

Abstract
The analysis of rare cells is not an easy task. This is especially true when cells representing a fetal microchimerism are to be utilized for the purpose of non-invasive prenatal diagnosis because it is both imperative and difficult to avoid contaminating the minority of fetal cells with maternal ones. Under these conditions, even highly specific biochemical markers are not perfectly reliable. We have developed a method to verify the genomic identity of rare cells that combines automatic screening for enriched target cells (based on immunofluorescence labelling) with isolation of single candidate microchimeric cells (by laser microdissection and subsequent laser catapulting) and low-volume on-chip multiplex PCR for DNA fingerprint analysis. The power of the method was tested using samples containing mixed cells of related and non-related individuals. Single-cell DNA fingerprinting was successful in 74% of the cells analysed (55/74), with a PCR efficiency of 59.2% (860/1452) for heterozygous loci. The identification of cells by means of DNA profiling was achieved in 100% (12/12) of non-related cells in artificial mixtures and in 86% (37/43) of cells sharing a haploid set of chromosomes and was performed on cells enriched from blood and cells isolated from tissue. We suggest DNA profiling as a standard for the identification of microchimerism on a single-cell basis.
Keywords: microchimerism, prenatal diagnosis, rare cell analysis, single-cell PCR
Introduction
Microchimerism constitutes the presence of a small number of cells that are genetically distinct from the host individual [1]. Examples have been reported of maternal cells present in the tissue of offspring (maternal microchimerism) [2] and, conversely, of fetal cells present in maternal tissues (fetal microchimerism) [3], in which these cells may survive even for years after delivery [4]. This phenomenon has been linked to autoimmune diseases [2, 5, 6], but it is also the basis of long-standing efforts to implement non-invasive prenatal diagnosis (NIPD) based on molecular genetic analysis of fetal cells present in the circulation of pregnant women. Unfortunately, because these cells are very rare [7, 8], single-cell PCR in rare cell analysis faces additional problems when compared with similar applications in pre-implantation genomic diagnosis (PGD). It is obviously necessary to enrich the rare cells and discriminate them from the vast majority of maternal cells. This is primarily done by sorting and staining for biochemical parameters, which introduces the basic problem of population overlap even when highly specific markers are used [9]. The use of fluorescence in situ hybridization (FISH) analysis presents similar challenges, because false-positive FISH results are common and it is even more difficult to screen large numbers of cells for rare FISH-positive cells than for positive cells in immunocytochemical staining.
During the last two decades, efforts directed towards cell-based NIPD have focused mainly on fetal erythroblasts and trophoblast cells (which are not expected to persist in the maternal circulation in subsequent pregnancies) for the purpose of analysis of fetal sex [10, 11], aneuploidy [12, 13] or single-gene disorders such as cystic fibrosis [14] and haemoglobinopathies, including thalassemia [15–17]. Fetal erythroblasts have turned out to be difficult to handle, as they show evidence of apoptosis [18, 19] and nucleic shrinking when exposed to the pO2 of maternal blood, leading to low FISH efficiency [19]. Furthermore, only a minor fraction express the ɛ chain of haemoglobin (Hbɛ), a specific marker for discrimination of embryonic and early fetal erythroblasts from maternal ones [16, 20]. In the 15th (mean) week of pregnancy, approximately half of the erythroblasts in the maternal circulation were proved to be of fetal origin [21]. Thus, pooling of fetal cells to increase the efficiency of PCR analysis can result in contamination with maternal cells.
The trophoblast cell, which originates from the placenta rather than from the foetus, still carries the fetal genome. This cell type can be expanded after enrichment by subsequent short-term culture [22]. Although biochemical markers exist for specific labelling of trophoblast cells and Hbɛ-positive erythroblasts, allowing them to be allocated to a candidate fetal cell status under the conditions of rare cell analysis, the identification of the fetal character of other interesting target cells such as fetal stem cells or progenitor cells [23–25] relies almost exclusively on a molecular genetic basis, using Y-FISH or multiplex PCR of polymorphic small tandem repeat (STR) loci. FISH has been optimized to fit rare cell conditions using two different Y probes [26] and reverse XY-FISH [27] but the identification of fetal cells based on Y-FISH does not allow for a diagnosis in the case of female foetuses. Multiplex PCR using microsatellite loci is most promising, as it allows for sex-independent identification of cells [28] and, in combination, for molecular genetic diagnosis [29].
Although PCR on single unfixed cells has been established, the analysis of fixed and stained rare cells remains a challenge [30]. In addition to procedure-related DNA degradation due to fixation and staining, single-cell PCR is prone to PCR failure, allele drop-out (ADO) and the appearance of artificial alleles (allele drop-in [ADI]) [10, 28, 30, 31]. DNA fingerprinting should be set to improve the identification of single cells; however, the costs of using commercially available kits should not be overlooked. Recently, low-volume PCR carried out on a DNA dilution series showed that DNA fingerprinting yields a full profile from as little as 32 pg of DNA [32]. This technique allows cell lysis and subsequent DNA amplification from end volumes of 1.5 μl on a chemically modified chip that is designed for optimal control of microdissected cells.
In order to improve the identification of microchimeric cells and, at the same time, to address the economics of genetic screening during pregnancy, we developed a method combining automated cell detection based on immunofluorescently labelled cells with laser microdissection and subsequent low-volume on-chip PCR. In two experimental settings, we first evaluated our method of single rare cell analysis by spiking peripheral blood mononucleated cells (PBMNCs) with trophoblast-like JAR cells (‘non-related’ individuals). This was done through automated detection of labelled cells by means of immunofluorescence, followed by laser microdissection and DNA fingerprinting using low-volume on-slide PCR technology. Second, we tested whether 16-plex PCR of highly variable loci would allow us to identify single cells derived from individuals sharing a haploid set of chromosomes, as this is the case for fetal and maternal cells. For this purpose, single cells prepared from placental villi (most of which carry the fetal genome) and maternal decidua (representing a mixture of maternal and genomically fetal trophoblast cells) were assigned candidate fetal or maternal origin based on the staining of biochemical markers using anti-Hbɛ and anti-trophoblast antibodies. In the second step, this assignment was confirmed or disproved by DNA profiling. We show that in this way it is possible to verify the genomic identity of the cells analysed.
Materials and methods
Ethics
The present study was approved by the Ethics Committee of the Medical University of Graz, Austria (no. 16–187 ex 04/05) and Sheba Medical Center’s Ethics Board (Israel, exp. no. 97426). Informed consent was obtained from all participants, according to the World Medical Association Declaration of Helsinki.
Antibodies
To enrich and label trophoblast cells, we used the mouse monoclonal antibodies GZ 112 and GZ 158, both of IgG1 isotype. GZ 112 has been described previously [33]. GZ 158 was generated from a fusion of P3-NS1-Ag4-1 myeloma cells with spleen cells from a BALB/c mouse immunized with homogenized Jeg-3 human choriocarcinoma cells according to the method described by Goding [34]. The hybridoma supernatants were tested for binding specificity to trophoblast cells by immunohistochemistry on frozen sections of first trimester placenta and decidua and on cytospin preparations of PBMNCs from a healthy individual. In addition, in order to determine whether these antibodies bind to the cell surface, FACS analysis was performed on purified (90%) trophoblast cells prepared from placental villi of elective abortions during the 9th and 10th weeks of pregnancy [35]. The antibodies were then produced on a larger scale, were purified (BioGenes, Berlin, Germany) and then were used for immunocytochemical staining at final concentrations of 2.24 μg/ml (GZ 112) and 2.76 μg/ml (GZ 158). Anti-Hbγ (both unlabelled and FITC-labelled) and anti-Hbɛ were purchased from Europa Bioproducts (Ely, UK) and used at 5 μg/ml concentration. As the second-step antibody, we utilized goat-antimouse F(ab′)2-FITC (Dako, Vienna, Austria) at 5 μg/ml concentration.
Cell lines
We used the human Y chromosome-positive choriocarcinoma cell line JAR (American Tissue Type Collection, HBT-144, LGC Standards, Wesel, Germany). The cells were cultured in RPMI 1640 medium (Biological Industries, Beit Haemek, Israel) supplemented with 10% FBS (Biological Industries), 2 mM L-glutamine (Biological Industries) and 1% penicillin/ streptomycin (Biological Industries). The cell line was maintained at 37°C in a humidified atmosphere of 5% CO2 for 7 days.
Preparation of blood samples, spiking with JAR cells and enrichment by magnetic cell sorting
In order to mimic microchimerism, contamination experiments were performed in which JAR cells were added to PBMNCs at a low concentration and a trophoblast enrichment protocol was applied [22]. At the Danek Gertner Institute of Human Genetics (Israel), blood samples from three non-pregnant women were drawn into heparin-washed tubes and separated by density-gradient centrifugation (1.077 g/ml) in Unisep tubes (Novamed, Jerusalem, Israel) for 20 min. at 1000 ×g at room temperature. From each sample, 1.5 × 107 PBMNCs were then mixed with 1500 JAR cells, resulting in a quantitative relation of 104:1. The cell preparation was incubated with the antibody GZ 158 (final concentration 96 μg/ml in PBS containing 0.1% bovine serum albumin [BSA]) for 30 min. After washing the cells with 500 μl of MACS buffer (PBS containing 2 mM ethylenediaminetetraacetic acid [EDTA] and 0.5% BSA), the pellet was incubated for 30 min. with 200 μl of FITC-labelled donkey-antimouse F(ab′)2 (1:50 in PBS/0.1% BSA; Jackson Immuno Research Europe, Suffolk, UK), washed and re-suspended in 20 μl of anti-FITC magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) diluted in 80 μl of MACS buffer. Following incubation for 15 min. at 4°C on a shaker and another washing step, the cells were re-suspended in 500 μl of MACS buffer and loaded onto a MiniMACS column (Miltenyi Biotec). MACS-positive cells were eluted according to the manufacturer’s protocol. Approximately 4 × 104 of the positive cells were loaded onto eight polyethylene-napthalate (PEN) membrane-coated glass slides (PALM Microlaser Technologies, Bernried, Germany) using cytocentrifugation (Shandon Cytospin 3; Thermo Fisher Scientific, Waltham, MA, USA). The cells on the slides were fixed in 1% formaldehyde (Sigma-Aldrich, Vienna, Austria) or methanol for 15 min., respectively, and rinsed twice for 5 min. in PBS. The air-dried slides were then shipped to Austria where nuclei were counterstained with 2 μM of the monomeric cyanine nucleic acid stain TO-PRO-3 [36] (Invitrogen, Lofer, Austria) in PBS for 10 min. and mounted in VectaShield (Vector Laboratories, Orton Southgate, Peterborough, UK). The slides were stored in the dark at room temperature before automated cell detection.
Preparation and staining of cells from placental villi and decidua of first trimester pregnancy
Cell suspensions from first trimester decidual and chorionic villous tissue were prepared from the tissue material from three terminated healthy pregnancies (7th–10th weeks of pregnancy). The tissue pieces were rinsed with PBS, separated under optical control and disaggregated using a MediMachine (Dako). The preparations were filtered through a 40-μm nylon filter (Falcon; Becton Dickinson, Erembodegem, Belgium) and approximately 1–2 × 105 cells in 200 μl of PBS were cytocentrifuged onto 60 mm2 of poly-L-lysine (Sigma-Aldrich)-coated polyethylene-naphthalate (PEN) membrane slides (PALM) for 5 min. at 270 ×g, followed by a 1-min. dry spin at 1170 ×g using a Hettich Centrifuge and Cytobuckets (Universal 32, Andreas Hettich, Tuttlingen, Germany). Before further processing, the cells were allowed to air-dry overnight at ambient temperature.
The cytospin preparations were then fixed and permeabilized using the Fix&Perm kit (An der Grub, Kaumberg, Austria). For this purpose, 100 μl of Reagent A (Fix&Perm, An der Grub) were applied for 5 min. onto the cytospin for fixation. To label fetal erythroblasts or trophoblast cells from placental villi, 100 μl of anti-Hbɛ antibody or a cocktail of GZ 158 and GZ 112 anti-trophoblast cell antibodies followed by goat-antimouse F(ab′)2-FITC was used. Cells from decidua basalis were labelled using a combination of anti-Hbɛ antibody and the GZ 158/112 cocktail in order to exclude fetal erythroblasts and trophoblast cells, respectively, from the analysis of maternal cells. All antibodies were used at a concentration of 5 μg/ml either in Reagent B (Fix&Perm) or in antibody diluent (Dako) plus 20% human AB serum. The cells were counterstained using 2 μM TO-PRO-3 in PBS for 10 min. All incubations were performed in a humidified chamber, and cover glasses were used in order to distribute the minute solution volumes evenly across the whole cytospin area. After counterstaining, the slides were mounted in VectaShield (Vector Laboratories), covered with a cover glass and stored in the dark at room temperature.
Automated cell detection, relocation, laser microdissection and laser pressure catapulting (LMPC) of cells
A scheme of the workflow is shown in Fig. 1. Automated cell detection was performed using a laser microdissection device based on an Axiovert 200 M fluorescence microscope (PALM MicroLaser Systems, Carl Zeiss GmbH, Vienna, Austria, MicroImaging, Munich, Germany) and equipped with a Metafer P (Metasystems, Altlussheim, Germany) module and the RCDetect software (MetaSystems, Altlussheim, Germany) [37]. Prior to the analysis, the software was trained to detect fetal erythroblasts (preparation described in Ref. [38]) and cytotrophoblast cells as specific fetal candidate cells. Optimized parameter settings retrieved from training data were stored as classifiers and used for scanning. Scanning of the cytospin was carried out with the 10× objective. Coordinates of positive events identified during the scans were stored and their images forwarded to an image gallery. Through visual examination of morphology and staining characteristics, potential (‘candidate’) fetal or maternal cells were selected from the image gallery for subsequent LMPC. For comparison, unstained PBMNCs or candidate maternal cells were also selected.
Fig 1.
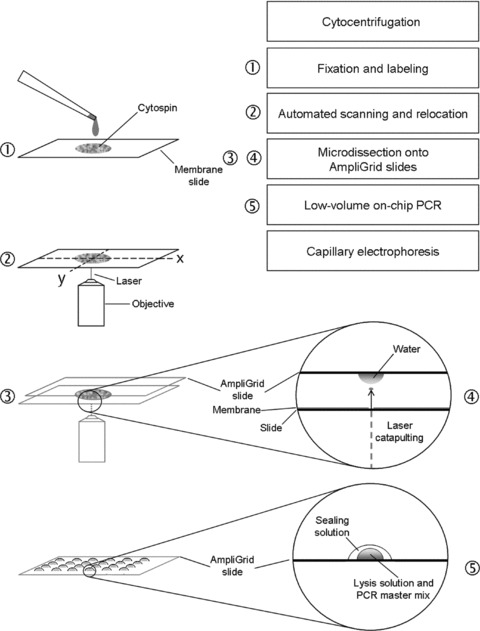
Workflow of the procedure. Briefly, cytospin preparations on membrane-coated slides are fixed and stained using immunocytochemistry and DNA counterstaining. Automated scanning and eventual relocation of positive candidate cells facilitate their microdissection and laser catapulting onto water droplets on anchors of AmpliGrid slides. After evaporation of the water, the cells are lysed and multiplex PCR is performed in droplets on the slide anchors. Finally, the amplification products are forwarded to analysis by capillary electrophoresis.
The mounted membrane slides were placed in a coplin jar containing PBS to allow the cover glass to slide off the membrane, rinsed with distilled water and allowed to air-dry. The membrane slides were then re-inserted onto the microscope stage. Upon relocation of the cells of interest, PALMRobo software (PALM MicroLaser Systems) was used to laser-microdissect the PEN membrane around the selected cells and catapult them onto the reaction sites of AmpliGrid slides (Olympus Life Science Research Europe, Munich, Germany) [32].
For cell collection, the hydrophilic cores of the reaction sites (anchors) on these slides were charged with 1 μl of nuclease-free water and placed upside down on the microscope stage so that the recovering droplets were located exactly above the cells of interest before catapulting. To prevent contact between the recovering droplet and the sample slide, a 1-mm spacer was located laterally between the sample slide and the AmpliGrid slides. Upon cell collection, the AmpliGrid slides were stored at ambient temperature to allow evaporation of the droplets. The anchors were checked for the presence or absence of microdissected cells and forwarded to cell lysis, followed by single and pooled cell DNA fingerprinting using a commercial multiplex PCR kit (PowerPlex 16 System Kit; Promega, Mannheim, Germany) that co-amplifies 15 polymorphic STR loci and the amelogenin locus.
On-chip cell lysis and DNA fingerprinting PCR
Molecular genetic analysis of pooled and single cells was performed on the AmpliGrid slides. Microdissected samples were lysed on the anchors of the AmpliGrid slides using a Cell Lysis and Extraction Kit (Olympus Life Science Research Europe). The cell lysis mix was prepared from 53 μl of nuclease-free water, 6 μl of 10× lysis buffer and 1 μl of lysis enzyme. Of this solution, 0.75 μl was applied onto each anchor containing microdissected cells, negative controls (nuclease-free water) or 100 pg ‘9947A’ female DNA positive control (Promega). The lysis mix was covered with 5 μl of sealing solution (supplied with the AmpliGrid slides) to prevent evaporation. For lysis, the AmpliGrid slide was placed in a slide cycler (AmpliSpeed 400D; Olympus Life Science Research Europe), incubated at 75°C for 5 min. and at 95°C for 3 min. and then allowed to cool down. As the samples reached room temperature, 0.75 μl of a PowerPlex 16 Master Mix (4 μl of Gold Star 10× buffer, 4 μl of PowerPlex 16 10× Primer Pair Mix, 1.28 μl of AmpliTaq Gold DNA Polymerase [ABI Austria, Brunn am Gebirge, Austria] and 10.72 μl of nuclease-free water) was added to each sample and amplification was performed according to the manufacturers’ recommendations. The amplification started with denaturation steps at 95°C for 11 min. and 96°C for 1 min. The first 10 cycles consisted of 30 sec. of denaturation at 94°C, 0.5°C/sec ramping to 60°C, primer annealing for 1 min. at 60°C, followed by another 0.3°C/sec ramping to 70°C and elongation at 70°C for 45 sec. The next 20 cycles started with 30 sec. of incubation at 90°C, followed by 0.5°C/sec ramping to 60°C, annealing for 1 min. at 60°C and another 0.3°C/sec ramping to 70°C and elongation at 70°C for 45 sec. Terminal elongation was performed for 30 min. at 60°C. After the amplification, both the sealing solution and the aqueous phase were recovered from the anchors and forwarded to DNA purification using Wizard SV Gel and PCR Clean-Up System (Promega). The purified samples were stored at 4°C in the dark until being analysed using capillary electrophoresis (3730 DNA Analyzer; ABI, Austria).
DNA profiles and PCR efficiency
PCR profiles were assessed by combining all DNA profiles from one and the same sample. Samples yielding at least one amplification product (‘successful PCR’) were included in the analysis. The lack of PCR fragments at single heterozygous or homozygous loci (no call) was defined as ‘amplification failure’. This was calculated from the number of failed loci divided by the total number of loci. ADO and heterozygous patterns were calculated from the results given by heterozygous loci only. ADI was defined as extra PCR fragments that either did not match both respective PCR profiles or matched the other (PBMNC or JAR, maternal or fetal) profile in cases in which contamination was unlikely (single-cell PCR). ADI was also calculated from heterozygous and homozygous loci. PCR efficiency is expressed as successfully amplified alleles at heterozygous loci based on the theoretically maximal number of PCR fragments at heterozygous loci.
Results
Specificity and binding characteristics of the anti-trophoblast antibodies
Immunohistochemical staining of frozen sections of first trimester placenta and decidua using GZ 112 resulted in positivity of extravillous cytotrophoblast cells in cell columns and of interstitial extravillous cytotrophoblast. Endovascular trophoblast showed weak staining. In placental villi, cell islands were positive, villous cytotrophoblast was weakly positive and the syncytiotrophoblast was negative. In addition to the trophoblast, GZ 112 stained uterine glandular epithelium and some smooth muscle cells in the tunica media of blood vessels. GZ 158 stained all types of trophoblast, uterine glandular epithelium and villous stroma of stem villi (Fig. 2). Neither of these antibodies bound to PBMNCs or vascular endothelium. In FACS analysis, isolated trophoblast cells showed surface staining for GZ 112 and GZ 158 (Fig. 3).
Fig 2.
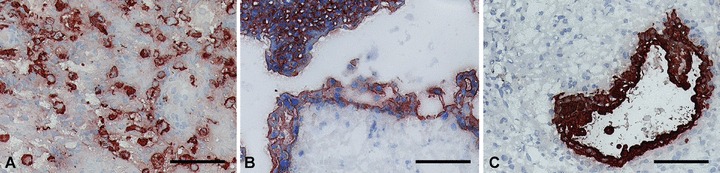
Immunohistochemical staining of first trimester placenta and decidua with the antibody GZ 158 shows reactivity with invasive extravillous cytotrophoblast in decidua basalis (A) with villous trophoblast (B) and glandular epithelium in decidua parietalis (C). Scale bar: 100 μm.
Fig 3.
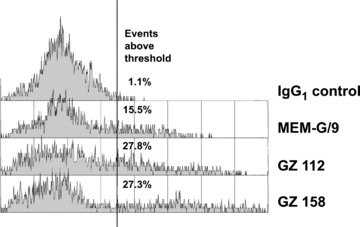
Flow cytometry of isolated trophoblast cells stained with the antibodies GZ 112 and GZ 158 in comparison to the negative control antibody and the positive control, the anti-HLA-G antibody MEM/G9 (Exbio Praha, Vestec, Czech Republic), which is known to bind to the surface of extravillous trophoblast and cell islands of villous trophoblast. The histograms are gated on viable cells based on light scatter. All antibodies are of IgG1 isotype.
Automatic cell detection
To train the RCDetect software to detect double-positive (FITC/TO-PRO-3) cells, cytospin slides with cord blood erythroblasts or tissue from terminations of pregnancies (early fetal erythroblasts) were initially used. The cells were labelled with antibodies against Hbɛ or Hbγ chains of fetal or embryonic haemoglobin and counterstained with TO-PRO-3. Scan fields showing 1–2 double-positive cells were chosen from these scans and forwarded to parameter optimization for correct detection of candidate cells. Optimized parameter settings were stored as a classifier. To test the power of the scanning software, we compared the performance of the screening software with a visual scan of two individuals using various samples containing different numbers of positive cells. In scans of 15 (14 Hbγ+ erythroblasts) and 122 (535 Hb+ erythroblasts) scan fields, the same numbers of labelled cells were detected. In a third comparison of 1867 scan fields, the screening software correctly scored 36 Hb+ erythroblasts, whereas visual scanning yielded 35 positive cells. The classifier was further optimized using both FITC-labelled (GZ 158+) trophoblast cells and FITC-labelled (Hb+) erythroblasts, resulting in a cell-type-independent detection protocol. Figure 4 gives an example of automatically retrieved JAR cells.
Performance of single rare cell analysis
In order to assess the feasibility of the method, pooled and single cells in both experimental settings were microdissected and amplified and then the PCR products were analysed (example in Fig. 5). The rate of successfully microdissected cells exceeded 95% (175 of 184 catapulting events). Both analysed pooled PBMNC samples from PBMNC/JAR sample 1 showed PCR failure. Thus, because PCR profiles could not be assessed, PBMNC/JAR sample 1 was excluded from analysis. DNA profiles from cells of tissue samples from terminations of pregnancies were obtained in two of three samples (IR 1 and 3). All three pooled samples from the decidua of IR 2 lacked PCR fragments in 10 of 16 loci (D21S11, D18S51, Penta E, D7S820, D16S539, CSF1PO, Penta D, D8S1179, TPOX and FGA). Here, a complete DNA profile could not be assessed either; therefore, IR 2 was excluded from analysis. The percentages of detected PCR fragments at heterozygous loci were used to assess the compatibility of the procedure involving fixation and staining with subsequent PCR. PCR success in pooled and single-cell samples from two different experimental settings is shown in Table 1. Compared PCR efficiency based on cell pools and single cells (irrespective of the cell type) is given in Table 2. A survey of all loci of the respective cells indicates whether or not cells can be allocated to their origin based on their unambiguous allelic pattern.
Fig 5.
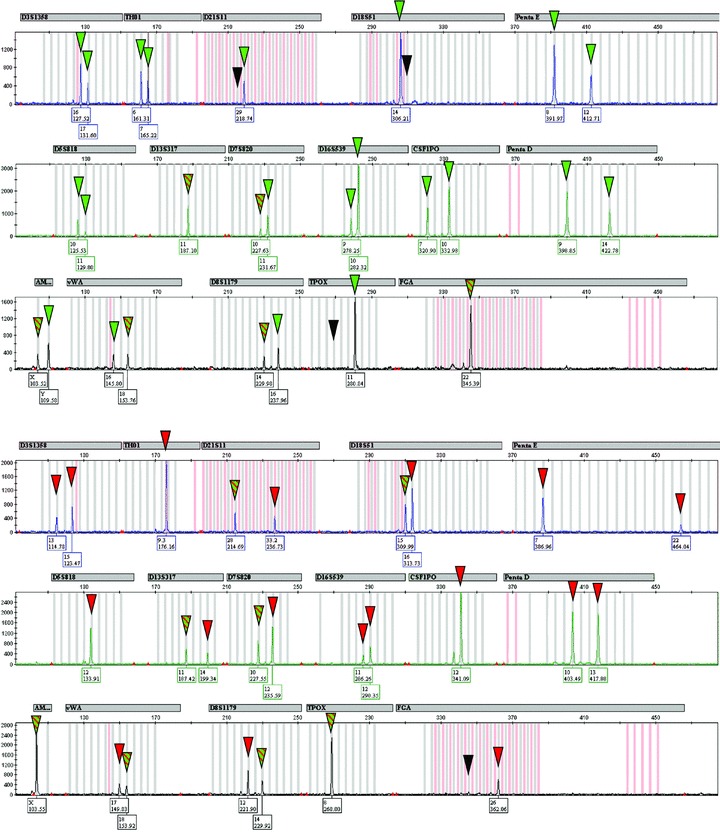
DNA profiles amplified from cells microdissected from sample 3. Top: single GZ 158-positive JAR cell (as shown in Fig. 4, top right). Bottom: cell pool of PBMNC to which the anti-trophoblast antibody GZ 158 did not bind. PCR products allowing unambiguous allocation of cells are highlighted with red (PBMNC) or green (JAR cell) triangles. Loci that show uninformative PCR products matching both DNA profiles are indicated with red-green striped triangles. Black triangles indicate allele drop-out at the respective loci as compared with DNA profiles derived from the summary of individual DNA fingerprinting from the respective individuals/samples.
Table 1.
PCR success in pooled and single-cell samples from two different experimental settings
| Female PBMNC of two samples | JAR | Interruption tissue | Interruption tissue | |||||
|---|---|---|---|---|---|---|---|---|
| Pooled cells (≤10) | Single cells | Pooled cells (5) | Single cells | |||||
| (n/total) | (%) | (n/total) | (%) | (n/total) | (%) | (n/total) | (%) | |
| Successful PCR* | 4/4 | 100 | 12/18 | 66.7 | 10/13 | 76.9 | 43/56 | 76.8 |
| No. of heterozygous loci† | Varying | 14 | Varying | Varying | ||||
| Amplification failure‡ | 1/64 | 1.6 | 52/192 | 27.1 | 10/160 | 6.3 | 190/688 | 27.6 |
| Allele drop-in§ (all loci) | 0/64 | 0 | 0/192 | 0 | 2/160 | 1.3 | 10/688 | 1.5 |
| Allele drop-out** (heterozygous loci) | 6/48 | 12.5 | 50/168 | 29.8 | 10/129 | 7.8 | 150/558 | 26.9 |
| Heterozygous pattern†† (heterozygous loci) | 41/48 | 85.4 | 78/168 | 46.4 | 109/129 | 84.5 | 252/558 | 45.2 |
| PCR efficiency‡‡ (heterozygous loci) | 88/96 | 91.7 | 206/336 | 61.3 | 228/258 | 88.4 | 654/1116 | 58.6 |
PCR yielding at least one amplification product.
Heterozygous loci of the respective samples as seen from PCR profiles (used for calculating ADO and PCR efficiency).
Number and percentage of loci yielding no PCR fragment.
Extra peaks matching no profile. **Heterozygous loci yielding only one fragment (heterozygous loci only).
Loci showing two allele repeats (heterozygous loci only).
Total number of PCR fragments calculated from heterozygous loci.
Table 2.
PCR efficiency based on the number of cells used as template
| Cell pools (≤10) | Cell pools (5) | Single cells | Overall | |||||
|---|---|---|---|---|---|---|---|---|
| (n/total) | (%) | (n/total) | (%) | (n/total) | (%) | (n/total) | (%) | |
| Successful PCR* | 4/4 | 100 | 10/13 | 76.9 | 55/74 | 74.3 | 69/91 | 75.8 |
| Amplification failure† | 1/64 | 1.6 | 10/160 | 6.3 | 242/880 | 27.5 | 253/1104 | 22.9 |
| Allele drop-in‡ (all loci) | 0/64 | 0 | 2/160 | 1.3 | 10/880 | 1.1 | 12/1104 | 1.1 |
| Allele drop-out§ (heterozygous loci) | 6/48 | 12.5 | 10/129 | 7.8 | 200/726 | 27.5 | 216/903 | 23.9 |
| Heterozygous pattern** (heterozygous loci) | 41/48 | 85.4 | 109/129 | 84.5 | 330/726 | 45.5 | 480/903 | 53.2 |
| PCR efficiency†† (heterozygous loci) | 88/96 | 91.7 | 228/258 | 88.4 | 860/1452 | 59.2 | 1176/1806 | 65.1 |
*PCR yielding at least one amplification product.
Number and percentage of loci yielding no PCR fragment.
Extra peaks matching no profile.
Heterozygous loci yielding only one fragment (heterozygous loci only).
*Loci showing two allele repeats (heterozygous loci only).
Total number of PCR fragments calculated from heterozygous loci.
Fig 4.
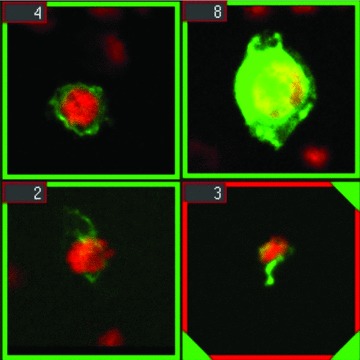
Artificially microchimeric JAR choriocarcinoma cells from a spiking experiment, as rendered by the gallery of the RCDetect software. The cells were fixed with methanol (top) or paraformaldehyde (bottom) and stained with the antibody GZ 158. Nuclear counterstaining with TO-PRO-3.
Contamination experiments of JAR cells in PBMNC
In order to obtain a complete DNA profile for an optimal contrast to the ‘rare’ JAR cells, we pooled FITC−/TO-PRO-3+ PBMNCs (10 cells as a maximum in each pool, depending on the efficiency of laser catapulting) as models of non-fetal cells that make up the majority. All four PBMNC pools yielded PCR fragments. ADO occurred in 12.5% (6/48 of heterozygous loci) and amplification failure in 1.6% (1/64 heterozygous and homozygous loci). A heterozygous pattern was detected in 41 of 48 heterozygous loci (85.4%). The PCR efficiency, based on the calculation of all PCR fragments detected at heterozygous loci, turned out to be 91.7% (88 of 96 possible PCR fragments). Single JAR cells failed to amplify 6 of 18 times (33.3%). In the remaining 12 amplified cells, ADO was present in 29.8% (50/168) of all heterozygous loci and amplification failure in 27.1% (52/192) of all loci. A heterozygous pattern was found in 78 of 168 loci (46.4%). PCR efficiency of single JAR cells was determined to be 61.3% (206/336).
Placental villi and decidua from termination of first trimester pregnancy
Amplifications of pools of five FITC−/TO-PRO-3+ cells from the decidua showed PCR failure in 3 of 13 (23.1%) cases. Amplification failure and ADO accounted for 6.3% (10/160) and 7.8% of cases (10/129), respectively. A heterozygous pattern was detected in 84.5% (109 of 129 heterozygous loci). PCR efficiency was calculated to be 88.4% (228 of 258 PCR fragments).
Of 56 single cells (Hbɛ-positive erythroblasts, trophoblast cells or FITC-negative candidate maternal cells), 13 (23.2%) failed to amplify at all. In the remaining 43 single-cell amplifications, failure was seen in 190 of 688 loci (27.6%). ADO occurred in 26.9% (150 of 558 loci) and extra alleles (ADI) were detected in 1.5% (10/688). Of 558 heterozygous loci, 252 (45.2%) showed two PCR products, resulting in a PCR efficiency of 58.6% (654/1116).
Attribution of genomic identity to cell pools and single cells by means of DNA profiling
DNA profiles of pooled and single cells were compared with full DNA profiles by means of allelic repeats (Tables 3–5).
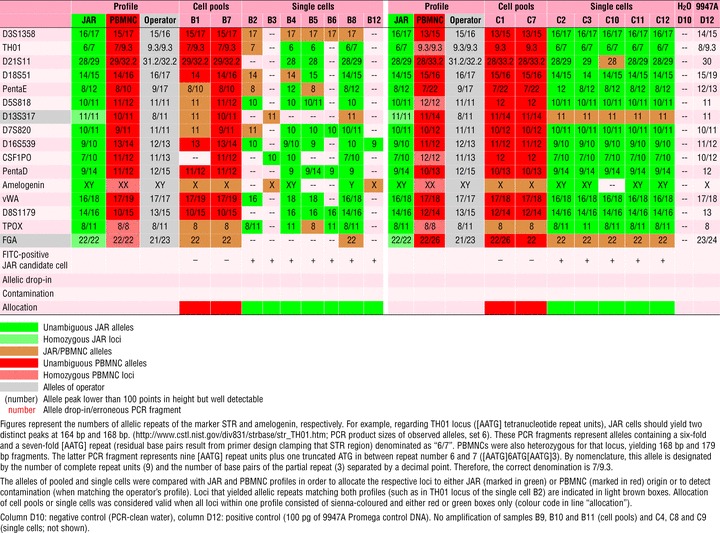
Table 3.
DNA profiles of JAR cells and PBMNC
 |
PBMNC spiked with JAR cells
All amplified samples yielding PCR fragments showed unambiguous PCR profiles and could be allocated to either JAR or PBMNC origin (16/16, 100%), as shown in Table 3.
Discrimination of maternal cells from fetal or placental (trophoblast) cells in the decidua
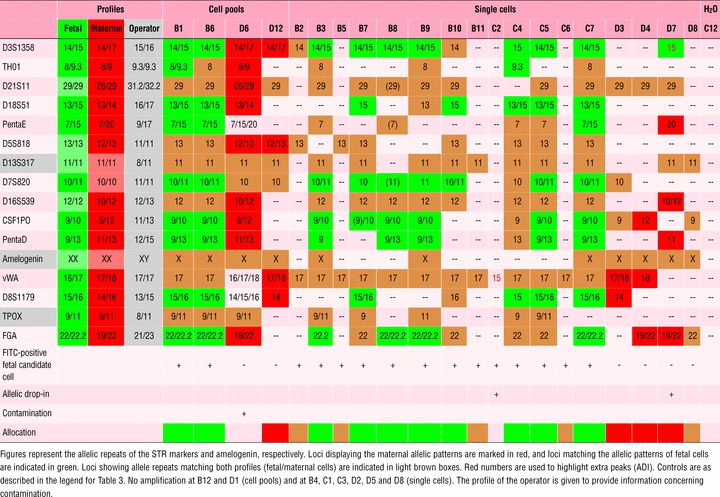
Two cell pools analysed from IR 1 (Table 4) consisting of five FITC+/TO-PRO-3+ candidate fetal cells could be allocated unambiguously to fetal origin by DNA profiling. One out of two cell pools consisting of unlabelled cells (thereby representing candidate maternal cells) gave the maternal DNA profile in the majority of loci analysed but presented evidence of contamination with fetal cells in three loci by showing a triallelic pattern in 3 of 13 informative patterns (Penta E, vWA and D8S1179). Another four loci either did not exclude fetal contamination because in these loci, the sample yielded a heterozygous pattern, whereas the fetal pattern was homozygous (D21S11, D5S818 and D16S539) or the data were uninformative (D7S820). The second pool of candidate maternal cells rendered a DNA profile compatible with the maternal but not with the fetal pattern. Furthermore, we analysed 17 single cells. Out of 13 FITC-labelled fetal candidate cells, DNA profiling confirmed the fetal identity unequivocally in eight instances. In five cases, the discrimination was not possible because only alleles shared by mother and foetus were found or, in one sample (C2), only a single PCR drop-in fragment was displayed. Out of four FITC-negative, presumably maternal cells, DNA profiling confirmed the presence of a maternal genome in two cases; the third one showed unambiguous loci for both profiles (1 fetal, 4 maternal and 3 inconclusive). In this case, it is very likely that allele drop-in occurred in one locus (D3S1358) mimicking fetal identity. The fourth cell yielded inconclusive results.
Table 4.
DNA profiles of fetal and maternal cells obtained from tissue from termination of pregnancy (IR 1)
 |
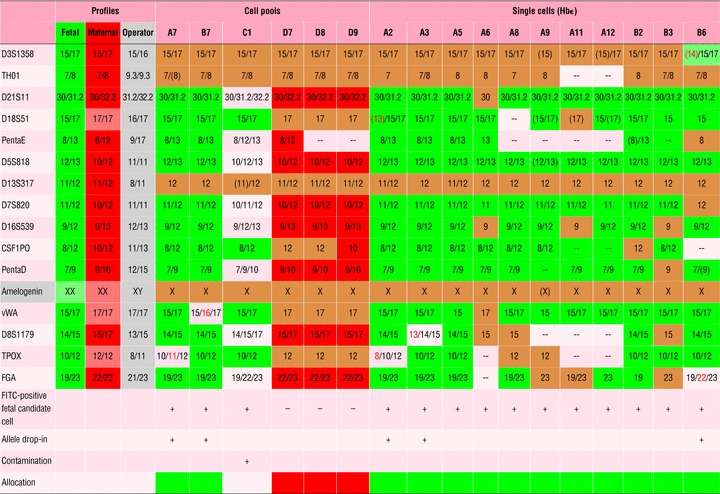
We analysed three pools of five each Hb+/TO-PRO-3+ erythroblasts (as fetal candidate cells, Table 5) from IR 3. In two pools, fetal identity was confirmed by DNA profiling; ADI occurred once in each of these two cases. The third pool was obviously contaminated with at least one maternal cell, as a triallelic pattern emerged for eight loci, combining the maternal and the fetal profile. Three pools of FITC-negative cells resulted in a maternal DNA profile. Eleven single Hb+/TO-PRO-3+ erythroblasts all gave the fetal genomic profile. Three of them showed ADI, fitting the operator’s profile in two instances. For all six single cells positive for staining with the cocktail of anti-trophoblast antibodies, the fetal genomic profile was detected. Unrelated ADI occurred in one case, and an ADI fitting the operator’s profile in the other case. In nine single cells negative for both anti-Hbɛ and anti-trophoblast staining (maternal candidate cells), the maternal genomic profile was detected. In two cases, the profile contained one fetal-specific allele (one of these ADI fit the fetal as well as the operator’s profile). Because these were single cells, it is likely that ADI mimicking fetal pattern occurred. ADI in a third case was unrelated. There were no profiles showing only inconclusive loci.
Table 5.
DNA profiles of fetal and maternal cells obtained from tissue from termination of pregnancy (IR 3)
 |
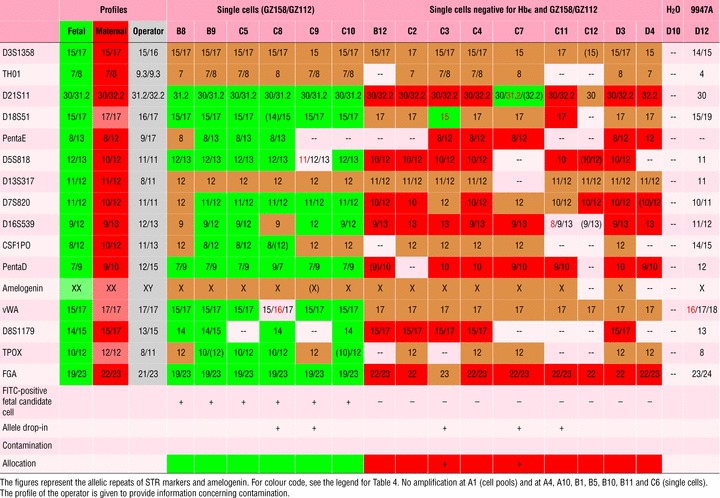 |
For non-related individuals, allocation to the genomic identity of cells was possible in 100% of the cells, whereas in the mixture of related cells, allocation to either mother or foetus was possible in 86% of single cells.
Discussion
The novelty of the method described is that it links automated cell detection based on automatic screening of cells labelled by immunofluorescence with laser catapulting of candidate target cells to reaction sites of slides designed for low-volume on-chip PCR. DNA fingerprint analysis performed in a volume of less than 2 μl is shown to be compatible with the preceding processing of cells for pre-identification (establishment of candidate status), making use of DNA profiles as powerful sex-independent markers in single-cell analysis.
NIPD based on cells circulating in the blood of pregnant women requires that cells carrying the fetal genome be reliably identified. Some of our pools of cells collected on the basis of immunofluorescence staining show signs of contamination with maternal cells even for target cells that were not rare, such as in the case of the analysis of fetal and maternal cells from the placenta. Because of the even more limited reliability of biochemical markers or FISH in a setting of rare cell analysis, we suggest single-cell DNA profiling as a standard for the identification of these rare cells.
Our approach consists basically of two steps: (i) definition of a group of candidate cells by a biochemical marker that need not be perfectly specific but of relevant sensitivity for the detection of fetal cells, and (ii) confirmation of the genomic identity of target cells based on DNA profile. Our data confirm the feasibility of this method on single immunocytochemically stained and laser-catapulted cells, proving the discriminative power between ‘rare’ JAR choriocarcinoma cells and contaminated PBMNCs and also between single cells that share a haploid set of chromosomes.
The multiplex PCR used for the identification might be combined with an analysis of specific monogenetic genomic disease markers. Work is under way to implement whole-genome amplification of single cells so that part of the expanded genome may be used for the identification on the basis of multiplex PCR and the rest of the DNA of confirmed fetal cells may be pooled and used for molecular genetic analysis of, for example, monogenetic diseases or chromosome aberrations by array comparative genomic hybridization (CGH).
A pre-condition for a diagnostic test is cost-effectiveness, with NIPD not being an exception. An important step in this direction is the automation of fetal candidate cell location using a slide screening device; in this way, a virtual enrichment step is added to antecedent physical enrichment steps, which we found satisfactorily efficient. For LMPC, automatic relocation of the cells of interest eliminates the labour-intensive step of cell retrieval.
In previous work, multiplex PCR of STR markers and amelogenin was used with low-volume on-chip PCR, demonstrating its feasibility for minute amounts of DNA. Full DNA profiles were achieved from as little as 32 pg of DNA [32]. An analysis of single cells without extraction was shown recently by Hagen-Mann et al.[39]. So far, however, it has not been known how fixed, labelled and lysed single cells would perform. In our approach, it has been necessary not only to adapt various steps in the procedure such as cell detection and relocation but also to optimize single-cell harvesting. Potentially harmful protocol steps such as fixation have been kept to a minimum and volumes have been adjusted to meet low-volume PCR compatibility. Down-scaling of PCR volumes has reduced the cost of reagents and has made it possible to control quality visually by seeing whether laser-microdissected cells have in fact landed on the chip anchor.
Our PCR performance data are slightly better than the low-volume on-chip PCR performance data reported by Schmidt et al.[32] who used dilution series of genomic DNA. They reported PCR failure in 9.1%, 42.7% and 50.0% at DNA template amounts of 63 pg, 32 pg and 8 pg, respectively, whereas we had no PCR failure using 10 pooled cells (corresponding to 60 pg of DNA) and no PCR products in 23.1% and 25.7% at the five-cell and single-cell level, respectively. Although it seems that fixed and labelled microdissected cells perform better than purified genomic DNA, it must be taken into account that dilution series contain calculated means of very low amounts of DNA with an unknown deviation. Thus, dilution series down to a few genome equivalents reflect neither calculated quantity nor equally distributed DNA, which may account for the difference. We observed quite a high range of PCR performance between our samples and could identify various factors that may have caused amplification failure specifically in relation to the on-chip approach. Occasionally, during microdissection, dust particles were attracted to the AmpliGrid slides, causing small air reservoirs to be trapped during subsequent charging of the anchors (not shown). This entrapment resulted in extensive evaporation at the aqueous/gaseous interphase underneath the sealing solution and led to an impairment of the PCR assembly. Minor changes in the workflow of LMPC resulted in less exposure to air, causing less difficulty for the PCR assembly. However, in some cases, PCR failure could not be traced back to either cause and remained unexplained.
ADO occurred in pooled samples of 10 cells in 12.5% of the cases studied. Although Schmidt et al. [32] detected no signs of ADO using 63 pg of genomic DNA (corresponding to 10 cells), they found a higher incidence of ADO at 32 pg (28.6%) and 8 pg (100%) concentrations than when we analysed pools of five cells (7.8%) and single cells (27.5%). ADO may be because of both cell quality and processing. In DNA profiles of unfixed cells stored at 4°C for approximately 1 week, we noticed lower PCR efficiency (obviously because of DNA degradation) in comparison to cells fixed on day 2 after sampling (data not shown). Thus, the time between sample collection or cell culture and fixation needs to be kept to a minimum, especially for analysis of single cells. Further factors accounting for ADO may include steric blocking of DNA polymerase to DNA because of inefficient cell lysis, strong DNA–histone interaction or DNA sticking to the hydrophilic surface of the chip anchor.
ADI was detected in 12 of 1104 (1.1%) analysed loci and occurred maximally once per multiplex PCR. In our study, more than three-fourths of the artefacts occurred in a single-tissue sample from a pregnancy termination. One positive control, amplified together with the cells from the respective sample, also showed ADI in three loci (TH01, CSF1PO and vWA), indicating that carry-over has taken place during handling. Fixation and staining procedures may also increase the frequency of ADI. Although ADI was not reported to occur with diluted genomic DNA, Dietmaier et al. observed non-specific PCR fragments in PCR not only from a few unfixed but also from pooled fixed cells [30]. However, in our hands, ADI did not interfere with the allocation of cells to their respective origin. Although some of the PCR products matched individual DNA profiles (Table 5: A2, A3, B6 and C9), contamination was largely ruled out, as PCR was done on single cells. Even when ADI occurred in pooled cell samples (Table 5: A7 and B7), it was easily distinguished from the contaminated samples (Table 4: D6 and Table 5: C1) because of differences in the number of triallelic STR pattern (1 versus 3 and 8 triallelic pattern, respectively). Three samples (Table 4: D7 and Table 5: C3 and C7) yielded DNA profiles inconsistent at one locus each. In these cells, specific fetal alleles were detected at loci D3S1358, D21S11 and D18S51, respectively, whereas the residual loci were either uninformative or of maternal origin. Although DNA profiles showed fetal alleles, maternal alleles were 4–6 times more frequent. Contamination by fetal cells or cells of the operator could be excluded because single cells were used and alleles accounting for ADI differed from those of the operator.
Cell allocation could be successful on the basis of even a single informative locus (Table 3: B3 and B12) but could also fail based on up to four loci (Table 4: D8). Uninformative PCR accounted for up to 70% of the DNA profile (10 of 14 loci; Table 4: C4). These data reflect the difference between both experimental settings used. It is easier to distinguish between cells at low levels of successful amplification when the cells are derived from two non-related individuals. The allocation was successful in more than 88% of cells with 16-plex PCR.
Many approaches described in the literature have used histochemical staining methods that convey little specificity for the detection of fetal cells [10, 12, 15, 21, 28, 31, 40]. PCR performance after histochemical stainings such as May–Grünwald Giemsa, haematoxylin or Wright staining are reported to result in amplification failure from 10% to 58% in cases in which erythroblasts were analysed [10, 15, 21, 28]. ADO or contamination occurred in 25–62% of the cases; uninformative loci were observed in 12.5–62.5% of the cases in which STR markers were used [15, 28]. Apart from a few samples, the high number of uninformative cases of the latter resulted from a low number of STR markers used for identification purposes [15]. We assume that 16-plex PCR increases the rate of successful confirmation of fetal candidate cells because of a higher number of loci analysed.
This approach may even be more crucial when using FISH in rare cell diagnosis. Kolvraa et al. could identify false-positive XY cells using reverse-colour XY-FISH, indicating that FISH is error-prone, especially when applied in rare cell diagnosis [16]. A serious handicap to using Y-FISH is that it is unable to detect female fetal cells. However, probes to detect trisomies (chromosomes 13, 18 and 21) might be used for the discovery of trisomic (fetal) cells. When examining cells derived from normal pregnancies, we observed split FISH signals and nuclei showing trisomic FISH pattern (data not shown). Preliminary data indicate that processing of nuclei for FISH does not interfere with subsequent DNA profiling (not shown), thus allowing for combinatory use to improve single-cell analysis.
Enrichment for trophoblast cells is promising using the isolation by size of epithelial tumor cells (ISET) technique or MACS combined with subsequent short-term culture [22, 40]. Both methods try to overcome fetal cell shortage based on conventional enrichment methods. We presume that a combination of one of these methods with whole-genome amplification and DNA fingerprinting is a promising avenue for cell-based NIPD.
We have shown that DNA profiling of single candidate cells representing microchimerism allows for the verification or falsification of their genetic origin in the majority of cases. When rare cells were compared with host cells from unrelated persons, the power of assignment amounted to 100%. We assume that this method will also be helpful in recognizing microchimerism from previous pregnancies, provided the DNA is available (from live children or aborted foetuses). Beyond NIPD, the applications may include the analysis of microchimerism in tissues, including the analysis of the effect of stem cell therapy.
Acknowledgments
We appreciate the excellent technical assistance of Gertrud Havlicek, Christine Daxböck, Astrid Blaschitz and Rudolf Schmied. Purified trophoblast cells were provided by Gernot Desoye and Renate Michlmaier, Department of Obstetrics and Gynecology, Medical University of Graz. The assistance of Karin Meister, Center for Medical Research of the Medical Unversity of Graz, with capillary electrophoresis is gratefully acknowledged. We are grateful for helpful discussions with Gábor Méhes (presently at the Department of Pathology of the University of Debrecen, Hungary), Wolfgang Mann (Olympus Life Sciences Europe, Munich, Germany), Renate Burgemeister, Gabriele Friedemann and Rainer Gangnus (Carl Zeiss MicroImaging, Munich, Germany) and Tilman Johannes (MetaSystems, Altlussheim, Germany). This study was financially supported by the European Commission (Network of Excellence ‘Special Non-Invasive Advances in Fetal and Neonatal Evaluation [SAFE]’, FP6–503243, lead researchers involved E.G and P.S.) and the Jubliäumsfonds of the Austrian National Bank (project no. 9995, to P.S.). T.K. is a recipient of a SAFE PhD bursary.
Conflict of Interest
The authors Petra Hartmann and Marianna Alunni-Fabbroni are employees of Olympus Life Science Research Europe. Peter Sedlmayr and Gottfried Dohr have filed the patent EP 1 367 380 A1 (pending).
References
- 1.Nelson JL. Your cells are my cells. Sci Am. 2008;298:64–71. [PubMed] [Google Scholar]
- 2.Nelson JL, Gillespie KM, Lambert NC, et al. Maternal microchimerism in peripheral blood in type 1 diabetes and pancreatic islet beta cell microchimerism. Proc Natl Acad Sci U S A. 2007;104:1637–42. doi: 10.1073/pnas.0606169104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artlett CM. Pathophysiology of fetal microchimeric cells. Clin Chim Acta. 2005;360:1–8. doi: 10.1016/j.cccn.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 4.Srivatsa B, Srivatsa S, Johnson KL, et al. Microchimerism of presumed fetal origin in thyroid specimens from women: a case-control study. Lancet. 2001;358:2034–8. doi: 10.1016/S0140-6736(01)07099-4. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez SA, Artlett CM. Microchimerism and systemic sclerosis. Curr Opin Rheumatol. 2005;17:86–90. doi: 10.1097/01.bor.0000145516.45854.7b. [DOI] [PubMed] [Google Scholar]
- 6.Reed AM, McNallan K, Wettstein P, et al. Does HLA-dependent chimerism underlie the pathogenesis of juvenile dermatomyositis? J Immunol. 2004;172:5041–6. doi: 10.4049/jimmunol.172.8.5041. [DOI] [PubMed] [Google Scholar]
- 7.Krabchi K, Gadji M, Forest JC, et al. Quantification of all fetal nucleated cells in maternal blood in different cases of aneuploidies. Clin Genet. 2006;69:145–54. doi: 10.1111/j.1399-0004.2005.00564.x. [DOI] [PubMed] [Google Scholar]
- 8.Krabchi K, Gros-Louis F, Yan J, et al. Quantification of all fetal nucleated cells in maternal blood between the 18th and 22nd weeks of pregnancy using molecular cytogenetic techniques. Clin Genet. 2001;60:145–50. doi: 10.1034/j.1399-0004.2001.600209.x. [DOI] [PubMed] [Google Scholar]
- 9.Weichel W, Irlenbusch S, Kato K. Sorting of rare cells. In: Radbruch A, et al., editors. Flow cytometry and cell sorting. Berlin, Heidelberg, Germany: Springer-Verlag; 1992. pp. 159–67. [Google Scholar]
- 10.Chen HP, Wang TR, Xu JP, et al. Fetal origin of single nucleated erythroblasts and free DNA in the peripheral blood of pregnant women. Int J Gynaecol Obstet. 2004;85:1–5. doi: 10.1016/j.ijgo.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Wang T, Chen H, Ma T. Noninvasive prenatal diagnosis of fetal sex by single-cell PEP-PCR method. J Huazhong Univ Sci Technolog Med Sci. 2004;24:66–7. doi: 10.1007/BF02830709. , 78. [DOI] [PubMed] [Google Scholar]
- 12.Yang YH, Yang ES, Kwon JY, et al. Prenatal diagnosis of trisomy 21 with fetal cells in maternal blood using comparative genomic hybridization. Fetal Diagn Ther. 2006;21:125–33. doi: 10.1159/000089062. [DOI] [PubMed] [Google Scholar]
- 13.Wei HY, Long GF, Lin WX, et al. Identification of fetal nucleated erythrocytes in maternal blood using short tandem repeat typing after primer extension preamplification. Zhonghua Xue Ye Xue Za Zhi. 2006;27:687–9. [PubMed] [Google Scholar]
- 14.Saker A, Benachi A, Bonnefont JP, et al. Genetic characterisation of circulating fetal cells allows non-invasive prenatal diagnosis of cystic fibrosis. Prenat Diagn. 2006;26:906–16. doi: 10.1002/pd.1524. [DOI] [PubMed] [Google Scholar]
- 15.Kolialexi A, Vrettou C, Traeger-Synodinos J, et al. Noninvasive prenatal diagnosis of beta-thalassaemia using individual fetal erythroblasts isolated from maternal blood after enrichment. Prenat Diagn. 2007;27:1228–32. doi: 10.1002/pd.1881. [DOI] [PubMed] [Google Scholar]
- 16.Kolvraa S, Christensen B, Lykke-Hansen L, et al. The fetal erythroblast is not the optimal target for non-invasive prenatal diagnosis: preliminary results. J Histochem Cytochem. 2005;53:331–6. doi: 10.1369/jhc.4A6396.2005. [DOI] [PubMed] [Google Scholar]
- 17.Mavrou A, Kouvidi E, Antsaklis A, et al. Identification of nucleated red blood cells in maternal circulation: a second step in screening for fetal aneuploidies and pregnancy complications. Prenat Diagn. 2007;27:150–3. doi: 10.1002/pd.1640. [DOI] [PubMed] [Google Scholar]
- 18.Kolialexi A, Tsangaris GT, Antsaklis A, et al. Fetal cells in maternal plasma are found in a late state of apoptosis. Prenat Diagn. 2004;24:719–21. doi: 10.1002/pd.861. [DOI] [PubMed] [Google Scholar]
- 19.Babochkina T, Mergenthaler S, De Napoli G, et al. Numerous erythroblasts in maternal blood are impervious to fluorescent in situ hybridization analysis, a feature related to a dense compact nucleus with apoptotic character. Haematologica. 2005;90:740–5. [PubMed] [Google Scholar]
- 20.Mavrou A, Kolialexi A, Antsaklis A, et al. Identification of fetal nucleated red blood cells in the maternal circulation during pregnancy using anti-hemoglobin-epsilon antibody. Fetal Diagn Ther. 2003;18:309–13. doi: 10.1159/000071971. [DOI] [PubMed] [Google Scholar]
- 21.Troeger C, Zhong XY, Burgemeister R, et al. Approximately half of the erythroblasts in maternal blood are of fetal origin. Mol Hum Reprod. 1999;5:1162–5. doi: 10.1093/molehr/5.12.1162. [DOI] [PubMed] [Google Scholar]
- 22.Guetta E, Gutstein-Abo L, Barkai G. Trophoblasts isolated from the maternal circulation: in vitro expansion and potential application in non-invasive prenatal diagnosis. J Histochem Cytochem. 2005;53:337–9. doi: 10.1369/jhc.4B6426.2005. [DOI] [PubMed] [Google Scholar]
- 23.Coata G, Tilesi F, Fizzotti M, et al. Prenatal diagnosis of genetic abnormalities using fetal CD34+ stem cells in maternal circulation and evidence they do not affect diagnosis in later pregnancies. Stem Cells. 2001;19:534–42. doi: 10.1634/stemcells.19-6-534. [DOI] [PubMed] [Google Scholar]
- 24.Guetta E, Gordon D, Simchen MJ, et al. Hematopoietic progenitor cells as targets for non-invasive prenatal diagnosis: detection of fetal CD34+ cells and assessment of post-delivery persistence in the maternal circulation. Blood Cells Mol Dis. 2003;30:13–21. doi: 10.1016/s1079-9796(03)00008-1. [DOI] [PubMed] [Google Scholar]
- 25.Khosrotehrani K, Johnson KL, Cha DH, et al. Transfer of fetal cells with multilineage potential to maternal tissue. JAMA. 2004;292:75–80. doi: 10.1001/jama.292.1.75. [DOI] [PubMed] [Google Scholar]
- 26.Mergenthaler S, Babochkina T, Kiefer V, et al. FISH analysis of all fetal nucleated cells in maternal whole blood: improved specificity by the use of two Y-chromosome probes. J Histochem Cytochem. 2005;53:319–22. doi: 10.1369/jhc.4A6404.2005. [DOI] [PubMed] [Google Scholar]
- 27.Christensen B, Philip J, Kolvraa S, et al. Fetal cells in maternal blood: a comparison of methods for cell isolation and identification. Fetal Diagn Ther. 2005;20:106–12. doi: 10.1159/000082432. [DOI] [PubMed] [Google Scholar]
- 28.Garvin AM, Holzgreve W, Hahn S. Highly accurate analysis of heterozygous loci by single cell PCR. Nucleic Acids Res. 1998;26:3468–72. doi: 10.1093/nar/26.15.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osborne EC, Trounson AO, Cram DS. A QF-PCR system to detect chromosome 13 aneuploidy from as few as ten cells. Am J Med Genet A. 2005;134A:33–8. doi: 10.1002/ajmg.a.30428. [DOI] [PubMed] [Google Scholar]
- 30.Dietmaier W, Hartmann A, Wallinger S, et al. Multiple mutation analyses in single tumor cells with improved whole genome amplification. Am J Pathol. 1999;154:83–95. doi: 10.1016/S0002-9440(10)65254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hahn S, Zhong XY, Troeger C, et al. Current applications of single-cell PCR. Cell Mol Life Sci. 2000;57:96–105. doi: 10.1007/s000180050501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt U, Lutz-Bonengel S, Weisser HJ, et al. Low-volume amplification on chemically structured chips using the PowerPlex16 DNA amplification kit. Int J Legal Med. 2006;120:42–8. doi: 10.1007/s00414-005-0041-2. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann M, Blaschitz A, Hammer A, et al. Immunohistochemical examination of trophoblast populations in human first trimester and term placentae and of first trimester spiral arteries with the monoclonal antibody GZ 112. Placenta. 1997;18:481–9. doi: 10.1016/0143-4004(77)90001-7. [DOI] [PubMed] [Google Scholar]
- 34.Goding JW. Production of monoclonal antibodies. 2nd ed. London: Academic Press; 1983. [Google Scholar]
- 35.Hiden U, Maier A, Bilban M, et al. Insulin control of placental gene expression shifts from mother to foetus over the course of pregnancy. Diabetologia. 2006;49:123–31. doi: 10.1007/s00125-005-0054-x. [DOI] [PubMed] [Google Scholar]
- 36.Bink K, Walch A, Feuchtinger A, et al. TO-PRO-3 is an optimal fluorescent dye for nuclear counterstaining in dual-colour FISH on paraffin sections. Histochem Cell Biol. 2001;115:293–9. doi: 10.1007/s004180100254. [DOI] [PubMed] [Google Scholar]
- 37.Mehes G, Lorch T, Ambros PF. Quantitative analysis of disseminated tumor cells in the bone marrow by automated fluorescence image analysis. Cytometry. 2000;42:357–62. doi: 10.1002/1097-0320(20001215)42:6<357::aid-cyto1002>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Hennerbichler S, Schmied R, Petek E, et al. Detection and relocation of cord blood nucleated red blood cells by laser scanning cytometry. Cytometry. 2002;48:87–92. doi: 10.1002/cyto.10112. [DOI] [PubMed] [Google Scholar]
- 39.Hagen-Mann K, Zacher T, Khanaga O, et al. Molekularbiologische Analyse von Einzelzellen. Medizinischegenetik. 2005;17:53–9. [Google Scholar]
- 40.Vona G, Beroud C, Benachi A, et al. Enrichment, immunomorphological, and genetic characterization of fetal cells circulating in maternal blood. Am J Pathol. 2002;160:51–8. doi: 10.1016/S0002-9440(10)64348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]