

Abstract
Among the multitude of dysregulated signalling mechanisms that comprise insulin resistance in divergent organs, the primary events in the development of type 2 diabetes are not well established. As protein kinase C (PKC) activation is consistently present in skeletal muscle of obese and insulin resistant subjects, we generated a transgenic mouse model that overexpresses constitutively active PKC-β2 in skeletal muscle to test whether activation of PKC is sufficient to cause an aversive whole-body phenotype. Upon this genetic modification, increased serine phosphorylation in Irs1 was observed and followed by impaired 3H-deoxy-glucose uptake and muscle glycogen content, and transgenic mice exhibited insulin and glucose intolerance as they age. Muscle histochemistry revealed an increase in lipid deposition (intramyocellular lipids), and transgenic mice displayed impaired expression of transcriptional regulators of genes involved in fatty acid oxidation (peroxisome proliferator-activated receptor-γ, PGC-1β, acyl-CoA oxidase) and lipolysis (hormone-sensitive lipase). In this regard, muscle of transgenic mice exhibited a reduced capacity to oxidize palmitate and contained less mitochondria as determined by citrate synthase activity. Moreover, the phenotype included a profound decrease in the daily running distance, intra-abdominal and hepatic fat accumulation and impaired insulin action in the brain. Together, our data suggest that activation of a classical PKC in skeletal muscle as present in the pre-diabetic state is sufficient to cause disturbances in whole-body glucose and lipid metabolism followed by profound alterations in oxidative capacity, ectopic fat deposition and physical activity.
Keywords: protein kinase C, insulin action, NAFLD, intramyocellular lipid deposition, fatty acid oxidation, lipolysis, physical activity
Introduction
In recent years, a large number of studies defined the characteristics of the pre-diabetic state that are predictive for the development of type 2 diabetes including central obesity, disturbances in triglyceride and cholesterol metabolism, and raised fasting glucose concentrations. However, due to the complexity of the metabolic phenotype and the presence of tissue crosstalk, there is still debate about the primary defect and the contribution of tissues such as liver, brain and skeletal muscle to cause the aversive whole-body phenotype.
In particular, insulin signalling in liver tissues of overweight and diabetic subjects is greatly diminished and therefore drives gluconeogenesis and triglyceride storage [1]. The significance of this finding was confirmed using liver-specific insulin receptor (IR) knockout mice that displayed severe insulin resistance and glucose intolerance in the face of elevated insulin levels to compensate for increased demand [2]. Moreover, insulin resistance of the liver is sufficient to cause dyslipidaemia and an increased risk for atherosclerosis [3].
The contribution of insulin signalling in the brain to maintain normoglycaemia and weight stability is supported by studies in human beings, where insulin activated cerebrocortical activity in lean subjects, but not in obese, indicating cerebral insulin resistance [4]. These studies in human beings therefore confirmed the relevance of the results obtained in neuron-specific IR knockout mice where female mice showed increased food intake, and both male and female developed diet-sensitive obesity that was accompanied by elevated body fat and plasma leptin levels, insulin resistance and hypertriglyceridaemia [5]. Thus, IR signalling in the central nervous system plays a critical role in the regulation of glucose and lipid disposal.
It became clear from a large number of human and animal studies that insulin action is greatly impaired in skeletal muscle of pre-diabetic and overweight subjects, and it was therefore suggested that skeletal muscle plays an important role in the beginning of the disease. Surprisingly, a knockout approach using muscle-specific IR knockout mice did not provide strong evidence for a key role of primary insulin resistance in skeletal muscle in the development of a diabetic phenotype [6, 7]. Thus, an almost complete loss of IRs in skeletal muscle was associated with alterations in lipid metabolism and fat accumulation, but was not sufficient to cause dramatic alterations in glucose metabolism whereas disruption of the glucose transporter GLUT-4 exerted marked glucose intolerance [8].
Together, these studies in human beings and animals clearly implicate that primary defects in insulin action in liver and brain tissues cause alterations in whole-body glucose metabolism. On the other hand, a large number of data strongly suggest that insulin resistance in skeletal muscle serves as a primary cause in the development of the impaired glucose tolerance and diabetes in human beings; however, the impact of alterations in skeletal muscle in the beginning of the disease and its role for alterations in other tissues like liver, fat and the brain is not well supported by the present animal models [7, 9].
We therefore addressed this issue in an alternative transgenic approach using a common modulator downstream of the insulin and IGF-1 receptor, protein kinase C (PKC) [10]. PKC isoforms are activated by elevated glucose, insulin and fatty acid concentrations; and studies using skeletal muscle of insulin resistant human beings revealed that divergent PKC isoforms are chronically up-regulated and phosphorylated in this state [11, 12].
Among the multitude of PKC isoforms, PKC-β2 was shown to be highly expressed in obese and diabetic mouse models [13, 14], and several studies demonstrated that PKC-β2 is able to disrupt the signal at the level of the IR itself as well as at the level of IR substrate (Irs) proteins through serine phosphorylation [15, 16]. We and others recently demonstrated in an in vitro approach that serine 318 in Irs1 is a specific phosphorylation site for PKC-β2[17], and is related to insulin resistance in vitro and in vivo[13, 17, 18, 19]. In addition, serine 307 in Irs1 is phosphorylated by the JNK and PI 3-kinase/mTor signalling pathways [20], and in vivo, enhanced levels of serine 307 phosphorylation were found in skeletal muscle biopsies of obese, insulin-resistant non-diabetic subjects, and accompanied by a decrease in Irs1 tyrosine phosphorylation as well as reduced insulin-dependent activation of Akt [11].
To resemble these alterations present in an obese and diabetic state, a transgenic mouse model overexpressing constitutively active PKC-β2 in skeletal muscle was generated and characterized for metabolic alterations, fatty acid oxidation, physical activity and ectopic fat deposition.
Our data strongly suggest that a sole genetic alteration at the level of a well-characterized modulator within the insulin signalling cascade in skeletal muscle is sufficient to induce aversive effects on whole-body glucose and lipid homeostasis that are followed by physical inactivity, impaired oxidative capacity and disproportionate fat storage.
Materials and methods
Plasmid and animals
To generate a constitutively active PKC-β2, the cDNA was mutated to contain Glu at position 25 instead of Ala. This cDNA was cloned into pMDAF2, which contains a 1.5-kb fragment of the MLC1 promoter and 0.9-kb fragment of the MLC1/3, muscle-specific enhancer [21–23]. The MLC/PKC-β2 chimeric gene was excised from the plasmid, purified, and microinjected into fertilized eggs. The general procedures for microinjection of the chimeric gene were as described [24].
Two founder animals that overexpress PKC-β2 in skeletal muscle (#2, ∼6-fold and #25, ∼2-fold) were selected and backcrossed on a C57Bl/6 background until the F6 generation and male mice were used for experiments. All of the wild-type controls were littermates of the transgenic mice. Total RNA was obtained from skeletal muscle by the guanidine isothiocyanate method, and RNA samples (30 μg) were electrophoresed on a 1% agarose gel containing 2.2 M formaldehyde. Northern blots were hybridized to 32P-labelled PKC-β2 cDNA probe [25]. Equal loading of the agarose gel was judged by staining with ethidium bromide.
When stated in the text, 4-week-old C57Bl/6 mice were fed a high-fat diet (HFD) or chow for 8 weeks (D12451, Research Diet, Inc., New Brunswick, NJ, USA) and liver tissue was removed for cDNA preparation as described earlier [26].
All animal experiments were done in accordance with the accepted standard of humane animal care and were approved by the local Animal Care and Use Committee.
Metabolic studies
Mice were fed a standard diet and kept under a light/dark cycle of 12 hrs. Blood glucose was determined in overnight fasted mice using a Glucometer Elite (Bayer Corp, Elkhart, IN, USA). Plasma insulin concentrations were measured by RIA (Linco Research, St. Charles, MO, USA).
Glucose tolerance tests were performed in overnight fasted male mice. Animals were intraperitoneally injected with a single dose of D-glucose (2 g/kg body weight), and blood glucose concentrations were detected at the indicated times. To determine insulin tolerance, a bolus of human insulin (1 unit/kg body weight) was injected intraperitoneally into fed mice and glucose concentrations were determined. The results were expressed as percent of the initial glucose levels.
Free fatty acid and triglyceride concentrations were detected by an enzymatic method (ADVIA 1650®, Siemens Medical Solutions Diagnostics GmbH, Fernwald, Germany), leptin levels by ELISA (Crystal Chem, Inc., Chicago, IL, USA), and serum insulin, glucagon, and adiponectin levels by RIA (Linco Research).
In vivo stimulation and Western blot analysis
For in vivo stimulation, a bolus of human insulin (1unit/mouse for 5 min.) was injected into the inferior Vena cava of overnight fasted mice. Controls received a comparable amount of diluent. Muscle (EDL), and brain tissues were removed and homogenized at 4°C (25 mM Tris-HCl, pH 7.4, 50 mM sodium pyrophosphate, 100 mM sodium fluoride, 10 mM EDTA, 1% NP-40, 1 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml leupeptin). Homogenates were allowed to solubilize for 20 min. on ice and clarified by centrifugation at 12,000 ×g for 15 min. Equal amounts of total protein were incubated with anti-IR [27], Irs1 or Irs2 antibodies (Upstate, Charlottesville, VA, USA), precipitated on immobilized protein G, resolved by SDS-PAGE and immunoblotted with PY-20 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Membranes were stripped and reprobed with the respective antibodies.
For detection of PKC-β2 overexpression, Akt phosphorylation, and serine318 and serine307 phosphorylation in Irs1, lysates containing 0.1 mg of total protein were used. Visualization after gel-electrophoresis and Western blotting with the anti-PKC-β2 (Santa Cruz Biotechnology, Inc.), anti-phospho-PKC-β2, anti-phospho serine473-Akt and anti-Akt antibody (Upstate), anti-phosphoserine318 antibody [17], and anti-phosphoserine307 Irs1 antibody (Upstate) was performed using ECL.
Determination of 2-deoxyglucose uptake in skeletal muscle
In vivo tissue glucose uptake during a glucose tolerance test was determined in 12-week-old male mice that had been fasted for 16 hrs. 2-deoxy-D[1,2–3H]glucose was mixed with D-glucose (10 μCi/mouse, 2 g/kg body weight) and injected intraperitoneally. Blood glucose levels were determined to ensure adequate injection. After 120 min., mice were killed and 100 mg of tissue were homogenized in 1 ml of water. Seven-percent ice-cold perchloric acid was added to the homogenate. The sample was then cleared by centrifugation, and 1 ml of supernatant was neutralized for 30 min. with KHCO3. The precipitate was removed by centrifugation, and the supernatant was used to determine total 3H-radioactivity by liquid scintillation counting.
Assay of PI 3-kinase activity
EDL muscle tissue lysates were immunopurified with anti-PY20 antibodies and immunocomplexes were absorbed to Protein A-Sepharose for 12 hrs. Immunoprecipitates were washed and pellets were directly incubated with L-α-phosphatidylinositol (Sigma-Aldrich, Taufkirchen, Germany) and [γ32P] adenosine triphosphate at room temperature for 10 min. After addition of HCl, lipids were extracted twice with chloroform/methanol and products were separated by thin layer chromatography as described earlier [28]. 32P-labelled phospholipids were detected by autoradiography. To ensure comparable protein concentration, tissue lysates were blotted with p85 antibodies (BD Bioscience, Rockville, MD, USA).
Real-time RT-PCR
For quantification of mRNA expression in skeletal muscle and liver tissues of mice, samples were stored for 16 hrs in RNAlater (Ambion, Huntington, UK) at 4°C, and RNA was isolated with peqGOLD TriFast according to the manufactor’s instructions (Peqlab, Erlangen, Germany). Total RNA was treated with RNase-free DNase I and transcribed into cDNA using AMV reverse transcriptase and the first strand cDNA kit (Roche Diagnostics, Mannheim, Germany). Quantitative PCR was performed with SYBR Green I dye on a high speed thermal cycler with integrated microvolume fluorometer. Primers were obtained from Invitrogen (Karlsruhe, Germany). Primer sequences can be provided upon request. Measurements were performed in duplicate and RNA content was normalized to 28S-rRNA. Cellular mRNA content is given in relative arbitrary units.
Fatty acid oxidation in skeletal muscle
Tibialis muscles were incubated in α-MEM containing 5.5 mM glucose, 3.3% fatty-acid free bovine serum albumin, 2 μCi/ml 3H-palmitate, and 60 μM unlabelled palmitate for 1 hr at 37°C. Production of tritiated water was determinated after solid-phase extraction of the supernatant using Oasis HLB cartridges (Waters, Milford, MA, USA) in a scintillation counter.
Citrate synthase activity in skeletal muscle
Muscle tissue was harvested from fed mice and lysed in CelLyticMT Cell lysis reagent (Sigma-Aldrich, Saint Louis, MI, USA). Protein concentration was determined by Bradford assay and 16 μg of total protein was used for further analysis. Measurement of citrate synthase activity (Citrate Synthase Assay Kit, Sigma-Aldrich) was performed according to the manufactures protocol and activity is depicted in μmol/ml/min.
Glycogen content in skeletal muscle
Glycogen levels were determined by a modification of the procedure described by Chan and Exton [29]. Muscle tissue was weighted and solubilized for 10 min. with 250 μl 30% KOH at 90°C. Glycogen was precipitated by centrifugation (30 min. at 10,000 ×g and 4°C) after adding 0.2 volumes 1 M Na2SO4 and 3 volumes 100% ice-cold ethanol. The precipitate was washed twice with 70% ethanol, dried and hydrolysed with 250 μl 1 M HCl at 90°C for 1 hr. After neutralization with 1 M NaOH, glucose was determined enzymatically using Hexokinase in an Advia 1650 system.
Running wheel
Male, single-housed mice were provided with solid-floor running wheels with a diameter of 20 cm where running distance and time were monitored with a bicycle computer. Littermates at the age of 12-weeks (transgenic and their respective wild-type controls) were compared, and data were collected for a 4-week period and represent a daily average.
Morphological studies
Liver tissue was taken from 6-month-old male mice and histological analysis was performed. Steatosis was assessed in haematoxylin and eosin stained liver sections (5–8-μm thick) and blindly scored by an experienced pathologist. Hepatic steatosis was graded according to the percentage of lipid-laden hepatocytes.
Extensor digitorum longus and quadriceps muscles were taken out of 6-month-old male mice. Muscles were kept at in situ length and frozen in liquid nitrogen. Cryostat sections, 8-μm thick, were stained with Oil Red O.
Magnetic resonance imaging in mice
Magnetic resonance imaging (MRI) was performed on a 3T whole body imager (Magnetom Trio, Siemens Healthcare, Erlangen Germany) applying a T1-weighted spin-echo technique. Mice were placed in supine position in the wrist-coil of the manufacturer. Twenty axial images with a slice thickness of 2 mm and an interslice gap of 0.6 mm were recorded with an echo time of 10 ms and a repetition time of 400 ms with an in-plane resolution of 0.2 mm from the abdominal region to visualize abdominal fat distribution.
Statistical analysis
Results are expressed as mean ± S.E.M. For comparison between the groups, the unpaired Student’s t-test was used. P-values less than 0.05 were considered as statistically different.
Results
Up-regulation of PKC-β2 in skeletal muscle
To mimic insulin resistance primarily in skeletal muscle, we generated two MLC-PKC-β2 transgenic mouse lines that overexpress constitutively active PKC-β2 specifically in skeletal muscle. Mice were backcrossed into a C57Bl/6 background and displayed normal fertility and growth (data not shown).
PKC-β2 overexpression was measured by Northern blot (Fig. 1A, upper panel) as well as by specific immunoblotting of skeletal muscle extracts containing equal amounts of total protein (Fig. 1A, middle and lower panel). The data indicate a low overexpression of constitutively active PKC-β2 in the transgenic line 25 (tg 25), and a high expression in line 2 (tg 2). Tissue specificity was verified by immunoblotting divergent tissue lysates of wild-type and PKC-β2 transgenic mice for PKC-β2 expression. Thereby, no alterations in PKC-β2 protein levels were observed in brain, fat, kidney, liver, lung and heart tissues (Fig. 1B).
Fig 1.
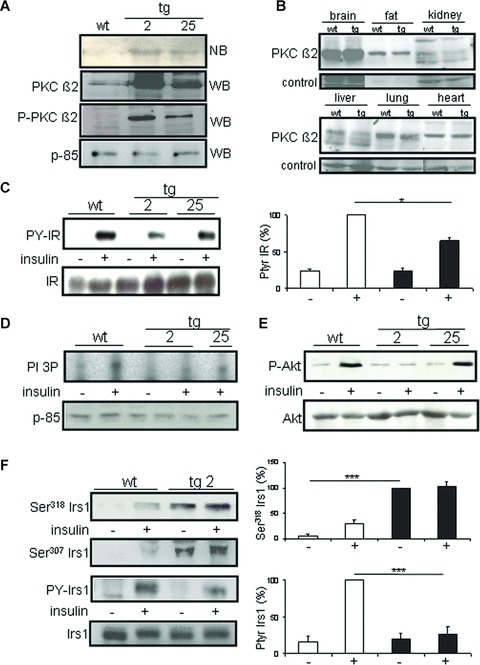
Protein kinase C (PKC)-β2 overexpression and insulin signalling in skeletal muscle of MLC-PKC-β2 transgenic mice. (A) Total cellular RNA was obtained from non-transgenic (wt) and transgenic (tg) mice (line #2 and #25) and analysed by Northern blot (NB, top panel). PKC-β2 protein levels and phosphorylated PKC-β2 detected by specific immunoblotting of total protein from muscle extracts of 12-week-old male mice (transgenic line #2 and #25) (WB, middle and bottom panel). Equal loading was verified by blotting p-85. (B) Western blot analysis of PKC-β2 expression on distinct tissues of wild-type (wt) and transgenic (tg) mice of the high expressing line 2. (C) Western blot analysis of IR tyrosine phosphorylation and expression in muscle lysates of PKC-β2 and wild-type mice. Animals were stimulated intravenously for 5 min. with insulin (+) or saline (–) as a control. Quantification for wt and the transgenic line 2 is given on the right, *P < 0.05. (D) PI 3-kinase activity and p-85 expression in skeletal muscle lysates of PKC-β2 transgenic and wild-type animals. (E) Phosphorylation of Akt on serine 473 and Akt expression in mouse skeletal muscle lysates of PKC-β2 transgenic and wild-type animals. Two transgenic lines (#2 and #25) were used. Each lane was loaded with muscle protein lysate from one animal and is representative of three independent experiments. (F) Mice were stimulated intravenously with insulin (+) or saline (−) as a control, and Irs1 immunoprecipitates from skeletal muscle lysates were immunoblotted with serine 318 (upper panel), serine 307 (middle panel), anti-phosphotyrosine (PY) and Irs1 (lower panels) antibodies in PKC-β2 transgenic and wild-type mice. Each lane was loaded with protein from one animal, and is representative of three independent experiments. Quantification is given on the right, ***P < 0.001.
PKC-β2 interferes with the insulin signalling cascade
To investigate the role of PKC-β2 on the insulin signalling cascade at the molecular level, we detected IR tyrosine phosphorylation in skeletal muscle tissue extracts of wild-type and PKC-β2 transgenic mice. Twelve-week-old mice were injected intravenously with a single bolus of human insulin and tyrosine phosphorylation of the IR in skeletal muscle was determined. As expected, overexpression of constitutively active PKC-β2 led to a diminished tyrosine phosphorylation following insulin bolus injection (Fig. 1C, P < 0.05). Moreover, insulin-induced PI 3-kinase activity was reduced in the PKC-β2 high expressing line (Fig. 1D), and consequently led to a diminished phosphorylation of Akt (Fig. 1E), whereas in line 25, the low expression level of PKC-β2 was not sufficient to cause alterations downstream at the level of Akt.
Previous data show that serine and tyrosine phosphorylation of Irs proteins are important modulators of the insulin signal in skeletal muscle [30–32]. We therefore determined phosphorylation of Irs1 on the two major serine phosphorylation sites 318 and 307 and on tyrosine residues in skeletal muscle of PKC-β2 transgenic and wild-type mice. Twelve-week-old mice were injected with human insulin, and phosphorylation was detected in muscle extracts. Looking at serine residues in Irs1, serine 318 phosphorylation was absent in fasted wild-type animals while it was clearly detectable following insulin stimulation. By contrast, serine 318 phosphorylation levels were already up-regulated in PKC-β2 transgenic animals in the basal state, and further stimulation was absent following insulin administration (Fig. 1F, upper panels). Identical results were obtained for serine 307 in Irs1 (Fig. 1F, middle panel), suggesting that PKC-β2 interferes with the insulin signalling cascade at the level of serine phosphorylation in Irs1 to down-regulate the insulin signal. While insulin stimulates tyrosine phosphorylation of Irs1 in wild-type animals, overexpression of constitutively active PKC-β2 leads to a diminished insulin-induced tyrosine phosphorylation of Irs1 (Fig. 1F, lower panels).
Metabolic effects of PKC-β2 overexpression in skeletal muscle
To define the impact of insulin resistance in skeletal muscle on whole-body metabolism, metabolic markers in PKC-β2 transgenic mice and their wild-type littermate controls were determined.
Male mice of the transgenic line 2 tended to be insulin resistant beginning at 12 weeks of age as determined by an intraperitoneal insulin tolerance test (Fig. 2A). This was accompanied by a 34% reduction in 2-deoxyglucose uptake into skeletal muscle (Fig. 2B). By contrast, 2-deoxyglucose uptake was not statistically different in the low expressing transgenic line 25, revealing a dose effect of PKC-β2 on glucose homeostasis (Fig. 2B).
Fig 2.
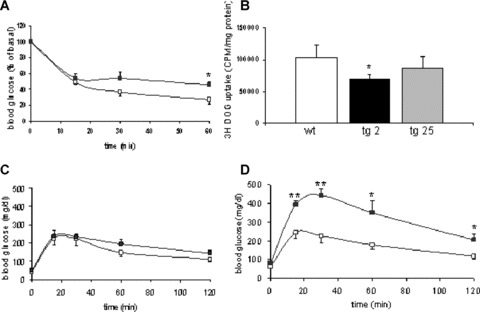
Insulin tolerance, glucose uptake and glucose tolerance test in PKC-β2 transgenic and wild-type mice. (A) Insulin tolerance test in 12-week-old PKC-β2 transgenic (black squares) and wild-type mice (white squares) after intraperitoneal injection of 1 unit/kg body weight of human regular insulin. Means ± S.E.M., n= 4. *P < 0.05. (B) 2-DOG uptake into skeletal muscle following intraperitoneal glucose injection in wild-type and PKC-β2 transgenic mice. Means ± S.E.M., n= 6. *P < 0.05. (C) Glucose tolerance test in 12-week-old PKC-β2 transgenic (black squares) and wild-type mice (white squares) after intraperitoneal injection of glucose. Means ± S.E.M., n= 4. (D) Glucose tolerance test in 6-month-old PKC-β2 transgenic (black squares) and wild-type mice (white squares) after intraperitoneal injection of glucose. Means ± S.E.M., n= 4. *P < 0.05, **P < 0.01.
As a result of PKC-β2 overexpression, transgenic animals of the high expressing line displayed slightly increased fasting glucose concentrations (52 ± 5 mg/dl versus 43 ± 2 mg/dl) in the presence of elevated plasma insulin levels (3.7 ± 0.6 ng/ml versus 2.2 ± 0.4 ng/dl) to compensate for peripheral insulin resistance; but overall glucose homeostasis determined by fasting blood glucose concentrations or intraperitoneal glucose tolerance test was not significantly different in 12-week-old mice (Fig. 2C). Insulin resistance progresses in skeletal muscle of aging C57Bl/6 mice [33], and hyperinsulinemia was not sufficient to fully compensate for increasing insulin resistance in PKC-β2 transgenic mice by 6 months of age as determined by elevated fasting glucose levels (82 ± 6 mg/dl versus 62 ± 1 mg/dl, P < 0.01). Serum insulin levels in the fed state declined in 6-month-old transgenic mice (1.44 ± 0.5 ng/dl versus 3.4 ± 0.4 ng/dl, n= 8, P < 0.05) and were accompanied with impaired intraperitoneal glucose tolerance tests (Fig. 2D) while body weight (wt: 30.0 ± 2.6 g versus tg: 31.0 ± 2.7 g) and food intake (wt: 3.9 g/day ± 0.3 g/day versus tg: 4.2 ± 0.6 g/day) were indistinguishable between groups.
Alterations in running capacity, lipid oxidation, and fat deposition in PKC-β2 transgenic mice
Based on our finding that PKC-β2 is sufficient to cause impaired insulin action, we went on to define the consequences for glycogen content in skeletal muscle. Thereby, overexpression of PKC-β2 led to impaired levels of muscle glycogen (wt: 2.07 ± 0.09 mg/g tissue versus tg: 1.51 ± 0.17 mg/g tissue, P < 0.05, n= 6), and compared to their littermate controls, transgenic mice were less physically active as determined by voluntary running using a running wheel (3.41 ± 0.32 km/day versus 6.08 ± 0.96 km/day, P < 0.005). While the duration of exercise was greatly diminished in the insulin resistant model compared to their wild-type littermate controls (3.25 ± 0.56 hr/day versus 5.07 ± 0.8 hr/day, P < 0.05), the speed was indistinguishable (P= 0.45).
As physical activity is tightly linked to the expression of oxidative genes and fat deposition in skeletal muscle, mRNA levels were determined in PKC transgenic mice. Thus, insulin resistant PKC-β2 transgenic animals displayed diminished expression of the nuclear hormone receptor peroxisome proliferator-activated receptor (PPAR)-γ and PPAR-γ coactivator-1β (PGC-1β) in skeletal muscle that are associated with alterations in genes involved in fatty acid oxidation (acyl-CoA oxidase, ACO) as well as lower expression of the intracellular hormone-sensitive lipase (HSL) that plays a pivotal role in lipolysis. Whereas adipose triglyceride lipase (ATGL) and CD36 were not significantly altered, sterol regulatory element-binding protein 1c (SREBP1c) involved in lipogenesis was up-regulated (Table 1). Moreover, palmitate oxidation was diminished in skeletal muscle of transgenic mice compared to controls (wt: 34.7 ± 10.0 cpm/mg tissue versus tg: 19.7 ± 8.6 cpm/mg tissue, P= 0.06, n= 3), suggesting that overexpression of PKC-β2 is sufficient to reduce the capacity to oxidize palmitate in skeletal muscle. As a consequence of the observed alterations in lipid oxidation, lipolysis and lipogenesis, profound intramyocellular fat deposition in skeletal muscle of PKC-β2 transgenic mice was revealed by histochemistry (Fig. 3A), and transgenic mice tend to display lower mitochondrial mass as determined by citrate synthase activity (wt: 0.025 ± 0.004 μmol/ml/min. versus tg: 0.015 ± 0.003 μmol/ml/min., P= 0.08, n= 4).
Table 1.
Gene expression in skeletal muscle and liver tissue of wild-type and PKC-β2 transgenic mice. Expression of genes were measured in 6-month-old male wild-type and PKC-β2 transgenic mice (n= 4–8) and liver tissue from 3-month-old male mice fed chow or high-fat diet (HFD), n= 3. Data are given as relative arbitrary units ± S.E.M. See methods for diet details and text for gene names.
| Skeletal muscle | Wild-type | PKC-β2 tg | P-value |
|---|---|---|---|
| PPAR delta/28S | 4.1 ± 0.5 | 2.7 ± 0.1 | <0.05 |
| PGC-lbeta/28S | 1.6 ± 0.06 | 0.8 ± 0.1 | <0.001 |
| HSL/28S | 3.6 ± 0.5 | 2.4 ± 0.2 | <0.01 |
| ATGL/28S | 4.9 ± 0.4 | 4.8 ± 0.1 | =0.84 |
| SREBPlc/28S | 1.4 ± 0.1 | 2.5 ± 0.2 | <0.05 |
| CD36/28S | 0.54 ± 0.05 | 0.55 ± 0.04 | 0.81 |
| ACO/28S | 0.1 ± 0.06 | 0.06 ± 0.007 | <0.01 |
| Liver | Wild-type | PKC-β2 tg | P-value |
| G6Pase/28S | 3.1 ± 0.9 | 2.1 ± 0.3 | =0.48 |
| Fetuin A/28S | 2.90 ± 0.50 | 2.60 ± 0.48 | =0.49 |
| SREBPlc/28S | 2.30 ± 1.2 | 2.3 ± 0.2 | =0.97 |
| Interleukin-6/28S | 2.0 ± 0.5 | 2.7 ± 0.4 | =0.25 |
| TNF-α/28S | 1.10 ± 0.3 | 0.78 ± 0.07 | =0.25 |
| Liver | Chow | HFD | P-value |
| G6Pase/28S | 1.38 ± 0.07 | 0.96 ± 0.04 | <0.001 |
| Fetuin A/28S | 1.36 ± 0.32 | 3.66 ± 0.73 | <0.001 |
| SREBPlc/28S | 1.7 ± 0.22 | 4.0 ± 0.93 | <0.001 |
Fig 3.
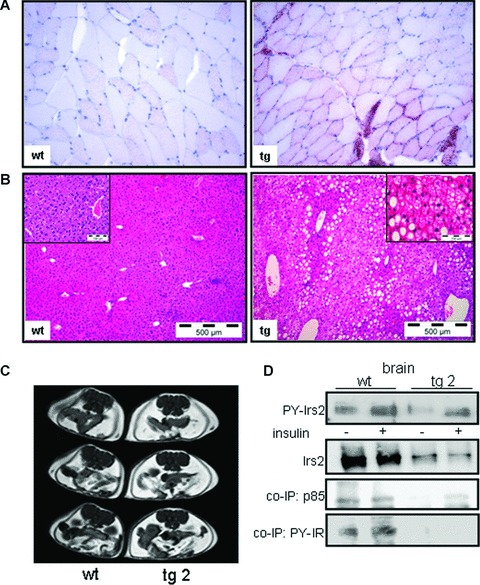
Histological analysis in muscle and liver tissues, MRT and insulin signalling in brain tissues in PKC-β2 transgenic and wild-type mice. (A) Muscle tissue was taken from wild-type (wt) and transgenic (tg) mice. Some fibres of the transgenic animal show intense lipid droplet accumulation using Oil Red O staining. Representative slides out of three animals are shown. (B) Histological analysis in liver tissues by haematoxylin and eosin stained liver sections in 6-month-old wt and tg mice. Hepatic steatosis was graded by lipid-loaded hepatocytes in three animals per genotype. (C) T1-weighted MR images of a 6-month-old wild-type (wt) or PKC-β2 transgenic (tg) mouse to detect intra-abdominal fat mass. (D) Western blot analysis of Irs2 immunoprecipitates detecting tyrosine phosphorylation (PY-Irs2) and expression in brain tissues of PKC-β2 and wild-type mice. Animals were stimulated intravenously for 5 min. with insulin or saline as a control. Tyrosine phosphorylation of Irs2 co-immunoprecipitated IR (PY-IR) and of p-85 was detected by using the respective antibodies. A representative immunoblot is shown out of three independent experiments, for quantification see result section.
Fat distribution and impaired insulin action and brain tissues
Since metabolic alterations related to impaired oxidative capacity are not restricted to skeletal muscle itself, we went on to define the acquired defects in individual tissues that are secondary to this phenotype. Thereby, Oil Red staining in liver tissue displayed that the metabolic phenotype in PKC-β2 transgenic mice was accompanied by fat accumulation in the liver (Fig. 3B) to the same degree as it was observed in mice fed a HFD where >90% of the hepatocytes were lipid-laden [26]. However, markers for non-alcoholic fatty liver disease (NAFLD) like glucose 6-phosphatase, fetuin-A [34], SREBP1c, IL-6 and TNF-α failed to be statistically different upon the sole genetic alteration in skeletal muscle. By comparison, we determined expression of glucose 6-phosphatase (G6Pase), fetuin-A [26] and SREBP1c in liver tissue of HFD fed animals that displayed comparable fat accumulation and found significant regulation of these genes, suggesting divergent quality of fat storage in liver tissues (Table 1).
As the pre-diabetic state is characterized by increased fat mass, we made use of the magnetic resonance imaging technique to determine whether primary insulin resistance in skeletal muscle drives abdominal fat accumulation. Thereby, transgenic mice displayed elevated fat mass compared to wild-type controls (Fig. 3C), and showed significantly higher levels of triglycerides (wt: 53.3 ± 4.6 mg/dl versus tg: 80.0 ± 7.2 mg/dl, n= 12, P > 0.001). Leptin concentrations were markedly increased in the fasted state (tg: 34.14 ± 4.9 ng/ml versus wt: 16.88 ± 6.3 ng/ml, P < 0.05), while adiponectin levels in wild-type and PKC-β2 transgenic mice displayed no significant differences (wt: 13.945 ± 1576 ng/ml versus tg: 12.728 ± 1311 ng/ml, n= 10, P= 0.58). Moreover, the phenotype included significantly elevated free fatty acid concentrations in the fasted state (1755 ± 250 μmol/l versus 1264 ± 176 μmol/l, P < 0.05). While insulin injection was able to suppress FFA levels in the wild-type animals by approximately 20% (P < 0.05), insulin completely failed to suppress lipolysis in PKC-β2 transgenic mice, suggesting that insulin-mediated suppression of lipolysis is disturbed in PKC-β2 transgenic mice.
To determine the consequences of this whole-body metabolic phenotype on insulin action in the brain, wild-type and transgenic mice were injected intravenously with insulin and brain tissue was harvested. As Irs2 is the most prominent post-receptor molecule in the insulin signalling pathway in the brain, we detected tyrosine phosphorylation of Irs2 in brain tissues following intravenous insulin stimulation. Thereby, insulin resistance primarily induced in skeletal muscle was sufficient to diminish insulin action in brain tissues at the level of Irs2 by 75%± 11% (P < 0.01, n= 3), as tyrosine phosphorylation as well as the amount of Irs2 co-immunoprecipitated p85 and phosphorylated IRs were greatly impaired (Fig. 3D). Of note, Irs2 expression was lowered by 83%± 3% (P < 0.001) in the brain of insulin resistant transgenic mice, suggesting that the aversive metabolic phenotype facilitates insulin resistance at the level of Irs2 expression in brain tissues.
Discussion
Impaired insulin action is a pivotal element in the development of type 2 diabetes, and among others, PKC isoforms are known to interfere with the insulin signalling cascade to cause such resistance. Based on these data, we set out to generate a mouse model that selectively overexpresses constitutively active PKC-β2 in skeletal muscle of mice to resemble a permanent defect primarily in skeletal muscle. Thereby, insulin action was inhibited at the level of the IR and Irs1 through phosphorylation at multiple serine sites [15, 16, 35, 36] and the overexpression therefore mimics an early pathophysiological state that is consistently found in obese and pre-diabetic subjects.
In our mouse model, disruption of the insulin signal in skeletal muscle resulted in disturbances in whole-body glucose homeostasis, fatty acid metabolism, and fat accumulation. The underlying mechanisms by which insulin resistance in skeletal muscle drives fat deposition are difficult to establish, however, data from muscle-specific IR knockout mice suggested that reduced glucose uptake into skeletal muscle is in part compensated by an increased uptake into fat tissue [37]. Moreover, muscle specific IR knockout animals displayed even elevated adiponectin levels in the presence of an increased number of adipocytes [38], while in our model, adiponectin levels were unaltered suggesting that adiponectin is not exclusively regulated by visceral adipose tissue mass.
In addition, our mouse model demonstrated that impaired insulin action in skeletal muscle is one of the primary causes of fat accumulation in tissues such as liver and skeletal muscle that in turn worsens the metabolic phenotype. Intramyocellular lipids (IMCL) are lipid droplets that are located in the sarcoplasm and are predominantly found in the vicinity of mitochondria, suggesting that they may serve as a rapidly available energy source that drives muscle fatty acid oxidation. In healthy subjects, physical training increased the capacity of fat oxidation, whereas consumption of a HFD favours fat storage in muscle rather than oxidation [39], and this is also true for our insulin resistant mouse model. Therefore, an increase in IMCL requires sufficient fat oxidative capacity to lower lipid metabolites [40]. This hypothesis is further supported by data showing a strong negative correlation between IMCL and insulin sensitivity in untrained subjects [41], and levels of IMCL are elevated in first-degree relatives of patients that are insulin resistant [42]. Thus, the absence of adaptation in oxidative capacity in a (pre)diabetic milieu as present in our mouse model may account for high IMCL levels.
Many of the oxidative genes are under transcriptional control of PPAR-γ and PGC-1β that stimulate oxidative phosphorylation and mitochondrial biogenesis. Similar to the observation in our transgenic mouse model, PGC-1 gene expression was repressed in patients with type 2 diabetes [43], and in turn, PGC-1β transgenic muscle fibres are mitochondria-rich and highly oxidative [44]. Whether PGC-1 down-regulation in PKC-β2 transgenic mice is a consequence of physical inactivity or directly regulated by PKC-β2 and/or impaired insulin action needs to be determined. However, given the fact that lipid infusion into healthy subjects resulted in an increase in free fatty acid concentrations, and is followed by decreased PGC-1 levels [45], it is likely that the metabolic environment causes a down-regulation of PGC-1 in PKC-β2 transgenic mice.
Similarly, the PPAR-γ target gene HSL plays a crucial role in lipid metabolism as it mobilizes fatty acids in multiple tissues. For example, HSL is dramatically decreased in fat tissue of obese subjects but can be up-regulated with PPAR-γ agonists and therefore modulate triglyceride storage [46]. Even more, SREBP1c mRNA expression in skeletal muscle of PKC-β2 transgenic mice was up-regulated as in morbid obese subjects [47] where massive lipid malabsorption after bariatric surgery resulted in a significant reduction in SREBP-1c mRNA expression in skeletal muscle, suggesting that this transcription factor might be involved in the accumulation of triglycerides in muscle cells of obese [48].
In addition, besides alterations in lipid deposition in skeletal muscle, patients with type 2 diabetes exhibit severe abnormalities in the disposal of liver triglycerides termed hepatic steatosis and elevated hepatocellular lipid levels mainly account for hepatic insulin resistance [1]. In contrast to previous animal models that were not effective in clarifying the consequences of insulin resistance in skeletal muscle on insulin action in liver tissue, our approach using constitutively active PKC-β2 clearly demonstrated that impaired insulin action in skeletal muscle is sufficient to mediate a fatty liver phenotype. Therefore, it can be speculated that an increase in the free fatty acid flux to the liver and other tissues, where FACoA favours triglyceride synthesis and fat deposition.
Based on the observation that insulin resistance in the brain resulted in an obese and inactive phenotype [49], and that chronically elevated insulin, leptin and free fatty acid concentrations as present in insulin resistant animals and human beings down-regulate the insulin signal, we went on to define the consequences of primary insulin resistance in skeletal muscle on insulin action in brain tissues. Obviously, the metabolic phenotype as observed in the PKC-β2 transgenic mouse model acts as a repressor of the insulin signal in the brain and might therefore worsen physical activity and fat deposition while it was not sufficient to increase food intake. In this respect, further experiments using distinct metabolites as present in obesity and insulin resistance are needed to address the underlying mechanisms.
In summary, our data demonstrate that PKC-β2 overexpression in skeletal muscle is sufficient to down-regulate the insulin signalling cascade and to drive alterations in whole-body glucose and lipid metabolism, physical activity, fat accumulation, and is able to communicate with and regulate insulin action in other tissues such as the brain.
Therefore, insulin resistance in skeletal muscle is the basis for disturbances in glucose and lipid metabolism that are accompanied by disproportionate fat storage and impaired physical fitness that are in turn harmful to the metabolic state.
Acknowledgments
This work was in part supported by the European Community’s FP6Eugene2 (LSHM-CT-2004–512013 to H.-U.H.) program, the Deutsche Forschungsgemeinschaft (KFO 114/2, HE 3653/3–1 to A.M.H.) and the Deutsche Diabetes Gesellschaft.
Conflict of interest
The authors have no conflict of interest related to the present study.
References
- 1.Roden M. Mechanisms of Disease: hepatic steatosis in type 2 diabetes-pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab. 2006;2:335–48. doi: 10.1038/ncpendmet0190. [DOI] [PubMed] [Google Scholar]
- 2.Michael MD, Kulkarni RN, Postic C, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 3.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–34. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tschritter O, Preissl H, Hennige AM, et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: a magnetoencephalographic study. Proc Natl Acad Sci USA. 2006;103:12103–8. doi: 10.1073/pnas.0604404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruning JC, Gautam D, Burks DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–25. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 6.Nandi A, Kitamura Y, Kahn CR, et al. Mouse models of insulin resistance. Physiol Rev. 2004;84:623–47. doi: 10.1152/physrev.00032.2003. [DOI] [PubMed] [Google Scholar]
- 7.Bruning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–69. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 8.Charron MJ, Gorovits N, Laidlaw JS, et al. Use of GLUT-4 null mice to study skeletal muscle glucose uptake. Clin Exp Pharmacol Physiol. 2005;32:308–13. doi: 10.1111/j.1440-1681.2005.04189.x. [DOI] [PubMed] [Google Scholar]
- 9.Cariou B, Postic C, Boudou P, et al. Cellular and molecular mechanisms of adipose tissue plasticity in muscle insulin receptor knockout mice. Endocrinology. 2004;145:1926–32. doi: 10.1210/en.2003-0882. [DOI] [PubMed] [Google Scholar]
- 10.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandyopadhyay GK, Yu JG, Ofrecio J, et al. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes. 2005;54:2351–59. doi: 10.2337/diabetes.54.8.2351. [DOI] [PubMed] [Google Scholar]
- 12.Farese RV, Sajan MP, Standaert ML. Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans. 2005;33:350–3. doi: 10.1042/BST0330350. [DOI] [PubMed] [Google Scholar]
- 13.Liberman Z, Plotkin B, Tennenbaum T, et al. Coordinated phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 and protein kinase C{beta}II in the diabetic fat tissue. Am J Physiol Endocrinol Metab. 2008;294:E1169–77. doi: 10.1152/ajpendo.00050.2008. [DOI] [PubMed] [Google Scholar]
- 14.Rajasekar P, Palanisamy N, Anuradha CV. Increase in nitric oxide and reductions in blood pressure, protein kinase C beta II and oxidative stress by L-carnitine: a study in the fructose-fed hypertensive rat. Clin Exp Hypertens. 2007;29:517–30. doi: 10.1080/10641960701743998. [DOI] [PubMed] [Google Scholar]
- 15.Strack V, Hennige AM, Krutzfeldt J, et al. Serine residues 994 and 1023/25 are important for insulin receptor kinase inhibition by protein kinase C isoforms beta2 and theta. Diabetologia. 2000;43:443–9. doi: 10.1007/s001250051327. [DOI] [PubMed] [Google Scholar]
- 16.Bossenmaier B, Mosthaf L, Mischak H, et al. Protein kinase C isoforms beta 1 and beta 2 inhibit the tyrosine kinase activity of the insulin receptor. Diabetologia. 1997;40:863–6. doi: 10.1007/s001250050761. [DOI] [PubMed] [Google Scholar]
- 17.Moeschel K, Beck A, Weigert C, et al. Protein kinase C-zeta-induced phosphorylation of Ser318 in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem. 2004;279:25157–63. doi: 10.1074/jbc.M402477200. [DOI] [PubMed] [Google Scholar]
- 18.Nawaratne R, Gray A, Jorgensen CH, et al. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: role of serine 24 phosphorylation. Mol Endocrinol. 2006;20:1838–52. doi: 10.1210/me.2005-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hennige AM, Stefan N, Kapp K, et al. Leptin down-regulates insulin action through phosphorylation of serine-318 in insulin receptor substrate 1. FASEB J. 2006;20:1206–8. doi: 10.1096/fj.05-4635fje. [DOI] [PubMed] [Google Scholar]
- 20.Mussig K, Staiger H, Fiedler H, et al. Shp2 is required for protein kinase c-dependent phosphorylation of serine 307 in insulin receptor substrate-1. J Biol Chem. 2005;280:32693–9. doi: 10.1074/jbc.M506549200. [DOI] [PubMed] [Google Scholar]
- 21.Rosenthal N, Kornhauser JM, Donoghue M, et al. Myosin light chain enhancer activates muscle-specific, developmentally regulated gene expression in transgenic mice. Proc Natl Acad Sci USA. 1989;86:7780–4. doi: 10.1073/pnas.86.20.7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donoghue MJ, Merlie JP, Rosenthal N, et al. Rostrocaudal gradient of transgene expression in adult skeletal muscle. Proc Natl Acad Sci USA. 1991;88:5847–51. doi: 10.1073/pnas.88.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otaegui PJ, Ferre T, Pujol A, et al. Expression of glucokinase in skeletal muscle: a new approach to counteract diabetic hyperglycemia. Hum Gene Ther. 2000;20:1543–52. doi: 10.1089/10430340050083270. [DOI] [PubMed] [Google Scholar]
- 24.Hogan B, Costantini F, Lacy E. A laboratory manual. 2006. Manipulating the mouse embryo. Cold Spring Harbor, New York; [Google Scholar]
- 25.Chirgwin JM, Przybyla AE, MacDonald RJ, et al. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–99. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 26.Stefan N, Hennige AM, Staiger H, et al. Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care. 2006;29:853–7. doi: 10.2337/diacare.29.04.06.dc05-1938. [DOI] [PubMed] [Google Scholar]
- 27.Hennige AM, Lehmann R, Weigert C, et al. Insulin glulisine: insulin receptor signaling characteristics in vivo. Diabetes. 2005;54:361–6. doi: 10.2337/diabetes.54.2.361. [DOI] [PubMed] [Google Scholar]
- 28.Kellerer M, Koch M, Metzinger E, et al. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2)dependent pathways. Diabetologia. 1997;40:1358–62. doi: 10.1007/s001250050832. [DOI] [PubMed] [Google Scholar]
- 29.Chan TM, Exton JH. A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal Biochem. 1976;71:96–105. doi: 10.1016/0003-2697(76)90014-2. [DOI] [PubMed] [Google Scholar]
- 30.Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia. 2004;47:170–84. doi: 10.1007/s00125-003-1313-3. [DOI] [PubMed] [Google Scholar]
- 31.Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Arch Pharm Res. 2004;27:361–70. doi: 10.1007/BF02980074. [DOI] [PubMed] [Google Scholar]
- 32.Hribal ML, Tornei F, Pujol A, et al. Transgenic mice overexpressing human G972R IRS-1 show impaired insulin action and insulin secretion. J Cell Mol Med. 2008;12:2096–106. doi: 10.1111/j.1582-4934.2008.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh YS, Khil LY, Cho KA, et al. A potential role for skeletal muscle caveolin-1 as an insulin sensitivity modulator in ageing-dependent non-obese type 2 diabetes: studies in a new mouse model. Diabetologia. 2008;51:1025–34. doi: 10.1007/s00125-008-0993-0. [DOI] [PubMed] [Google Scholar]
- 34.Hennige AM, Staiger H, Wicke C, et al. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS ONE. 2008;3:e1765. doi: 10.1371/journal.pone.0001765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chin JE, Liu F, Roth RA. Activation of protein kinase C alpha inhibits insulin-stimulated tyrosine phosphorylation of insulin receptor substrate-1. Mol Endocrinol. 1994;8:51–8. doi: 10.1210/mend.8.1.7512195. [DOI] [PubMed] [Google Scholar]
- 36.Formisano P, Oriente F, Fiory F, et al. Insulin-activated protein kinase Cbeta bypasses Ras and stimulates mitogen-activated protein kinase activity and cell proliferation in muscle cells. Mol Cell Biol. 2000;20:6323–33. doi: 10.1128/mcb.20.17.6323-6333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JK, Michael MD, Previs SF, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–7. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cariou B, Postic C, Boudou P, et al. Cellular and molecular mechanisms of adipose tissue plasticity in muscle insulin receptor knockout mice. Endocrinology. 2004;145:1926–32. doi: 10.1210/en.2003-0882. [DOI] [PubMed] [Google Scholar]
- 39.Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, et al. Intramyocellular lipid content and molecular adaptations in response to a 1-week high-fat diet. Obes Res. 2005;13:2088–94. doi: 10.1038/oby.2005.259. [DOI] [PubMed] [Google Scholar]
- 40.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–6. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perseghin G, Scifo P, De Cobelli F, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48:1600–6. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- 42.Jacob S, Machann J, Rett K, et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–9. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 43.Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–71. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arany Z, Lebrasseur N, Morris C, et al. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Richardson DK, Kashyap S, Bajaj M, et al. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–7. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 46.Deng T, Shan S, Li PP, et al. Peroxisome proliferator-activated receptor-gamma transcriptionally up-regulates hormone-sensitive lipase via the involvement of specificity protein-1. Endocrinology. 2006;147:875–84. doi: 10.1210/en.2005-0623. [DOI] [PubMed] [Google Scholar]
- 47.Kamei Y, Miura S, Suganami T, et al. Regulation of SREBP1c gene expression in skeletal muscle: role of RXR/LXR and FOXO1. Endocrinology. 2008;149:2293–330. doi: 10.1210/en.2007-1461. [DOI] [PubMed] [Google Scholar]
- 48.Mingrone G, Rosa G, Greco AV, et al. Intramyocitic lipid accumulation and SREBP-1c expression are related to insulin resistance and cardiovascular risk in morbid obesity. Atherosclerosis. 2003;170:155–61. doi: 10.1016/s0021-9150(03)00254-5. [DOI] [PubMed] [Google Scholar]
- 49.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–72. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]