

Abstract
To improve regeneration of the injured myocardium, cardiomyocyte progenitor cells (CMPCs) have been put forward as a potential cell source for transplantation therapy. Although cell transplantation therapy displayed promising results, many issues need to be addressed before fully appreciating their impact. One of the hurdles is poor graft-cell survival upon injection, thereby limiting potential beneficial effects. Here, we attempt to improve CMPCs survival by increasing microRNA-155 (miR-155) levels, potentially to improve engraftment upon transplantation. Using quantitative PCR, we observed a 4-fold increase of miR-155 when CMPCs were exposed to hydrogen-peroxide stimulation. Flow cytometric analysis of cell viability, apoptosis and necrosis showed that necrosis is the main cause of cell death. Overexpressing miR-155 in CMPCs revealed that miR-155 attenuated necrotic cell death by 40 ± 2.3%via targeting receptor interacting protein 1 (RIP1). In addition, inhibiting RIP1, either by pre-incubating the cells with a RIP1 specific inhibitor, Necrostatin-1 or siRNA mediated knockdown, reduced necrosis by 38 ± 2.5% and 33 ± 1.9%, respectively. Interestingly, analysing gene expression using a PCR-array showed that increased miR-155 levels did not change cell survival and apoptotic related gene expression. By targeting RIP1, miR-155 repressed necrotic cell death of CMPCs, independent of activation of Akt pro-survival pathway. MiR-155 provides the opportunity to block necrosis, a conventionally thought non-regulated process, and might be a potential novel approach to improve cell engraftment for cell therapy.
Keywords: cardiac progenitor cells, microRNA, RIP1, cell death, necrosis
Cell replacement therapy offers a potential novel approach to repair the injured heart. For this, different cell types are considered to be transplanted to replace damaged tissue. To be able to regenerate the heart, cells should have the capability of differentiating into all cell types required for cardiac repair: functional cardiomyocytes, endothelial cells and smooth muscle cells. In the last decade, it was discovered that the heart was not a terminally differentiated organ. Additionally, progenitor cells could be isolated from the myocardium. It was suggested that these progenitors are an ideal candidate to regenerate the heart due to their localization. Many different types of cardiac progenitor cells (CPCs) or cardiac stem cells (CSCs) have been isolated from the heart, based on the selection of Sca-1+, c-kit+, Islet-1+, side population (SP) and the formation of cardiospheres [1–5]. CPCs are lineage negative and negative for haematopoietic markers as CD34 and CD45.
Previously, we reported the isolation of cardiomyocyte progenitor cells (CMPCs) from the human heart that are able to proliferate and efficiently differentiate into functional cardiomyocytes without requiring co-culture with neonatal myocytes [6, 7]. Three months after transplantation of CMPCs in a mouse model of myocardial infarction, we observed less outward remodelling and improved cardiac function as compared to control injections [8]. Although high numbers of cells were injected in several studies, few implanted cells survived, limiting their potential contribution for myocardial repair. Most of the engrafted cells died in the first 48 hrs after transplantation, partially due to the hostile microenvironment of the ischemic myocardium [9–11]. Therefore there is a lot to gain if we can find a strategy to improve cell survival after implantation.
MicroRNAs (miRNAs) are small, 22 nucleotide long, endogenous present RNA molecules that post-translationally target mRNA by imperfect binding to its target sequence. Hereby translation is blocked and protein production is prevented. miRNAs are involved in regulating various cellular processes, such as cell proliferation, differentiation and migration. Accumulating evidence has suggested that miRNAs also play an important role in cell survival [12–14]. miR-155 is demonstrated to be involved in cell death in several cell lines [15–17], e.g. macrophages and pancreatic cells. Recently, we observed that miR-155 is expressed in growing CMPCs, and demonstrated that miR-155 is not involved in cellular proliferation [18]. Here we show that miR-155 efficiently enhanced CMPCs survival upon oxidative stress, by targeting receptor interacting protein 1 (RIP1), a death domain protein required for activation of necrosis [19, 20] This study demonstrates that miR-155 efficiently inhibited necrosis in CMPCs and suggests that the efficiency of cell-based therapy for cardiac regeneration can potentially be promoted by modulating miR-155 levels in CMPCs.
Methods
CMPC isolation and culture
CMPCs were isolated and propagated as previously described [6]. Briefly, human foetal heart tissue was collected after elective abortion and individual permission using standard informed consent procedures and prior approval of the ethics committee of the University Medical Center Utrecht were obtained. The heart was cut into small pieces and digested by collagenase, followed by passing through a cell strainer to get a single cell suspension. CMPCs were further isolated via magnetic cell sorting using an iron-labelled mouse anti-Sca-1 antibody and characterized as described [6, 7, 21]. CMPCs differentiation experiment was performed as previously reported [6]. For our experiments we used six individual isolated CMPC cell-lines.
Quantitative RT-PCR for miRNAs expression
Total DNA-free RNA was isolated with Tripure isolation reagent (Roche Applied Science, Indianapolis, IN, USA). A total of 3.3 ng RNA were used for reverse transcription (Taqman® MicroRNA Reverse Transcriptase Kit, Applied Biosystems) followed by Taqman® MicroRNA Assays for quantification of miR-155 and RNU19 control transcripts (Applied Biosystems, Carlsbad, CA, USA: 4373124, 4373378, respectively), according to the manufacturers conditions. Amplification and detection of specific PCR products was performed in a MyIQ single-colour real-time PCR system (Bio-Rad, Hercules, CA, USA) at 95°C for 10 min., followed by 40 cycles of 95°C for 15 sec. and 60°C for 60 sec. The expression level of miR-155 was calculated (ΔΔCt) and presented as fold induction (2−ΔΔCt).
Quantitative RT-PCR
Total DNA-free RNA was isolated with TriPure reagent (Roche, Basel, Switzerland). cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR (qRT-PCR) amplification was performed with Sybr-green mastermix (Bio-Rad) in a MyIQ single-colour real-time PCR system (Bio-Rad) as described before [22]. RIP1 primer sequences used are Forward: 5′-AGTCCTGGTTTGCTCCTTCCC-3′; Reverse: 5′-GCGTCTCCTTTCCTCCTCTCTG-3′, with 63.9°C annealing temperature. Fold induction values were normalized for GAPDH (Forward: 5′-ACAGTCAGCCGCATCTTC-3′; Reverse: 5′-GCCCAATACGACCAAATCC-3′, with 56.1°C annealing temperature). TroponinT (TNNT2) and myosin light chain (MYL)2 expression are detected by using customized PCR array from SABiosciences (Frederick, MD, USA; CAPH09443) and the relative expression was calculated by ΔΔCt.
Small RNA transfection
Pre-miR™ precursor molecules for miR-155 (PM 12601) (pre-miR-155), anti-miR™ inhibitor for miR-155 (AM 12601) (anti-miR-155) and a scrambled miR control (AM 17121) (scr-miR) were obtained from Ambion (Austin, TX, USA). SiRIP1 duplex (sense strand 5′- GGAGCAAACUGAAUAAUGAUU-3′) and siNS (not significant) duplex (sense strand 5′- CAGAGAGGAGGAAAGGAGAUU-3′) were synthesized by Dharmacon RNAi Technologies (Lafayette, CO, USA). CMPCs were transfected with 30 nM of appropriate miRNAs or 100 nM of siRNA with siPORT™ NeoFX™ Transfection Agent (Ambion), according to the manufacturer. Transfection efficiency of miRNAs or siRNAs was confirmed by RT-PCR and Western blot.
Luciferase experiments
In a 12-well plate, HEK 293 cells were transfected with 400 ng luciferase-reporter plasmid, containing the 3′-untranslated region (UTR) of Bach1 which is a validated target of miR-155 [23] (a generous gift from Dr. Erik Flemington), with 30 nM of miR-155, a combination of miR-155 and anti-miR-155, or scr-miR. A total of 400 ng cytomegalovirus (CMV)-β-galactosidase plasmid was co-transfected as an internal transfection efficiency control. After 48 hrs of transfection, cells were lysed and luciferase and β-galactosidase activities were measured with the Luciferase Assay System and β-galactosidase Enzyme Assay System (Promega, Madison, WI, USA), respectively, as described [18].
Cell viability assay
CMPCs were seeded and transfected with 30 nM of appropriate miRNAs in a 96-well plate. Twenty-four hours after transfection, CMPCs were challenged with 100 μM H2O2 in serum-free medium for 16–18 hrs. Subsequently, 10 μl WST-1 reagent (Roche 11644807001) was added to each well according to the manufacturer. The optical density was measured at different time-points by colorimetric multiscan FC (Thermo Scientific, Lafayette, CO, USA) with absorption at 450 nm and reference at 750 nm.
Flow cytometric analysis of apoptosis and necrosis
CMPCs were seeded and transfected with 30 nM of the appropriate miRNAs in a 12-well plate. Twenty-four hours after transfection, CMPCs were challenged with 50 μM H2O2 in serum-free medium for 16–20 hrs. All cells were collected and subsequently washed with phosphate-buffered solution (PBS). To detect necrosis only, cells were incubated with 3 μl propidium iodide (PI, 556463; BD Pharmingen, San Diego, CA, USA) in 1× binding buffer for 25 min. in dark container. For apoptosis and necrosis detection, cells were incubated with 5 μl AnnexinV-PE (AnnV) and 5 μl 7-Amino-actinomycin-D (7-AAD) in 1× binding buffer (BD Pharmingen, apoptosis and necrosis detection kit I, 559763) for 25 min. in a dark container. After incubation, apoptotic, necrotic and live cell populations were detected by flow cytometric analysis.
Western blot
Seventy-two hours after miRNAs or siRNA transfection, total protein lysate was extracted. Samples were reduced with 0.5%β-mercaptoethanol, separated by PAGE, and transferred to a nitrocellulose membrane (Schleicher & Schuell, Keene, NH, USA). Membranes were blocked (5% non-fat-dry milk, PBS-0.1% Tween), probed with RIP1 antibody (1:500, rabbit polyclonal, sc-7881, Santa Cruz, Santa Cruz, CA, USA) and β-tubulin antibody (1:2000, rabbit polyclonal, #2146, Cell Signaling, Danvers, MA, USA), followed by goat anti-rabbit IgG secondary antibody (P0448; Sigma-Aldrich, St. Louis, MO, USA) incubation. Signal was visualized with enhanced chemiluminescence (Amersham, South Plainfield, NJ, USA) and detected by using the ChemiDoc XRS system (Bio-Rad).
Necrostatin-1 (Nec-1) treatment
CMPCs are pre-incubated with 30 μM Nec-1 (Calbiochem, San Diego, CA, USA; 480065) in serum-free medium for 30 min. followed by stimulation with 50 μM H2O2. After 16–18 hrs, cell viability, apoptosis and necrosis were analysed by flow cytometry as described above.
Human pro-survival and apoptosis pathway super-arrays
CMPCs are transfected with pre-miR-155 or scr-miR. After 48 hrs, total RNA was isolated and cDNA was synthesized as described above. A total of 84 key genes, involved in apoptosis and PI3K-AKT pro-survival pathways (SABiosciences, PAHS-012A and PAHS-058A), respectively, were detected using qRT-PCR, followed by analysis using SABiosciences online RT2 Profiler™ PCR Array Data Analysis software.
Statistical analysis
Data is presented as mean ± S.E.M. of at least three independent experiments and were compared using the two-tailed paired Student’s t-test or one-way ANOVA, a difference with a P < 0.05 was considered to be statistically significant.
Results
Increased expression of miR-155 in CMPCs
Because miR-155 overexpression did not alter cell proliferation [18], we investigated if it could have a function in cell survival, as was demonstrated before in other cell lines [15–17]. After stimulation with H2O2, miR-155 levels increased 4-fold (Fig. 1A), suggesting a role of miR-155 in stress response. Next we modulated miR-155 levels in CMPCs. Transfection of pre-miR155 resulted in a 280-fold increase of miR155 level (Fig. 1B) compared to anti-miR155 or scr-miR transfection. Pre-mir-155 overexpression significantly reduced the activity of a miR-155 luciferase reporter plasmid, containing a 3′-UTR sequence of Bach1, a known target of miR-155 [23], which could be abolished by addition of anti-miR155 (Fig. 1C).
Fig 1.
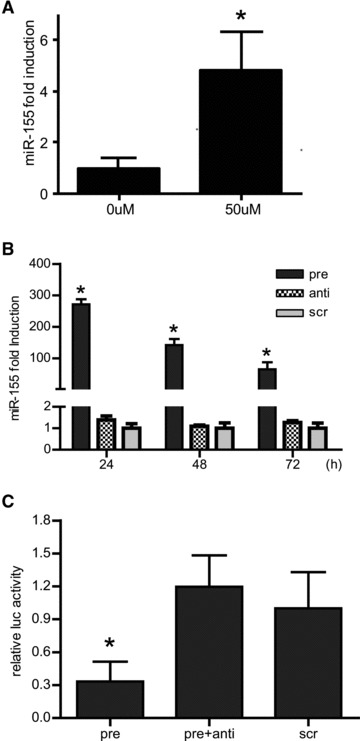
(A) miR-155 expression was increased in CMPCs upon 16 hrs of 50 μM H2O2 stimulation, (B) CMPCs were transfected with 30 nM pre-miR-155, anti-miR-155 or scr-miR. MiR-155 expression was detected by Taqman MicroRNA real-time PCR 24, 48 or 72 hrs after transfection. (C) The functional effectiveness of miRNA molecules was confirmed by luciferase activity assay. CMPCs were co-transfected with luciferase reporter plasmid containing the 3′-UTR of a miR-155 target and with different combinations of appropriate miRNAs. Data are presented as mean ± S.E.M., N= 3 and *P < 0.05.
MiR-155 overexpression augments cell survival during oxidative-stress stimulation
Exposure of miR-155 overexpressing CMPCs to oxidative stress resulted in less cell death (Fig. 2A). Both flow cytometric analysis (Fig. 2B) and cell counting (Fig. S1) confirmed this observation and displayed a 42 ± 3.1% improvement of cell survival. More detailed analysis revealed that the increase in cell number was achieved via a 40 ± 2.3% reduction in necrosis, the predominant type of cell death, as compared to scr-miR transfection control and affirmed by a diminished number of PI+ cells (Fig. 2D, E). Improved cell survival was also found when performing a cell viability assay (Fig. 2C). Interestingly, no significant difference in the number of apoptotic cells was found when comparing CMPCs overexpressing pre-mir-155, anti-mir155 or scr-mir.
Fig 2.
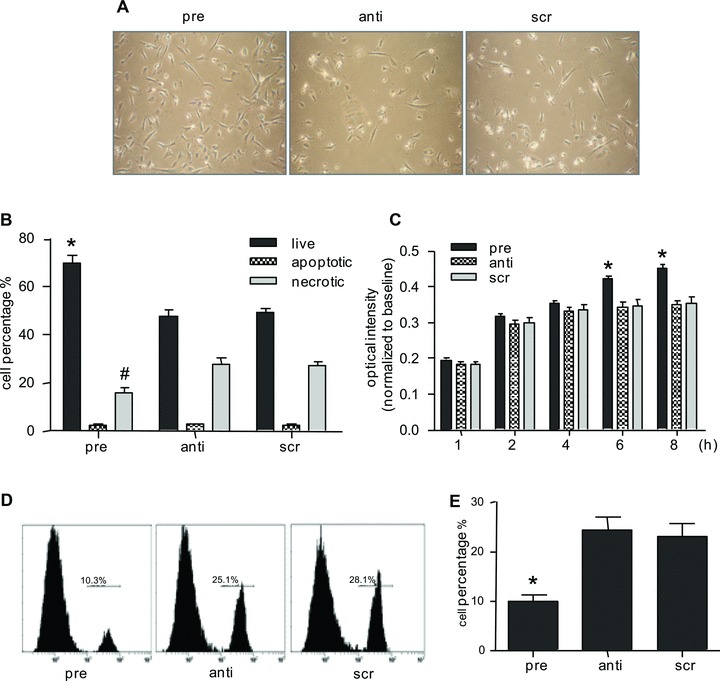
(A) CMPCs were transfected with 30 nM pre-miR-155, anti-miR-155 or scr-miR and challenged by 50 μM H2O2 in serum-free medium for 16 hrs. (B) Live, apoptotic and necrotic cells were detected by AnnV/7-AAD staining using flow cytometric analysis. (C) A cell viability assay was performed on appropriate miRNAs transfected CMPCs, followed by exposure to 100 μM H2O2. (D) Histogram of PI staining showed a reduced necrotic cell population in CMPCs upon miR-155 transfection. (E) Quantification of PI+ cells in (D). Data are presented as mean ± S.E.M., N= 4 and *P < 0.05.
To investigate whether necrosis was a primary result or a secondary event of apoptosis, we performed a time course experiment. In time, apoptosis remained a minor type of cell death, whereas, necrosis increased dramatically after 12 hrs of stimulation, being the predominant type of cell death (Fig. S2). Additionally, we tested the activities of caspases in CMPCs upon stimulation by a homogeneous caspases assay, but we did not observe the presence of any enzymatic activity (data not shown). Taking together, our observations show that in CMPCs miR-155 increases cell survival upon oxidative stress by reducing necrotic cell death.
To test whether changing miR-155 levels affect CMPC cardiomyogenic differentiation potential, we transfected pre-miR-155, anti-miR-155 or scr-miR into CMPCs and started differentiation experiments as previously reported [6]. We observed increased levels of TNNT2 and ventricular MYL2 1 week after the initiation of differentiation, similar as non-transfected CMPCs, indicating that miR-155 transfection did not affect cardiomyogenic differentiation potential (Fig. S3).
MiR-155 reduces necrotic cell death via targeting RIP1
RIP1 is a validated target for mir-155 in the response of macrophages to endotoxin shock [15], required for death-receptor agonists activated necrosis [20]. Therefore, we tested whether miR-155 might inhibit necrosis by repressing RIP1.
We validated this in CMPCs and we showed that miR-155 repressed RIP1 expression both at the RNA and protein levels (Fig. 3A–C). To test the functional relevance of RIP1 in the observed necrotic cell death, we used Nec-1, a specific inhibitor of RIP1 [24], in oxidative-stress stimulated CMPCs. In Nec-1 treated CMPCs, cell survival improved and necrosis diminished to a similar extent as miR-155 overexpression (Fig. 3D), with no changes in apoptosis. In addition, knockdown of RIP1 expression by siRNA treatment efficiently repressed RIP1 mRNA and protein levels (Fig. 3E–G), thereby preventing necrotic cell death and improving cell survival (Fig. 3H). Our results suggest that miR-155 represses RIP1, a key effector in necrosis, thereby attenuating necrotic cell death induced by oxidative stress.
Fig 3.
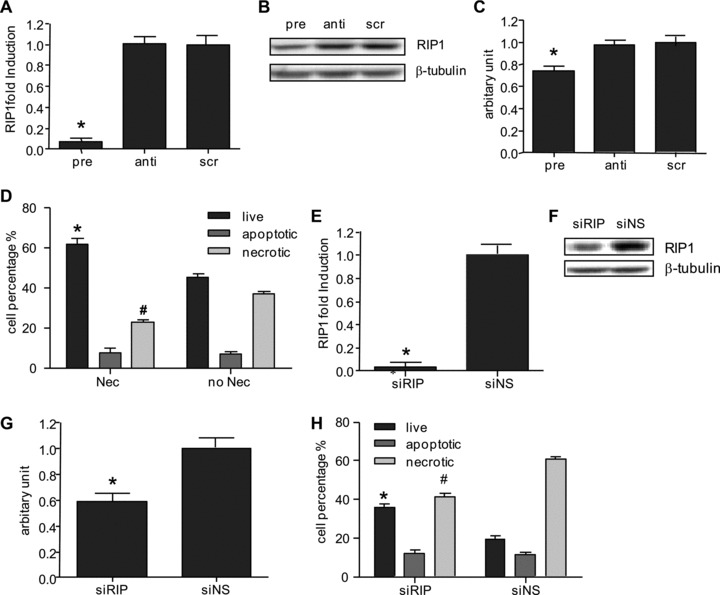
(A) RIP1 mRNA expression in miR-155 overexpressing CMPCs, (B) RIP1 protein expression detected by Western blot and (C) Quantification of Western blot. (D) CMPCs were pre-incubated with 30 μM Nec-1, inhibitor of RIP1, followed by 50 μM H2O2 stimulation. Live, apoptotic and necrotic cells were detected by AnnV/7-AAD staining using flow cytometric analysis. (E) RIP1 mRNA expression after transfections of small interference RNA for RIP1 (siRIP)/or a not significant control sequence (siNS) in CMPCs as detected by RT-qPCR. (F) RIP1 protein expression detected by Western blot and (G) quantification of Western blot. (H) Live, apoptotic, and necrotic cell populations were assessed by flow cytometric analysis in siRIP or siNS transfected CMPCs. Data are presented as mean ± S.E.M., N= 3 and *P < 0.05.
MiR-155 inhibits necrosis, independent of activation of Akt pro-survival pathway
Because cell death and survival is a tightly regulated process involving many different pathways, we analysed whether other pathways that can regulate cell survival are affected upon miR-155 modulation. Using PCR-arrays, we analysed the expression of 84 key genes, involved in the PI3K/Akt pro-survival pathway and apoptosis pathway. We found that increased miR-155 levels did not change the expression patterns of cell survival and apoptotic related genes. In the Akt-survival pathway related genes only a 4-fold down-regulation of NF-κB could be observed (Fig. 4A, B). Similarly, in the array focused on apoptosis-related genes, only BIRC3 and CASP1 showed a decrease of more than 4-fold (Fig. 4C, D). These data confirm that cell survival and apoptosis pathways are not widely affected upon miR-155 overexpression.
Fig 4.
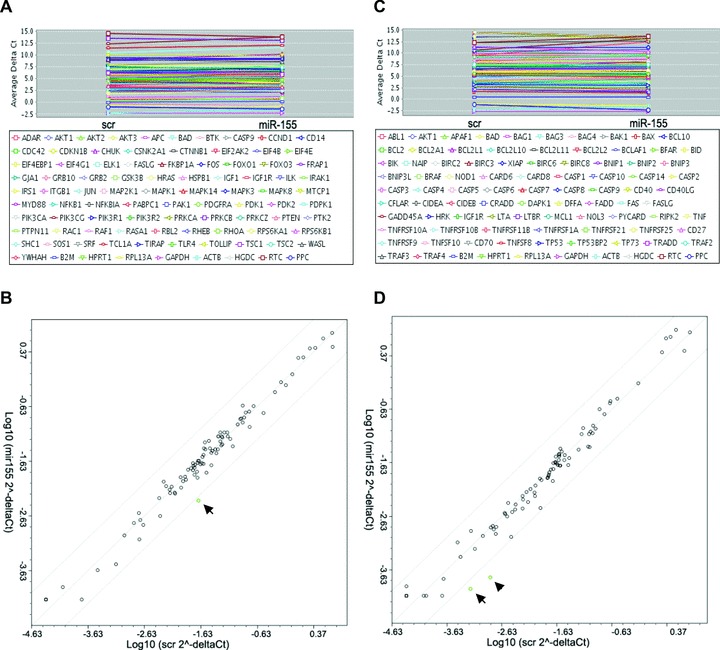
A total of 84 genes involved in PI3K-Akt pro-survival pathways in pre-miR-155/scr-miR transfected CMPCs were detected by RT-qPCR and normalized for four housekeeping genes: (A) Gene names and average ΔCt are shown, (B) the relative expression was plotted in a scatter plot, NF-κB was indicated by black arrow. Eighty-four genes involved in apoptosis pathways in pre-miR-155/scr-miR transfected CMPCs were detected by RT-qPCR and normalized for four housekeeping genes: (C) Gene names and average ΔCt are shown, (D) the relative expression was plotted in a scatter plot, BIRC3 was indicated by black arrow, CASP1 was indicated by black arrow head.
Discussion
Cell-based therapy has emerged as a promising, alternative strategy to regenerate or improve function upon myocardial infarction (MI) and possibly for chronic heart failure. The feasibility of various cell sources and different cell delivery methods have been extensively explored [21, 25–29]. The recognition of resident CPCs that could contribute to both myocardial regeneration and vascularization has provided an ideal candidate for cardiac repair. Previously we reported that upon transplantation CMPCs reduced the extent of left ventricular remodelling up to 3 months after MI [8], thereby improving cardiac output. Due to the hostile environment of the ischemic heart, however, the survival of newly implanted cells remains problematic [12, 30]. Several strategies of preconditioning or stimulations are studied [31–35]. Although very promising and effective, the direct mechanisms of these strategies are not clear, because paracrine effects of transplanted cells and local environmental cues also stimulate the recipient tissue. To be able to understand the role of transplanted cells and improve only their survival rate, new strategies are required to enhance graft cell survival.
MiRNAs are newly identified, non-coding small RNAs, known to be involved in multiple cellular processes, including cell survival, Their endogenous presence provides a novel platform for cell survival modification and improvements. Several studies have shown the cyto-protective effect of different miRNAs in cardiac tissue [36–39]. In the present study, we demonstrated that miR-155 efficiently prevent oxidative-stress induced necrosis, the dominant type of cell death in CMPCs. Even detailed follow-up experiments demonstrated that apoptosis has a minor contribution whereas necrosis increased dramatically in time. In addition, no active caspase activities were observed upon oxidative stress, suggesting the apoptosis pathway not being activated.
RIP1 emerged as a potential target candidate for miR-155 regulated inhibition of necrosis. RIP1, a death domain protein, is dispensable for the induction of death-receptor-mediated apoptosis [39]. In apoptosis-deficient conditions, however, RIP1 has been found to be required for the activation of necrosis by death-receptor agonists [20]. This process is termed necroptosis by Degterev et al. and they identified RIP1 as a key universal effector molecule, potentially followed by divergent downstream execution steps, depending on the cellular context [24]. Interestingly, RIP1 was described as a target for miR-155 in macrophages in their response to endotoxin shock [15]. In CMPCs we could observe that miR-155 did reduce RIP1 at RNA and protein levels and we confirmed a direct effect of RIP1 on necrosis by using a specific inhibitor of RIP1, Nec-1, or via siRNA treatment for RIP1, efficiently attenuating necrosis without affecting apoptosis. To study whether pro-survival pathways or apoptosis-related genes are also involved, we analysed over 160 different genes upon miR-155 transfection. We observed that gene expression pattern was not altered generally, indicating that miR-155 specifically blocks necrosis, independently of pro-survival and apoptosis pathways. This was in line with our finding that miR-155 attenuated necrosis induced by oxidative stress without affecting apoptosis.
Although increased expression of miR-155 could efficiently blocks necrosis, we could not induce necrotic cell death by inhibiting endogenous miR-155. This suggests that the biological role of miR-155 in CMPCs may not be anti-necrotic. However, because pre-miR155 transfection only transiently increased miR-155 levels and did not affect cell proliferation and cardiomyogenic differentiation potential, preconditioning CMPCs with miR-155 provides a novel opportunity to improve cell survival after transplantation from a therapeutic point of view.
In conclusion, the current study demonstrates that oxidative-stress induced cell death in CMPCs is mainly mediated by necrosis. By increasing miR-155 levels, necrosis could be inhibited and cell survival is efficiently improved without affecting cardiomyogenic differentiation potential. This is mediated by repressing the receptor interacting protein1, RIP1. Hereby, we provide data which suggest that miR-155 is a potential novel alternative for improving graft-cell survival and suggests the broader possibility of specifically targeting necrosis as a therapeutic strategy.
Acknowledgments
This work was supported by the BSIK program ‘Dutch Program for Tissue Engineering’, grants 6746 (J.P.G.S., A.v.M.), and a Bekalis price (P.A.D.). This research forms part of the Project P1.04 SMARTCARE of the research program of the BioMedical Materials institute, co-funded by the Dutch Ministry of Economic Affairs (K.V.). The financial contribution of the Nederlandse Hartstichting is gratefully acknowledged. This work was sponsored by China Scholarship Council: Grant number 2007–3020.
Conflict of interest
The authors confirm that there are no conflicts of interest to declare.
Supporting Information
Trypan blue cell counting for viable cells upontransfection with appropriate miRNAs, followed by 50 μMH2O2 stimulation. Data are presented as mean± S.E.M., N = 4 and *P < 0.05.
Time course of live (A), apoptotic(B) and necrotic (C) cells as detected by AnnV/7-AADstaining using flow cytometric analysis inH2O2 stimulated, appropriate miRNAstransfected cells. Data are presented as mean ± S.E.M.,N = 3 and *P < 0.05.
CMPCs were transfected with pre-miR-155,anti-miR-155 or scr-miR, followed by stimulation with 5-aza andTGF-β to induce cardiac differentiation. TNNT2 and ventricularMYL2 mRNA expression were analysed before (undiff.) and 1 weekafter differentiation (diff.). Non-transfected CMPCs before andafter differentiation were used as a negative and positive control,respectively. Data are presented as mean ± S.E.M., N= 3 and *P < 0.05.
References
- 1.Hierlihy AM, Seale P, Lobe CG, et al. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002;530:239–43. doi: 10.1016/s0014-5793(02)03477-4. [DOI] [PubMed] [Google Scholar]
- 2.Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 3.Oh H, Chi X, Bradfute SB, et al. Cardiac muscle plasticity in adult and embryo by heart-derived progenitor cells. Ann NY Acad Sci. 2004;1015:182–9. doi: 10.1196/annals.1302.015. [DOI] [PubMed] [Google Scholar]
- 4.Laugwitz KL, Moretti A, Lam J, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–53. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith RR, Barile L, Cho HC, et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 6.Smits AM, van Vliet P, Metz CH, et al. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: an in vitro model for studying human cardiac physiology and pathophysiology. Nat Protoc. 2009;4:232–43. doi: 10.1038/nprot.2008.229. [DOI] [PubMed] [Google Scholar]
- 7.Goumans MJ, de Boer TP, Smits AM, et al. TGF-beta1 induces efficient differentiation of human cardiomyocyte progenitor cells into functional cardiomyocytes in vitro. Stem Cell Res. 2007;1:138–49. doi: 10.1016/j.scr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Smits AM, van Laake LW, den OK, et al. Human cardiomyocyte progenitor cell transplantation preserves long-term function of the infarcted mouse myocardium. Cardiovasc Res. 2009;83:527–35. doi: 10.1093/cvr/cvp146. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki K, Murtuza B, Beauchamp JR, et al. Role of interleukin-1beta in acute inflammation and graft death after cell transplantation to the heart. Circulation. 2004;110:II219–24. doi: 10.1161/01.CIR.0000138388.55416.06. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Methot D, Poppa V, et al. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol. 2001;33:907–21. doi: 10.1006/jmcc.2001.1367. [DOI] [PubMed] [Google Scholar]
- 11.Muller-Ehmsen J, Whittaker P, Kloner RA, et al. Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol. 2002;34:107–16. doi: 10.1006/jmcc.2001.1491. [DOI] [PubMed] [Google Scholar]
- 12.Ostenfeld MS, Bramsen JB, Lamy P, et al. miR-145 induces caspase-dependent and -independent cell death in urothelial cancer cell lines with targeting of an expression signature present in Ta bladder tumors. Oncogene. 2010;29:1073–84. doi: 10.1038/onc.2009.395. [DOI] [PubMed] [Google Scholar]
- 13.Borralho PM, Kren BT, Castro RE, et al. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J. 2009;276:6689–700. doi: 10.1111/j.1742-4658.2009.07383.x. [DOI] [PubMed] [Google Scholar]
- 14.Subramanian S, Thayanithy V, West RB, et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J Pathol. 2010;220:58–70. doi: 10.1002/path.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 16.Gironella M, Seux M, Xie MJ, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci USA. 2007;104:16170–5. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajanca F, Luz M, Duxson MJ, et al. Integrins in the mouse myotome: developmental changes and differences between the epaxial and hypaxial lineage. Dev Dyn. 2004;231:402–15. doi: 10.1002/dvdy.20136. [DOI] [PubMed] [Google Scholar]
- 18.Sluijter JP, van Mil A, van Vliet P, et al. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol. 2010;30:859–68. doi: 10.1161/ATVBAHA.109.197434. [DOI] [PubMed] [Google Scholar]
- 19.Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 21.van Vliet P, Roccio M, Smits AM, et al. Progenitor cells isolated from the human heart: a potential cell source for regenerative therapy. Neth Heart J. 2008;16:163–9. doi: 10.1007/BF03086138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sluijter JP, Smeets MB, Velema E, et al. Increased collagen turnover is only partly associated with collagen fiber deposition in the arterial response to injury. Cardiovasc Res. 2004;61:186–95. doi: 10.1016/j.cardiores.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 23.Yin Q, McBride J, Fewell C, et al. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J Virol. 2008;82:5295–306. doi: 10.1128/JVI.02380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 26.Hassink RJ, Brutel dlR, Mummery CL, et al. Transplantation of cells for cardiac repair. J Am Coll Cardiol. 2003;41:711–7. doi: 10.1016/s0735-1097(02)02933-9. [DOI] [PubMed] [Google Scholar]
- 27.Wollert KC, Drexler H. Cell-based therapy for heart failure. Curr Opin Cardiol. 2006;21:234–9. doi: 10.1097/01.hco.0000221586.94490.d2. [DOI] [PubMed] [Google Scholar]
- 28.Zwaginga JJ, Doevendans P. Stem cell-derived angiogenic/vasculogenic cells: possible therapies for tissue repair and tissue engineering. Clin Exp Pharmacol Physiol. 2003;30:900–8. doi: 10.1046/j.1440-1681.2003.03931.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Sluijter JP, Goumans MJ, et al. Cell therapy for myocardial regeneration. Curr Mol Med. 2009;9:287–98. doi: 10.2174/156652409787847218. [DOI] [PubMed] [Google Scholar]
- 30.Del Re DP, Sadoshima J. Optimizing cell-based therapy for cardiac regeneration. Circulation. 2009;120:831–4. doi: 10.1161/CIRCULATIONAHA.109.887836. [DOI] [PubMed] [Google Scholar]
- 31.Pasha Z, Wang Y, Sheikh R, et al. Preconditioning enhances cell survival and differentiation of stem cells during transplantation in infarcted myocardium. Cardiovasc Res. 2008;77:134–42. doi: 10.1093/cvr/cvm025. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Zingarelli B, O’Connor M, et al. Overexpression of Hsp20 prevents endotoxin-induced myocardial dysfunction and apoptosis via inhibition of NF-kappaB activation. J Mol Cell Cardiol. 2009;47:382–90. doi: 10.1016/j.yjmcc.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou Y, Zhu W, Sakamoto M, et al. Heat shock transcription factor 1 protects cardiomyocytes from ischemia/reperfusion injury. Circulation. 2003;108:3024–30. doi: 10.1161/01.CIR.0000101923.54751.77. [DOI] [PubMed] [Google Scholar]
- 34.Padin-Iruegas ME, Misao Y, Davis ME, et al. Cardiac progenitor cells and biotinylated insulin-like growth factor-1 nanofibers improve endogenous and exogenous myocardial regeneration after infarction. Circulation. 2009;120:876–87. doi: 10.1161/CIRCULATIONAHA.109.852285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gnecchi M, He H, Liang OD, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11:367–8. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 36.Won KH, Haider HK, Jiang S, et al. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J Biol Chem. 2009;284:33161–8. doi: 10.1074/jbc.M109.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren XP, Wu J, Wang X, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–66. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang Y, Zheng J, Sun Y, et al. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J. 2009;50:377–87. doi: 10.1536/ihj.50.377. [DOI] [PubMed] [Google Scholar]
- 39.Grimm S, Stanger BZ, Leder P. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci USA. 1996;93:10923–7. doi: 10.1073/pnas.93.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trypan blue cell counting for viable cells upontransfection with appropriate miRNAs, followed by 50 μMH2O2 stimulation. Data are presented as mean± S.E.M., N = 4 and *P < 0.05.
Time course of live (A), apoptotic(B) and necrotic (C) cells as detected by AnnV/7-AADstaining using flow cytometric analysis inH2O2 stimulated, appropriate miRNAstransfected cells. Data are presented as mean ± S.E.M.,N = 3 and *P < 0.05.
CMPCs were transfected with pre-miR-155,anti-miR-155 or scr-miR, followed by stimulation with 5-aza andTGF-β to induce cardiac differentiation. TNNT2 and ventricularMYL2 mRNA expression were analysed before (undiff.) and 1 weekafter differentiation (diff.). Non-transfected CMPCs before andafter differentiation were used as a negative and positive control,respectively. Data are presented as mean ± S.E.M., N= 3 and *P < 0.05.