

Abstract
The anticancer drug doxorubicin induces the synthesis of nitric oxide, a small molecule that enhances the drug cytotoxicity and reduces the drug efflux through the membrane pump P-glycoprotein (Pgp). Doxorubicin also induces the translocation on the plasma membrane of the protein calreticulin (CRT), which allows tumour cells to be phagocytized by dendritic cells. We have shown that doxorubicin elicits nitric oxide synthesis and CRT exposure only in drug-sensitive cells, not in drug-resistant ones, which are indeed chemo-immunoresistant. In this work, we investigate the mechanisms by which nitric oxide induces the translocation of CRT and the molecular basis of this chemo-immunoresistance. In the drug-sensitive colon cancer HT29 cells doxorubicin increased nitric oxide synthesis, CRT exposure and cells phagocytosis. Nitric oxide promoted the translocation of CRT in a guanosine monophosphate (cGMP) and actin cytoskeleton-dependent way. CRT translocation did not occur in drug-resistant HT29-dx cells, where the doxorubicin-induced nitric oxide synthesis was absent. By increasing nitric oxide with stimuli other than doxorubicin, the CRT exposure was obtained also in HT29-dx cells. Although in sensitive cells the CRT translocation was followed by the phagocytosis, in drug-resistant cells the phagocytosis did not occur despite the CRT exposure. In HT29-dx cells CRT was bound to Pgp and only by silencing the latter the CRT-operated phagocytosis was restored, suggesting that Pgp impairs the functional activity of CRT and the tumour cells phagocytosis. Our work suggests that the levels of nitric oxide and Pgp critically modulate the recognition of the tumour cells by dendritic cells, and proposes a new potential therapeutic approach against chemo-immunoresistant tumours.
Keywords: doxorubicin, nitric oxide, P-glycoprotein, drug resistance, calreticulin, phagocytosis, dendritic cells, colon cancer
Introduction
The anthracycline doxorubicin has the peculiarity of inducing the synthesis of nitric oxide, a small signalling molecule which plays an important role in cell growth, differentiation and apoptosis [1]. The effect of doxorubicin is mediated by the drug-induced activation of the transcriptional factor NF-κB, which in turn up-regulates the transcription of the inducible nitric oxide synthase (iNOS; EC 1.14.13.39) gene, leading to the production of huge amounts of nitric oxide [2]. It has been suggested that at least part of the cytotoxic effects elicited by doxorubicin are due to the increased nitric oxide synthesis [3, 4]. Nitric oxide also reduces the rate of doxorubicin efflux through ATP-binding cassette (ABC) membrane pumps, such as P-glycoprotein (Pgp) and multidrug resistance related protein 3 (MRP3), two transporters that are responsible for the resistance towards doxorubicin in cancer cells [5]. By nitrating these proteins on tyrosine residues, nitric oxide reduces Pgp and MRP3 activity, reversing doxorubicin-resistance in solid tumours [4, 6]. Notably, in doxorubicin-resistant cells the induction of iNOS gene is absent, in consequence of the fast drug extrusion; however, when the nitric oxide levels are increased in resistant cells by agents other than doxorubicin, the drug efflux is reduced and the cytotoxicity is restored [4].
A great interest has been raised by the discovery that, besides exerting a direct anticancer effect, anthracyclines also stimulate the host immune response against the tumour [7, 8]. It has been discovered that the anthracyclines immunogenicity relies on their ability to induce the tumour cells phagocytosis by immature dendritic cells (iDCs). After this step, iDCs are stimulated to further maturate and to raise a complete immune response against transformed cells [8]. Following doxorubicin exposure, a change in tumour cell plasma membrane occurs, leading to the exposure of intracellular proteins, such as calreticulin (CRT), which is recognized by iDCs and triggers the iDCs-mediated phagocytosis [8, 9]. CRT is usually present in the endoplasmic reticulum (ER), where it acts as a chaperon and a Ca2+ sensor protein [8, 10].
We have recently demonstrated that doxorubicin mediates the exposure of CRT and the phagocytosis by iDCs thanks to the induction of iNOS in drug-sensitive cells: indeed neither the translocation of CRT nor the phagocytosis occurred in the doxorubicin-sensitive cells silenced for iNOS gene. Doxorubicin was devoid of pro-immunogenic effects also in drug-resistant cells, where the anthracycline was not able to accumulate at a sufficient extent to increase the synthesis of nitric oxide [6]. These results suggested that chemo- and immunoresistance to doxorubicin are strictly associated, and may both depend on the lack of nitric oxide synthesis in drug-resistant cells.
In the present work we first investigated the molecular mechanisms by which high levels of nitric oxide induce the translocation of CRT to the cell surface, comparing the doxorubicin-sensitive human colon cancer HT29 cells and the doxorubicin-resistant HT29-dx cells. Moreover we observed that nitric oxide was sufficient to promote the translocation of CRT followed by the phagocytosis in drug-sensitive cells and was necessary to elicit the CRT exposure also in drug-resistant cells, but surprisingly the drug-resistant cells remained poorly phagocitized even in the presence of CRT levels superimposable to sensitive cells. We thus focused on the molecular basis of this strong association between chemo- and immunoresistance, and we analysed whether the increased expression of Pgp in drug-resistant cells may affect the CRT-mediated phagocytosis, thus contributing to their immunoresistant phenotype.
Materials and methods
Materials
Foetal bovine serum (FBS), penicillin-streptomycin (PS) and Roswell Park Memorial Institute (RPMI) 1640 were supplied by Sigma Chemical Co. (St. Louis, MO, USA), plasticware for cell culture was from Falcon (BD Biosciences, Bedford, MA, USA). Electrophoresis reagents were obtained from Bio-Rad Laboratories (Hercules, CA, USA), the protein content of cell monolayers and cell lysates was assessed with the bicinchoninic acid kit from Sigma Chemical Co. Recombinant human tumour necrosis factor-α (TNF-α) was obtained from R&D Systems (Minneapolis, MN, USA), 8-bromoguanosine-3′:5′-cyclic monophosphorothioate, Rp-isomer (Rp-8-Br-cGMPS) from Calbiochem (San Diego, CA, USA), latrunculin A was from Enzo Life Sciences International, Inc. (Plymouth Meeting, PA, USA). When not otherwise specified, all the other reagents were purchased from Sigma Chemical Co.
Cells
Human colon cancer cells (HT29 cell line, provided by Istituto Zooprofilattico Sperimentale ‘Bruno Umbertini’, Brescia, Italy) were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% PS and 1% L-glutamine, and maintained in a humidified atmosphere at 37°C and 5% CO2. A subpopulation of HT29 cells, named HT29-dx, characterized by resistance to doxorubicin, was generated as previously described [4]. In the present study the drug resistance was previously assessed by measuring the Pgp/MRP3 expression, the intracellular doxorubicin accumulation and the drug-induced cytotoxicity, by fluorescence-activated cell sorting (FACS) analysis of cells positive for annexin V-fluorescein isothiocyanate (FITC) as described [2] (data not shown).
Nitrite production
Then nitrite production was measured by adding 0.15 ml of cell culture medium to 0.15 ml of Griess reagent in a 96-well plate, and after 10 min. incubation at 37°C in the dark, the absorbance was measured at 540 nm with a Packard EL340 microplate reader (Bio-Tek Instruments, Winooski, VT, USA). A blank was prepared for each experimental condition in the absence of cells, and its absorbance was subtracted from the one obtained in the presence of cells. Nitrite concentration was expressed as nanomoles of nitrite/mg cell proteins.
Measurement of NOS activity
The procedure described in Ghigo et al.[11] was followed. The cells were detached with 0.05% v/v trypsin, washed with phosphate-buffered solution (PBS), re-suspended at the concentration of 100 μg cell proteins in 0.6 ml of 20 mmol/l Hepes buffer (pH 7.2) and sonicated with one 10 sec. burst, using a Labsonic Sonicator (Hielscher, Teltow, Germany). A total of 15 μl of lysate were withdrawn to assess the protein content. The protease inhibitors pepstatin (75 μmol/l) and leupeptin (20 μmol/l) were added to the cell lysate, which was then mixed with the following reagents in a 500 μl final volume: 0.2 mmol/l NADP+, 360 μmol/l L-arginine, 2 μmol/l tetrahydrobiopterin, 0.3 mmol/l CaCl2, 0.2 mmol/l dithiothreitol, 1.8 mmol/l MgCl2, 0.17 mmol/l glucose 6-phosphate and 40 mU/ml glucose 6-phosphate dehydrogenase (G6PD; from Saccaromyces cerevisiae; EC 1.1.1.49). The NOS/G6PD reaction mixture was incubated at 37°C for 3 hrs, then heated at 100°C for 5 min. to inactivate G6PD. To oxidize the remaining reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), which might interfere with the subsequent Griess reaction, the mixture was incubated with 10 mU/ml L-lactate dehydrogenase (from pig muscle; EC 1.1.1.27) and 300 μmol/l sodium pyruvate at 37°C for 5 min. The nitrite production was measured using the Griess reagent as described. Blanks were prepared by replacing the cell lysate with Hepes solution, in the NOS/G6PD reaction mix, which was subjected to the same experimental procedure used for samples containing lysate. The NOS activity was expressed as nanomoles nitrites/min./mg protein.
Analysis of cell surface CRT
For flow cytometry analysis, cells were washed twice with PBS, rinsed with 1 ml of 0.25% w/v PBS-bovine serum albumin (BSA) and centrifuged at 10,000 ×g for 5 min. The pelleted cells were incubated for 45 min. (4°C) with an anti-CRT rabbit polyclonal antibody (Affinity Bioreagents, Rockford, IL, USA), diluted 1:500 in 100 μl of PBS-BSA, then washed and incubated with 100 μl anti-rabbit FITC-conjugated antibody (diluted 1:50 in PBS-BSA) for 30 min. at 4°C in the dark. After fixation in paraformaldehyde 2%, cells were re-suspended in 500 μl PBS-BSA and analysed using a FACS-Calibur system (BD Biosciences). For each analysis 10,000 events were collected. The percentage of fluorescent cells was calculated by the Cell Quest software (BD Biosciences). Control experiments included incubation of cells with non-immune isotypic antibodies followed by the appropriate labelled secondary antibody. Because doxorubicin has a strong autofluorescence which might lead to misinterpret flow cytometry results, we validated FACS experiments by measuring the CRT exposure in biotinylation assays, using the Cell Surface Protein isolation kit from Thermo Fisher Scientific, Inc. (Rockford, IL, USA), as previously reported [6]. A total of 25 μg of the biotinylated proteins were separated by 10% SDS-PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane sheet (Immobilon-P. Millipore, Billerica, MA, USA) and probed with the anti-CRT rabbit polyclonal antibody (diluted 1:1000 in PBS-BSA 1%). The whole cellular content of CRT was measured after ultracentrifugation of the cell lysates at 100,000 ×g for 1 hr. A total of 10 μg of the pelleted proteins were separated by 10% SDS-PAGE, transferred to PVDF membrane sheet and probed with the anti-CRT antibody. The membranes were washed with PBS-Tween 0.1% and subjected for 1 hr to a horseradish peroxidase-conjugated anti-rabbit antibody (diluted 1:3000 in PBS-Tween 0.1% with blocker non-fat dry milk 5%, Bio-Rad Laboratories). The membranes were washed again and the proteins were detected by enhanced chemiluminescence (PerkinElmer, Waltham, MA, USA).
In vitro phagocytosis assay
iDCs were generated as described [12]. The tumour cells were green-stained with PKH2-FITC (Sigma Chemical Co.), washed twice and incubated for 20 hrs at 37°C with 1 × 105 iDC at a 1:1 ratio, and the mixed culture was stained with the phycoerythrin (PE)-conjuganted anti-CD80 antibody (BD Bioscience) for 20 min. at 4°C. Phagocytosis of tumour cells by iDC was assessed by flow cytometric analysis as the percentage of double-stained (FITC plus PE) versus red (PE) stained cells on a total of 10,000 events, using the DIVA software (FacsCanto system, BD Biosciences). A phagocytosis assay was performed by co-incubating iDC and tumour cells at 4°C, instead of 37°C, and the percentage of double-stained cells obtained after the incubation at 4°C was subtracted from the one obtained after a 37°C incubation. The phagocytosis rate was expressed as phagocytic index, calculated as previously reported [9].
Immunofluorescence microscopy
Cells were seeded on sterile glass cover slips and incubated under the experimental conditions indicated in ‘Results’, then rinsed with PBS and fixed with 4% w/v paraformaldehyde for 15 min. To visualize intracellular CRT, the samples were permeabilized with 0.1% v/v Triton-×100 for 5 min. on ice. Both permeabilized and non-permeabilized cells (used to detect surface CRT) were washed three times with PBS and stained with a rabbit polyclonal anti-CRT antibody (diluted 1:100 in PBS containing 1% FBS) for 1 hr at room temperature. After washing, the samples were incubated with a biotinylated goat anti-rabbit IgG antibody (diluted 1:50; Vector Laboratories, Burlingame, CA, USA) for 30 min. at room temperature, washed and incubated with fluorescein avidin D (diluted 1:300, Vector Laboratories) for further 30 min. at room temperature. To visualize actin cytoskeleton, cells were co-incubated with phalloidin-tetramethylrhodamine B isothiocyanate (diluted 1:1000). Finally all samples were washed with PBS, stained with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI; diluted 1:20,000) and washed again. The cover slips were mounted with 4 μl of Gel Mount Aqueous Mounting and examined with a Leica DC100 fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany). For each experimental point, a minimum of five microscopic fields were examined.
Western blot analysis
Cells were washed twice with PBS, then lysed in heated lysis buffer (25 mmol/l Hepes, 135 mmol/l NaCl, 1% v/v Nonidet P-40, 5 mmol/l ethylenediaminetetraacetic acid (EDTA), 1 mmol/l ethylene glycol tetraacetic acid (EGTA), 1 mmol/l ZnCl2 and 10% v/v glycerol) and sonicated with one 10 sec. burst, using a Labsonic Sonicator (Hielscher). After centrifugation (13,000 ×g for 15 min.) the protease inhibitor cocktail III (100 mmol/l 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 80 μmol/l aprotinin, 5 mmol/l bestatin, 1.5 mmol/l E-64, 2 mmol/l leupeptin, and 1 mmol/l pepstatin; Calbiochem), 2 mmol/l phenylmethylsulfonyl fluoride and 1 mmol/l NaVO4 were added to the supernatant. A total of 25 μg cell proteins were separated by SDS-PAGE, transferred to PVDF membrane sheets and probed with an anti-vasodilator-stimulated phosphoprotein (VASP) (rabbit polyclonal, diluted 1:500 in PBS-BSA 1%; Cell Signaling Technology, Inc., Danvers, MA, USA) or an anti-phospho(Ser 239)-VASP (mouse monoclonal, diluted 1:500 in PBS non-fat dry milk 3%, Millipore) antibody.
In co-immunoprecipitation experiments, 100 μg of total cell proteins or biotinylated proteins, re-suspended in 200 μl TEEN-Triton buffer (Tris 50 mmol/l, EDTA 1 mmol/l, EGTA 1 mmol/l, NaCl 150 mmol/l, Triton X-100 1% v/v), were immunoprecipitated overnight with the anti-CRT antibody (diluted 1:100), then subjected to 7% SDS-PAGE and probed for Pgp (with a rabbit polyclonal antibody, diluted 1:250 in PBS non-fat dry milk 5%, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The proteins were detected by enhanced chemiluminescence as described above. The densitometric analysis of Western blots was performed with the ImageJ software (http://rsb.info.nih.gov/ij/) and expressed as arbitrary units, where ‘1 unit’ is the mean band density of the experimental condition ‘HT29 CTRL’.
Small interfering RNA (siRNA)
A total of 200,000 cells were plated and cultured in RPMI 1640 containing 10% FBS. After 24 hrs, cells were washed with 2 ml siRNA transfection medium (Santa Cruz Biotechnology, Inc.) and incubated for 6 hrs with 1 ml siRNA transfection medium containing 5 μl of siRNA transfection reagent (Santa Cruz Biotechnology, Inc.) and 50 pmol of Pgp specific siRNA (Santa Cruz Biotechnology, Inc.). In each set of experiments, one dish was treated with 50 pmol of Control siRNA-A (Santa Cruz Biotechnology, Inc.), a scrambled non targeting 20-to 25-nucleotide siRNA, designed as a negative control to assess siRNA specificity. At the end of the incubation, 1 ml of RPMI 1640 containing 1% PS and 20% FBS was added for 24 hrs; cells were washed and cultured for 72 hrs in RPMI 1640 with 1% PS and 10% FBS. To verify the siRNA efficacy, the expression of Pgp protein was analysed by Western blotting as reported above. The expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), chosen as a housekeeping gene, was checked with a specific antibody (from rabbit, diluted 1:500 in PBS-BSA 1%, Santa Cruz Biotechnology, Inc.).
Intracellular doxorubicin accumulation
Cells were grown in 35-mm-diameter Petri dishes and incubated in RPMI 1640 for 3 hrs with 5 μmol/l doxorubicin, and then the intracellular content of the drug was detected fluorimetrically as previously reported [4].
Statistical analysis
All data in text and figures are provided as mean ± S.E. The results were analysed by a one-way ANOVA and Tukey’s test. P < 0.05 was considered significant.
Results
Doxorubicin fails to increase CRT exposure and HT29 cells phagocytosis when nitric oxide synthesis is inhibited
Doxorubicin increased the activity of NOS enzyme (Fig. 1A) and the synthesis of nitrite (Fig. 1B) in HT29 cells; both the events were reversed by the NOS inhibitor NG-monomethyl-L-arginine (L-NMMA). The nitric oxide scavenger 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), which per se did not affect the enzymatic activity of NOS (Fig. 1A), effectively reduced the increase of nitrite elicited by the anthracycline (Fig. 1B). By labelling the surface proteins of HT29 cells with biotin (Fig. 1C) and by employing flow cytometry (Fig. 1D), we showed that doxorubicin increased the amount of plasma membrane associated CRT in HT29 cells and that this response was lowered by PTIO and L-NMMA. No change in total CRT was detected in the presence of doxorubicin, PTIO and L-NMMA (Fig. 1C). Doxorubicin also enhanced the phagocytosis of HT29 cells by iDCs, but when the nitric oxide scavenger or the NOS inhibitor were co-incubated with the drug, the phagocytosis was lowered (Fig. 1E).
Fig 1.
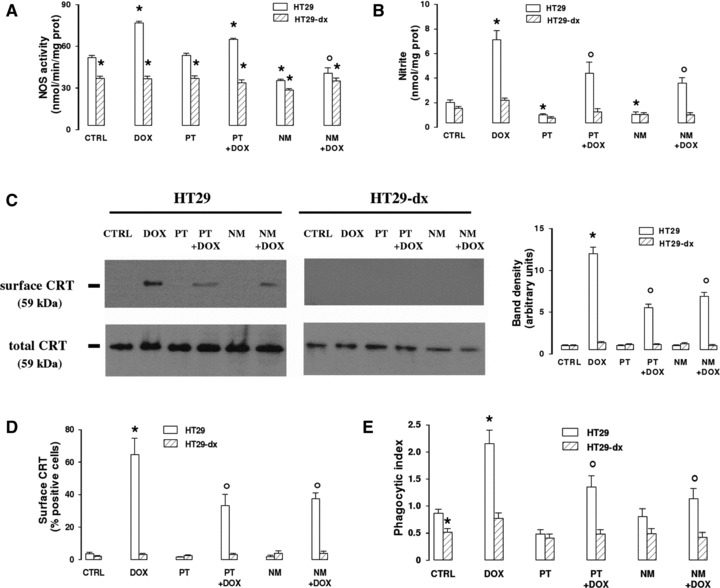
Effects of PTIO and L-NMMA on doxorubicin-induced nitric oxide synthesis, CRT translocation and tumour cells phagocytosis. HT29 and HT29-dx cells were cultured for 6 hrs in the absence (CTRL) or in the presence of doxorubicin (5 μmol/l, DOX), alone or together with the nitric oxide scavenger PTIO (100 μmol/l, PT) or the NOS inhibitor L-NMMA (1 mmol/l, NM). Then the following assays were performed. (A) The NOS activity was measured in the cell lysates by a spectrophotometric assay. Measurements were performed in triplicate. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.01; versus DOX: °P < 0.001. (B) Colorimetric detection of nitrite amounts in cell culture medium was performed in duplicate. Data are presented as means ± S.E. (n= 4). Vs CTRL HT29: *P < 0.001; versus DOX: °P < 0.05. (C) Western blot analysis of biotinylated surface CRT and of total CRT was performed as reported under ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between surface and total CRT was expressed as arbitrary units. Vs CTRL HT29: *P < 0.001; versus DOX: °P < 0.01. (D) Flow cytometry analysis of cells positive for surface CRT was performed in duplicate as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.001; versus DOX: °P < 0.02. (E) The phagocytosis of HT29 and HT29-dx cells by iDCs was measured in duplicate by flow cytometry (see ‘Materials and methods’ for details). Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.05; versus DOX: °P < 0.05.
On the contrary, doxorubicin, which was less accumulated in HT29-dx cells [6], did not elicit any increase of nitric oxide synthesis (Fig. 1A and B), CRT translocation on plasma membrane (Fig. 1C and D) and phagocytosis (Fig. 1E) in drug-resistant cells. PTIO and L-NMMA were devoid of effects on all these parameters in HT29-dx cells (Fig. 1A–E). HT29-dx cells exhibited a basally lower activity of NOS enzyme (Fig. 1A), a result already reported in this experimental model [6]. Unexpectedly, we observed that the rate of phagocytosis in untreated HT29-dx cells was lower than in HT29 cells (Fig. 1E).
Nitric oxide promotes the translocation of CRT in a cGMP-dependent way, via the actin cytoskeleton remodelling
Amongst its several target enzymes, nitric oxide may activate a soluble guanylate cyclase (sGC) and increase the synthesis of cGMP, which in turn promotes the activation of a cGMP-dependent protein kinase (PKG). This event accounts for several effects of nitric oxide such as smooth muscle relaxation, platelet aggregation and cytoskeleton remodelling [13]. Therefore we measured the expression of cell surface CRT in the presence of the nitric oxide donor S-nitrosopenicillamine (SNAP), of the stable cGMP analogue 8-Br-cGMP, of the sGC inhibitor 1H-[1, 2, 4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ) and of the PKG inhibitor Rp-8-Br-cGMPS. 8-Br-cGMP was as effective as SNAP in inducing the translocation of CRT (Fig. 2A and B). On the contrary, ODQ and Rp-8-Br-cGMPS greatly reduced the effect of SNAP (Fig. 2A and B). Remarkably, the response to cGMP pathway modulators was similar in doxorubicin-sensitive and doxorubicin-resistant cells.
Fig 2.
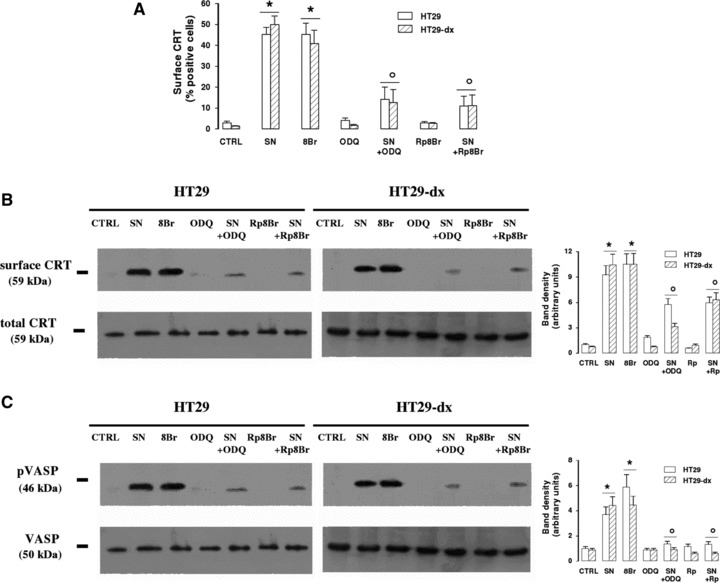
Effect of nitric oxide/cGMP pathway on CRT translocation. HT29 and HT29-dx cells were treated for 1 hr with fresh medium (CTRL), SNAP (100 μmol/l, SN), 8-Br-cGMP (10 μmol/l, 8Br), ODQ (10 μmol/l, ODQ) and Rp-8-Br-cGMPS (10 μmol/l, Rp8Br), in different combinations, then subjected to the following investigations. (A) The percentage of cells positive for surface CRT was measured in duplicate by FACS analysis as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.001; versus SN: °P < 0.002. (B) Biotinylated surface CRT and total CRT were measured in Western blotting experiments, as reported in ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between surface and total CRT was expressed as arbitrary units. Vs CTRL HT29: *P < 0.001; versus SN: °P < 0.02. (C) Western blot analysis of VASP and phospho(Ser239)-VASP was performed on cytosolic extracts as described under ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between phospho(Ser239)-VASP and total VASP was expressed as arbitrary units. Vs CTRL HT29: *P < 0.01; versus SN: °P < 0.02.
Immunofluorescence analysis revealed that untreated HT29 and HT29-dx cells had no detectable amounts of CRT on plasma membrane, whereas the great majority of the protein was contained within cells (Fig. S1). Nitric oxide promoted the translocation of CRT in a time-dependent way: indeed CRT was nearly undetectable 10 min. after the application of SNAP, but became more appreciable after 30 min. At 1 hr the fluorescence was more intense and detected on larger portions of cell surface. Again, the response to the nitric oxide donor was superimposable in chemo-sensitive and chemo-resistant cells (Fig. S1), as observed in flow cytometry analysis (Fig. 2A) and biotinylation assay (Fig. 2B).
A downstream effector of PKG is the VASP, which, after being phosphorylated on serine by PKG, has a prominent role in actin cytoskeleton remodelling [14]: in HT29 and HT29-dx cells the phosphorylation of VASP was absent under basal conditions, became detectable in the presence of SNAP and 8-Br-cGMP, and was reduced when SNAP was co-incubated with ODQ and Rp-8-Br-cGMPS (Fig. 2C). Total VASP did not vary in any experimental condition (Fig. 2C).
To evaluate the involvement of actin cytoskeleton in the nitric oxide/cGMP-mediated translocation of CRT, we analysed the effect of latrunculin A, a specific inhibitor of G-actin monomers assembly: interestingly, SNAP and 8-Br-cGMP were not able to elicit any detectable exposure of CRT in both HT29 and HT29-dx cells, when co-incubated with latrunculin A (Fig. 3A and B). Untreated HT29 and HT29-dx cells showed a widespread intracellular actin distribution, with a continuous ring in the sub-plasma membrane region (Fig. 3C). Such a distribution was not modified by the SNAP, whereas latrunculin, both alone and in the presence of the nitric oxide donor, induced a pronounced change in cell morphology, disrupted the sub-plasma membrane ring and condensed actin in selective areas of cells surface. Moreover when co-incubated with latrunculin, SNAP was not able to elicit the translocation of CRT on cell plasma membrane (Fig. 3C).
Fig 3.
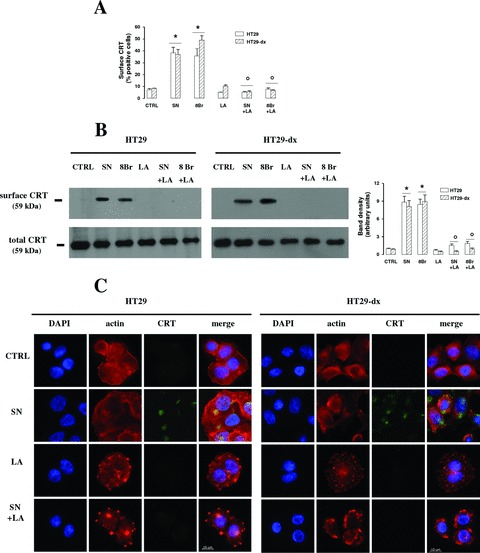
Effect of cytoskeleton remodelling in the nitric oxide/cGMP-mediated translocation of CRT. HT29 and HT29-dx cells were treated for 1 hr with fresh medium (CTRL), SNAP (100 μmol/l, SN), 8-Br-cGMP (10 μmol/l, 8Br), latrunculin A (200 nmol/L, LA), in different combinations, then the following experiments were performed. (A) The percentage of cells positive for surface CRT, obtained by FACS analysis, was calculated in duplicate as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.02 versus SN: °P < 0.02. (B) Biotinylated surface CRT and total CRT were measured by Western blot analysis as reported in ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between surface CRT and total CRT was expressed as arbitrary units. Vs CTRL HT29: *P < 0.001; versus SN: °P < 0.01. (C) Cells were stained with phalloidin-tetramethylrhodamine B isothiocyanate to visualize actin fibres; surface CRT was detected by an anti-CRT antibody, as specified in ‘Materials and methods’. The micrographs are representative of three experiments with similar results.
Taken together these results indicate that nitric oxide elicits the translocation of CRT on plasma membrane, via the cGMP/PKG/VASP activation and the remodelling of actin cytoskeleton. This pathway is as active in chemosensitive as in chemoresistant cells.
CRT exposure is increased by nitric oxide in both HT29 and HT29-dx cells, but is followed by phagocytosis only in doxorubicin-sensitive cells
TNF-α, a potent inducer of iNOS, significantly increased the NOS activity (Fig. 4A), the nitrite levels (Fig. 4B) and the CRT translocation (Fig. 4C and D) in both doxorubicin-sensitive and doxorubicin-resistant cells. PTIO, which lowered the levels of nitrite (Fig. 4B) without affecting per se NOS activity (Fig. 4A), also decreased the exposure of CRT (Fig. 4C and D). In parallel TNF-α induced, whereas PTIO reduced, the index of phagocytosis of HT29 cells by iDCs (Fig. 4E). Despite the increase of nitrite and CRT, TNF-α did not induce the iDCs-mediated uptake of HT29-dx cells, which were less phagocytized than drug-sensitive cells in each experimental condition (Fig. 4E).
Fig 4.
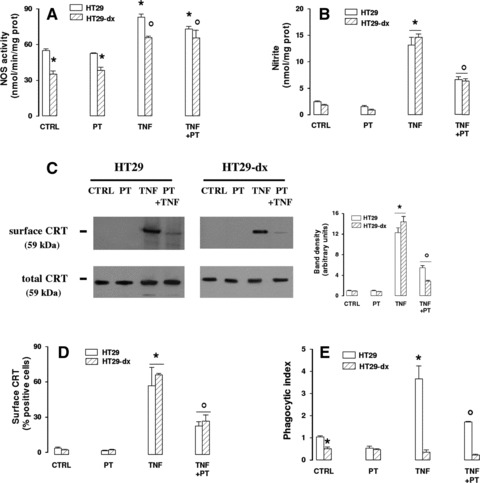
Effects of TNF-α on doxorubicin-induced nitric oxide synthesis, CRT translocation and tumour cells phagocytosis. HT29 and HT29-dx cells were cultured for 6 hrs in the absence (CTRL) or presence of the iNOS inducer TNF-α (50 ng/ml, TNF), alone or together with the nitric oxide scavenger PTIO (100 μmol/l, PT), then cells were subjected to the following investigations. (A) Spectrophotometric detection of the NOS activity in cell lysates. Measurements were performed in triplicate. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.001; versus CTRL HT29-dx: °P < 0.005. (B) The nitrite levels in the culture supernatant, assessed by Griess method, were measured in duplicate. Data are presented as means ± S.E. (n= 4). Vs CTRL HT29: *P < 0.01; versus TNF: °P < 0.005. (C) Biotinylated surface CRT and total cell CRT expression was measured by Western blot experiments, as reported under ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between surface and total CRT was expressed as arbitrary units. Vs CTRL HT29: *P < 0.01; versus TNF: °P < 0.005. (D) The percentage of cells positive for surface CRT was performed in duplicate by FACS analysis as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.05; versus TNF: °P < 0.05. (E) The rate of HT29 and HT29-dx cells phagocytized by iDCs was measured in duplicate by flow cytometry (see ‘Materials and methods’ for details). Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.05; versus TNF: °P < 0.05.
Doxorubicin and the nitric oxide donor SNAP per se increased the nitrite levels (Fig. 5A) and the CRT exposure (Fig. 5C and D) in HT29 cells, displaying an additive effect when co-incubated (Fig. 5A, C and D). SNAP did not modify the activity of NOS, which was raised by doxorubicin in sensitive cells and remained superimposable to the control cells in all the other conditions (Fig. 5B), but increased the nitrite amounts by releasing nitric oxide (Fig. 5A). When SNAP was co-incubated with doxorubicin in doxorubicin-resistant cells, where the drug alone was devoid of effect, the nitrite amount was significantly increased (Fig. 5A) and the CRT exposure was induced (Fig. 5C and D). Cisplatin, an anticancer agent which did not affect the nitric oxide synthesis (Fig. 5A and B) was not able to elicit any translocation of CRT (Fig. 5C and D) or to promote the phagocytosis (Fig. 5E). However, when SNAP was added to cisplatin, the CRT expression became detectable in plasma membrane (Fig. 5C and D) and the uptake of HT29 cells by iDCs was significantly raised (Fig. 5E). Again, a dissociation between the CRT exposure and the phagocytosis was observed in HT29-dx cells, where in the presence of cisplatin plus SNAP, the CRT translocation on cell surface increased (Fig. 5C and D), but the phagocytosis did not (Fig. 5E).
Fig 5.
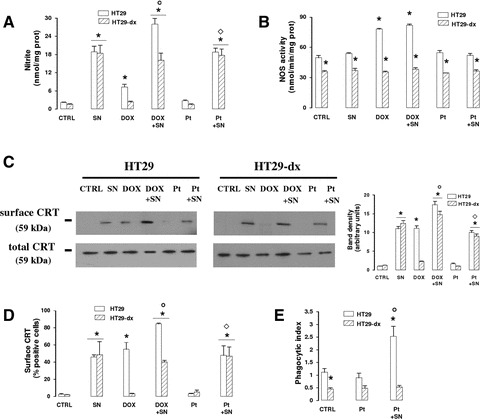
Effect of SNAP and anticancer drugs on CRT translocation and phagocytosis in HT29 and HT29-dx cells. HT29 and HT29-dx cells were incubated for 6 hrs in the absence (CTRL) or presence of the nitric oxide donor SNAP (100 μmol/l, SN), doxorubicin (5 μmol/l, DOX) or cisplatin (10 μmol/l, Pt), in different combinations. Then the following investigations were performed. (A) The nitrite amount was detected in the supernatant of cell cultures in duplicate, as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 4). Vs CTRL HT29: *P < 0.005; versus DOX: °P < 0.001; versus Pt: ⋄P < 0.001. (B) The NOS activity was measured spectrophotometrically in the cell lysates. Measurements were performed in triplicate. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.005. (C) Western blot analysis of biotinylated surface CRT and of total cell CRT was performed as reported under ‘Materials and methods’. The figure is representative of three experiments with similar results. The band density ratio between surface and total CRT was expressed as arbitrary units. Vs CTRL HT29: *P < 0.001; versus DOX: °P < 0.002; versus Pt: ⋄P < 0.02. (D) The surface expression of CRT was assessed by flow cytometry analysis (see ‘Materials and methods’ for details). Data are presented as means ± S.E. (n= 4). Vs CTRL HT29: *P < 0.001; versus DOX: °P < 0.01; versus Pt: ⋄P < 0.02. (E) The uptake of HT29 and HT29-dx cells by iDCs was evaluated in duplicate by flow cytometry, as reported in ‘Materials and methods’. Data are presented as means ± S.E. (n= 3). Vs CTRL HT29: *P < 0.01; versus Pt: °P < 0.02.
These data gave us further evidence that nitric oxide, generated by an iNOS inducer as well as released by a nitric oxide donor, was necessary to promote the CRT translocation in both doxorubicin-sensitive and doxorubicin-resistant tumour cells, but it was sufficient to induce phagocytosis by iDCs only in the drug-sensitive ones.
In HT29-dx cells CRT is associated to Pgp, which exerts an inhibitory effect on the phagocytosis
To analyse why the increased amount of surface CRT was not relatable to an increased phagocytosis in HT29-dx cells, we investigated whether Pgp, a membrane protein which is overexpressed in HT29-dx cells, but not in HT29 cells [4, 6], might interfere with the functional activity of the surface CRT or with the phagocytosis of drug-resistant cells. We first assessed if Pgp was physically associated to CRT: we immunoprecipitated with an anti-CRT antibody the cell extracts enriched with the ER membranes (where the vast majority of CRT resides) or containing the cell surface biotinylated proteins; then we probed the immunoprecipitated samples with an anti-Pgp antibody (Fig. 6). In HT29 cells, where the expression of Pgp was low (Fig. 7A), we did not detect any association between Pgp and CRT (Fig. 6A). On the contrary, in ER extracts derived from HT29-dx cells, Pgp co-immunoprecipitated with CRT (Fig. 6A). Such an association was detected in untreated cells and was not affected by doxorubicin or SNAP (Fig. 6A). The nitric oxide donor SNAP was the only agent able to induce the exposure of CRT in the plasma membrane in HT29-dx cells (Figs 5B, C and 6B): following SNAP incubation, we observed a co-immunoprecipitation of CRT and Pgp also in the cell extracts containing surface biotinylated proteins (Fig. 6B). This phenomenon was absent in untreated cells and in HT29-dx cells exposed to doxorubicin (Fig. 6B). Such a result suggests that Pgp was bound to CRT in ER and that this association was not lost during the translocation on the plasma membrane of doxorubicin-resistant cells.
Fig 6.
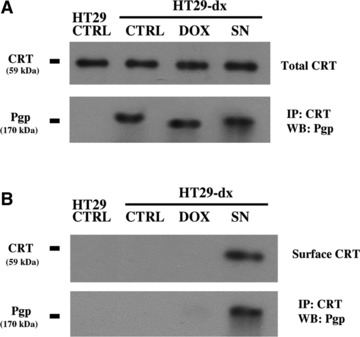
Co-immunoprecipitation of CRT and Pgp in HT29 and HT29-dx cells. Cells were incubated for 6 hrs in the absence (CTRL) or presence of doxorubicin (5 μmol/l, DOX) or SNAP (100 μmol/l, SN), then they were lysed and subjected to ultracentrifugation to isolate the ER-enriched fraction (A). In parallel, in an aliquot of cells incubated under the same experimental conditions, the surface proteins were isolated by a biotinylation assay (B). Each sample was immunoprecipitated (IP) with an anti-CRT antibody and probed with an anti-Pgp or an anti-CRT antibody by Western blotting (WB), as reported in ‘Materials and methods’. The results shown here are representative of two similar experiments, giving superimposable results.
Fig 7.
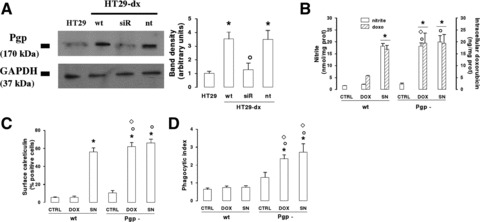
Effect of Pgp knocking-down on CRT exposure and HT29-dx cell phagocytosis. Non-silenced HT29-dx cells (wt) or HT29-dx cells silenced for Pgp (Pgp-) were incubated for 6 hrs with fresh medium (CTRL), doxorubicin (5 μmol/l, DOX) or SNAP (100 μmol/l, SN), then subjected to the following investigations. (A) Western blot detection of Pgp. HT29-dx cells were grown for 72 hrs in the absence (wt) or presence of siRNA sequences (siR) targeting Pgp gene, as reported in ‘Materials and methods’. When indicated, a non-targeting scrambled siRNA sequence (nt) was added instead of siRNA for Pgp. The expression of Pgp was detected by Western blot analysis, comparing HT29-dx with untreated HT29 cells. The expression of the housekeeping protein GAPDH was measured as equal control loading. The the band density ratio between Pgp and GAPDH was expressed as arbitrary units. Vs HT29: *P < 0.05; versus HT29-dx wt: °P < 0.05. (B) The nitrite synthesis in the culture supernatant (open bars) and the intracellular doxorubicin accumulation (hatched bars) were measured in duplicate, as described in ‘Materials and methods’. Data are presented as means ± S.E. (n= 4). For nitrite: versus CTRL wt: *P < 0.05; versus CTRL Pgp-: °P < 0.05; versus DOX wt: ⋄P < 0.01. For doxorubicin: versus DOX wt: *P < 0.05. (C) The surface expression of CRT, assessed by flow cytometry analysis as reported under ‘Materials and methods’, was measured in duplicate. Data are presented as means ± S.E. (n= 4). Vs CTRL wt: *P < 0.01; versus CTRL Pgp-: °P < 0.01; versus DOX wt: ⋄P < 0.002. (D) The phagocytosis rate of HT29-dx cells by iDCs was evaluated in duplicate by flow cytometry (see ‘Materials and methods’). Data are presented as means ± S.E. (n= 3). Vs CTRL wt: *P < 0.05; versus CTRL Pgp-: °P < 0.05; versus DOX or SN wt, respectively: ⋄P < 0.02.
To further clarify whether the interaction between CRT and Pgp has a functional role in the immunoresistance of HT29-dx cells, we knocked down Pgp with a siRNA approach. Besides reducing the protein expression levels in HT29-dx cells (Fig. 7A), the silencing of Pgp raised the accumulation of doxorubicin to a level superimposable to that of not-silenced HT29-dx cells treated with SNAP (Fig. 7B).
Wild-type HT29-dx and HT29-dx Pgp– cells did not differ in nitrite levels (Fig. 7B) and surface CRT amounts (Fig. 7C) under basal conditions and after the incubation with SNAP. On the contrary, doxorubicin was able to increase the nitric oxide synthesis (Fig. 7B) and the CRT exposure on the plasma membrane (Fig. 7C) only in HT29-dx cells silenced for Pgp, where the drug intracellular content was significantly higher (Fig. 7B). As far as the uptake by iDCs is concerned, untreated HT29-dx Pgp–, which exhibited a low exposure of CRT (Fig. 7C), were poorly phagocytized, similar to HT29-dx cells (Fig. 7D). However, in the presence of doxorubicin or SNAP, which both increased the surface CRT levels in HT29-dx Pgp– cells (Fig. 7C), the silencing of Pgp induced the uptake by iDCs, suggesting that the overexpression of Pgp exerts a negative control on the CRT-dependent phagocytosis of drug-resistant cells (Fig. 7D).
Discussion
The combination of conventional anticancer drugs to immunostimulating agents has recently obtained promising results in tumour mice models and in clinical trials [15, 16]. Anthracyclines, like doxorubicin, are useful tools in the chemoimmunotherapy, because they are the only anticancer drugs with a proved stimulating effect on macrophages [17], lymphocytes [18] and DCs [8]. Most anticancer drugs elicit in tumour cells an apoptotic death, which is considered not immunogenic, in opposition to a necrotic death, which releases in the microenvironment several tumour antigens, recognized by iDCs [8]. Doxorubicin exerts both an apoptotic and an immunogenic cell death: indeed following the drug administration, several intracellular proteins, like CRT, are exposed on the plasma membrane and trigger the tumour cells uptake by iDCs [8]. Besides inducing CRT translocation, doxorubicin is also able to increase the synthesis of nitric oxide, which may act as a tumoricidal agent, as an immunomodulator agent [19] and as an inhibitor of the doxorubicin efflux through ABC transporters [4, 6]. We have recently demonstrated that nitric oxide is also the mediator of the CRT translocation in the HT29 doxorubicin-sensitive colon cancer cells: after this event, tumour cells are phagocytized by iDCs, which acquired the competence to prime alloantigen reactive lymphocytes [6]. No translocation of CRT is elicited by doxorubicin when the synthesis of nitric oxide is lacking, like in HT29 cells silenced for iNOS gene, or in doxorubicin-resistant HT29-dx cells, where the drug is actively extruded by Pgp and is not accumulated at a sufficient amount to raise the nitric oxide synthesis [6].
In the present work we first investigated by which mechanisms nitric oxide could exert such an effect. In chemosensitive HT29 cells incubated with doxorubicin, in the presence of a nitric oxide scavenger or a NOS inhibitor, the nitrite increase elicited by the anthracycline, the CRT translocation and the cells phagocytosis by iDCs were all reduced. On the contrary, in drug-resistant HT29-dx cells, where nitric oxide levels were not modulated by doxorubicin, PTIO and NMMA, CRT translocation and phagocytosis by iDCs remained low. Interestingly, NOS activity was lower in untreated HT29-dx cells than in HT29 cells, although no significant difference was detected in the nitrite levels between these cell populations: such a discrepancy is likely due to the higher sensitivity of the spectrophotometric assay of NOS activity towards the colorimetric Griess method for nitrite detection [11]. We did not observe any variation in the total CRT expression, despite significant changes in the CRT levels on the plasma membrane. Most CRT usually resides in ER and is absent on plasma membrane, where only small amounts of CRT translocate; it has been suggested that, although the fraction of CRT in ER does not change, small variations in the plasma membrane fraction may be sensed as an effective ‘eat me’ signal by iDCs [9]. The appearance of small but detectable amounts of CRT was a powerful eating marker also in our experimental model. This event has been reported to be the first step a more complex activation of the host immune system against the tumour.
For example the exposure of CRT on the mouse CT26 colon cells surface has been shown to trigger the activation of immune system cells in mice xenografts [9] and the up-regulation of CRT gene in colon cancers with microsatellite instability has been associated with a more pronounced lymphocytic infiltrate, a more active immune response and a better prognosis of patients [20]. In keeping with these observations, we have previously observed that in HT29 cells the CRT exposure increased the tumour cell uptake and the progression of immature DCs to an antigen-presenting mature phenotype [6].
Several mechanisms have been invoked to explain how CRT appears on the plasma membrane following anthracyclines administration. Because such translocation occurs in both enucleated and nucleated tumour cells, the increase of CRT on the cell surface is not attributable to augmented gene transcription [9]. Some experimental evidence show that agents preventing the ER stress, such as the inhibitors of PP1/GADD34 complex, facilitate the CRT translocation. However the inhibition of PP1/GADD34 per se is not sufficient to evoke an immunogenic death of tumour cells [9]. The human neuroblastoma SH-SY5Y cell line is completely refractory to translocate CRT in response to mitoxantrone, but the depletion of Ca++ in ER partially restores this CRT translocation [21]. Nitric oxide has already been reported to lower the Ca++ level in ER [22] and we cannot exclude that this mechanism could be implicated in the translocation of CRT from ER to the plasma membrane. However, our results suggest an involvement of the cGMP/PKG pathway in such a translocation in colon cancer cells: indeed both the nitric oxide donor SNAP and the sGC activator 8-Br-cGMP increased CRT exposure, which was prevented by inhibiting sGC or PKG. When PKG is activated it can in turn phosphorylate and activate the serine–threonine kinase VASP [14], an event which was observed in both HT29 and HT29-dx cells stimulated by SNAP or 8-Br-cGMP and was reduced when the sGC/PKG pathway was blocked. Active VASP deeply affects the activity and localization of intercellular junction proteins and integrins, owing to its effect on actin cytoskeleton [13]. Interestingly, the anthracycline-mediated exposure of CRT in mouse colon cells was reduced by inhibiting the actin fibres remodelling, whereas was unaffected by agents disrupting microtubules [9]. In our experimental conditions, latrunculin A, a toxin inhibiting the polymerization of G-actin, blocked the CRT translocation elicited by SNAP or 8-Br-cGMP. Therefore we can suggest that nitric oxide promotes the exposure of CRT by activating the cGMP/PKG/VASP pathway and by inducing a remodelling of the actin fibres. Nitric oxide promoted a time-dependent translocation on plasma membrane of CRT, which was concentrated in selective portions of cell surface. Such a patchy distribution was observed also in CT26 colon cancer cells exposed to mitoxantrone [23], suggesting a preferential localization of CRT in specific microdomains of plasma membrane. In CT26 cells CRT co-localizes with ERp57, another protein of ER, which translocates on cell surface and becomes an integral component of the major histocompatibility complex class I system [23]. We cannot exclude that nitric oxide promotes a co-translocation of CRT and ERp57 from ER to plasma membrane also in HT29 colon cancer cells.
Notably no differences in the translocation of CRT induced by the nitric oxide/cGMP-pathway were detected between doxorubicin-sensitive and doxorubicin-resistant cells, suggesting that the downstream effectors of nitric oxide worked at the same extent in the two populations.
Similarly, if we increased the nitric oxide levels with a stimulus different from doxorubicin, such as the iNOS inducer TNF-α, the synthesis of nitrites and the translocation of CRT occurred in HT29 and in HT29-dx cells with no appreciable differences, suggesting that the lack of nitric oxide production in doxorubicin-treated HT29-dx was not due to a defect in the iNOS induction pathway, but rather to the inability to accumulate enough doxorubicin to activate the NF-κB signalling transduction.
Not all chemotherapeutic drugs exert pro-immunogenic effects; some non-immunogenic drugs, such as etoposide, camptothecine and mitomycin C, acquire pro-immunogenic properties only when co-administrated with recombinant CRT [9]. We show in the present work that the co-incubation with a nitric oxide donor may produce the same effect: indeed cisplatin did not increase nitric oxide levels and CRT exposure in HT29 and HT29-dx cells, but all these events were induced when cisplatin was associated to SNAP. Moreover when SNAP and doxorubicin were added together, they had a small but detectable additive effect on CRT translocation in HT29 cells, suggesting that 5 μmol/l doxorubicin did not exert a maximal translocation and that nitric oxide may further increase the exposure of CRT elicited by doxorubicin in sensitive cells. On the other hand, when doxorubicin was devoid of effect, as it occurred in HT29-dx cells, the addition of SNAP restored the translocation of CRT.
Every time we detected an increase of CRT translocation in chemosensitive cells, we also observed an increased phagocytosis by iDCs. On the contrary, although the CRT density was as high on the HT29-dx cells surface as in HT29 cells (e.g. following TNF-α or SNAP), the phagocytosis of chemoresistant cells did not increase. Our results suggest that nitric oxide was sufficient to promote the CRT translocation followed by the phagocytosis in doxorubicin-sensitive cells. The situation looks different in drug-resistant cells, where nitric oxide was necessary to induce the exposure of CRT, but this event was not sufficient to induce the uptake by iDCs. Interestingly also untreated HT29-dx cells were less phagocytized than HT29 cells: CRT was low in both the populations in this experimental condition, so we wondered if other additional features made doxorubicin-sensitive and doxorubicin-resistant cells different as to their phagocytosis by iDCs. A striking difference between HT29 and HT29-dx cells is the overexpression in the latter population of Pgp, which is responsible of the drug-resistant phenotype. We therefore evaluated whether this protein could play a role also in the immunoresistance of HT29-dx cells.
Surprisingly we found that CRT was bound to Pgp, in a manner dependent on the expression level of the latter. Indeed in HT29 cells Pgp was not co-immunoprecipitated with CRT, probably because of the very low amounts of this ABC transporter. On the other hand, in HT29-dx cells, where the efflux pump is abundant, CRT clearly interacted with Pgp. Our results on the ER-enriched fraction and on the plasma membrane fraction suggest that such an association started in the ER membrane, where both CRT and Pgp are present [24], and was not lost during the movement to the cell surface. Nitric oxide did not alter the binding of CRT with Pgp, since the interaction between them persisted in the presence of SNAP. It is conceivable that the activity of the nitric oxide/cGMP pathway is not specific for a single ER protein like CRT, but generically facilitates the traffic of microsomal vesicles from ER to plasma membrane. Acting as a chaperon protein, CRT may bind many other proteins synthesized in ER and help them in the intracellular processing and folding: for example it has been recently demonstrated that CRT forms complexes with collagen type I in the ER of fibroblasts. This association plays a crucial role in the trafficking of collagen from ER to Golgi apparatus and is important to target the collagen fibres to their final destination in the extracellular environment [25]. Because Pgp is actively synthesized in drug-resistant cells, we may suppose that the ABC transporter interacts with CRT during the quality control step in the ER and that CRT acts as an escort protein for Pgp, accompanying the latter to its definitive localization on the plasma membrane. However, following this interaction, CRT looses at least one of its functional properties, that is the peculiarity to function as a docking protein for iDCs on tumour surface.
When the expression of Pgp was knocked down by specific siRNAs, the CRT-operated phagocytosis was restored in HT29-dx cells: in silenced cells doxorubicin was able again to accumulate, to induce nitric oxide synthesis and CRT translocation and to promote phagocytosis by iDCs. Also SNAP, which was capable to induce the CRT exposure but not the phagocytosis in HT29-dx cells, became an ‘eat me’ agent in Pgp-silenced HT29-dx cells, reproducing all the effects exhibited in drug-sensitive HT29 cells. These data show that Pgp, when overexpressed, inhibit the tumour cells phagocytosis and play a role not only in the chemoresistance, but also in the immunoresistance. At present we do not know the nature of the binding between CRT and Pgp, and the mechanism of the Pgp-mediated inhibition of phagocytosis. The ABC transporter might exert a steric hindrance, due to its high molecular weight and abundance on cells surface of resistant cells, impairing the correct membrane presentation or clusterization of CRT and altering the recognition of tumour cells by iDCs.
Chemo-immunotherapy is a very promising approach in the treatment of the most aggressive cancers, refractory to the conventional anticancer drugs [16]; a better knowledge of the mechanisms which regulate the chemo-immunoresistance to doxorubicin in human cancer cells is certainly useful to optimize the chemo-immunotherapeutic protocols. In our work we have demonstrated that the association of nitric oxide releasing compounds, which are widely used in the management of cardiovascular diseases, to conventional anticancer drugs, may represent a novel approach of chemo-immunotherapy in chemosensitive tumours. As for the chemoresistant tumours, which we showed are also immunoresistant, nitric oxide was able to restore the sensitivity to the doxorubicin cytotoxic effects and the translocation of CRT [4, 6], but was not sufficient to trigger the activation of iDCs against the tumour. However the refractoriness of drug-resistant cells to be phagocytized may be circumvented by down-regulating the Pgp protein. Recently the selective silencing of Pgp, followed by the administration of anticancer drugs, has successfully arrested the growth of chemoresistant tumour in mice xenografts, prolonging the overall survival of the animals and showing new possibilities of combination therapies for aggressive cancers [26]. Our work may suggest that the combination of nitric oxide releasing agents with therapeutic tools down-regulating Pgp fully restores the functional role of CRT in drug-resistant cells and might be successful in obtaining a reversion of chemo-and immunoresistance in these cells.
Acknowledgments
We are grateful to Mr. Costanzo Costamagna for the technical assistance provided and to Dr. Silvia Peola for the help with the flow cytometry analysis. This work has been supported with grants from Fondazione Internazionale Ricerche Medicina Sperimentale (FIRMS), Compagnia di San Paolo, Regione Piemonte (Ricerca Sanitaria Finalizzata 2008 and 2009) and Ministero dell’Università e della Ricerca (Rome, Italy).
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Kinetics of CRT translocation on cell surfaceelicited by nitric oxide. HT29 and HT29-dx cells were treated for 1hr with fresh medium (CTRL) or SNAP (100 μmol/l for 10,30 and 60 min., SN), then washed and stained for CRT. TotalCRT was detected in untreated cells after fixation andpermeabilization; CRT present on cell surface was evaluated innon-permeabilized cells. The micrographs are representative ofthree experiments with similar results.
References
- 1.Murphy MP. Nitric oxide and cell death. Biochim Biophys Acta. 1999;1411:401–14. doi: 10.1016/s0005-2728(99)00029-8. [DOI] [PubMed] [Google Scholar]
- 2.Riganti C, Doublier S, Costamagna C, et al. Activation of nuclear factor-kB pathway by simvastatin and RhoA silencing increases doxorubicin cytotoxicity in human colon cancer HT29 cells. Mol Pharmacol. 2008;74:476–84. doi: 10.1124/mol.108.045286. [DOI] [PubMed] [Google Scholar]
- 3.Lind DS, Kontaridis MI, Edwards PD, et al. Nitric oxide contributes to adriamycin’s antitumor effect. J Surg Res. 1997;69:283–7. doi: 10.1006/jsre.1997.5015. [DOI] [PubMed] [Google Scholar]
- 4.Riganti C, Miraglia E, Viarisio D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005;65:516–25. [PubMed] [Google Scholar]
- 5.Takara K, Sakaeda T, Okumura K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr Pharm Des. 2006;12:273–86. doi: 10.2174/138161206775201965. [DOI] [PubMed] [Google Scholar]
- 6.De Boo S, Kopecka J, Brusa D, et al. iNOS activity is necessary for the cytotoxic and immunogenic effects of doxorubicin in human colon cancer cells. Mol Cancer. 2009;8:108. doi: 10.1186/1476-4598-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaput N, De Botton S, Obeid M, et al. Molecular determinants of immunogenic cell death: surface exposure of calreticulin makes the difference. J Mol Med. 2007;85:1069–76. doi: 10.1007/s00109-007-0214-1. [DOI] [PubMed] [Google Scholar]
- 8.Apetoh L, Mignot G, Panaretakis T, et al. Immunogenicity of anthracyclines: moving towards more personalized medicine. Trends Mol Med. 2008;14:141–51. doi: 10.1016/j.molmed.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Obeid M, Testiere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 10.Obeid M, Tesniere A, Panaretakis T, et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev. 2007;220:22–34. doi: 10.1111/j.1600-065X.2007.00567.x. [DOI] [PubMed] [Google Scholar]
- 11.Ghigo D, Riganti C, Gazzano E, et al. Cycling of NADPH by glucose 6-phosphate dehydrogenase optimizes the spectrophotometric assay of nitric oxide synthase activity in cell lysates. Nitric Oxide. 2006;15:148–53. doi: 10.1016/j.niox.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Brusa D, Garetto S, Chiorino G, et al. Post-apoptotic tumors are more palatable to dendritic cells and enhance their antigen cross-presentation activity. Vaccine. 2008;26:6422–32. doi: 10.1016/j.vaccine.2008.08.063. [DOI] [PubMed] [Google Scholar]
- 13.Wojciak-Stothard B, Torondel B, Zhao L, et al. Modulation of Rac1 activity by ADMA/DDAH regulates pulmonary endothelial barrier function. Mol Biol Cell. 2009;20:33–42. doi: 10.1091/mbc.E08-04-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Levine YC, Golan DE, et al. Atrial natriuretic peptide-initiated cGMP pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J Biol Chem. 2008;283:4439–47. doi: 10.1074/jbc.M709439200. [DOI] [PubMed] [Google Scholar]
- 15.Pratesi G, Petrangolini G, Tortoreto M, et al. Therapeutic synergism of gemcitabine and CpG-oligodeoxynucleotides in an orthotopic human pancreatic carcinoma xenograft. Cancer Res. 2005;65:6388–93. doi: 10.1158/0008-5472.CAN-05-0602. [DOI] [PubMed] [Google Scholar]
- 16.Braly P, Nicodemus CF, Chu C, et al. The immune adjuvant properties of front-line carboplatin-paclitaxel: a randomized phase 2 study of alternative schedules of intravenous oregovomab chemoimmunotherapy in advanced ovarian cancer. J Immunother. 2009;32:54–65. doi: 10.1097/CJI.0b013e31818b3dad. [DOI] [PubMed] [Google Scholar]
- 17.Stoychkov JN, Schultz RM, Chirigos MA, et al. Effects of adriamycin and cyclophosphamide treatment on induction of macrophage cytotoxic function in mice. Cancer Res. 1979;39:3014–17. [PubMed] [Google Scholar]
- 18.Ho RL, Maccubbin D, Zaleskis G, et al. Development of a safe and effective adriamycin plus interleukin 2 therapy against both adriamycin-sensitive and -resistant lymphomas. Oncol Res. 1993;5:373–81. [PubMed] [Google Scholar]
- 19.Weigert A, Brüne B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide. 2008;19:95–102. doi: 10.1016/j.niox.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Banerjea A, Ahmed S, Hands RE, et al. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol Cancer. 2004;3:21–32. doi: 10.1186/1476-4598-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tufi R, Panaretakis T, Bianchi K, et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008;15:274–82. doi: 10.1038/sj.cdd.4402275. [DOI] [PubMed] [Google Scholar]
- 22.Oyadomari S, Takeda K, Takiguchi M, et al. Nitric oxide-induced apoptosis in pancreatic b cells is mediated by the endoplasmic reticulum stress pathway. Proc Nat Acad Sci. 2001;98:10845–50. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panaretakis T, Joza N, Modjtahedi N, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499–509. doi: 10.1038/cdd.2008.67. [DOI] [PubMed] [Google Scholar]
- 24.Molinari A, Calcabrini A, Meschini S, et al. Subcellular detection and localization of the drug transporter P-glycoprotein in cultured tumor cells. Curr Protein Pept Sci. 2002;3:653–70. doi: 10.2174/1389203023380413. [DOI] [PubMed] [Google Scholar]
- 25.Van Duyn Graham L, Sweetwyne MT, Pallero MA, et al. Intracellular calreticulin regulates multiple steps in fibrillar collagen expression, trafficking, and processing into the extracellular matrix. J Biol Chem. 2010;285:7067–78. doi: 10.1074/jbc.M109.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, et al. Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug. Nat Biotech. 2009;27:643–54. doi: 10.1038/nbt.1547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinetics of CRT translocation on cell surfaceelicited by nitric oxide. HT29 and HT29-dx cells were treated for 1hr with fresh medium (CTRL) or SNAP (100 μmol/l for 10,30 and 60 min., SN), then washed and stained for CRT. TotalCRT was detected in untreated cells after fixation andpermeabilization; CRT present on cell surface was evaluated innon-permeabilized cells. The micrographs are representative ofthree experiments with similar results.