

Abstract
Inhibitor of κB kinase (IKK) gamma (IKKγ), also known as nuclear factor κB (NF-κB) essential modulator (NEMO), is a component of the IKK complex that is essential for the activation of the NF-κB pathway. The NF-κB pathway plays a major role in the regulation of the expression of genes that are involved in immune response, inflammation, cell adhesion, cell survival and development. As part of the IKK complex, IKKγ plays a regulatory role by linking the complex to upstream signalling molecules. IKKγ contains two coiled-coil regions, a leucine zipper domain and a highly conserved zinc finger domain. Mutations affecting IKKγ have been associated with X-linked hypohidrotic ectodermal dysplasia with immune deficiency (HED-ID), with the majority of these mutations affecting the C-terminal region of the protein where the zinc finger is located. The zinc finger of IKKγ is needed for NF-κB activation in a cell- and stimulus-specific manner. The major mechanism by which the zinc finger plays this role appears to be the recognition of polyubiquitinated upstream signalling intermediates. This assertion reinforces the current notion that ubiquitination plays a major role in mediating protein–protein interactions in the NF-κB signalling pathway. Because the zinc finger domain of IKKγ is very likely involved in mediating interactions with ubiquitinated proteins, investigations that look for upstream activators or inhibitors of the IKK complex that bind to and interact with the zinc finger of IKKγ are required to gain a better insight into the exact roles of this domain and into the pathogenesis of HED-ID.
Keywords: IKKgamma, NEMO, zinc finger, ubiquitination, ectodermal dysplasia, hyper IgM syndrome, immune deficiency
Inhibitor of κB kinase gamma (IKKγ) protein, a key component of the NF-κB pathway
Inhibitor of κB kinase (IKK) gamma (IKKγ), also commonly referred to as nuclear factor κB (NF-κB) essential modulator (NEMO), was first reported independently by four groups of investigators during the period 1998–1999 [1–4]. The discovery of IKKγ followed the identification of IKKα and IKKβ as two catalytic components of a high-molecular-weight complex (the IKK complex) involved in the inducible phosphorylation of the inhibitor of κB (IκB) proteins [5–10]. IκB phosphorylation, which leads its degradative ubiquitination, is a key step in the activation of the NF-κB proteins. The NF-κB proteins normally exist sequestered in the cytoplasm and hence inactive due to their binding to the IκB proteins. These proteins, once released from the IκB proteins following the stimulus-induced degradation of the latter, form homodimers and heterodimers that function as transcription factors. The dimers bind to the consensus DNA sequence 5′-GGGGYNNCCY-3′, where Y is a pyrimidine and N is any nucleotide [11]. Five different proteins have been identified as having NF-κB activity: p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), RelB and c-Rel.
The NF-κB proteins regulate the expression of a large number of cytokines and acute phase proteins such as tumour necrosis factor-α (TNFα), IL-1, IL-2, IL-6, interferon γ (IFNγ) and C3 complement, thus playing various roles in immunity and inflammation, as well as the expression of cell adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) [11–14]. They are also important in the expression of proteins that are involved in anti-apoptotic processes, such as inhibitor of apoptosis (IAP) proteins, cellular FLICE inhibitory protein (c-FLIP) and Bcl-2 [13]. Furthermore, they are also involved in the expression of some proteins that play roles in development, such as Wnt10a and Notch1 [15, 16]. Therefore, the activation of the NF-κB signalling pathway modulates the expression of genes whose protein products modulate inflammation, immune response, cell adhesion, cell survival and development.
The NF-κB pathway can be activated by a plethora of stimuli, such as TNFα, IL-1, lipopolysaccharide (LPS), ultraviolet (UV) radiation, phorbol 12-myristate 13-acetate (PMA), ligation of the T cell receptor (TCR), double strand RNA, reactive oxygen intermediates and the human T-cell lymphotropic virus type 1 (HTLV-1) Tax protein [11, 12, 17]. There are two pathways that lead to NF-κB activation, namely the canonical or classical pathway and the non-canonical or alternative pathway. The canonical pathway is the major pathway for the activation of NF-κB and is the route employed by most of the NF-κB-inducing stimuli. It involves the induced degradation of IκB proteins (mainly IκBα) and it requires both IKKα and IKKβ[13, 17]. In this pathway, the stimuli trigger cytoplasmic signalling cascades that result in the activation of the IKK complex (Fig. 1). The IKK complex phosphorylates IκBα at two key serine residues. Phosphorylated IκBα undergoes ubiquitination and is then rapidly degraded by the proteasome, thus releasing the NF-κB proteins and allowing them to translocate to the nucleus. The non-canonical pathway is a minor pathway that is induced by a few stimuli such as lymphotoxin β. In this pathway, IKKβ, IKKγ and the degradation of IκBα are not required. Instead, activation takes place via the IKKα-mediated processing of p100, which allows it to form a dimer with RelB and function as a transcription factor [13, 18].
Fig 1.
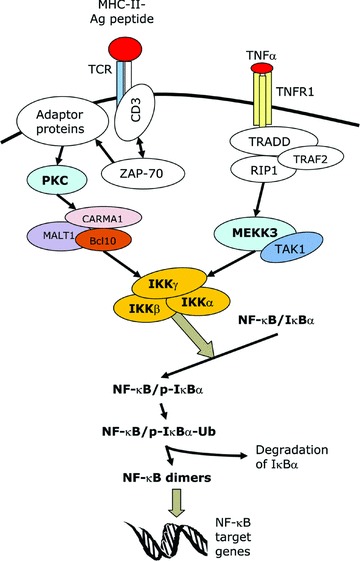
The canonical NF-κB signalling pathway. This is a schematic representation of the signalling pathways that lead to the activation of NF-κB following stimulation by two of the major NF-κB-inducing stimuli, namely the triggering of the TCR and treatment with TNFα. MHC-II, major histocompatibility complex II; Ag, antigen; CD, cluster of differentiation; ZAP-70, zeta-associated protein of 70 kD; PKC, protein kinase C; CARMA1, CARD-MAGUK protein 1; MALT1, mucosa-associated lymphoid tissue lymphoma translocation gene 1; Bcl10, B-cell CLL/lymphoma 10; TNFR1, TNF receptor-1; RIP1, receptor interacting protein 1; TRADD, TNF receptor- associated death domain protein; TRAF2, TNF receptor-associated factor-2; TAK1, TGF-beta activated-kinase 1; MEKK3, MAPK-ERK kinase kinase-3; p-IκBα, phosphorylated IκBα; Ub, ubiquitin chain.
IKKγ has been shown to be required for the activation of NF-κB by a variety of stimuli. Using an immune complex assay, Rothwarf et al. showed that IKKγ is essential for the phosphorylation of IκBα and hence for NF-κB activation in response to TNFα and IL-1 in HeLa cells [2]. In addition, Yamaoka et al. showed that IKKγ is essential for the activation of NF-κB by the HTLV-1 Tax protein, LPS, IL-1, double strand RNA and PMA [1]. IKKγ is also required for the activation of the canonical NF-κB pathway that is caused by the Epstein-Barr virus (EBV)-encoded latent membrane protein-1 (LMP1) [19]. Triggering the TCR in a T cell line lacking IKKγ failed to increase NF-κB activity and IL-2 secretion demonstrating that IKKγ is essential for TCR-mediated NF-κB activation [20]. The exact functions of IKKγ as part of the IKK complex have not been fully determined. However, recently a large number of studies have demonstrated that ubiquitination plays a key role in the functions of IKKγ pertaining to NF-κB activation [21]. The roles of ubiquitination relate either to the recognition of ubiquiti nated upstream signalling intermediates by IKKγ or to the induced ubiquitination of IKKγ itself. Several groups have demonstrated that IKKγ can recognize certain ubiquitinated proteins involved in the NF-κB pathway and the ubiquitin-binding domains of IKKγ have been mapped [21, 22]. In addition, IKKγ has been shown to undergo non-degradative ubiquitination in the presence of certain NF-κB-inducing stimuli. The ubiquitination of IKKγ involves the attachment of either branched (K63-linked) or linear polyubiquitin chains [21, 23].
Jin and Jeang isolated the full-length human IKKγ cDNA and, by sequence alignment, they mapped the gene to locus Xq28 [24]. The human IKKγ gene is located in a head-to-head orientation in relation to the glucose 6-phosphate dehydrogenase (G6PD) gene and the two genes overlap by about 800 bp with IKKγ transcribed toward the telomere. The mouse IKKγ gene is also located on the X chromosome in close proximity to the G6PD gene [25]. The 23-kb human IKKγ gene is composed of 10 exons, with four alternative non-coding first exons that are independently spliced to exon 2 and with the coding region consisting of the distal portion of exon 2, the whole segments of exons 3–9 and the proximal portion of exon 10 [26, 27]. Expression analysis of human and mouse IKKγ showed that the gene becomes active early during embryogenesis and is expressed ubiquitously [28]. By performing Northern blot analysis, two groups have reported that IKKγ is widely expressed in human tissues, including the heart, brain, placenta, lung, liver, skeletal muscle, kidney, pancreas, bone marrow, lymph nodes, adrenals, spinal cord, thyroid, thymus, spleen, breast, and prostate [4, 28]. In addition, Makris et al., using immunohistochemical assays, showed the expression of IKKγ in the human epidermis [25]. A low level of expression of IKKγ was reported in the stomach, small intestine, colon, trachea, bladder and uterus [28].
Three groups of investigators have generated IKKγ-knockout mice and they have demonstrated that the complete loss of IKKγ function is incompatible with life [25, 29, 30]. IKKγ-null male mice died in utero from severe liver damage due to apoptosis [25, 30]. Rudolph et al. reported that murine embryonic fibroblasts (MEFs) derived from IKKγ-null mice failed to exhibit induction of NF-κB activity as evidenced by the lack of the ability to induce IκBα phosphorylation, IκBα degradation and NF-κB binding to DNA in response to treatment with TNFα, LPS, IL-1 or polyriboinosinic: polyribocytidylic acid (poly IC) [29]. Also the IKKγ-null MEFs were susceptible to undergo apoptosis following treatment with TNFα. According to Makris et al. and Schmidt-Supprian and colleagues, female mice heterozygous for the IKKγ deficiency (IKKγ+/−) survived but developed a developmental abnormality of the skin that resembled the human disease referred to as incontinentia pigmenti in which the keratinocytes are highly susceptible to undergo apoptosis [25, 30]. Makris et al. reported that by day 9 after birth, 55% of the heterozygous female mice were dead, but by 1 month, the surviving female mice had recovered [25].
It appears that IKKγ is particularly essential for the generation and/or survival of lymphocytes. Schmidt-Supprian et al. reported that chimeric mice generated from IKKγ-knockout embryonic stem (ES) cells did not have any ES cell-derived T and B lymphocytes, and suggested that IKKγ-null lymphocytes either do not develop or are counter-selected [30]. Also, according to Makris et al., IKKγ-deficient female mice examined at age of 9 days had very small spleens and thymuses associated with apoptotic changes [25]. In another study, Schmidt-Supprian et al. showed that T cell-specific ablation of IKKγ in mice prevented the development of peripheral T cells, indicating that IKKγ is essential for the generation and survival of mature T cells [31]. Evidence that IKKγ is essential for T cell development and/or survival in human beings was provided by the very low number of naïve-phenotype T cells in a male child who had a markedly reduced expression of IKKγ due to a 4.4-kb duplication within the IKKγ gene [32]. Pasparakis et al. performed B lineage-specific disruption of IKKγ and showed that this leads to the disappearance of mature B lymphocytes in adult mice and they concluded that IKKγ is necessary for the maintenance of mature B cells [33]. However, on the contrary, using IKKγ(–/–) ES cells from IKKγ(–/–) mice and an in vitro differentiation system, Kim et al. demonstrated that IKKγ is not required for B cell development but plays an important role in B-cell survival [34].
The domain structure of IKKγ protein
Human IKKγ is composed of 419 amino acid residues, whereas the murine IKKγ has 412 amino acid residues [4, 24]. The number of amino acid residues in the bovine IKKγ is 419 [35]. The human protein shares 87.9% identity and 90.5% similarity with its mouse homologue and 86% identity and 91% similarity with the bovine IKKγ. It has a predicted molecular weight of 48 kD. Human IKKγ has two coiled-coil regions, a leucine zipper (LZ) domain and a zinc finger domain, which are separated by α helical regions (Fig. 2A). The first coiled coil (CC1) is located in the region of amino acid residues 63–193 and the second coiled coil (CC2) is situated in the region encompassing amino acid residues 258–298; the leucine zipper spans amino acid residues 319–346 [26]. Two pseudo-leucine zipper domains have also been identified in the middle portion of the protein [36, 37]. The zinc finger domain is located at the C-terminus and spans the region between residues 397 and 419 in the human IKKγ (Fig. 2B). The sequence of the zinc finger domain is identical among the human, mouse and rat IKKγ, whereas only one amino acid residue is different in the zinc finger of the bovine IKKγ as compared to the human IKKγ (Fig. 3) [24, 35]. The last 36 amino acid residues of IKKγ, encompassing the zinc finger domain, are identical among human, rat and mouse IKKγ, whereas three residues in this region of human IKKγ are different from those in the bovine IKKγ (Fig. 3).
Fig 2.
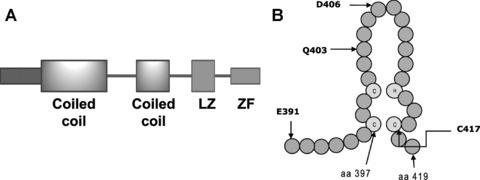
The domain structure of IKKγ and its zinc finger. (A) The positions of the two coiled-coil regions, the leucine zipper (LZ) domain and the zinc finger (ZF) domain are depicted. (B) A schematic representation of the zinc finger domain of IKKγ, which extends from amino acid (aa) residues 397 to 419, is shown. Common mutation sites in the region are also indicated with the letter symbol of the amino acid followed by the residue number. The three cysteine residues and the single histidine residue that coordinate a zinc ion are indicated by the lettered circles.
Fig 3.
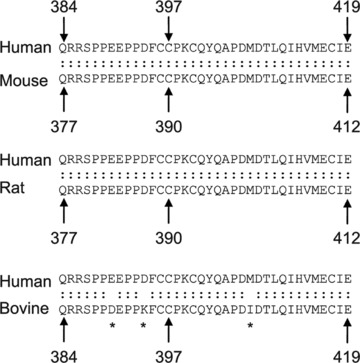
A comparative sequence analysis of the C-terminus of IKKγ. Alignment of the amino acid sequence from the C-terminus of the human IKKγ against that from the mouse, rat and bovine IKKγ is presented. The numbers indicate the sequence number of the amino acid residue. An asterisk indicates a position of mismatch.
A few studies have demonstrated that some of the domains of IKKγ have stimulus-specific roles with regards to the activation of NF-κB. For example, it was reported that the first 245 amino acids of human IKKγ are needed for NF-κB activation by Tax, TNFα or IL-1 but the zinc finger is not needed by Tax [38]. In support of this finding, Chu et al. showed that amino acid residues 1–100 of murine IKKγ are required for the binding of Tax to IKKγ[39]. However, on the contrary, Harhaj and Sun reported that the C-terminal region of IKKγ is crucial for strong binding to Tax [40].
Moreover, the various domains of IKKγ appear to have unique roles in the interactions of the protein with the two catalytic components of the IKK complex, namely IKKα and IKKβ, and with other IKKγ molecules. The N-terminus of IKKγ is responsible for the interaction with IKKα and IKKβ[36, 41]. This region has also been reported to be required for the stimulation of IKKβ kinase activity [41]. According to Ye et al., the middle region of the protein is the self-association region and also interacts with receptor interacting protein 1 (RIP1) [36]. Marienfeld et al. mapped the IKK-binding domain of IKKγ to the region between amino acid residues 80 and 120; but, in contrast to the findings of Ye et al., they identified the region between amino acids 40 and 80 as the IKKγ dimerization domain [42].
Mutations involving the zinc finger of IKKγ
Three categories of mutations affecting IKKγ have been reported in human beings [43–49]. The first category of these mutations consists of hypomorphic mutations typically involving the zinc finger domain and nearby C-terminal regions and causing hypohidrotic ectodermal dysplasia with immune deficiency (HED-ID) in males. The second class consists of amorphic mutations causing incontinentia pigmenti (IP) in females and, generally, pre-natal death in males. The third category is composed of hypomorphic mutations involving the stop codon causing ectodermal dysplasia, anhidrotic, with immunodeficiency, osteopetrosis and lymphedema (OL-EDA-ID) in males.
Hypohidrotic ectodermal dysplasia with immune deficiency (HED-ID), which is also known as ectodermal dysplasia, anhidrotic with immune deficiency (EDA-ID), is characterized by an abnormal development of ectodermal appendages and abnormalities of immune function [50]. Patients characteristically display the features of two syndromes in combination: hypohidrotic ectodermal dysplasia and hyper-IgM syndrome. Hypohidrotic (anhidrotic) ectodermal dysplasia (Christ-Siemens-Touraine syndrome) is the result of an abnormal development of ectodermal appendages [51]. It is characterized by the absence or deficiency of at least two derivatives of the ectoderm: teeth, hair, sweat glands or nails. Hyper IgM (HIGM) syndrome is characterized by a defective antibody heavy chain class switching and an impaired somatic hypermutation. Clinically the patients have low levels of serum IgG and IgA and normal or elevated IgM, leading to an increased susceptibility to infections [47].
HED-ID has been associated with mutations affecting three proteins, namely IKKγ, IκBα and interleukin-1 receptor-associated kinase-4 (IRAK-4) [52]. A hypermorphic mutation in the IκBα gene has been reported to be associated with autosomal dominant HED-ID and mutations in the IRAK-4 gene (which codes for a protein involved in IL-1 signalling) have been associated with autosomal recessive HED-ID. Hypomorphic mutations in the IKKγ gene are associated with X-linked recessive HED-ID and account for the majority of cases of HED-ID. The estimated incidence of X-linked HED-ID is 1:250,000 live male births [53]. The disease affects males whereas females serve as carriers and usually show a skewed X-inactivation [54]. However, for some of the mutations Aradhya et al. reported the existence of only a slight skewing or random X-inactivation in females [55]. The mutations in IKKγ associated with X-linked HED-ID were first discovered by Zonana et al. who, following the discovery of amorphic mutations of IKKγ as the cause of incontinentia pigmenti [26], hypothesized that milder mutations of IKKγ could be the cause of HED-ID [56]. At least 47 patients have been reported in the Medline literature so far to have HED-ID.
The IKKγ mutations that have been associated with HED-ID include insertions, deletions or substitutions and generally occur in exons 4–10. The majority of these mutations affect the C-terminal region of IKKγ, where the zinc finger domain is located (Table 1). So far, seven different mutations (two single nucleotide insertions and five missense substitutions) within the zinc finger have been reported from 13 patients [53–62]. Most of those patients had the onset of HED-ID during their early childhood years. Nine of those patients had mutations that involved the C417 residue; the cysteine residue was replaced by an arginine residue in six cases, by a phenyalanine residue in two cases and by a tyrosine residue in one case. One patient had a D406V mutation and another one had a frameshift mutation at D406 that added 13 new amino acid residues. There was a patient with a non-sense mutation at residue Q403 and there was another patient with a frameshift mutation at residue I412, which added seven new amino acid residues. In addition to the mutations that occur within the zinc finger, there were several patients who had mutations in the region just proximal to the beginning of the zinc finger domain and those mutations resulted in the loss of the whole zinc finger domain [53, 56, 60–64]. Also, a missense mutation (1259A>G or X420W) at the stop codon that results in the addition of 27 novel amino acid residues has been associated with OL-EDA-ID, a novel syndrome related to HED-ID and for which there are two patients reported [26, 28]. Those patients had osteopetrosis and lymphedema as well as the manifestations of HED-ID.
Table 1.
Mutations in the C-terminus of IKKγ associated with HED-ID
| Change in DNA | Change in protein | Novel amino acid residues | No. of patients | References |
|---|---|---|---|---|
| dupC1161a | P390fsX394b | RGATx | 1 | [55] |
| 1167insC | E390fsX394b | RGATx | 6 | [53, 56, 60–64] |
| 1167insC | E390fsX394b | RGATx | 1c | [58] |
| 1171G>T | E391Xb | 3 | [56] | |
| 1207C>T | Q403X | 1 | [53, 59] | |
| 1217A>T | D406V | 1 | [57] | |
| 1218insA | D406fsX419 | EYGHPADTCHGVHx | 1 | [54] |
| 1235insC | I412fsX418 | TTCHGVHx | 1 | [60] |
| 1249T>C | C417R | 6 | [53, 54, 56, 57, 59] | |
| 1250G>A | C417Y | 1 | [53] | |
| 1250G>T | C417F | 2 | [54, 56] | |
| 1259A>G | 419fsX27d | 27 extra residues | 2 | [26, 28] |
Symbols used to designate sequence changes: dup, duplication; ins, insertion; >, substitution; fs, frameshift; X, translation termination site.
These mutations result in the complete loss of the zinc finger domain.
The patient had normal levels of IgM.
These patients had the OL-EDA-ID syndrome.
In addition to exhibiting the features of ectodermal dysplasia, patients with HED-ID show immunologic abnormalities and as a result suffer from recurrent and sometimes disseminated infections caused by a variety of infectious organisms beginning in infancy. The reported infectious agents include: bacteria such as Staphylococcus, Streptococcus, Listeria, Klebsiella, Haemophilus and Pseudomonas species and atypical mycobacteria; viruses such as cytomegalovirus, Epstein-Barr virus, herpesvirus, varicella virus, molluscum contagiosum virus and human papilloma virus; fungi such as Candida albicans; protozoa such as Pneumocystis carini and Giardia lamblia[50, 53]. The affected organs include the skin, bones, meninges, intestines and lungs but the infections can also involve multiple organs. Most patients die during early childhood but a few cases have survived well into the second decade [54, 56, 59, 65].
The immunologic lesions of HED-ID are predominantly those of HIGM syndrome [53, 57]. The patients usually have hypogammaglobulinemia with high IgM, low IgG and low IgA levels, but this is highly variable and there are patients with normal IgM levels, normal IgG levels or high IgA levels. There may be a deficiency in the generation of antibodies to specific antigens. The numbers of T and B cells are usually normal but this is also variable. In some cases, the CD40-induced proliferation of B cells may be impaired. Some patients show a decrease in NK cell cytotoxicity [59]. Probably the variability of the immunologic features is related to the particular mutation involved since HED-ID has been associated with a variety of mutations involving IKKγ.
The biochemical deficits caused by the mutations associated with HED-ID vary depending on the nature of the particular mutation and the kind of cell stimulation used. It appears that there is some residual NF-κB-activating ability since total loss of IKKγ function is incompatible with life. In this regard, some of the mutations have been shown to impair but not to abolish NF-κB signalling [54, 66]. Some of the mutations impair NF-κB activation by certain stimuli, but not by others [57, 59, 67]. For instance, Aradhya et al. tested some of the mutations for their ability to complement for the deficient NF-κB activity in an IKKγ-null cell line: the ΔC1161 mutation showed no induction of NF-κB activity and the dupC1161 and dup1166–1178 mutations showed a markedly reduced induction compared to the wild-type protein [55].
The roles of the zinc finger domain of IKKγ
The classic zinc finger is a structural motif that forms the DNA interacting domain of certain DNA-binding proteins. In the original zinc finger model, which was proposed for the protein TFIIIA and which consists of about 30 residues, two invariant pairs of cysteines and histidines (C2H2) constitute a tetrahedral coordination site for a zinc ion, and the amino acid residues between these coordination sites protrude as a finger [68]. Subsequently, zinc finger domains with C2HC and C2C2 sites have been reported [69]. The number of potential zinc finger domains in proteins ranges from one to 30 [69]. Zinc finger proteins that bind to DNA usually contain two or more zinc finger motifs often arranged in tandem arrays [70]. Even though zinc finger proteins have been commonly recognized as DNA binding proteins, some zinc finger proteins have also been shown to bind to RNA or protein [69, 71].
Zinc finger proteins are abundant in the genomes of eukaryotes; in addition to their role in DNA recognition, they have been implicated to play roles in RNA packaging, in the regulation of apoptosis, in protein folding and assembly and in lipid binding [72]. A number of zinc finger proteins play key roles during development and mutations in some zinc finger proteins have been associated with human disease [73]. IKKγ contains a single zinc finger domain which is located at its C-terminus and that has the C2HC zinc coordination site. A number of groups have examined the roles of the zinc finger domain of IKKγ in cells derived from patients harbouring mutations in this region of the protein or in various cell lines transfected with the mutant forms of the protein (either zinc finger deletion mutants or reconstructions of mutations that have been found in patients). The lessons learned from those mutational and other in vitro studies regarding the roles of the zinc finger in the functions of IKKγ are discussed below.
Activation of NF-κB
The results of published studies that examined the effects of IKKγ zinc finger mutations on NF-κB activity are summarized in Table 2. The need for an intact zinc finger domain appears to depend on the particular cell type and the nature of the stimulus. In dendritic cells, the zinc finger of IKKγ appears to be required for NF-κB activation by CD154 but not by LPS [74]. In monocytes, the zinc finger does not appear to be essential for NF-κB activation by TNFα or LPS, but is needed for NF-κB activation by CD154 [57]. However, in a human monocyte cell line that had an endogenous expression of IKKγ, overexpression of the C417R mutant IKKγ inhibited NF-κB activation in response to TNFα or LPS [75]. In B cells, according to studies reported by two groups, the zinc finger is essential for NF-κB activation by CD154, LPS or IL-1β[67, 76]. However, according to another report, in B cells, the zinc finger domain is not needed for the activation of NF-κB by fast activators such as TNFα and LPS but is essential for the activation of NF-κB by slow activators such as UV light and the topoisomerase inhibitor etoposide [77]. In T cells, the zinc finger is required for the activation of NF-κB by treatment with TNFα or PMA/ionomycin or following overexpression of TRAF2 or TRAF6 [75, 76, 78, 79].
Table 2.
A summary of the effects of IKKγ zinc finger mutations on NF-κB activity
| IKKγ mutation | Cell type | Source of cells | Stimulus | Effect on NF-κB activation | References |
|---|---|---|---|---|---|
| C417R | Dendritic cells | Patient | CD154 | Decrease | [74] |
| LPS | No effect | ||||
| C417R or D406V | Monocytes | Patient | CD154 | Decrease | [57] |
| LPS | No effect | ||||
| TNFα | No effect | ||||
| C417R | B cells | Patient | CD154 | Decrease | [67] |
| ZFDa, b | Fibroblasts | Patient | TNFα | Decrease | [80] |
| IL-1 | Decrease | ||||
| C389S/C393Sc | IKKγ-null mouse fibroblasts | Cell line | TNFα | Decrease | [80] |
| IL-1 | No effect | ||||
| C417R, D406V or ZFD | IKKγ-null mouse B cells | Cell line | UV | Decrease | [77] |
| Etoposide | Decrease | ||||
| LPS | No effect | ||||
| TNFα | No effect | ||||
| C417R | IKKγ-null mouse B cells | Cell line | LPS | Slight decrease | [76] |
| IL-1β | Decrease | ||||
| C417R | Human monocytes | Cell line | LPS | Decrease | [75] |
| TNFα | Decrease | ||||
| IKKγ-null human T cells | Cell line | TRAF2d | Decrease | ||
| TRAF6 | Decrease | ||||
| C417R or ZFD | IKKγ-null human T cells | Cell line | TNFα | Decrease | [79] |
| C417R or D406V | IKKγ-null human T cells | Cell line | TNFα/ | Decrease | [78] |
| PMA/ionomycin | Decrease | ||||
| C417R | IKKγ-null human T cells | Cell line | TNFα | Decrease | [76] |
ZFD, zinc finger deletion.
Caused by a duplication of nucleotides 1166–1178, which produced a frameshift at P393 residue and then truncated the protein after the addition of four new amino acid residues.
This sequence number refers to the murine IKKγ, whereas all the other sequence numbers in this table refer to the human IKKγ.
TRAF, tumour necrosis factor receptor-associated factor.
Schmid et al. carried out experiments on lymphocytes obtained from a patient who harboured the E390fsX394 mutation that resulted in the loss of the zinc finger domain of IKKγ[61]. They reported that lymphocyte proliferation in response to anti-CD3 was poor; there was also a poor proliferative response to the mitogens phytohemagglutinin (PHA), staphylococcal enterotoxin B or PMA/ionomycin, even though that effect was transient. They also reported that there was normal CD154 and CD69 expression following mitogenic stimulation but PHA failed to induce IL-2 secretion. Zonana et al. studied T cells from two patients with a similar mutation (E391X), which also eliminates the zinc finger domain, and showed that the expression of CD154 and CD69 in stimulated lymphocytes was normal; they also reported that the lymphocytes initially showed poor proliferative responses which, however, subsequently normalized [56]. Shifera and Horwtiz stably expressed the C417R and D407V mutations in a Jurkat cell line that was null for IKKγ and showed that either mutation blocked the PMA/ionomycin induction of IL-2 [78]. According to Jain et al., T cells from patients the C417R or D407V mutation secreted normal amounts of TNFα and IFNγ when stimulated with anti-CD3 antibodies or with PHA [57]. Therefore, the zinc finger appears to be involved in the proliferation of T cells and in some aspects of T-cell activation.
Jain et al. showed that B cells derived from patients with the C417R or D406V mutation co-expressed IgM and IgD on their surfaces but they failed, in spite of a normal expression of CD40, to secrete IgG and IgA in response to stimulation by a combination of CD154 trimer, IL-4, IL-2 and IL-10 [57]. Orange et al. showed that peripheral blood mononuclear cells (PBMCs) from patients with the C417R or Q403X mutation proliferated poorly when stimulated with CD40 ligation and that the CD40-mediated up-regulation of CD23 and CD54 surface expression in B cells was impaired. In vitro IgE synthesis by PBMCs was low with the C417R mutation but was normal with the Q403X mutation [59]. Therefore, the zinc finger of IKKγ also seems to play a role in some aspects of B cell activation.
Makris et al. examined the role of the zinc finger on NF-κB activity in primary fibroblasts derived from aborted male foetuses that carried the dup1166–1178 mutation in IKKγ[80]. This mutation produced a frameshift at the P393 residue and then truncated the protein after the addition of four new amino acid residues, thus resulting in a complete loss of the zinc finger. They found that there was an induction of IκBα degradation and NF-κB DNA binding activity following stimulation with TNFα or IL-1 but the responses were lower than those in wild-type fibroblasts, especially with TNFα, indicating that the zinc finger domain is needed for full activation [80]. In contrast, when the same group expressed the murine IKKγ mutated at cysteine residues within the zinc finger domain (C389S and C393S double mutant) in IKKγ-null MEFs they found that the mutations inhibited IKK activation by TNFα but not by IL-1 [80].
Phosphorylation of IKKγ
Post-translational modification, including phosphorylation, has been reported to play a role in regulating some of the functions of IKKγ. Carter et al. showed that Tax protein induced constitutive phosphorylation of murine IKKγ as a result of Tax-induced phosphorylation of IKKβ, which in turn is required for the activation of the IKK complex [81]. They demonstrated that in the presence of Tax protein or TNFα stimulation, both of which triggered NF-κB, IKKβ caused the phosphorylation of human IKKγ at S31, S43 and S376 [82]. In addition, they showed that IKKγ phosphorylation was decreased when its zinc finger was deleted indicating that the zinc finger is important in the phosphorylation of IKKγ. However, the functional consequence of IKKγ phosphorylation is not clear.
Ubiquitination of IKKγ
Tang et al. showed that TNFα induced the non-degradative ubiquitination of IKKγ mediated by inhibitor of apoptosis protein-1 (c-IAP1), a protein which is part of the tumour necrosis factor receptor-1 (TNFR1) complex [79]. The ubiquitination of IKKγ induced by TNFα treatment of T cells was inhibited when the zinc finger of IKKγ was deleted or in the presence of the C417R mutation in the zinc finger [79]. The K399 residue, which is within the zinc finger of IKKγ, was suggested as a possible ubiquitination site [79]. Temmerman et al. examined dendritic cells derived from two patients with the C417R mutation and they showed that there was a lack of IKKγ ubiquitination following stimulation via CD40, associated with normal p65 but absent c-Rel activity; however, there was a normal degree of IKKγ ubiquitination and NF-κB activation when the cells were stimulated with LPS [74]. Therefore, the zinc finger seems to be needed in the induced ubiquitination of IKKγ during the activation of NF-κB by certain stimuli.
Recognition of ubiquitinated proteins by IKKγ
It also appears that the zinc finger of IKKγ plays a role in the recognition of ubiquitinated proteins. Cordier and colleagues examined the solution structure of the zinc finger of IKKγ by nuclear magnetic resonance [76]. They found that both the wild-type and the C417R mutant exhibited a global ββα fold and both bound zinc with a similar affinity but the mutant protein exhibited a destabilization of two potential-protein binding surfaces [76]. The same group also demonstrated that the zinc finger of IKKγ binds to ubiquitin and also showed that the recognition of ubiquitin chains by the zinc finger of IKKγ is required for the activation of NF-κB by TNFα or PMA/ionomycin in T cells [22, 83]. Laplantine and colleagues reported that the sole function of the zinc finger of IKKγ is to recognize K63-linked ubiquitin chains [83]. According to their observations, the zinc finger together with the ubiquitin-binding domain in the CC2-LZ region of IKKγ forms a bipartite ubiquitin-binding domain that interacts with ubiquitin with a high affinity [83]. In addition, they reported that the zinc finger is not involved in the recognition of linear polyubiquitin chains [83], which are rather recognized by the CC2-LZ region, in a manner that is required for NF-κB activation by stimuli such as TNFα, IL-1, LPS and PMA/ionomycin [84]. It is very interesting to note that the zinc fingers of other proteins involved in the NF-κB pathway have also been reported to bind to K63-linked ubiquitin. For example, the single zinc fingers of the TAK1 binding (TAB) proteins TAB2 and TAB3, adaptor proteins that link the IKK complex to proximal signalling components, bind to K63-linked diubiquitin [85, 86]. Therefore, according to the observations mentioned above, the zinc finger of IKKγ, in cooperation with the CC2-LZ region of IKKγ, is very likely involved in the recognition of polyubiquitinated upstream signalling intermediates. IKKγ is known to recognize and interact with ubiquitinated proteins, such as ubiquitinated RIP1 in the TNFR1 signalling cascade [87, 88] and ubiquitinated B cell CLL/lymphoma 10 (Bcl10) in the TCR signalling cascade [89]. It is possible that the zinc finger mutations associated with HED-ID interfere with the recognition of these polyubiquitinated intermediates.
Conclusions and perspectives
IKKγ contains a highly conserved single zinc finger domain at its C-terminus. A number of mutations affecting this region have been associated with a human disease involving abnormalities of ectodermal development and defects of immune functions. Such mutations are not embryonic lethal but lead to an early death due to impaired resistance to diseases caused by microbial pathogens. The zinc finger of IKKγ is likely engaged in mediating the interactions of IKKγ with other proteins, such as upstream activators or inhibitors of the IKK complex. This role could involve the recognition of the upstream regulators of IKK that have undergone non-degradative ubiquitination [21, 88, 89]. Also, the zinc finger itself could be required for the ubiquitination of IKKγ, a modification known to regulate the functioning of IKKγ.
The zinc finger is essential for NF-κB activation in various immune cell types but this requirement is related to the particular cell type and stimulus used. It is possible that the differential need for a functional zinc finger in NF-κB activation is related to whether or not the zinc finger is involved in the recognition of ubiquitinated upstream regulators following exposure of a particular cell type to a specific stimulus. Since ubiquitination is a recurrent theme in the regulation of the NF-κB pathway and because zinc fingers have been shown to be engaged in ubiquitin recognition and binding, it is worth investigating the ubiquitin binding role of the IKKγ zinc finger in various immune and ectodermal cell types in response to different stimuli. In addition, the roles of the IKKγ zinc finger on NF-κB activation in cells derived from ectodermal tissues have not been examined in detail and this area also deserves to be investigated in the future.
Acknowledgments
The author is supported by a grant from Thomas J. Long Foundation to Dr. Jorge A. Alvarado at the University of California, San Francisco, CA, USA.
References
- 1.Yamaoka S, Courtois G, Bessia C, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–40. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 2.Rothwarf DM, Zandi E, Natoli G, et al. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 3.Mercurio F, Murray BW, Shevchenko A, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–38. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Kang J, Friedman J, et al. Identification of a cell protein (FIP-3) as a modulator of NF-kappaB activity and as a target of an adenovirus inhibitor of tumor necrosis factor alpha-induced apoptosis. Proc Natl Acad Sci USA. 1999;96:1042–7. doi: 10.1073/pnas.96.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiDonato JA, Hayakawa M, Rothwarf DM, et al. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 6.Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–6. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 7.Zandi E, Rothwarf DM, Delhase M, et al. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–52. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 8.Regnier CH, Song HY, Gao X, et al. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–83. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 9.Woronicz JD, Gao X, Cao Z, et al. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–9. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 10.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–62. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 12.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 13.Delhalle S, Blasius R, Dicato M, et al. A beginner’s guide to NF-kappaB signaling pathways. Ann N Y Acad Sci. 2004;1030:1–13. doi: 10.1196/annals.1329.002. [DOI] [PubMed] [Google Scholar]
- 14.Huang H, Calderon TM, Berman JW, et al. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun. 1999;67:5434–40. doi: 10.1128/iai.67.10.5434-5440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Stark GR. NFkappaB-dependent signaling pathways. Exp Hematol. 2002;30:285–96. doi: 10.1016/s0301-472x(02)00777-4. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Peet GW, Balzarano D, et al. Novel NEMO/IkappaB kinase and NF-kappa B target genes at the pre-B to immature B cell transition. J Biol Chem. 2001;276:18579–90. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 18.Dejardin E, Droin NM, Delhase M, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–35. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 19.Eliopoulos AG, Caamano JH, Flavell J, et al. Epstein-Barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-kappaB2 to p52 via an IKKgamma/NEMO-independent signalling pathway. Oncogene. 2003;22:7557–69. doi: 10.1038/sj.onc.1207120. [DOI] [PubMed] [Google Scholar]
- 20.He KL, Ting AT. Essential role for IKKgamma/NEMO in TCR-induced IL-2 expression in Jurkat T cells. Eur J Immunol. 2003;33:1917–24. doi: 10.1002/eji.200323650. [DOI] [PubMed] [Google Scholar]
- 21.Chen F, Bhatia D, Chang Q, et al. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 2006;13:1835–8. doi: 10.1038/sj.cdd.4402014. [DOI] [PubMed] [Google Scholar]
- 22.Cordier F, Grubisha O, Traincard F, et al. The zinc finger of NEMO is a functional ubiquitin-binding domain. J Biol Chem. 2009;284:2902–7. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 23.Tokunaga F, Sakata S, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–32. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 24.Jin DY, Jeang KT. Isolation of full-length cDNA and chromosomal localization of human NF-kappaB modulator NEMO to Xq28. J Biomed Sci. 1999;6:115–20. doi: 10.1007/BF02256442. [DOI] [PubMed] [Google Scholar]
- 25.Makris C, Godfrey VL, Krahn-Senftleben G, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–79. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 26.Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000;405:466–72. doi: 10.1038/35013114. [DOI] [PubMed] [Google Scholar]
- 27.Fusco F, Mercadante V, Miano MG, et al. Multiple regulatory regions and tissue-specific transcription initiation mediate the expression of NEMO/IKKgamma gene. Gene. 2006;383:99–107. doi: 10.1016/j.gene.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 28.Aradhya S, Woffendin H, Jakins T, et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet. 2001;10:2171–9. doi: 10.1093/hmg/10.19.2171. [DOI] [PubMed] [Google Scholar]
- 29.Rudolph D, Yeh WC, Wakeham A, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–62. [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt-Supprian M, Bloch W, Courtois G, et al. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:981–92. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt-Supprian M, Courtois G, Tian J, et al. Mature T cells depend on signaling through the IKK complex. Immunity. 2003;19:377–89. doi: 10.1016/s1074-7613(03)00237-1. [DOI] [PubMed] [Google Scholar]
- 32.Nishikomori R, Akutagawa H, Maruyama K, et al. X-linked ectodermal dysplasia and immunodeficiency caused by reversion mosaicism of NEMO reveals a critical role for NEMO in human T-cell development and/or survival. Blood. 2004;103:4565–72. doi: 10.1182/blood-2003-10-3655. [DOI] [PubMed] [Google Scholar]
- 33.Pasparakis M, Schmidt-Supprian M, Rajewsky K. IkappaB kinase signaling is essential for maintenance of mature B cells. J Exp Med. 2002;196:743–52. doi: 10.1084/jem.20020907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S, La Motte-Mohs RN, Rudolph D, et al. The role of nuclear factor-kappaB essential modulator (NEMO) in B cell development and survival. Proc Natl Acad Sci USA. 2003;100:1203–8. doi: 10.1073/pnas.0337707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rottenberg S, Schmuckli-Maurer J, Grimm S, et al. Characterization of the bovine IkappaB kinases (IKK)alpha and IKKbeta, the regulatory subunit NEMO and their substrate IkappaBalpha. Gene. 2002;299:293–300. doi: 10.1016/s0378-1119(02)01011-9. [DOI] [PubMed] [Google Scholar]
- 36.Ye J, Xie X, Tarassishin L, et al. Regulation of the NF-kappaB activation pathway by isolated domains of FIP3/ IKKgamma, a component of the IkappaB-alpha kinase complex. J Biol Chem. 2000;275:9882–9. doi: 10.1074/jbc.275.13.9882. [DOI] [PubMed] [Google Scholar]
- 37.Sun SC, Harhaj EW, Xiao G, et al. Activation of I-kappaB kinase by the HTLV type 1 Tax protein: mechanistic insights into the adaptor function of IKKgamma. AIDS Res Hum Retroviruses. 2000;16:1591–6. doi: 10.1089/08892220050193001. [DOI] [PubMed] [Google Scholar]
- 38.Iha H, Kibler KV, Yedavalli VR, et al. Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: implications for stimulus-specific interruption of oncogenic signaling. Oncogene. 2003;22:8912–23. doi: 10.1038/sj.onc.1207058. [DOI] [PubMed] [Google Scholar]
- 39.Chu ZL, Shin YA, Yang JM, et al. IKKgamma mediates the interaction of cellular IkappaB kinases with the Tax transforming protein of human T cell leukemia virus type 1. J Biol Chem. 1999;274:15297–300. doi: 10.1074/jbc.274.22.15297. [DOI] [PubMed] [Google Scholar]
- 40.Harhaj EW, Sun SC. IKKgamma serves as a docking subunit of the IkappaB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J Biol Chem. 1999;274:22911–4. doi: 10.1074/jbc.274.33.22911. [DOI] [PubMed] [Google Scholar]
- 41.Li XH, Fang X, Gaynor RB. Role of IKKgamma/nemo in assembly of the Ikappa B kinase complex. J Biol Chem. 2001;276:4494–500. doi: 10.1074/jbc.M008353200. [DOI] [PubMed] [Google Scholar]
- 42.Marienfeld RB, Palkowitsch L, Ghosh S. Dimerization of the I kappa B kinase-binding domain of NEMO is required for tumor necrosis factor alpha-induced NF-kappa B activity. Mol Cell Biol. 2006;26:9209–19. doi: 10.1128/MCB.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371–5. doi: 10.1093/hmg/11.20.2371. [DOI] [PubMed] [Google Scholar]
- 44.Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002;47:169–87. doi: 10.1067/mjd.2002.125949. [DOI] [PubMed] [Google Scholar]
- 45.Courtois G, Smahi A, Israel A. NEMO/IKK gamma: linking NF-kappa B to human disease. Trends Mol Med. 2001;7:427–30. doi: 10.1016/s1471-4914(01)02154-2. [DOI] [PubMed] [Google Scholar]
- 46.Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–43. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 47.Durandy A, Peron S, Fischer A. Hyper-IgM syndromes. Curr Opin Rheumatol. 2006;18:369–76. doi: 10.1097/01.bor.0000231905.12172.b5. [DOI] [PubMed] [Google Scholar]
- 48.Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006;16:282–8. doi: 10.1016/j.gde.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 49.Uzel G. The range of defects associated with nuclear factor kappaB essential modulator. Curr Opin Allergy Clin Immunol. 2005;5:513–8. doi: 10.1097/01.all.0000191241.66373.74. [DOI] [PubMed] [Google Scholar]
- 50.McDonald DR, Janssen R, Geha R. Lessons learned from molecular defects in nuclear factor kappaB dependent signaling. Microbes Infect. 2006;8:1151–6. doi: 10.1016/j.micinf.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 51.Wisniewski SA, Kobielak A, Trzeciak WH, et al. Recent advances in understanding of the molecular basis of anhidrotic ectodermal dysplasia: discovery of a ligand, ectodysplasin A and its two receptors. J Appl Genet. 2002;43:97–107. [PubMed] [Google Scholar]
- 52.Puel A, Picard C, Ku CL, et al. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41. doi: 10.1016/j.coi.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 53.Orange JS, Jain A, Ballas ZK, et al. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol. 2004;113:725–33. doi: 10.1016/j.jaci.2004.01.762. [DOI] [PubMed] [Google Scholar]
- 54.Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 55.Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma) Am J Hum Genet. 2001;68:765–71. doi: 10.1086/318806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zonana J, Elder ME, Schneider LC, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–62. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain A, Ma CA, Liu S, et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–8. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 58.Lee WI, Torgerson TR, Schumacher MJ, et al. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105:1881–90. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 59.Orange JS, Brodeur SR, Jain A, et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest. 2002;109:1501–9. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orange JS, Levy O, Geha RS. Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kappaB activation. Immunol Rev. 2005;203:21–37. doi: 10.1111/j.0105-2896.2005.00221.x. [DOI] [PubMed] [Google Scholar]
- 61.Schmid JM, Junge SA, Hossle JP, et al. Transient hemophagocytosis with deficient cellular cytotoxicity, monoclonal immunoglobulin M gammopathy, increased T-cell numbers, and hypomorphic NEMO mutation. Pediatrics. 2006;117:1049–56. doi: 10.1542/peds.2005-2062. [DOI] [PubMed] [Google Scholar]
- 62.Sitton JE, Reimund EL. Extramedullary hematopoiesis of the cranial dura and anhidrotic ectodermal dysplasia. Neuropediatrics. 1992;23:108–10. doi: 10.1055/s-2008-1071322. [DOI] [PubMed] [Google Scholar]
- 63.Chang TT, Behshad R, Brodell RT, et al. A male infant with anhidrotic ectodermal dysplasia/immunodeficiency accompanied by incontinentia pigmenti and a mutation in the NEMO pathway. J Am Acad Dermatol. 2008;58:316–20. doi: 10.1016/j.jaad.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 64.Mancini AJ, Lawley LP, Uzel G. X-linked ectodermal dysplasia with immunodeficiency caused by NEMO mutation: early recognition and diagnosis. Arch Dermatol. 2008;144:342–6. doi: 10.1001/archderm.144.3.342. [DOI] [PubMed] [Google Scholar]
- 65.Niehues T, Reichenbach J, Neubert J, et al. Nuclear factor kappaB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:1456–62. doi: 10.1016/j.jaci.2004.08.047. [DOI] [PubMed] [Google Scholar]
- 66.Orstavik KH, Kristiansen M, Knudsen GP, et al. Novel splicing mutation in the NEMO (IKK-gamma) gene with severe immunodeficiency and heterogeneity of X-chromosome inactivation. Am J Med Genet A. 2006;140:31–9. doi: 10.1002/ajmg.a.31026. [DOI] [PubMed] [Google Scholar]
- 67.Jain A, Ma CA, Lopez-Granados E, et al. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest. 2004;114:1593–602. doi: 10.1172/JCI21345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller J, McLachlan AD, Klug A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985;4:1609–14. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iuchi S. Three classes of C2H2 zinc finger proteins. Cell Mol Life Sci. 2001;58:625–35. doi: 10.1007/PL00000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 71.Gamsjaeger R, Liew CK, Loughlin FE, et al. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem Sci. 2007;32:63–70. doi: 10.1016/j.tibs.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 72.Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11:39–46. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 73.Ladomery M, Dellaire G. Multifunctional zinc finger proteins in development and disease. Ann Hum Genet. 2002;66:331–42. doi: 10.1017/S0003480002001215. [DOI] [PubMed] [Google Scholar]
- 74.Temmerman ST, Ma CA, Borges L, et al. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood. 2006;108:2324–31. doi: 10.1182/blood-2006-04-017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang F, Yamashita J, Tang E, et al. The zinc finger mutation C417R of I-kappa B kinase gamma impairs lipopolysaccharide- and TNF-mediated NF-kappa B activation through inhibiting phosphorylation of the I-kappa B kinase beta activation loop. J Immunol. 2004;172:2446–52. doi: 10.4049/jimmunol.172.4.2446. [DOI] [PubMed] [Google Scholar]
- 76.Cordier F, Vinolo E, Veron M, et al. Solution structure of NEMO zinc finger and impact of an anhidrotic ectodermal dysplasia with immunodeficiency-related point mutation. J Mol Biol. 2008;377:1419–32. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 77.Huang TT, Feinberg SL, Suryanarayanan S, et al. The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol Cell Biol. 2002;22:5813–25. doi: 10.1128/MCB.22.16.5813-5825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shifera AS, Horwitz MS. Mutations in the zinc finger domain of IKK gamma block the activation of NF-kappa B and the induction of IL-2 in stimulated T lymphocytes. Mol Immunol. 2008;45:1633–45. doi: 10.1016/j.molimm.2007.09.036. [DOI] [PubMed] [Google Scholar]
- 79.Tang ED, Wang CY, Xiong Y, et al. A role for NF-kappaB essential modifier/IkappaB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IkappaB kinase complex by tumor necrosis factor-alpha. J Biol Chem. 2003;278:37297–305. doi: 10.1074/jbc.M303389200. [DOI] [PubMed] [Google Scholar]
- 80.Makris C, Roberts JL, Karin M. The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol Cell Biol. 2002;22:6573–81. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carter RS, Geyer BC, Xie M, et al. Persistent activation of NF-kappa B by the Tax transforming protein involves chronic phosphorylation of IkappaB kinase subunits IKKbeta and IKKgamma. J Biol Chem. 2001;276:24445–8. doi: 10.1074/jbc.C000777200. [DOI] [PubMed] [Google Scholar]
- 82.Carter RS, Pennington KN, Ungurait BJ, et al. In vivo identification of inducible phosphoacceptors in the IKKgamma/ NEMO subunit of human IkappaB kinase. J Biol Chem. 2003;278:19642–8. doi: 10.1074/jbc.M301705200. [DOI] [PubMed] [Google Scholar]
- 83.Laplantine E, Fontan E, Chiaravalli J, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28:2885–95. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rahighi S, Ikeda F, Kawasaki M, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 85.Kulathu Y, Akutsu M, Bremm A, et al. Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat Struct Mol Biol. 2009;16:1328–30. doi: 10.1038/nsmb.1731. [DOI] [PubMed] [Google Scholar]
- 86.Sato Y, Yoshikawa A, Yamashita M, et al. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by NZF domains of TAB2 and TAB3. EMBO J. 2009;28:3903–9. doi: 10.1038/emboj.2009.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu CJ, Conze DB, Li T, et al. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 88.Ea CK, Deng L, Xia ZP, et al. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–57. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 89.Wu CJ, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105:3023–8. doi: 10.1073/pnas.0712313105. [DOI] [PMC free article] [PubMed] [Google Scholar]