expression of each MCT isoform in Xenopus oocytes demonstrated that MCT2 has a considerably higher affinity for lactate (Km = 0.7 mM) than MCT1 (Km = 3–5 mM) while MCT4 showed a much lower affinity (Km = 28 mM) [216, 221, 222]. Because of its low affinity for lactate MCT4 is adapted to extrude of lactate [216]. Moreover, the lactate transport activity through MCT1 was enhanced in the presence of cytosolic CAII in Xenopus oocytes[223, 224]. However, this physical interaction did not require the catalytic activity of CAII. This result suggested that both MCT1 and CAII are important components of the ‘metabolon’, a membrane protein complex that maximizes metabolitic (e.g. H+, lactate, pyruvate, HCO3−) flux and the transport rate to regulate their production [223, 224]. This finding may be of considerable significance in tumour pH-regulation since CAII and AE have been reported previously to form a ‘metabolon’[225, 226] as for CAIX/AE2 [202], and NHE-1/CAII [227, 228], which may interact to enhance the transport of metabolites in hypoxia (Fig. 7). MCT1 was recently proposed to be a crucial component of metabolic symbiosis based on lactate exchange between glycolytic/hypoxic and oxidative tumour cells within solid tumours [229]. The authors demonstrated that oxidative SiHa cells expressed higher levels of mct1 compared to mct4 mRNA and proteins and that MCT1 expression decreased in the hypoxic area of the tumour while MCT4 was induced. Thus, it was proposed that MCT1 captures the lactate produced by hypoxic/glycolytic cells and extruded it by MCT4, to fuel oxidative metabolism in oxygenated tumour cells. Moreover, the inhibition of MCT1 induced a shift from lactate-fuelled respiration to glycolysis in the oxidative part of the tumour making glucose unavailable for hypoxic/glycolytic cells, which die. Thus the authors concluded that specific inhibition of MCT1 revealed a substantial anti-tumour effect. Investigations into the contribution of lactic acid transport in tumour cells and stroma cells have so far concerned MCT1. The intracellular transport of lactate by endothelial cells that express active MCT1 in tumours may also modify the microenvironment through promoting angiogenesis [230, 231]. However, since hypoxic tumour cells induce MCT4, the functionality of MCT4 in a MCT1 background requires further investigation. In spite of the low-affinity of MCT4 for lactate transport compared to MCT1, we hypothesize that the function of MCT4 is of great importance in the context of in vivo metabolism, tumour pHi regulation and growth. Validation of both specific MCT1 and MCT4 as anticancer targets is presently in progress. Co-immunoprecipitation assays have revealed a selective interaction between β1-integrin and MCT4 in epithelial cell lines expressing MCT1 and MCT4. Thus, knockdown of MCT4 reduced cell migration while knockdown of MCT1 did not slow migration [217]. The knockdown of CD147, the ancillary protein required for the functional expression of MCT1 and MCT4 at the plasma membrane, reduced the level of expression of both MCT1 and MCT4, and led to an increase in intracellular lactate [232, 233], and a reduction in the rate of pancreatic tumour cell proliferation both in vitro and in vivo in a xenograft model. Inversely, silencing of MCT4 affected the maturation and trafficking of CD147 to the plasma membrane and decreased migration of a metastatic breast cancer cell line [233]. The synergistic relationship within the MCT/CD147 complex could facilitate the development of anticancer molecules.
Strategies taking advantage of changes in the oxygen level, energy balance and pH homeostasis to target primary tumours and metastases
Subsequent to Warburg’s observation of the up-regulation of glycolysis in tumours and the recognition that glucose addiction of transformed cells is linked to PI3K mutation or Akt activation, several types of inhibitors targeting oncogenes (such as Akt inhibitors) have been developed [234]. 2-deoxy-glucose, a non-metabolizable analogue of glucose that competes with glucose, is also a candidate molecule as an anticancer treatment and is in phase I clinical trials [235, 236]. Several inhibitors blocking glycolytic enzymes have been shown to have potent anti-tumour effects in vivo, as seen for 3-bromopyruvate, which inhibits HK activity, when in combination with cisplatin [237]. AMP, which is known to activate allosterically PFK, could be challenged by the quantity of ATP within the cells [238]. Thus, in hypoxia, the increase in the AMP/ATP ratio leads to allosteric activation of PFK and increased glycolysis. AMP activates the AMP-activated protein kinase (AMPK) that by phosphorylation of key metabolic enzymes reduces ATP consumption and modifies gene expression (GLUT-1 and GLUT-4 for example) [239–241], so AMPK might be a candidate target. Thus, a specific inhibitor of AMPK would act as an inhibitor of glycolytic enzymes and would target many steps of the glycolytic pathway, and thus, would help to increase rapid meta bolic deficiency. Another adenosine nucleotide that plays a crucial role in the hypoxia-sensitivity of glycolysis and the maintenance of ATP production from glycolysis is NADH. The step that regenerates NADH from NAD+ involves glyceraldehyde phosphate dehydrogenase (GAPDH). Interest in targeting GAPDH lies in the fact that it is the only enzyme that recycles NADH and inhibition of its regeneration would have a dramatic impact on ATP production and cell survival, as did the silencing of LDH-A. One of the most distinct hallmarks of cancer cells is the low pHo and the rather high pHi, particularly in hypoxic regions. The recognition of HIF induction of the pHi-regulating system and other proteins involved in key survival processes, has led to the development of HIF inhibitors as anticancer treatments [242–245]. However, this strategy may not be as beneficial as first thought since it affects the pleiotropic action of HIF-1 on pro-survival and pro-death genes and might be better directed to the inhibition of downstream HIF-1-target gene products, as we proposed for HIF-1-induced pHi-regulating systems [10, 24]. Indeed, by clamping their own intracellular acidity through inhibition of key hypoxia-inducible pHi-regulating systems, cells would collapse their energy production and multiple biological functions, thereby precipitating cells of the primary tumour into necrosis. Another novel and potentially efficient drug approach consists in increasing the pHo, to reduce metastatic formation and MDR and to stimulate NK cell activity. Many areas of research have yet to be explored within the context of cellular and microenvironmental pH regulation in cancer. For example little is known about the influence of pH on the emergence of cancer stem cells [246]. Yet a lot of interest has been paid recently to cancer stem cell research [247, 248], although the existence of such cells has been questioned [249]. However, hypoxia has been proposed to influence development of both tissue stem cells and cancer stem cells [250, 251].
Decreasing the pHi of hypoxic cells of the primary tumour by inhibiting key pHi-regulating systems to collapse ATP production
Hypoxic areas that evolve distant from blood vessels are highly resistant to chemotherapeutic agents and radiotherapy, thus a lot of interest is being paid to this part of the tumour and to understanding how hypoxic cells adapt to such a low oxygenated and acidic microenvironment. Thus, to precipitate hypoxic tumour cells into necrosis, targeting a pHi-regulating system in this region to produce a high amount of acid would be a selective and specific therapy. We are convinced that inhibition of acid extrusion is a selective strategy that would only target tumour cells. This specificity resides in the fact that tumour cells have an abnormal proliferation rate and cannot enter the G0 checkpoint. Thus, by poisoning cells with their own acid, the ATP level would drop and tumour cells would rapidly experience necrotic cell death. Several pHi-regulating systems have now been validated and represent potential targets for anticancer therapy. Inhibition of NHE-1 had an effect on in vivo growth of H-ras transformed fibroblasts [58] and on leukemic [216] and breast cancer cells [252]. In addition, long-term treatment of rats with a NHE-1 inhibitor (Cariporide, Sanofi-Aventis, Paris, France) extends their lifespan and reduces tumour take, which is consistent with data observed by Pouyssegur’s group. Inhibition of the recently validated pHi-regulating system CAIX and CAXII [24] represents a strong challenge for chemists and biologists who are now developing specific ‘CA-antagonist’ antibodies, cell impermeable drugs [253] and selective bioreductive sulfonamide inhibitors targeting both CAIX and CAXII [254]. A recent study revealed that treatment with albumin-binding acetazolamide derivatives reduced the rate of xenograft growth of human RCC SK-RC-52 cells [255]. Such inhibitors are being actively investigated at the molecular and cellular levels and may hold promise as effective anticancer treatments [143, 199]. Another approach consists in exploiting the strong expression of CAIX in hypoxic areas of the tumour to induce cell death. A monoclonal antibody (mAbG250) directed toward CAIX has entered phase I [256, 257]. It did not inhibit the function of CAIX but lead to internalization of the protein and induced cell death of hypoxic cells expressing CAIX. CO2, the substrate of carbonic anhydrase is believed to diffuse freely across cell membranes. Information on the water channel proteins aquaporins (AQP) that mediate CO2 and H2O flux has brought new insight into the understanding of cell physiology. More recently, the expression of AQPs such as AQP1 has been shown to correlate with HIF-1α expression within breast cancer tumours and human endothelial retinal cells [258]. In the context of tumour physiology and pH regulation, AQP1 might play a key role by potentially interacting with the cytosolic CAs and the membrane-associated CAIX and CAXII in hypoxia [259] (Fig. 8). AQP1-null mice show a dramatic reduction in the tumour growth rate and a decrease in cell migration and metastatic potential that suggest that AQP1 is a promising anticancer target [260]. To target lactic acid extrusion, and to collapse ATP production through glycolysis, a specific inhibitor of the common MCT1 and MCT4 chaperone CD147/basigin would also hold potential as an effective anticancer treatment [261].
Increasing pHo and the extracellular buffering capacity in targeting metastasis and reducing multidrug resistance
One of the major challenges in cancer therapy is to prevent tumour cells forming metastases. Acidosis of the tumour microenvironment has been reported to favour metastasis. Pre-treatment of tumour cells with acid before in vivo injection has shown an increase in metastases, suggesting that a low pH activates invasive and survival pathways [137, 262]. Extracellular acidosis itself may also explain MDR in a p38 MAPK-dependent manner [29]. Thus, a reduction in tumour extracellular acidification or inhibition of p38 MAPK would reduce MDR and sensitize the tumour to cytotoxic affects of chemotherapeutic agents. Recently, Gatenby and colleagues gave mice NaHCO3 in their drinking water and reported an alkalinized tumour microenvironment and reduce metastatic formation in tumour bearing mice [263]. They reported that NaHCO3 selectively and effectively increased the extracellular tumour pH without affecting the pHi and thus had no effect on the rate of growth of the primary tumours. However, NaHCO3-induced inhibition of external tumour acidity reduced the incidence of in vivo metastases of breast and prostate cancer cells and increased survival. They suggested that inhibition was due to an increase in the bicarbonate level in the tumour, which increased the bicarbonate buffering capacity. However, the mechanism involved is not completely resolved since it is not known whether addition of bicarbonate leads to a decrease in survival of circulating cells or to inhibition of colonization at the metastatic site. The drawback of this therapy concerns the balance between the acid load produced by the tumour and the quantity of bicarbonate that can be administrated. Thus the effectiveness of this therapy will be reduced in large tumours [264]. However, the strong advantage of this strategy would be to restore a more neutral tumour pHo that should slow down the metastatic process but also diminish drug resistance. Expression of the plasma-membrane V-ATPase was shown to be enhanced in multidrug resistant cells (cisplatin), thus, an increase in the pHi, decreases the pHo and avoids the cytotoxicity of cisplatin [265]. The V-ATPase that has a higher activity in an acidic extracellular microenvironment, is involved in invasion and metastasis. Inhibition of its activity through proton pump inhibitors is being validated and seems to reduce the metastatic capacity of tumours cells [266], and to induce apoptosis of tumour cells without influencing normal cells from the same tissue [267]. Thus V-ATPase is thought to be a potential target for anticancer treatment [268]. However, tumour cells in hypoxia have developed key strategies to reduce ATP consumption (reduction of proliferation, protein synthesis) thus, the V-ATPase that requires ATP to extrude H+, seems not be the preferential candidate for targeting tumour pHi regulation.
Conclusion
Since pHi regulation controls many cellular functions involved in energy production, cell survival, proliferation and migration, a strategy that would inhibit some of the major pHi-regulating systems of highly glycolytic cells, such as transformed cells and hypoxic tumour cells, is now being intensely investigated. Validation of the role of HIF-1-induced pHi-regulating systems such as CAIX, CAXII and MCT4 in tumorigenesis and tumour development is in progress. Thus, the challenge now consists in evaluating the most specific and promising drugs that target these systems, for possible combination with chemotherapeutic compounds.
Acknowledgments
The laboratory is funded by grants from the Ligue Nationale Contre le Cancer (Equipe labellisée), the Centre A. Lacassagne, the Centre National de la Recherche Scientifique (CNRS), the Ministère de l’Education, de la Recherche et de la Technologie, the Institut National de la Santé et de la Recherche Médicale (Inserm) and the Institut National du Cancer (INCA). The authors declare no conflict of interest. J.C. is enrolled at the Ecole Doctorale CBS de Montpellier and supported by the CNRS.
References
- 1.Lindner D, Raghavan D. Intra-tumoural extra-cellular pH: a useful parameter of response to chemotherapy in syngeneic tumour lines. Br J Cancer. 2009;100:1287–91. doi: 10.1038/sj.bjc.6605022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillies RJ, Alger JR, Den Hollander JA, et al. Intracellular pH measured by NMR: methods and results. Kroc Found Ser. 1981;15:79–104. [PubMed] [Google Scholar]
- 3.Gillies RJ, Raghunand N, Garcia-Martin ML, et al. pH imaging. A review of pH measurement methods and applications in cancers. IEEE Eng Med Biol Mag. 2004;23:57–64. doi: 10.1109/memb.2004.1360409. [DOI] [PubMed] [Google Scholar]
- 4.Gillies RJ, Liu Z, Bhujwalla Z. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am J Physiol. 1994;267:C195–203. doi: 10.1152/ajpcell.1994.267.1.C195. [DOI] [PubMed] [Google Scholar]
- 5.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49:24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 6.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 7.Stubbs M, McSheehy PM, Griffiths JR, et al. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today. 2000;6:15–9. doi: 10.1016/s1357-4310(99)01615-9. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths JR. Are cancer cells acidic? Br J Cancer. 1991;64:425–7. doi: 10.1038/bjc.1991.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–65. [PubMed] [Google Scholar]
- 10.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–43. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 12.Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 13.Pouyssegur J, Sardet C, Franchi A, et al. A specific mutation abolishing Na+/H+ antiport activity in hamster fibroblasts precludes growth at neutral and acidic pH. Proc Natl Acad Sci USA. 1984;81:4833–7. doi: 10.1073/pnas.81.15.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chambard JC, Pouyssegur J. Intracellular pH controls growth factor-induced ribosomal protein S6 phosphorylation and protein synthesis in the G0–G1 transition of fibroblasts. Exp Cell Res. 1986;164:282–94. doi: 10.1016/0014-4827(86)90029-7. [DOI] [PubMed] [Google Scholar]
- 15.Matsuyama S, Llopis J, Deveraux QL, et al. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol. 2000;2:318–25. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 16.Sardet C, Franchi A, Pouyssegur J. Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell. 1989;56:271–80. doi: 10.1016/0092-8674(89)90901-x. [DOI] [PubMed] [Google Scholar]
- 17.Counillon L, Pouyssegur J. The expanding family of eucaryotic Na(+)/H(+) exchangers. J Biol Chem. 2000;275:1–4. doi: 10.1074/jbc.275.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Zaguilan R, Lynch RM, Martinez GM, et al. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol. 1993;265:C1015–29. doi: 10.1152/ajpcell.1993.265.4.C1015. [DOI] [PubMed] [Google Scholar]
- 19.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–28. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 20.Alper SL. Molecular physiology of SLC4 anion exchangers. Exp Physiol. 2006;91:153–61. doi: 10.1113/expphysiol.2005.031765. [DOI] [PubMed] [Google Scholar]
- 21.Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO 3 – transporters. Pflugers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- 22.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030–7. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 23.Ivanov S, Liao SY, Ivanova A, et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol. 2001;158:905–19. doi: 10.1016/S0002-9440(10)64038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiche J, Ilc K, Laferriere J, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69:358–68. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- 25.Glunde K, Guggino SE, Solaiyappan M, et al. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia. 2003;5:533–45. doi: 10.1016/s1476-5586(03)80037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Zaguilan R, Seftor EA, Seftor RE, et al. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis. 1996;14:176–86. doi: 10.1007/BF00121214. [DOI] [PubMed] [Google Scholar]
- 27.Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. 2001;69:522–30. [PubMed] [Google Scholar]
- 28.Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 29.Sauvant C, Nowak M, Wirth C, et al. Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer. 2008;123:2532–42. doi: 10.1002/ijc.23818. [DOI] [PubMed] [Google Scholar]
- 30.Morita T. Low pH leads to sister-chromatid exchanges and chromosomal aberrations, and its clastogenicity is S-dependent. Mutat Res. 1995;334:301–8. doi: 10.1016/0165-1161(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 31.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194–8. [PubMed] [Google Scholar]
- 32.Gerweck LE. The pH difference between tumor and normal tissue offers a tumor specific target for the treatment of cancer. Drug Resist Updat. 2000;3:49–50. doi: 10.1054/drup.2000.0122. [DOI] [PubMed] [Google Scholar]
- 33.Chandrashekar J, Yarmolinsky D, Von Buchholtz L, et al. The taste of carbonation. Science. 2009;326:443–5. doi: 10.1126/science.1174601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wohlhueter RM, Plagemann PG. Hexose transport and phosphorylation by Novikoff rat hepatoma cells as function of extracellular pH. J Biol Chem. 1981;256:869–75. [PubMed] [Google Scholar]
- 35.Buckwitz D, Jacobasch G, Gerth C, et al. A kinetic model of phosphofructokinase from Plasmodium berghei. Influence of ATP and fructose-6-phosphate. Mol Biochem Parasitol. 1988;27:225–32. doi: 10.1016/0166-6851(88)90041-2. [DOI] [PubMed] [Google Scholar]
- 36.Spriet LL. Phosphofructokinase activity and acidosis during short-term tetanic contractions. Can J Physiol Pharmacol. 1991;69:298–304. doi: 10.1139/y91-046. [DOI] [PubMed] [Google Scholar]
- 37.Gluck S, Nelson R. The role of the V-ATPase in renal epithelial H+ transport. J Exp Biol. 1992;172:205–18. doi: 10.1242/jeb.172.1.205. [DOI] [PubMed] [Google Scholar]
- 38.Gluck SL, Nelson RD, Lee BS, et al. Biochemistry of the renal V-ATPase. J Exp Biol. 1992;172:219–29. doi: 10.1242/jeb.172.1.219. [DOI] [PubMed] [Google Scholar]
- 39.Merezhinskaya N, Ogunwuyi SA, Mullick FG, et al. Presence and localization of three lactic acid transporters (MCT1, -2, and -4) in separated human granulocytes, lymphocytes, and monocytes. J Histochem Cytochem. 2004;52:1483–93. doi: 10.1369/jhc.4A6306.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilegaard H, Domino K, Noland T, et al. Effect of high-intensity exercise training on lactate/H+ transport capacity in human skeletal muscle. Am J Physiol. 1999;276:E255–61. doi: 10.1152/ajpendo.1999.276.2.E255. [DOI] [PubMed] [Google Scholar]
- 41.Pilegaard H, Terzis G, Halestrap A, et al. Distribution of the lactate/H+ transporter isoforms MCT1 and MCT4 in human skeletal muscle. Am J Physiol. 1999;276:E843–8. doi: 10.1152/ajpendo.1999.276.5.E843. [DOI] [PubMed] [Google Scholar]
- 42.Boron WF. Sodium-coupled bicarbonate transporters. Jop. 2001;2:176–81. [PubMed] [Google Scholar]
- 43.Vaughan-Jones RD, Spitzer KW. Role of bicarbonate in the regulation of intracellular pH in the mammalian ventricular myocyte. Biochem Cell Biol. 2002;80:579–96. doi: 10.1139/o02-157. [DOI] [PubMed] [Google Scholar]
- 44.Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246:2561–73. [PubMed] [Google Scholar]
- 45.Brant SR, Yun CH, Donowitz M, et al. Cloning, tissue distribution, and functional analysis of the human Na+/N+ exchanger isoform, NHE3. Am J Physiol. 1995;269:C198–206. doi: 10.1152/ajpcell.1995.269.1.C198. [DOI] [PubMed] [Google Scholar]
- 46.Praetorius J, Andreasen D, Jensen BL, et al. NHE1, NHE2, and NHE3 contribute to regulation of intracellular pH in murine duodenal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G197–206. doi: 10.1152/ajpgi.2000.278.2.G197. [DOI] [PubMed] [Google Scholar]
- 47.Tse CM, Brant SR, Walker MS, et al. Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney-specific Na+/H+ exchanger isoform (NHE-3) J Biol Chem. 1992;267:9340–6. [PubMed] [Google Scholar]
- 48.Wang Z, Orlowski J, Shull GE. Primary structure and functional expression of a novel gastrointestinal isoform of the rat Na/H exchanger. J Biol Chem. 1993;268:11925–8. [PubMed] [Google Scholar]
- 49.Attaphitaya S, Park K, Melvin JE. Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J Biol Chem. 1999;274:4383–8. doi: 10.1074/jbc.274.7.4383. [DOI] [PubMed] [Google Scholar]
- 50.Goyal S, Vanden Heuvel G, Aronson PS. Renal expression of novel Na+/H+ exchanger isoform NHE8. Am J Physiol Renal Physiol. 2003;284:F467–73. doi: 10.1152/ajprenal.00352.2002. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura N, Tanaka S, Teko Y, et al. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005;280:1561–72. doi: 10.1074/jbc.M410041200. [DOI] [PubMed] [Google Scholar]
- 52.Numata M, Orlowski J. Molecular cloning and characterization of a novel (Na+,K+)/H+ exchanger localized to the trans-Golgi network. J Biol Chem. 2001;276:17387–94. doi: 10.1074/jbc.M101319200. [DOI] [PubMed] [Google Scholar]
- 53.Paris S, Pouyssegur J. Growth factors activate the Na+/H+ antiporter in quiescent fibroblasts by increasing its affinity for intracellular H+ J Biol Chem. 1984;259:10989–94. [PubMed] [Google Scholar]
- 54.Pouyssegur J, Chambard JC, Franchi A, et al. Growth factor activation of an amiloride-sensitive Na+/H+ exchange system in quiescent fibroblasts: coupling to ribosomal protein S6 phosphorylation. Proc Natl Acad Sci USA. 1982;79:3935–9. doi: 10.1073/pnas.79.13.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pouyssegur J, Franchi A, L’Allemain G, et al. Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett. 1985;190:115–9. doi: 10.1016/0014-5793(85)80439-7. [DOI] [PubMed] [Google Scholar]
- 56.Franchi A, Perucca-Lostanlen D, et al. Functional expression of a human Na+/H+ antiporter gene transfected into antiporter-deficient mouse L cells. Proc Natl Acad Sci USA. 1986;83:9388–92. doi: 10.1073/pnas.83.24.9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagarde AE, Franchi AJ, Paris S, et al. Effect of mutations affecting Na+: H+ antiport activity on tumorigenic potential of hamster lung fibroblasts. J Cell Biochem. 1988;36:249–60. doi: 10.1002/jcb.240360306. [DOI] [PubMed] [Google Scholar]
- 58.Pouyssegur J, Franchi A, Pages G. pHi, aerobic glycolysis and vascular endothelial growth factor in tumour growth. Novartis Found Symp. 2001;240:186–96. doi: 10.1002/0470868716.ch13. [DOI] [PubMed] [Google Scholar]
- 59.Rotin D, Steele-Norwood D, Grinstein S, et al. Requirement of the Na+/H+ exchanger for tumor growth. Cancer Res. 1989;49:205–11. [PubMed] [Google Scholar]
- 60.Franchi A, Silvestre P, Pouyssegur J. A genetic approach to the role of energy metabolism in the growth of tumor cells: tumorigenicity of fibroblast mutants deficient either in glycolysis or in respiration. Int J Cancer. 1981;27:819–27. doi: 10.1002/ijc.2910270614. [DOI] [PubMed] [Google Scholar]
- 61.Pouyssegur J, Franchi A, Salomon JC, et al. Isolation of a Chinese hamster fibroblast mutant defective in hexose transport and aerobic glycolysis: its use to dissect the malignant phenotype. Proc Natl Acad Sci USA. 1980;77:2698–701. doi: 10.1073/pnas.77.5.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Newell K, Franchi A, Pouyssegur J, et al. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci USA. 1993;90:1127–31. doi: 10.1073/pnas.90.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.L’Allemain G, Paris S, Pouyssegur J. Role of a Na+-dependent Cl-/HCO3- exchange in regulation of intracellular pH in fibroblasts. J Biol Chem. 1985;260:4877–83. [PubMed] [Google Scholar]
- 64.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 65.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 66.Larson SM. Positron emission tomography-based molecular imaging in human cancer: exploring the link between hypoxia and accelerated glucose metabolism. Clin Cancer Res. 2004;10:2203–4. doi: 10.1158/1078-0432.ccr-0002-4. [DOI] [PubMed] [Google Scholar]
- 67.Funes JM, Quintero M, Henderson S, et al. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA. 2007;104:6223–8. doi: 10.1073/pnas.0700690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fang JS, Gillies RD, Gatenby RA. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin Cancer Biol. 2008;18:330–7. doi: 10.1016/j.semcancer.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 70.Kondoh H, Lleonart ME, Gil J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–85. [PubMed] [Google Scholar]
- 71.Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 72.Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–3. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 73.Xu RH, Pelicano H, Zhang H, et al. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia. 2005;19:2153–8. doi: 10.1038/sj.leu.2403968. [DOI] [PubMed] [Google Scholar]
- 74.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–42. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 75.Rathmell JC, Fox CJ, Plas DR, et al. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–28. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blum R, Jacob-Hirsch J, Amariglio N, et al. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005;65:999–1006. [PubMed] [Google Scholar]
- 77.Osthus RC, Shim H, Kim S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 78.Shim H, Dolde C, Lewis BC, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA. 1997;94:6658–63. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122:501–14. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- 81.Chen MK, Espat NJ, Bland KI, et al. Influence of progressive tumor growth on glutamine metabolism in skeletal muscle and kidney. Ann Surg. 1993;217:655–66. doi: 10.1097/00000658-199306000-00007. ; discussion 66–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klimberg VS, McClellan JL. Claude H. Organ, Jr. Honorary Lectureship. Glutamine, cancer, and its therapy. Am J Surg. 1996;172:418–24. doi: 10.1016/s0002-9610(96)00217-6. [DOI] [PubMed] [Google Scholar]
- 83.DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaplan DL, Boron WF. Long-term expression of c-H-ras stimulates Na-H and Na(+)-dependent Cl-HCO3 exchange in NIH-3T3 fibroblasts. J Biol Chem. 1994;269:4116–24. [PubMed] [Google Scholar]
- 85.Gillies RJ, Martinez-Zaguilan R, Martinez GM, et al. Tumorigenic 3T3 cells maintain an alkaline intracellular pH under physiological conditions. Proc Natl Acad Sci USA. 1990;87:7414–8. doi: 10.1073/pnas.87.19.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Perona R, Serrano R. Increased pH and tumorigenicity of fibroblasts expressing a yeast proton pump. Nature. 1988;334:438–40. doi: 10.1038/334438a0. [DOI] [PubMed] [Google Scholar]
- 87.Reshkin SJ, Bellizzi A, Caldeira S, et al. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000;14:2185–97. doi: 10.1096/fj.00-0029com. [DOI] [PubMed] [Google Scholar]
- 88.Cardone RA, Busco G, Greco MR, et al. HPV16 E7-dependent transformation activates NHE1 through a PKA-RhoA-induced inhibition of p38alpha. PLoS One. 2008;3:e3529. doi: 10.1371/journal.pone.0003529. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Kunz M, Ibrahim SM. Molecular responses to hypoxia in tumor cells. Mol Cancer. 2003;2:23. doi: 10.1186/1476-4598-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brahimi-Horn MC, Pouyssegur J. Harnessing the hypoxia-inducible factor in cancer and ischemic disease. Biochem Pharmacol. 2007;73:450–7. doi: 10.1016/j.bcp.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 91.Brahimi-Horn MC, Pouyssegur J. The hypoxia-inducible factor and tumor progression along the angiogenic pathway. Int Rev Cytol. 2005;242:157–213. doi: 10.1016/S0074-7696(04)42004-X. [DOI] [PubMed] [Google Scholar]
- 92.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 93.Berra E, Richard DE, Gothie E, et al. HIF-1-dependent transcriptional activity is required for oxygen-mediated HIF-1alpha degradation. FEBS Lett. 2001;491:85–90. doi: 10.1016/s0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 94.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–26. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 95.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 96.Lando D, Peet DJ, Gorman JJ, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hewitson KS, McNeill LA, Riordan MV, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–5. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 98.Lee C, Kim SJ, Jeong DG, et al. Structure of human FIH-1 reveals a unique active site pocket and interaction sites for HIF-1 and von Hippel-Lindau. J Biol Chem. 2003;278:7558–63. doi: 10.1074/jbc.M210385200. [DOI] [PubMed] [Google Scholar]
- 99.Bilton R, Trottier E, Pouyssegur J, et al. ARDent about acetylation and deacetylation in hypoxia signalling. Trends Cell Biol. 2006;16:616–21. doi: 10.1016/j.tcb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 100.Brahimi-Horn C, Mazure N, Pouyssegur J. Signalling via the hypoxia-inducible factor-1alpha requires multiple posttranslational modifications. Cell Signal. 2005;17:1–9. doi: 10.1016/j.cellsig.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 101.Koivunen P, Hirsila M, Gunzler V, et al. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 102.Kallio PJ, Okamoto K, O’Brien S, et al. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17:6573–86. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Found Symp Proc. 2007:35–53. doi: 10.1007/2789_2008_088. [DOI] [PubMed] [Google Scholar]
- 104.Hammond EM, Giaccia AJ. The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Commun. 2005;331:718–25. doi: 10.1016/j.bbrc.2005.03.154. [DOI] [PubMed] [Google Scholar]
- 105.Shafee N, Kaluz S, Ru N, et al. PI3K/Akt activity has variable cell-specific effects on expression of HIF target genes, CA9 and VEGF, in human cancer cell lines. Cancer Lett. 2009;282:109–15. doi: 10.1016/j.canlet.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–69. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 107.Dayan F, Roux D, Brahimi-Horn MC, et al. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006;66:3688–98. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- 108.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–7. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–5. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–4. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 111.Bell EL, Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27. doi: 10.1042/BSE0430017. [DOI] [PubMed] [Google Scholar]
- 112.Chavez A, Miranda LF, Pichiule P, et al. Mitochondria and hypoxia-induced gene expression mediated by hypoxia-inducible factors. Ann N Y Acad Sci. 2008;1147:312–20. doi: 10.1196/annals.1427.021. [DOI] [PubMed] [Google Scholar]
- 113.Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–94. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 114.Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol. 2008;28:5106–19. doi: 10.1128/MCB.00060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Milner PI, Wilkins RJ, Gibson JS. The role of mitochondrial reactive oxygen species in pH regulation in articular chondrocytes. Osteoarthritis Cartilage. 2007;15:735–42. doi: 10.1016/j.joca.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 116.Shimoda LA, Fallon M, Pisarcik S, et al. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291:L941–9. doi: 10.1152/ajplung.00528.2005. [DOI] [PubMed] [Google Scholar]
- 117.Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 118.Papandreou I, Cairns RA, Fontana L, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 119.Fukuda R, Zhang H, Kim JW, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 120.Natsuizaka M, Ozasa M, Darmanin S, et al. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res. 2007;313:3337–48. doi: 10.1016/j.yexcr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 121.Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30:142–50. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 122.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 123.Pelicano H, Martin DS, Xu RH, et al. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–46. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 124.Gutierrez G. A mathematical model of tissue-blood carbon dioxide exchange during hypoxia. Am J Respir Crit Care Med. 2004;169:525–33. doi: 10.1164/rccm.200305-702OC. [DOI] [PubMed] [Google Scholar]
- 125.Svastova E, Hulikova A, Rafajova M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004;577:439–45. doi: 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 126.Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res. 2004;64:6160–5. doi: 10.1158/0008-5472.CAN-03-2224. [DOI] [PubMed] [Google Scholar]
- 127.Swietach P, Wigfield S, Cobden P, et al. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J Biol Chem. 2008;283:20473–83. doi: 10.1074/jbc.M801330200. [DOI] [PubMed] [Google Scholar]
- 128.Mekhail K, Gunaratnam L, Bonicalzi ME, et al. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat Cell Biol. 2004;6:642–7. doi: 10.1038/ncb1144. [DOI] [PubMed] [Google Scholar]
- 129.Willam C, Warnecke C, Schefold JC, et al. Inconsistent effects of acidosis on HIF-alpha protein and its target genes. Pflugers Arch. 2006;451:534–43. doi: 10.1007/s00424-005-1486-3. [DOI] [PubMed] [Google Scholar]
- 130.Xu L, Fidler IJ. Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res. 2000;60:4610–6. [PubMed] [Google Scholar]
- 131.Fukumura D, Xu L, Chen Y, et al. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001;61:6020–4. [PubMed] [Google Scholar]
- 132.Shi Q, Le X, Wang B, et al. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene. 2001;20:3751–6. doi: 10.1038/sj.onc.1204500. [DOI] [PubMed] [Google Scholar]
- 133.Bischoff DS, Zhu JH, Makhijani NS, et al. Acidic pH stimulates the production of the angiogenic CXC chemokine, CXCL8 (interleukin-8), in human adult mesenchymal stem cells via the extracellular signal-regulated kinase, p38 mitogen-activated protein kinase, and NF-kappaB pathways. J Cell Biochem. 2008;104:1378–92. doi: 10.1002/jcb.21714. [DOI] [PubMed] [Google Scholar]
- 134.Van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–84. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ryan S, McNicholas WT, Taylor CT. A critical role for p38 map kinase in NF-kappaB signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun. 2007;355:728–33. doi: 10.1016/j.bbrc.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 136.Barbera MJ, Puig I, Dominguez D, et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene. 2004;23:7345–54. doi: 10.1038/sj.onc.1207990. [DOI] [PubMed] [Google Scholar]
- 137.Moellering RE, Black KC, Krishnamurty C, et al. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25:411–25. doi: 10.1007/s10585-008-9145-7. [DOI] [PubMed] [Google Scholar]
- 138.Rios EJ, Fallon M, Wang J, et al. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L867–74. doi: 10.1152/ajplung.00455.2004. [DOI] [PubMed] [Google Scholar]
- 139.Gatenby RA, Smallbone K, Maini PK, et al. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer. 2007;97:646–53. doi: 10.1038/sj.bjc.6603922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reshkin SJ, Bellizzi A, Albarani V, et al. Phosphoinositide 3-kinase is involved in the tumor-specific activation of human breast cancer cell Na(+)/H(+) exchange, motility, and invasion induced by serum deprivation. J Biol Chem. 2000;275:5361–9. doi: 10.1074/jbc.275.8.5361. [DOI] [PubMed] [Google Scholar]
- 141.Stock C, Gassner B, Hauck CR, et al. Migration of human melanoma cells depends on extracellular pH and Na+/H+ exchange. J Physiol. 2005;567:225–38. doi: 10.1113/jphysiol.2005.088344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol. 2002;159:1087–96. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 144.Pastorek J, Pastorekova S, Callebaut I, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene. 1994;9:2877–88. [PubMed] [Google Scholar]
- 145.Wykoff CC, Beasley NJ, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83. [PubMed] [Google Scholar]
- 146.Opavsky R, Pastorekova S, Zelnik V, et al. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics. 1996;33:480–7. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 147.Barathova M, Takacova M, Holotnakova T, et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumour phenotype. Br J Cancer. 2008;98:129–36. doi: 10.1038/sj.bjc.6604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lau KW, Tian YM, Raval RR, et al. Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer. 2007;96:1284–92. doi: 10.1038/sj.bjc.6603675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jakubickova L, Biesova Z, Pastorekova S, et al. Methylation of the CA9 promoter can modulate expression of the tumor-associated carbonic anhydrase IX in dense carcinoma cell lines. Int J Oncol. 2005;26:1121–7. [PubMed] [Google Scholar]
- 150.Kaluz S, Kaluzova M, Liao SY, et al. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: a one transcription factor (HIF-1) show? Biochim Biophys Acta. 2009;1795:162–72. doi: 10.1016/j.bbcan.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kaluz S, Kaluzova M, Opavsky R, et al. Transcriptional regulation of the MN/CA 9 gene coding for the tumor-associated carbonic anhydrase IX. Identification and characterization of a proximal silencer element. J Biol Chem. 1999;274:32588–95. doi: 10.1074/jbc.274.46.32588. [DOI] [PubMed] [Google Scholar]
- 152.Kondo K, Kaelin WG., Jr The von Hippel-Lindau tumor suppressor gene. Exp Cell Res. 2001;264:117–25. doi: 10.1006/excr.2000.5139. [DOI] [PubMed] [Google Scholar]
- 153.Ivanov SV, Kuzmin I, Wei MH, et al. Down-regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc Natl Acad Sci USA. 1998;95:12596–601. doi: 10.1073/pnas.95.21.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Grabmaier K, AdW MC, Verhaegh GW, et al. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene. 2004;23:5624–31. doi: 10.1038/sj.onc.1207764. [DOI] [PubMed] [Google Scholar]
- 155.Kaluzova M, Kaluz S, Lerman MI, et al. DNA damage is a prerequisite for p53-mediated proteasomal degradation of HIF-1alpha in hypoxic cells and downregulation of the hypoxia marker carbonic anhydrase IX. Mol Cell Biol. 2004;24:5757–66. doi: 10.1128/MCB.24.13.5757-5766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kopacek J, Barathova M, Dequiedt F, et al. MAPK pathway contributes to density- and hypoxia-induced expression of the tumor-associated carbonic anhydrase IX. Biochim Biophys Acta. 2005;1729:41–9. doi: 10.1016/j.bbaexp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 157.Kaluz S, Kaluzova M, Stanbridge EJ. The role of extracellular signal-regulated protein kinase in transcriptional regulation of the hypoxia marker carbonic anhydrase IX. J Cell Biochem. 2006;97:207–16. doi: 10.1002/jcb.20633. [DOI] [PubMed] [Google Scholar]
- 158.Sang N, Stiehl DP, Bohensky J, et al. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–9. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kaluz S, Kaluzova M, Chrastina A, et al. Lowered oxygen tension induces expression of the hypoxia marker MN/carbonic anhydrase IX in the absence of hypoxia-inducible factor 1 alpha stabilization: a role for phosphatidylinositol 3’-kinase. Cancer Res. 2002;62:4469–77. [PubMed] [Google Scholar]
- 160.Dayan F, Bilton RL, Laferriere J, et al. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J Cell Physiol. 2009;218:167–74. doi: 10.1002/jcp.21584. [DOI] [PubMed] [Google Scholar]
- 161.Van Den Beucken T, Koritzinsky M, Niessen H, et al. Hypoxia-induced expression of carbonic anhydrase 9 is dependent on the unfolded protein response. J Biol Chem. 2009;284:24204–12. doi: 10.1074/jbc.M109.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ihnatko R, Kubes M, Takacova M, et al. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol. 2006;29:1025–33. [PubMed] [Google Scholar]
- 163.Rafajova M, Zatovicova M, Kettmann R, et al. Induction by hypoxia combined with low glucose or low bicarbonate and high posttranslational stability upon reoxygenation contribute to carbonic anhydrase IX expression in cancer cells. Int J Oncol. 2004;24:995–1004. [PubMed] [Google Scholar]
- 164.Pastorekova S, Parkkila S, Parkkila AK, et al. Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology. 1997;112:398–408. doi: 10.1053/gast.1997.v112.pm9024293. [DOI] [PubMed] [Google Scholar]
- 165.Hilvo M, Rafajova M, Pastorekova S, et al. Expression of carbonic anhydrase IX in mouse tissues. J Histochem Cytochem. 2004;52:1313–22. doi: 10.1177/002215540405201007. [DOI] [PubMed] [Google Scholar]
- 166.Kallio H, Pastorekova S, Pastorek J, et al. Expression of carbonic anhydrases IX and XII during mouse embryonic development. BMC Dev Biol. 2006;6:22. doi: 10.1186/1471-213X-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jarvela S, Parkkila S, Bragge H, et al. Carbonic anhydrase IX in oligodendroglial brain tumors. BMC Cancer. 2008;8:1. doi: 10.1186/1471-2407-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Niemela AM, Hynninen P, Mecklin JP, et al. Carbonic anhydrase IX is highly expressed in hereditary nonpolyposis colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:1760–6. doi: 10.1158/1055-9965.EPI-07-0080. [DOI] [PubMed] [Google Scholar]
- 169.Hynninen P, Vaskivuo L, Saarnio J, et al. Expression of transmembrane carbonic anhydrases IX and XII in ovarian tumours. Histopathology. 2006;49:594–602. doi: 10.1111/j.1365-2559.2006.02523.x. [DOI] [PubMed] [Google Scholar]
- 170.Chen J, Rocken C, Hoffmann J, et al. Expression of carbonic anhydrase 9 at the invasion front of gastric cancers. Gut. 2005;54:920–7. doi: 10.1136/gut.2004.047340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Juhasz M, Chen J, Lendeckel U, et al. Expression of carbonic anhydrase IX in human pancreatic cancer. Aliment Pharmacol Ther. 2003;18:837–46. doi: 10.1046/j.1365-2036.2003.01738.x. [DOI] [PubMed] [Google Scholar]
- 172.Bartosova M, Parkkila S, Pohlodek K, et al. Expression of carbonic anhydrase IX in breast is associated with malignant tissues and is related to overexpression of c-erbB2. J Pathol. 2002;197:314–21. doi: 10.1002/path.1120. [DOI] [PubMed] [Google Scholar]
- 173.Mayer A, Hockel M, Vaupel P. Carbonic anhydrase IX expression and tumor oxygenation status do not correlate at the microregional level in locally advanced cancers of the uterine cervix. Clin Cancer Res. 2005;11:7220–5. doi: 10.1158/1078-0432.CCR-05-0869. [DOI] [PubMed] [Google Scholar]
- 174.Pastorekova S, Parkkila S, Zavada J. Tumor-associated carbonic anhydrases and their clinical significance. Adv Clin Chem. 2006;42:167–216. [PubMed] [Google Scholar]
- 175.Maseide K, Kandel RA, Bell RS, et al. Carbonic anhydrase IX as a marker for poor prognosis in soft tissue sarcoma. Clin Cancer Res. 2004;10:4464–71. doi: 10.1158/1078-0432.CCR-03-0541. [DOI] [PubMed] [Google Scholar]
- 176.Trastour C, Benizri E, Ettore F, et al. HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer. 2007;120:1451–8. doi: 10.1002/ijc.22436. [DOI] [PubMed] [Google Scholar]
- 177.Loncaster JA, Harris AL, Davidson SE, et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res. 2001;61:6394–9. [PubMed] [Google Scholar]
- 178.Jonathan RA, Wijffels KI, Peeters W, et al. The prognostic value of endogenous hypoxia-related markers for head and neck squamous cell carcinomas treated with ARCON. Radiother Oncol. 2006;79:288–97. doi: 10.1016/j.radonc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 179.Chia SK, Wykoff CC, Watson PH, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol. 2001;19:3660–8. doi: 10.1200/JCO.2001.19.16.3660. [DOI] [PubMed] [Google Scholar]
- 180.Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell Cycle. 2004;3:164–7. [PubMed] [Google Scholar]
- 181.Haapasalo JA, Nordfors KM, Hilvo M, et al. Expression of carbonic anhydrase IX in astrocytic tumors predicts poor prognosis. Clin Cancer Res. 2006;12:473–7. doi: 10.1158/1078-0432.CCR-05-0848. [DOI] [PubMed] [Google Scholar]
- 182.Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Hypoxia-regulated carbonic anhydrase-9 (CA9) relates to poor vascularization and resistance of squamous cell head and neck cancer to chemoradiotherapy. Clin Cancer Res. 2001;7:3399–403. [PubMed] [Google Scholar]
- 183.Bui MH, Seligson D, Han KR, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res. 2003;9:802–11. [PubMed] [Google Scholar]
- 184.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tureci O, Sahin U, Vollmar E, et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci USA. 1998;95:7608–13. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Barnett DH, Sheng S, Charn TH, et al. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008;68:3505–15. doi: 10.1158/0008-5472.CAN-07-6151. [DOI] [PubMed] [Google Scholar]
- 187.Haapasalo J, Hilvo M, Nordfors K, et al. Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro Oncol. 2008;10:131–8. doi: 10.1215/15228517-2007-065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hynninen P, Hamalainen JM, Pastorekova S, et al. Transmembrane carbonic anhydrase isozymes IX and XII in the female mouse reproductive organs. Reprod Biol Endocrinol. 2004;2:73. doi: 10.1186/1477-7827-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Karhumaa P, Parkkila S, Tureci O, et al. Identification of carbonic anhydrase XII as the membrane isozyme expressed in the normal human endometrial epithelium. Mol Hum Reprod. 2000;6:68–74. doi: 10.1093/molehr/6.1.68. [DOI] [PubMed] [Google Scholar]
- 190.Kivela A, Parkkila S, Saarnio J, et al. Expression of a novel transmembrane carbonic anhydrase isozyme XII in normal human gut and colorectal tumors. Am J Pathol. 2000;156:577–84. doi: 10.1016/S0002-9440(10)64762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Parkkila S, Parkkila AK, Saarnio J, et al. Expression of the membrane-associated carbonic anhydrase isozyme XII in the human kidney and renal tumors. J Histochem Cytochem. 2000;48:1601–8. doi: 10.1177/002215540004801203. [DOI] [PubMed] [Google Scholar]
- 192.Kivela AJ, Parkkila S, Saarnio J, et al. Expression of transmembrane carbonic anhydrase isoenzymes IX and XII in normal human pancreas and pancreatic tumours. Histochem Cell Biol. 2000;114:197–204. doi: 10.1007/s004180000181. [DOI] [PubMed] [Google Scholar]
- 193.Kivela AJ, Parkkila S, Saarnio J, et al. Expression of von Hippel-Lindau tumor suppressor and tumor-associated carbonic anhydrases IX and XII in normal and neoplastic colorectal mucosa. World J Gastroenterol. 2005;11:2616–25. doi: 10.3748/wjg.v11.i17.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liao SY, Ivanov S, Ivanova A, et al. Expression of cell surface transmembrane carbonic anhydrase genes CA9 and CA12 in the human eye: overexpression of CA12 (CAXII) in glaucoma. J Med Genet. 2003;40:257–61. doi: 10.1136/jmg.40.4.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Leppilampi M, Saarnio J, Karttunen TJ, et al. Carbonic anhydrase isozymes IX and XII in gastric tumors. World J Gastroenterol. 2003;9:1398–403. doi: 10.3748/wjg.v9.i7.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hilvo M, Baranauskiene L, Salzano AM, et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem. 2008;283:27799–809. doi: 10.1074/jbc.M800938200. [DOI] [PubMed] [Google Scholar]
- 197.De Simone G, Supuran CT. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbapap.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 198.Whittington DA, Waheed A, Ulmasov B, et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA. 2001;98:9545–50. doi: 10.1073/pnas.161301298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des. 2008;14:685–98. doi: 10.2174/138161208783877893. [DOI] [PubMed] [Google Scholar]
- 200.Pastorekova S, Ratcliffe PJ, Pastorek J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int. 2008;101:8–15. doi: 10.1111/j.1464-410X.2008.07642.x. [DOI] [PubMed] [Google Scholar]
- 201.Swietach P, Patiar S, Supuran CT, et al. The Role of Carbonic Anhydrase 9 in Regulating Extracellular and Intracellular pH in Three-dimensional Tumor Cell Growths. J Biol Chem. 2009;284:20299–310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Morgan PE, Pastorekova S, Stuart-Tilley AK, et al. Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. Am J Physiol Cell Physiol. 2007;293:C738–48. doi: 10.1152/ajpcell.00157.2007. [DOI] [PubMed] [Google Scholar]
- 203.Svichar N, Waheed A, Sly WS, et al. Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl–HCO3- exchange in hippocampal neurons. J Neurosci. 2009;29:3252–8. doi: 10.1523/JNEUROSCI.0036-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gut MO, Parkkila S, Vernerova Z, et al. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology. 2002;123:1889–903. doi: 10.1053/gast.2002.37052. [DOI] [PubMed] [Google Scholar]
- 205.Leppilampi M, Karttunen TJ, Kivela J, et al. Gastric pit cell hyperplasia and glandular atrophy in carbonic anhydrase IX knockout mice: studies on two strains C57/BL6 and BALB/C. Transgenic Res. 2005;14:655–63. doi: 10.1007/s11248-005-7215-z. [DOI] [PubMed] [Google Scholar]
- 206.Pan P, Leppilampi M, Pastorekova S, et al. Carbonic anhydrase gene expression in CA II-deficient (Car2-/-) and CA IX-deficient (Car9-/-) mice. J Physiol. 2006;571:319–27. doi: 10.1113/jphysiol.2005.102590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kivela AJ, Saarnio J, Karttunen TJ, et al. Differential expression of cytoplasmic carbonic anhydrases, CA I and II, and membrane-associated isozymes, CA IX and XII, in normal mucosa of large intestine and in colorectal tumors. Dig Dis Sci. 2001;46:2179–86. doi: 10.1023/a:1011910931210. [DOI] [PubMed] [Google Scholar]
- 208.Dorai T, Sawczuk IS, Pastorek J, et al. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur J Cancer. 2005;41:2935–47. doi: 10.1016/j.ejca.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 209.Svastova E, Zilka N, Zat’ovicova M, et al. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with beta-catenin. Exp Cell Res. 2003;290:332–45. doi: 10.1016/s0014-4827(03)00351-3. [DOI] [PubMed] [Google Scholar]
- 210.Parkkila S, Rajaniemi H, Parkkila AK, et al. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc Natl Acad Sci USA. 2000;97:2220–4. doi: 10.1073/pnas.040554897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kim JY, Shin HJ, Kim TH, et al. Tumor-associated carbonic anhydrases are linked to metastases in primary cervical cancer. J Cancer Res Clin Oncol. 2006;132:302–8. doi: 10.1007/s00432-005-0068-2. [DOI] [PubMed] [Google Scholar]
- 212.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343:281–99. [PMC free article] [PubMed] [Google Scholar]
- 213.Kirk P, Wilson MC, Heddle C, et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19:3896–904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Su J, Chen X, Kanekura T. A CD147-targeting siRNA inhibits the proliferation, invasiveness, and VEGF production of human malignant melanoma cells by down-regulating glycolysis. Cancer Lett. 2009;273:140–7. doi: 10.1016/j.canlet.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 215.Poole RC, Sansom CE, Halestrap AP. Studies of the membrane topology of the rat erythrocyte H+/lactate cotransporter (MCT1) Biochem J. 1996;320:817–24. doi: 10.1042/bj3200817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Dimmer KS, Friedrich B, Lang F, et al. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350:219–27. [PMC free article] [PubMed] [Google Scholar]
- 217.Gallagher SM, Castorino JJ, Philp NJ. Interaction of monocarboxylate transporter 4 with beta1-integrin and its role in cell migration. Am J Physiol Cell Physiol. 2009;296:C414–21. doi: 10.1152/ajpcell.00430.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Liang YX, He HC, Han ZD, et al. CD147 and VEGF expression in advanced renal cell carcinoma and their prognostic value. Cancer Invest. 2009;27:788–93. doi: 10.1080/07357900802709167. [DOI] [PubMed] [Google Scholar]
- 219.Pinheiro C, Longatto-Filho A, Simoes K, et al. The prognostic value of CD147/ EMMPRIN is associated with monocarboxylate transporter 1 co-expression in gastric cancer. Eur J Cancer. 2009;45:2418–24. doi: 10.1016/j.ejca.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 220.Du ZM, Hu CF, Shao Q, et al. Upregulation of caveolin-1 and CD147 expression in nasopharyngeal carcinoma enhanced tumor cell migration and correlated with poor prognosis of the patients. Int J Cancer. 2009;125:1832–41. doi: 10.1002/ijc.24531. [DOI] [PubMed] [Google Scholar]
- 221.Broer S, Broer A, Schneider HP, et al. Characterization of the high-affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem J. 1999;341:529–35. doi: 10.1042/0264-6021:3410529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lin RY, Vera JC, Chaganti RS, et al. Human monocarboxylate transporter 2 (MCT2) is a high affinity pyruvate transporter. J Biol Chem. 1998;273:28959–65. doi: 10.1074/jbc.273.44.28959. [DOI] [PubMed] [Google Scholar]
- 223.Becker HM, Hirnet D, Fecher-Trost C, et al. Transport activity of MCT1 expressed in Xenopus oocytes is increased by interaction with carbonic anhydrase. J Biol Chem. 2005;280:39882–9. doi: 10.1074/jbc.M503081200. [DOI] [PubMed] [Google Scholar]
- 224.Becker HM, Deitmer JW. Nonenzymatic proton handling by carbonic anhydrase II during H+-lactate cotransport via monocarboxylate transporter 1. J Biol Chem. 2008;283:21655–67. doi: 10.1074/jbc.M802134200. [DOI] [PubMed] [Google Scholar]
- 225.Sterling D, Reithmeier RA, Casey JR. A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem. 2001;276:47886–94. doi: 10.1074/jbc.M105959200. [DOI] [PubMed] [Google Scholar]
- 226.Sterling D, Reithmeier RA, Casey JR. Carbonic anhydrase: in the driver’s seat for bicarbonate transport. Jop. 2001;2:165–70. [PubMed] [Google Scholar]
- 227.Li X, Alvarez B, Casey JR, et al. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002;277:36085–91. doi: 10.1074/jbc.M111952200. [DOI] [PubMed] [Google Scholar]
- 228.Li X, Liu Y, Alvarez BV, et al. A novel carbonic anhydrase II binding site regulates NHE1 activity. Biochemistry. 2006;45:2414–24. doi: 10.1021/bi051132d. [DOI] [PubMed] [Google Scholar]
- 229.Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hosoya K, Kondo T, Tomi M, et al. MCT1-mediated transport of L-lactic acid at the inner blood-retinal barrier: a possible route for delivery of monocarboxylic acid drugs to the retina. Pharm Res. 2001;18:1669–76. doi: 10.1023/a:1013310210710. [DOI] [PubMed] [Google Scholar]
- 231.Hunt TK, Aslam RS, Beckert S, et al. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid Redox Signal. 2007;9:1115–24. doi: 10.1089/ars.2007.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schneiderhan W, Scheler M, Holzmann KH, et al. CD147 Silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in-vivo and in-vitro models. Gut. 2009;58:1391–8. doi: 10.1136/gut.2009.181412. [DOI] [PubMed] [Google Scholar]
- 233.Gallagher SM, Castorino JJ, Wang D, et al. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007;67:4182–9. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]
- 234.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 235.Simons AL, Ahmad IM, Mattson DM, et al. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67:3364–70. doi: 10.1158/0008-5472.CAN-06-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Singh D, Banerji AK, Dwarakanath BS, et al. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol. 2005;181:507–14. doi: 10.1007/s00066-005-1320-z. [DOI] [PubMed] [Google Scholar]
- 237.Kim W, Yoon JH, Jeong JM, et al. Apoptosis-inducing antitumor efficacy of hexokinase II inhibitor in hepatocellular carcinoma. Mol Cancer Ther. 2007;6:2554–62. doi: 10.1158/1535-7163.MCT-07-0115. [DOI] [PubMed] [Google Scholar]
- 238.Passonneau JV, Lowry OH. Phosphofructokinase and the Pasteur effect. Biochem Biophys Res Commun. 1962;7:10–5. doi: 10.1016/0006-291x(62)90134-1. [DOI] [PubMed] [Google Scholar]
- 239.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 240.Hardie DG. Metabolic control: a new solution to an old problem. Curr Biol. 2000;10:R757–9. doi: 10.1016/s0960-9822(00)00744-2. [DOI] [PubMed] [Google Scholar]
- 241.Russell RR, 3rd, Bergeron R, Shulman GI, et al. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277:H643–9. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 242.Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007;26:341–52. doi: 10.1007/s10555-007-9059-x. [DOI] [PubMed] [Google Scholar]
- 243.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–9. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 244.Melillo G. Hypoxia-inducible factor 1 inhibitors. Methods Enzymol. 2007;435:385–402. doi: 10.1016/S0076-6879(07)35020-9. [DOI] [PubMed] [Google Scholar]
- 245.Semenza GL. HIF-1 Inhibitors for cancer therapy: from gene expression to drug discovery. Curr Pharm Des. 2009;15:3839–43. doi: 10.2174/138161209789649402. [DOI] [PubMed] [Google Scholar]
- 246.Anderson KM, Jajeh A, Guinan P, et al. Do intra-tumor alkaline micro-regions represent additional therapeutically privileged sites? Med Hypotheses. 2008;70:1193–6. doi: 10.1016/j.mehy.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 247.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 248.Shackleton M, Quintana E, Fearon ER, et al. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–9. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 249.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 250.Hill RP, Marie-Egyptienne DT, Hedley DW. Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol. 2009;19:106–11. doi: 10.1016/j.semradonc.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 251.Heddleston JM, Li Z, McLendon RE, et al. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–84. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wong P, Kleemann HW, Tannock IF. Cytostatic potential of novel agents that inhibit the regulation of intracellular pH. Br J Cancer. 2002;87:238–45. doi: 10.1038/sj.bjc.6600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Thiry A, Supuran CT, Masereel B, et al. Recent developments of carbonic anhydrase inhibitors as potential anticancer drugs. J Med Chem. 2008;51:3051–6. doi: 10.1021/jm701526d. [DOI] [PubMed] [Google Scholar]
- 254.D’Ambrosio K, Vitale RM, Dogne JM, et al. Carbonic anhydrase inhibitors: bioreductive nitro-containing sulfonamides with selectivity for targeting the tumor associated isoforms IX and XII. J Med Chem. 2008;51:3230–7. doi: 10.1021/jm800121c. [DOI] [PubMed] [Google Scholar]
- 255.Ahlskog JK, Dumelin CE, Trussel S, et al. In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives. Bioorg Med Chem Lett. 2009;19:4851–6. doi: 10.1016/j.bmcl.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 256.Oosterwijk E, Bander NH, Divgi CR, et al. Antibody localization in human renal cell carcinoma: a phase I study of monoclonal antibody G250. J Clin Oncol. 1993;11:738–50. doi: 10.1200/JCO.1993.11.4.738. [DOI] [PubMed] [Google Scholar]
- 257.Oosterwijk E. Carbonic anhydrase IX/G250/MN: a molecule too good to be true? BJU Int. 2008;101:527–8. doi: 10.1111/j.1464-410X.2007.07431.x. [DOI] [PubMed] [Google Scholar]
- 258.Yin T, Yu S, Xiao L, et al. Correlation between the expression of aquaporin 1 and hypoxia-inducible factor 1 in breast cancer tissues. J Huazhong Univ Sci Technolog Med Sci. 2008;28:346–8. doi: 10.1007/s11596-008-0327-y. [DOI] [PubMed] [Google Scholar]
- 259.Hayashi Y, Edwards NA, Proescholdt MA, et al. Regulation and function of aquaporin-1 in glioma cells. Neoplasia. 2007;9:777–87. doi: 10.1593/neo.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Saadoun S, Papadopoulos MC, Hara-Chikuma M, et al. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005;434:786–92. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- 261.Baba M, Inoue M, Itoh K, et al. Blocking CD147 induces cell death in cancer cells through impairment of glycolytic energy metabolism. Biochem Biophys Res Commun. 2008;374:111–6. doi: 10.1016/j.bbrc.2008.06.122. [DOI] [PubMed] [Google Scholar]
- 262.Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer. 1991;64:663–70. doi: 10.1038/bjc.1991.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Robey IF, Baggett BK, Kirkpatrick ND, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–8. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Silva AS, Yunes JA, Gillies RJ, et al. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69:2677–84. doi: 10.1158/0008-5472.CAN-08-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Murakami T, Shibuya I, Ise T, et al. Elevated expression of vacuolar proton pump genes and cellular PH in cisplatin resistance. Int J Cancer. 2001;93:869–74. doi: 10.1002/ijc.1418. [DOI] [PubMed] [Google Scholar]
- 266.Perez-Sayans M, Garcia-Garcia A, Reboiras-Lopez MD, et al. Role of V-ATPases in solid tumors: importance of the subunit C. Int J Oncol. 2009;34:1513–20. doi: 10.3892/ijo_00000280. [DOI] [PubMed] [Google Scholar]
- 267.Morimura T, Fujita K, Akita M, et al. The proton pump inhibitor inhibits cell growth and induces apoptosis in human hepatoblastoma. Pediatr Surg Int. 2008;24:1087–94. doi: 10.1007/s00383-008-2229-2. [DOI] [PubMed] [Google Scholar]
- 268.Harguindey S, Arranz JL, Wahl ML, et al. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. 2009;29:2127–36. [PubMed] [Google Scholar]