

Abstract
Besides the well-understood DNA damage response via establishment of G2 checkpoint arrest, novel studies focus on the recovery from arrest by checkpoint override to monitor cell cycle re-entry. The aim of this study was to investigate the role of Chk1 in the recovery from G2 checkpoint arrest in HCT116 (human colorectal cancer) wt, p53–/– and p21–/– cell lines following H2O2 treatment. Firstly, DNA damage caused G2 checkpoint activation via Chk1. Secondly, overriding G2 checkpoint led to (i) mitotic slippage, cell cycle re-entry in G1 and subsequent G1 arrest associated with senescence or (ii) premature mitotic entry in the absence of p53/p21WAF1 causing mitotic catastrophe. We revealed subtle differences in the initial Chk1-involved G2 arrest with respect to p53/p21WAF1: absence of either protein led to late G2 arrest instead of the classic G2 arrest during checkpoint initiation, and this impacted the release back into the cell cycle. Thus, G2 arrest correlated with downstream senescence, but late G2 arrest led to mitotic catastrophe, although both cell cycle re-entries were linked to upstream Chk1 signalling. Chk1 knockdown deciphered that Chk1 defines long-term DNA damage responses causing cell cycle re-entry. We propose that recovery from oxidative DNA damage-induced G2 arrest requires Chk1. It works as cutting edge and navigates cells to senescence or mitotic catastrophe. The decision, however, seems to depend on p53/p21WAF1. The general relevance of Chk1 as an important determinant of recovery from G2 checkpoint arrest was verified in HT29 colorectal cancer cells.
Keywords: DNA damage, G2 checkpoint, recovery, checkpoint-kinase 1 (Chk1), senescence, mitotic catastrophe
Introduction
The development of cancer results from dysregulated proliferation or an incompetence of cells to undergo apoptotic cell death. Hence, an important issue emerging in drug discovery is to target anticancer treatments on cell cycle checkpoints that are responsible for the control of cell cycle phase progression or on apoptosis, eliminating defective cells [1]. Because damage to DNA might be the common underlying mechanism for the positive outcome of chemotherapy, reactive oxygen species (ROS)-generating anticancer drugs have raised clinical interest [2, 3]. In response to DNA damage, the G2 checkpoint is activated to halt cell cycle progression, preventing cells from entering mitosis. Its activation proceeds through maintenance of the Thr14/Tyr15 inhibitory phosphorylations on cdc2, realized by the protein kinases Wee1 and Myt1, respectively [4, 5].
ATR indirectly modulates the phosphorylation status of these sites by activating the downstream protein kinase Chk1 via phosphorylation on Ser317 and Ser345 [6–9]. Activated Chk1 phosphorylates the dual specificity phosphatase cdc25C on Ser216, thus creating a binding site for 14–3-3 proteins [10]. The 14–3-3/cdc25C protein complexes are sequestered in the cytoplasm, thereby preventing cdc25C from activating cdc2 through removal of the inhibitory phosphorylations. This results in the maintenance of the cdc2-cyclin B1 complex in its inactive state and blocks entry into mitosis [11]. Conceptually, besides checkpoint initiation, the delay comprises two additional phases: maintenance during repair and termination to allow cell cycle progression. As much information has been gained regarding the signalling pathways involved in establishing a G2 checkpoint arrest, novel studies focus on the question of how checkpoint signalling is maintained [12] and overcome to allow cell cycle re-entry [13, 14], especially if the drug is removed. p53 plays a critical role in maintaining G2 checkpoint arrest. At least one half of the tumours are p53-deficient, and some also show mutations or altered expressions of other components of the G2 checkpoint [15]. With this, induction of mitotic catastrophe as a result of checkpoint deficiency appears to be a desirable goal in cancer treatment [16]. In addition, permanently arresting tumour cell growth through the induction of senescence also seems to be an attractive treatment approach [17, 18].
Collectively, senescence [19–21] and mitotic catastrophe [22, 23] are two major effects desired in drug treatment, although many studies report a separate role of Chk1 in DNA damage response [24, 25]. This has encouraged us to link the single findings to a general model regulating oxidative DNA damage in colorectal cancer cells, firstly using HCT116 wt, p53–/– and p21–/– cells. We intended (i) to highlight recovery from arrest monitoring cell cycle re-entry and (ii) to find an underlying signalling pathway upstream of the long-term DNA damage responses. Secondly, we confirmed the general relevance of Chk1 as an important determinant of recovery from G2 checkpoint arrest also in the HT29 colorectal cancer cell line.
We have recently shown that H2O2-induced DNA damage establishes a G2/M arrest in HCT116 cells through epigenetic p21WAF1 regulation [26]. To obtain information on how checkpoint signalling is overcome to initiate cell cycle re-entry, we again used short-term, high bolus H2O2 exposure to efficiently damage DNA, a prerequisite for monitoring long-term DNA damage responses. In the present study, we observed different desired cell fates that are in line with long-term signalling in therapy following Chk1-involved checkpoint initiation, including senescence or mitotic catastrophe. The decision, however, seems to depend upon balance on p53/p21WAF1. Importantly, we did not consider the cellular outcomes as single events, but we found them linked to one upstream signalling as the first response: an activation of the Chk1 pathway. Moreover, Chk1 knockdown showed that the upstream Chk1 signalling seems to navigate cells to cell cycle re-entry in the G1 or M phases, which finally led to senescence or mitotic catastrophe.
Material and methods
Cell culture and treatment
Colorectal cancer cell lines HCT116 wt, HCT116 p53–/–, HCT116 p21–/– and HT29 were maintained in RPMI or DMEM (cell culture media) with 10% foetal bovine serum, penicillin (100 U/ml) and streptomycin (100 μg/ml) in a humidified 5% CO2 atmosphere at 37°C. Cells were treated with 30 mM H2O2 for 3 min., because a majority of studies have reported that H2O2 was added directly to the cells as a bolus [27, 28]. Therefore, cells were initially exposed to high H2O2 concentrations. Cells were collected after 1, 6, 24, 48 and 72 hrs following treatment. More details regarding cell lines are given in the Supporting Information.
Flow cytometric analysis of DNA content
One day before treatment, cells were seeded into Petri dishes (90 mm diameter) at a density of 1.2–2.0 × 106 cells per dish. For cell cycle analysis following Chk1 siRNA transfection, cells were seeded in 6-well plates at a density of 1.5–2.4 × 105 cells per well. After the indicated times, the supernatants were collected and combined with cells that were harvested by trypsin, washed twice with phosphate-buffered saline, fixed with 70% ethanol, treated with 1% RNase and finally stained with a hypotonic propidium iodide solution (50 μg/ml). Distribution of cell cycle phases with different DNA contents was determined using a flow cytometer (Calibur, Becton-Dickinson, CA, USA). Cells whose DNA was less intensively stained than that of G1 cells (Pre-G1 cells) in flow cytometric histograms were considered apoptotic cells. Analysis of cell cycle distribution and the percentage of cells in the Pre-G1, G1, S and G2/M phase of the cell cycle were determined using the software CellQuest Pro (Becton-Dickinson).
Western blotting
Proteins were prepared as described previously [26]. For Western blot analysis, we used antibodies to the following proteins: Chk1, phospho-cdc25CSer216 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase 3, cdc2, phospho-cdc2Thr14, phospho-cdc2Tyr15, cyclin D1, phospho-H3Ser10 (Cell Signaling, Danvers, MA, USA), phospho-Chk1Ser317 (Novus, Littleton, CO, USA), cdc25C (Acris, Herford, Germany), cyclin B1 (Novo Castra, Newcastle upon Tyne, UK), p53, p21WAF1 (Calbiochem, Gibbstown, NJ, USA), phospho-p53Ser15 (Abcam, Cambridge, UK), phospho-H2AXSer139 (γ-H2AX, Millipore, Bedford, MA, USA), poly (ADP-ribose) polymerase (PARP) (Alexis Biochemicals, Lausen, Switzerland) and β-actin (Sigma-Aldrich, Munich, Germany). As Chk1 was activated 1 hr after H2O2 treatment in HCT116 wt, p53–/– and p21–/– cells, and HT29 cells, the 1 hr time-point was used as an internal control for investigations regarding long-term DNA damage responses following Chk1 activation.
Fluorescence immunostaining analysis
The γ-H2AX and cyclin B1 subcellular localization was investigated in isogenic HCT116 cells treated with 30 mM H2O2 for 3 min. and further recovery for 1 hr or 24 hrs on slides. γ-H2AX was stained with anti-H2AXSer139 (Millipore), cyclin B1 with anti-cyclin B1 (Novo Castra) and the nucleus with 4,6-diamidino-2-phenylindole (DAPI). Slides were examined under a fluorescence microscope Axioplan2 imaging from ZEISS (Jena, Germany) using Isis V 3.4.0 software and appropriate filters.
Comet assay
To estimate DNA damage, we performed the CometAssay (Trevigene, Gaithersburg, MD, USA), according to the protocol of the supplier. Evaluation was performed with a fluorescence microscope (Axioplan2, ZEISS) equipped with appropriate filter sets. Images were acquired using Isis V 3.4.0 software. More details are given in the Supporting Information.
Senescence β-galactosidase staining
The histochemical detection of senescence-associated β-galactosidase activity at pH 6.0 was performed with the senescence β-galactosidase staining kit (Cell Signaling) according to the instructions of the manufacturer.
siRNA transfection
Chk1 knockdown was performed according to the manufacturer’s instructions (Santa Cruz Biotechnology) as described previously [29], reaching a p-Chk1Ser317 protein down-regulation of at least 60%. More details are given in the Supporting Information.
Results
H2O2 induces establishment of Chk1-involved G2 checkpoint arrest
Recently, we have shown that short-term, high bolus H2O2 exposure induces G2/M arrest in HCT116 wt and p53–/– cells through epigenetic regulation of the p21WAF1 promoter [26]. Due to the known essential function of Chk1 in DNA damage response, we further investigated Chk1 signalling following H2O2 treatment in HCT116 wt and p53–/– cells. Here, we show that H2O2 also activated the G2 checkpoint (Fig. 1A): Chk1 was phosphorylated in both cell lines 1 hr after treatment. Subsequently, there was an increase in the Chk1 target p-cdc25CSer216 after 24 hrs. Interestingly, the extent of G2 arrest after 24 hrs via p-cdc25CSer216 did correlate with inhibitory phosphorylation on cdc2 on Thr14, but not with that on Tyr15. Chk1 knockdown showed 36% and 14% reduced G2 checkpoint arrest at 24 hrs in wt and p53–/– cells, respectively (Fig. 1B and C). Consequently, the establishment of G2 checkpoint arrest in HCT116 wt and p53–/– cells involves Chk1.
Fig 1.
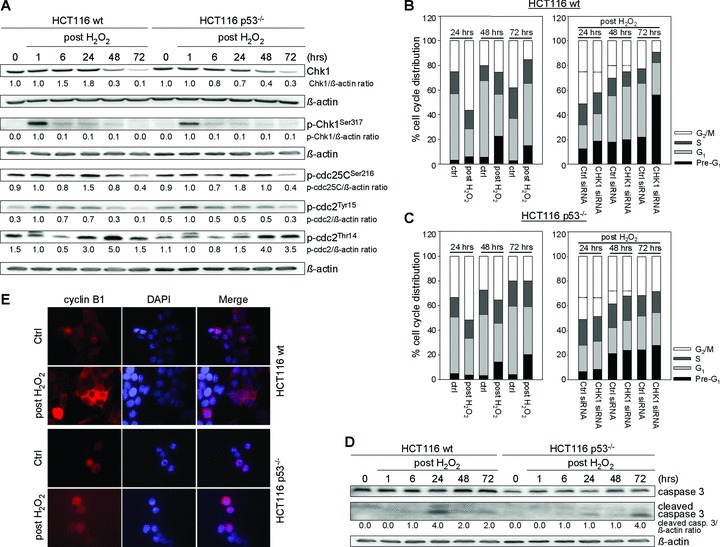
H2O2 treatment induces establishment and override of Chk1-involved G2 checkpoint arrest in HCT116 wt and p53–/– cells. (A) After H2O2 treatment (30 mM, 3 min.), G2 checkpoint arrest is established via Chk1 involvement reflected by the accumulation of active p-Chk1Ser317, inactive p-cdc25CSer216 and p-cdc2Thr14. Whole cell lysates were processed for Western blot analysis and probed with indicated antibodies. β-actin served as loading control. Fold expression changes are given below the blots. (B, C) G2 checkpoint override causes Chk1-dependent long-term DNA damage responses in HCT116 wt (B, G1 arrest) and p53–/– cells (C, apoptosis). FACS analyses were performed at 24, 48 and 72 hrs after treatment. Twenty-four hours after transfection with Chk1 siRNA, cells were treated with 30 mM H2O2 for 3 min. and grown for 24, 48 or 72 hrs. A control siRNA was used as a negative control for targeted siRNA transfection. Differentially gated cell populations were counted; their percentage in the total cell populations was calculated and presented in the diagram. Dashed lines contribute to cell cycle analysis without H2O2 treatment to mark H2O2-induced G2 arrest as well as reduced G2 arrest following Chk1 siRNA transfection. Data are means of three independent experiments. (D) Whole cell lysates were subjected to caspase 3 Western blot analysis. β-actin served as loading control. Fold expression changes are given below the blots. (E) Analysis of cyclin B1 localization 24 hrs after treatment revealed its dominant cytoplasmic localization in wt cells (G2 arrest) and its dominant nuclear localization in p53–/– cells (late G2 arrest). Cells were fixed, subsequently stained with anti-cyclin B1, and counterstained with DAPI.
G2 checkpoint override causes Chk1-dependent long-term DNA damage responses
To further study cell fates following H2O2-induced, Chk1-involved G2 checkpoint arrest, we elucidated long-term DNA damage signalling in HCT116 wt and p53–/– cells (48, 72 hrs). We found a regressive H2O2-induced G2 checkpoint arrest after 48 hrs in both cell lines, followed by G1 arrest exclusively in wt cells 72 hrs after treatment (Fig. 1B and C). In addition, wt cells underwent apoptosis starting at 24 hrs, although early apoptosis resistance in p53–/– cells could be overcome by prolongating the recovery phase up to 72 hrs (Fig. 1B–D). Thus, G2 checkpoint override caused both apoptosis and cell cycle re-entry, leading to G1 arrest, in wt cells or delayed apoptosis in p53–/– cells. Importantly, the results of Chk1 knockdown (Fig. 1B and C) were as follows: (i) 52% and 57% reduced G2 checkpoint arrest at 48 hrs in wt and p53–/– cells, respectively and (ii) abrogation of G1 arrest at 72 hrs in wt cells and 10% less cells in the G2/M phase at 72 hrs in p53–/– cells. Notably, a reduction in the cell numbers in the G1 or G2/M phases of HCT116 wt or p53–/– at 72 hrs following Chk1 siRNA transfection caused enhanced cell death reflected by increased Pre-G1 cell populations (Fig. 1B and C). In summary, the override of the G2 checkpoint arrest in HCT116 wt and p53–/– cells is Chk1 dependent.
Timing of Chk1-involved G2 arrest impacts on the release back into the cell cycle
To unravel subtle differences in the initial Chk1-involved cell cycle arrest with respect to the p53 status, we associated the subcellular localization of cyclin B1 with G2 arrest modality. Localization of cyclin B1 in the cytoplasm or the nucleus has been linked to G2 arrest or an arrest just at the onset of mitosis (late G2 arrest), respectively [30]. Immunofluorescence analysis of cyclin B1 in H2O2-treated wt cells confirmed that in 82% of cells cyclin B1 was located in the cytoplasm (cyt), indicating G2 arrest (Fig. 1E). In contrast, cyclin B1 was found in the nucleus in 90% of p53–/– cells, allowing for the conclusion that p53–/– cells accumulated in late G2 just before mitosis (Fig. 1E). To link the timing of G2 arrest to downstream cell cycle re-entry, we studied the expression of cell cycle regulators (Fig. 2). According to G2 arrest noticed in wt cells 24 hrs after H2O2 treatment, cyclin B1 and cdc2 were up-regulated. However, 72 hrs after H2O2 treatment, cells showed up-regulated G1 arrest marker cyclin D1, whereas the expression of key mitotic markers cyclin B1 and cdc25C was completely abolished, and cdc2 was markedly reduced. In addition, accumulation of p53, p-p53Ser15 and p21WAF1 especially after 48 and 72 hrs accounted for G1 arrest. Because we could not observe increased levels of mitosis-specific p-H3Ser10, we concluded that wt cells had not entered mitosis. These results suggest that after Chk1-involved G2 arrest, wt cells may have re-entered the cell cycle in G1 phase through mitotic slippage.
Fig 2.

H2O2 treatment alters expression of cell cycle regulatory proteins in HCT116 wt and p53–/– cells. Cells were treated with 30 mM H2O2 for 3 min. and further grown for 1 hr up to 72 hrs. Whole cell lysates were analysed by Western blot and probed with indicated antibodies. β-actin was used to control protein loading. Fold expression changes are given below the blots.
In contrast, p53–/– cells did not completely repress key mitotic regulators cyclin B1 and cdc25C at 72 hrs, indicating mitotic entry, which was confirmed by p-H3Ser10 immunoblotting (Fig. 2). Altogether, p53–/– cells recovered from Chk1-involved late G2 arrest by re-entering the cell cycle in the M phase, and damaged cells proceeded through mitosis to apoptosis, resulting in mitotic catastrophe. This idea is also supported by low levels of p21WAF1.
Role of p53 in Chk1-dependent long-term DNA damage responses
To link the observed long-term cellular responses to the extent of DNA damage, we placed emphasis on the question of how the damage sensor p53 deals with the initial signal over time. For this, H2O2-induced single strand DNA breaks (SSB) and double strand DNA breaks (DSB) were analysed in HCT116 wt and p53–/– cells using the comet assay. Cells treated with H2O2 showed clearly visible comet tails of DNA fragments indicative of broken DNA strands, independently of the p53 status as early as 1 and 24 hrs after H2O2 treatment (Fig. 3A). In addition, we observed remarkable nuclear γ-H2AX foci formation in both cell lines already after 1 hr [31, 32], (Fig. 3B). However, γ-H2AX immunoblotting showed accumulated DNA damage until 48 hrs in wt cells, whereas it decreased at 72 hrs, suggesting DNA repair (Fig. 3C). In accordance, we observed accumulation of p-p53Ser15 in wt cells in response to DNA damage [33], (Fig. 2).
Fig 3.
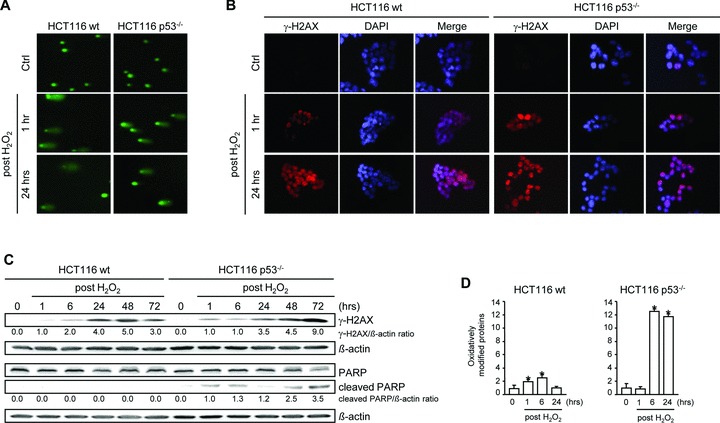
Role of p53 in Chk1-dependent long-term DNA damage responses. (A) Comet assay analysis of nuclear DNA in HCT116 wt and p53–/– cells revealed DNA strand breaks in cells exposed to 30 mM H2O2 for 3 min. and further grown for 1 hr and 24 hrs. (B) Formation of γ-H2AX foci in H2O2-treated wt and p53–/– cells. Cells were fixed and subsequently stained with anti-γ-H2AX and counterstained with DAPI. (C) Accumulation of γ-H2AX in H2O2-treated wt and p53–/– cells and PARP cleavage in p53–/– cells. Whole cell lysates were subjected to Western blot analysis. β-actin was used to control protein loading. Fold expression changes are given below the blots. (D) Effect of H2O2 treatment on protein modification in HCT116 wt and p53–/– cells. Cells were treated with 30 mM H2O2 for 3 min. Proteins having undergone oxidative modifications were detected after 1, 6 and 24 hrs after H2O2. Data are means ± S. D. of three independent experiments. *, P < 0.05 versus untreated cells.
In contrast, p53–/– cells accumulated DNA damage over time as indicated by processed expression of γ-H2AX (Fig. 3C). In this context, we also observed PARP cleavage (Fig. 3C), which reflects an inactivation of the enzyme, destroying its ability to respond to DNA strand breaks. Furthermore, we observed a dramatic increase in the amount of oxidative damage to proteins in p53–/– cells (Fig. 3D), which is in accordance with the observation that these cells failed to repair DNA damage. Overall, p53–/– cells undergo premature mitosis as their DNA is largely damaged, which drives these cells into mitotic catastrophe, whereas wt cells with repairable DNA damage slip through mitosis to finally arrest in G1.
Contribution of p21WAF1 to Chk1-dependent long-term DNA damage responses
To refer the G2 regulator p21WAF1 a role in recovery from G2 checkpoint arrest, we investigated H2O2-induced signalling in p21–/– cells. We also observed G2 checkpoint activation (Fig. 4A): Chk1 was activated 1 hr after treatment, cdc25C was phosphorylated at Ser216 at 24 hrs, and subsequently, pcdc2Tyr15 and pcdc2Thr14 accumulated at 24 hrs. Importantly, p21–/– cells did not arrest 48 hrs after H2O2 treatment (Fig. 4B), suggesting that p21WAF1 may retain cells in G2. Apoptosis induction could be observed from 24 to 72 hrs (Fig. 4A and B). Simultaneously, mitotic key regulators cdc2, cdc25C and cyclin B1 were not completely down-regulated at 72 hrs in p21–/– cells (Fig. 4C). Chk1 knockdown revealed abrogation of G2 checkpoint arrest at 24 hrs and 11% and 12% fewer cells in the G2/M phase along with increased Pre-G1 cell populations at 48 and 72 hrs (Fig. 4B).
Fig 4.
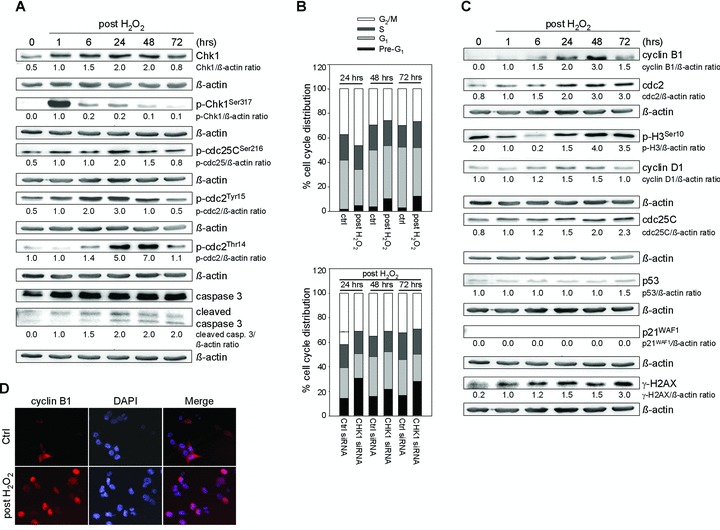
Analysis of establishment and override of Chk1-dependent G2 checkpoint arrest in HCT116 p21–/– cells. (A) H2O2 treatment (30 mM, 3 min.) induces establishment of G2 checkpoint arrest in p21–/– cells via the Chk1 pathway. H2O2 also induces apoptosis as indicated by the expression level of cleaved caspase 3. Whole cell lysates were subjected to Western blot analysis. β-actin was used to control protein loading. Fold expression changes are given below the blots. (B) G2 checkpoint override causes increased Chk1-dependent apoptosis as indicated by Pre-G1 cell population. FACS analyses were performed at 24, 48 and 72 hrs after treatment. Twenty-four hours after transfection with Chk1 siRNA, cells were treated with 30 mM H2O2 for 3 min. and grown for 24, 48 or 72 hrs. A control siRNA was used as a negative control for targeted siRNA transfection. Differentially gated cell populations were counted; their percentage in the total cell populations was calculated and presented in the diagram. The dashed line contributes to cell cycle analysis without H2O2 treatment to mark H2O2-induced G2 arrest as well as abrogation of G2 arrest following Chk1 siRNA transfection. Data are means of three independent experiments. (C) H2O2 treatment alters expression of cell cycle regulatory proteins in p21–/– cells. Whole cell lysates were subjected to Western blot analysis. β-actin was used to control protein loading. Fold expression changes are given below the blots. (D) Analysis of cyclin B1 localization 24 hrs after H2O2 revealed its dominant nuclear localization in p21–/– cells (late G2 arrest). Cells were fixed and subsequently stained with anti-cyclin B1 and counterstained with DAPI.
Investigating cyclin D1 and p-H3Ser10 levels, we confirmed that p21–/– cells did not enter G1 phase but mitotic prophase at 48 hrs and obviously stayed there, which might result in apoptotic mitosis (Fig. 4C). Immunofluorescence analysis of cyclin B1 revealed its dominant nuclear localization 24 hrs after H2O2 treatment (Fig. 4D, late G2 arrest). Collectively, these data demonstrate that premature mitotic entry cumulates in mitotic catastrophe on the basis of Chk1-dependent late G2 arrest in the presence of accumulated DNA damage as shown by γ-H2AX immunoblotting (Fig. 4C).
Chk1 navigates senescence and mitotic catastrophe during recovery from G2 checkpoint arrest
We found that wt cells establish a G1 arrest following Chk1-involved G2 arrest, which was associated with a senescent phenotype as shown by staining for β-galactosidase activity (Fig. 5A). In addition, wt cells showed the characteristic flattened and enlarged morphology in the majority of cells (Fig. 5A) together with prolonged expression of p53 and p21WAF1 (Fig. 2). In contrast, the absence of p53/p21WAF1 promotes premature mitosis after Chk1-involved or Chk1-dependent late G2 arrest, and therefore, cells entered apoptosis. According to this, microscopic examination revealed H2O2-induced formation of multinucleated p53–/– and p21–/– cells, which are characteristic of the mitotic catastrophic state (Fig. 5A). However, both cellular outcomes followed an upstream Chk1 activation, suggesting that mitotic catastrophe and senescence may be functionally linked with upstream Chk1 signalling. To mechanistically investigate if the upstream Chk1 pathway directs mitotic catastrophe and senescence, we performed knockdown experiments of Chk1 in all three isogenic cell lines. Our results show that Chk1 knockdown significantly decreased the expression of p-Chk1Ser317 in wt cells 1 hr after H2O2 treatment (Fig. 5B). Remarkably, the late expression (72 hrs) of G1 arrest-associated p-p53Ser15, p21WAF1 and cyclin D1 was significantly reduced following Chk1 knockdown (Fig. 5B). Consequently, as G1 arrest was shown to be Chk1 dependent (Fig. 1B), Chk1 knockdown reverses the senescent phenotype of wt cells (Fig. 5E). Interestingly, abrogation of G1 arrest at 72 hrs was accompanied by increased Pre-G1 cell population (Fig. 1B). However, as no increase in cleaved caspase 3 could be observed following Chk1 knockdown, we suggest caspase-independent cell death at 72 hrs (Fig. 5B). In summary, we speculate that it is Chk1 that determines downstream senescence in wt cells through activating p53, which induces p21WAF1.
Fig 5.
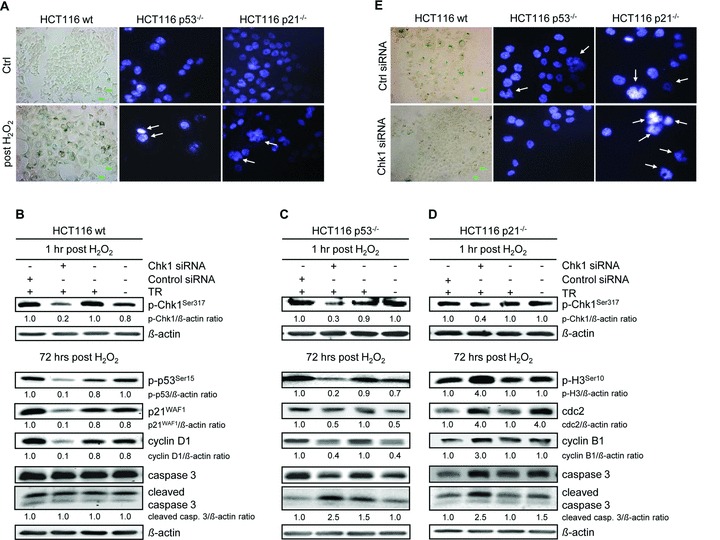
Chk1 directs downstream senescence and mitotic catastrophe in HCT116 wt and p53–/– or p21–/– cells after H2O2 treatment. (A) HCT116 wt cells became senescent after they were treated with 30 mM H2O2 for 3 min. and grown for 72 hrs. Cells were fixed and subsequently stained for β-galactosidase activity. Cells grew larger, assumed a flattened shape and expressed senescence-associated β-galactosidase (blue areas). In contrast, p53–/– and p21–/– cells went into mitotic catastrophe 72 hrs after treatment. Cells were fixed and stained with DAPI. Multinucleation is marked. (B)–(D) Twenty-four hours after transfection with Chk1 siRNA, cells were treated with H2O2 and further analysed 1 and 72 hrs after H2O2 treatment. (B) Chk1 navigates HCT116 wt cells to senescence. Down-regulation of p-Chk1Ser317 in wt cells causes time-delayed down-regulation of senescence-associated G1 arrest markers p-p53Ser15, p21WAF1 and cyclin D1 after 72 hrs. (C) Chk1 navigates HCT116 p53–/– cells to mitotic catastrophe. Down-regulation of p-Chk1Ser317 in p53–/– cells causes down-regulation of mitotic markers p-H3Ser10, cdc2 and cyclin B1 after 72 hrs. (D) Chk1 navigates HCT116 p21–/– cells to mitotic catastrophe. Down-regulation of p-Chk1Ser317 in p21–/– cells causes up-regulation of mitotic markers p-H3Ser10, cdc2 and cyclin B1 after 72 hrs. The transfection medium alone (TR) and a control siRNA were used as negative controls for targeted siRNA transfection. Whole cell lysates were subjected to Western blot analysis. Fold expression changes are given below the blots. β-actin was immunoblotted to control protein loading. (E) Chk1 knockdown affects cell morphology of H2O2-treated HCT116 cells. Twenty-four hours after transfection with Chk1 siRNA, cells were treated with 30 mM H2O2 for 3 min. and grown for 72 hrs. Cells were fixed and subsequently stained for β-galactosidase activity (blue areas: wt cells) or with DAPI (multinucleation is marked: p53–/–, p21–/– cells). A control siRNA was used as a negative control for targeted siRNA transfection.
Analogically, Chk1 knockdown also reduced the expression of p-Chk1Ser317 in p53–/– cells 1 hr after H2O2 treatment and caused time-delayed down-regulation of mitotic p-H3Ser10, cdc2 and cyclin B1 after 72 hrs (Fig. 5C). In this context, cell cycle analysis revealed fewer cells in the G2/M phase at 72 hrs following Chk1 siRNA transfection (Fig. 1C). In addition, inhibition of Chk1 by siRNA resulted in an increase in cell death as seen by 1.2-fold increase in Pre-G1 cell population (Fig. 1C) and by 2.5-fold increase in caspase 3 cleavage (Fig. 5C). In summary, we suppose that upstream Chk1 activation directs mitotic catastrophe in the absence of p53.
Interestingly, Chk1 knockdown reduced the expression of p-Chk1Ser317 in p21–/– cells, but significantly increased the expression of mitotic p-H3Ser10, cdc2, cyclin B1 and cleaved caspase 3 (Fig. 5D). Cell cycle analysis showed fewer cells in the G2/M phase, but 1.7-fold increased Pre-G1 cell population (Fig. 4B). Thus, we presume that upstream Chk1 activation protects cells lacking p21WAF1 from stronger mitotic catastrophe, which would result in increased cell death. Indeed, p21–/– cells showed increased multinucleation following Chk1 siRNA transfection, whereas Chk1 knockdown reverses the multinucleated phenotype of p53–/– cells (Fig. 5E).
Chk1 regulates long-term cell fate in recovery from G2 checkpoint arrest also in HT29 cells
In order to strengthen the conclusion that Chk1 directs recovery from G2 checkpoint arrest and therefore cell cycle re-entry, we performed key experiments also in the p53 mutant colorectal cancer cell line HT29. We found G2 checkpoint activation (Fig. 6A): Chk1 was activated 1 hr after treatment, and cdc25C was phosphorylated at Ser216 at 1, 24, 48 and 72 hrs. Consequently, we observed accumulation of pcdc2Tyr15 and pcdc2Thr14. Cell cycle analysis revealed the establishment of G2 checkpoint arrest at 24 hrs, which was regressive at 48 and 72 hrs, but was still maintained until 6 days (Fig. 6B). Thus, the override of the first G2 checkpoint arrest was not complete in HT29 cells, suggesting biphasic G2 checkpoint arrest. As Chk1 knockdown revealed no reduced G2 checkpoint arrest at 24 hrs (Fig. 6B), we presume that the establishment of the first G2 checkpoint arrest does not proceed under Chk1 participation despite Chk1 activation. Notably, Chk1 siRNA transfection showed 17%, 24% and 30% reduced G2 checkpoint arrest at later time-points (48, 72 hrs, 6 days), respectively (Fig. 6B). Thus, both incomplete recovery from first G2 checkpoint arrest and second, prolonged G2 checkpoint arrest are Chk1 dependent in HT29 cells. Focusing on recovery from first G2 checkpoint arrest, apoptosis induction (Fig. 6A and B) and multinucleation (Fig. 6C) could be observed at 72 hrs. In addition, mitotic key regulators cdc2, cyclin B1 and pH3Ser10 were not completely down-regulated at later time-points (Fig. 6A). Thus, we presume that DNA-damaged HT29 cells enter premature mitosis in recovery from first G2 checkpoint arrest, which cumulates in mitotic catastrophe. This process may also be directed through Chk1. Indeed, following Chk1 siRNA transfection, we observed that (i) multinucleation as a sign of mitotic catastrophe (Fig. 6C) and (ii) expression of mitotic markers (Fig. 6D) at 72 hrs were Chk1 dependent. In summary, in HT29 cells, Chk1 also directs recovery from G2 checkpoint arrest and therefore the long-term DNA damage response mitotic catastrophe.
Fig 6.
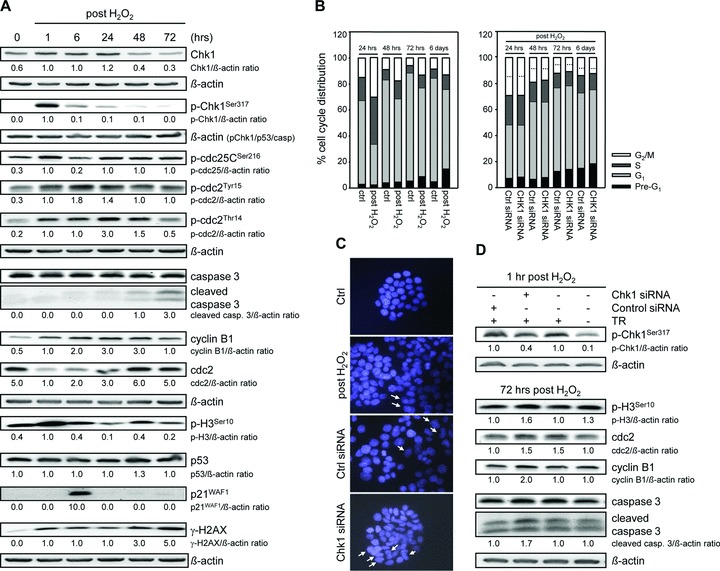
Chk1 regulates long-term DNA damage response in HT29 cells. (A) H2O2 treatment causes activation of the Chk1 pathway, apoptosis induction and altered expression of cell cycle regulatory proteins in HT29 cells. Whole cell lysates were processed for Western blot analysis and probed with indicated antibodies. β-actin served as loading control. Fold expression changes are given below the blots. (B) G2 checkpoint override causes Chk1-dependent apoptosis induction in HT29 cells. FACS analyses were performed at 24, 48, 72 hrs, and 6 days after treatment. Twenty-four hours after transfection with Chk1 siRNA, cells were treated with 30 mM H2O2 for 3 min. and grown until 6 days. A control siRNA was used as a negative control for targeted siRNA transfection. Differentially gated cell populations were counted; their percentage in the total cell populations was calculated and presented in the diagram. Dashed lines contribute to cell cycle analysis without H2O2 treatment to mark H2O2-induced G2 arrest as well as reduced G2 arrest following Chk1 siRNA transfection. Data are means of three independent experiments. (C) Chk1 knockdown affects cell morphology of H2O2-treated HT29 cells. Twenty-four hours after transfection with Chk1 siRNA, cells were treated with 30 mM H2O2 for 3 min. and grown for 72 hrs. Cells were fixed and subsequently stained with DAPI. Multinucleation is marked. A control siRNA was used as a negative control for targeted siRNA transfection. (D) Chk1 navigates HT29 cells to mitotic catastrophe. Down-regulation of p-Chk1Ser317 in HT29 cells causes up-regulation of mitotic markers p-H3Ser10, cdc2 and cyclin B1 after 72 hrs. The transfection medium alone (TR) and a control siRNA were used as negative controls for targeted siRNA transfection. Whole cell lysates were subjected to Western blot analysis. Fold expression changes are given below the blots. β-actin was immunoblotted to control protein loading.
Discussion
The cellular response to DNA damage involves both activation of signalling pathways, known as checkpoints, and repair of DNA lesions. The G2 checkpoint prevents mitotic entry in the presence of DNA damage. Conceptually, DNA damage checkpoint arrest can be divided into three phases: initiation, maintenance during repair and release back to the cell cycle. Although many studies concentrate on checkpoint initiation, there is increasing interest in the question of how the checkpoint is maintained and then switched off. Therefore, we focused on the role of Chk1 in recovery from DNA damage-induced G2 checkpoint arrest monitoring cell cycle re-entry. Our approach regarding short-term, high bolus H2O2 exposure allowed us, besides epigenetic investigations [26], to dissect and unravel cell responses, such as senescence and mitotic catastrophe, in recovery from G2 checkpoint arrest. At first glance similarly, exposure of HCT116 wt cells to the anticancer agents doxorubicin and irinotecan induced senescence, whereas absence of p53/p21WAF1 led to mitotic catastrophe [34, 35]. However, Chang et al. demonstrated the establishment of these treatment responses rather during drug exposure than after drug removal. One should keep in mind that long-term drug exposure without additional repeat could be equal to a catabolized substance, thus resembling the observation of recovery from checkpoint arrest. However, both studies did not link the observed treatment responses to Chk1 signalling. In addition, Macip et al. demonstrated a role of Chk1 in oxidative stress-induced G2 checkpoint arrest atypically associated with a senescent-like phenotype in p53-null human bladder cancer cells [36]. However, our data demonstrate a classic Chk1-mediated G1 arrest associated with senescence in recovery from G2 checkpoint arrest. Recently, it has been reported that inhibition of heat shock protein HSP90 by geldanamycin pushed irradiated p53 signalling-defective HCT116 cells into premature mitosis [37]. In this context, depletion of the major heat shock protein HSP72 led to defects in Chk1 activation and thus to induction of the senescence program [38]. However, here we show that both mitotic catastrophe and senescent signalling are Chk1 dependent.
Collectively, we could bridge single observations, such as senescence and mitotic catastrophe, in the process of DNA damage response in colorectal cancer cells. We linked them to functional upstream Chk1 activation, which suggests a comprehensive role of Chk1 also in recovery from checkpoint arrest. Moreover, we discovered a role of Chk1 as a ‘track builder’ for the long-term DNA damage responses, although levels of p53 and p21WAF1 finally decide about the fate of the cell. The absence of p21WAF1 had a marked effect on DNA damage response, because p21–/– cells fail to maintain G2 arrest. Therefore, the levels of p21WAF1 seem to arrange the duration of the G2/M arrest supported by p-Chk1Ser317. In addition, p53 plays a key role in DNA repair as shown by the reduced formation of γ-H2AX. According to Banáth et al.[39], the inability of the comet assay to detect significant DNA damage at 48 and 72 hrs after H2O2 (Fig. S1) suggests that many residual γ-H2AX foci may not be associated with a physical break.
Timing of the upstream Chk1-involved G2 arrest impacts on long-term DNA damage responses
The checkpoint effector kinase Chk1 mediates temporal cell cycle arrest and allows for successful completion of DNA repair before progressing into mitosis. Initially, in all three isogenic cell lines, H2O2 activated the upstream Chk1 pathway as indicated by phosphorylation of cdc25C, thus preventing dephosphorylation of cdc2, holding the cells in the G2 phase. Interestingly, cdc2 was maintained in its inactive state by Tyr15-phosphorylation, possibly by Wee1 kinase in p21–/– cells, whereas in wt and p53–/– cells, cdc2 was phosphorylated mainly at Thr14, indicating participation of the Myt1 pathway. In addition, we observed down-regulation of un-phosphorylated Chk1 in HCT116 wt and p53–/– cells at later time-points, whereas Chk1 protein levels retained nearly unchanged in p21–/– cells. Thus, we suggest a p21WAF1-dependent Chk1 down-regulation as it was reported from Gottifredi et al.[40]. Moreover, we observed a transient decrease of the levels in the Chk1 pathway proteins pcdc25CSer216, pcdc2Tyr15 and pcdc2Thr14 at 6 hrs in wt and p53–/– cells, and this was paralleled by increased p21WAF1 levels. As increased levels of Chk1 pathway proteins were found in p21–/– cells, we suggest that if G2 checkpoint activation proceeds via p21WAF1, it may negatively regulate not only Chk1 expression [40] but also Chk1 signalling. Indeed, initiating G2 checkpoint arrest seems to be exclusively mediated via p21WAF1 in wt cells [26], although we observed Chk1-dependent G2 checkpoint arrest at 6 hrs in p21–/– cells (data not shown).
Although Chk1 was activated rapidly in all three cell lines, the duration of activation was different. p21–/– cells showed sustained levels of key mitotic regulators cdc2 and cdc25C. As a consequence, cells re-entered the cell cycle in the M phase after Chk1-dependent late G2 arrest as indicated by nuclear cyclin B1 accumulation. p53–/– cells retained moderate levels of the key mitotic proteins until 72 hrs, and accumulated cyclin B1 in the nucleus, also indicating mitotic re-entry after Chk1-involved late G2 arrest. Obviously, mitotic catastrophe requires the activation of cdc2, and it is currently assumed that premature entry of active cdc2-cyclin B1 complex into the nucleus suffices to cause premature chromatin condensation and apoptosis [41, 42]. In accordance with this, our data imply that promotion of cyclin B1 nuclear localization after H2O2-induced DNA damage resulting in late G2 arrest is one of the mechanisms responsible for the recovery from G2 checkpoint arrest causing re-entry into mitosis. Here, we link this knowledge to upstream Chk1 activation, which directs cells into mitotic catastrophe following premature mitotic re-entry. In contrast, wt cells nearly completely repressed key mitotic regulators at later time-points and did not show p-H3Ser10 protein accumulation. This suggests that they did not enter mitosis, but re-entered and arrested in G1 in association with senescence, although this long-term DNA damage response was shown to be also Chk1 dependent. Collectively, the G2 arrest modality during checkpoint initiation had an impact on the subsequent release back into the cell cycle, whereas classic G2 arrest correlated with senescence, and late G2 arrest caused mitotic catastrophe. Thus, the timing of Chk1-involved G2 arrest determines recovery from oxidative DNA damage-induced G2 checkpoint arrest to allow cell cycle re-entry in the G1 or M phases, which finally led to senescence or mitotic catastrophe.
Recovery from oxidative DNA damage-induced G2 arrest requires Chk1
Performing Chk1 knockdown, we revealed that Chk1 defines long-term DNA damage responses. In the case of senescence, Chk1 is required for recovery and re-entry to the cell cycle following H2O2-induced G2 arrest, and the further establishment of the classic senescent arrest in G1 also seems to be dependent at least in part on an intact Chk1-dependent checkpoint, because p-Chk1Ser317 down-regulation markedly reduced G1-associated arrest markers p-p53Ser15, p21WAF1 and cyclin D1. Consequently, as G1 arrest is associated with senescence, cell morphology of wt cells could be restored following Chk1 siRNA transfection. Therefore, we presume that Chk1 may determine senescence in wt cells through transmitting the DNA damage signal to downstream G1 checkpoint by phosphorylating p53 at Ser15, which induces p21WAF1, thereby inhibiting cdk2-cyclin E complexes.
As p53 function is often lost in human cancers, it still needs to be clarified which role Chk1 plays in the scenario of long-term DNA damage responses following G2 checkpoint override in the absence of p53/p21WAF1 after upstream late G2 arrest. Here, we show that the upstream Chk1 activation protects p53–/– cells from increased cell death by navigating cells into mitotic catastrophe, as p-Chk1Ser317 down-regulation markedly reduced the expression of mitotic markers p-H3Ser10, cyclin B1 and cdc2. Consistently, Chk1 knockdown reversed the multinucleated phenotype of p53–/– cells, whereas cleavage of caspase 3 was increased. Interestingly, p-Chk1Ser317 down-regulation caused increased expression of p-H3Ser10, cyclin B1, cdc2 and caspase 3 in p21–/– cells. In accordance with this, p21–/– cells showed increased multinucleation following Chk1 knockdown. Therefore, upstream Chk1 activation seems to protect p21–/– cells from stronger mitotic catastrophe.
Collectively, the last few years have seen the development of a large variety of chemical inhibitors to the checkpoint kinase Chk1, reflecting its central function in DNA damage response. Because H2O2 mimics ROS-generating DNA-damaging anticancer drugs, it may serve as a basal model for chemotherapy studies. In fact, here we show that knockdown of Chk1 resulted in increased cell death in colorectal cancer cell lines HCT116 wt, p53–/– and p21–/–, and HT29 after H2O2. In this context, we have recently shown that the pro-oxidant, plant-derived drug thymoquinone induces ROS generation and DNA damage in HCT116 cells, which contributed to apoptosis [29]. In accordance, Chk1 knockdown in HCT116 p53–/– cells sensitized them to thymoquinone-induced apoptosis [29]. This supports the conclusion that Chk1 inhibition is a promising strategy to improve chemotherapy treatment. In addition, we could show that HCT116 wt, p53–/– and p21–/– cells, as well as the p53 mutant HT29 cell line, responded to the cytostatic drug 5-fluorouracil (5-FU) by activating Chk1 (Fig. S2A). γ-H2AX immunoblotting confirmed DNA damage caused by 5-FU. Apoptosis induction could be observed in each cell line (Fig. S2A). As DNA damage is the underlying mechanism of 5-FU’s impact, Chk1 knockdown caused increased apoptosis induction (Fig. S2B). Therefore, Chk1 inhibition is a powerful tool to improve also the efficiency of 5-FU.
Proposed model
Recently, we have investigated the establishment of G2 checkpoint arrest in HCT116 wt and p53–/– cells [26]. In this study, greater emphasis was placed on recovery from arrest by checkpoint override to monitor cell cycle re-entry. The results presented here may answer the question of how Chk1 regulates even long-term DNA damage response in colorectal cancer cells (Fig. 7A and B). H2O2-induced DNA damage causes upstream Chk1 stimulation, which activates three DNA damage signalling axes: (i) Chk1-involved G2 arrest, (ii) apoptosis induction and (iii) a classic senescent arrest in G1. Importantly, p53 and p21WAF1 are not necessary for the G2 arrest to occur, but p53 and p21WAF1 play an important role in (i) the blockage of the cell cycle either in G2 or late G2, (ii) the repair of DNA damage and (iii) the fate of long-term DNA damage responses in recovery from G2 checkpoint arrest, namely mitotic catastrophe or senescence. It is significant that Chk1 activation is the underlying signalling pathway upstream of the long-term DNA damage responses senescence (Fig. 7A) and mitotic catastrophe (Fig. 7B). Chk1 knockdown experiments have shown that re-entry of DNA-damaged cells into the cell cycle is Chk1 dependent, whereas the phase of re-entry is determined by the levels of p53/p21WAF1 as follows:
Fig 7.
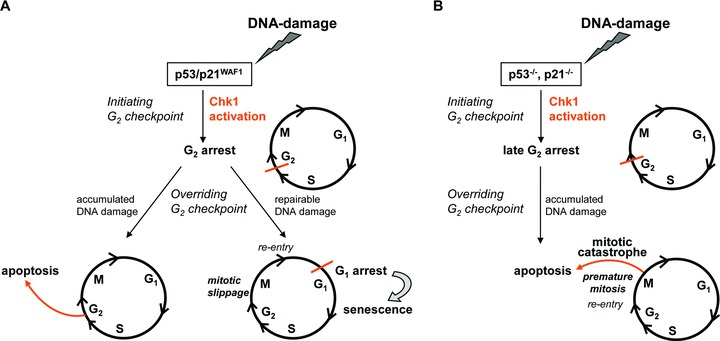
Proposed model of how Chk1 regulates oxidative DNA damage in HCT116 colorectal cancer cells. (A) H2O2-induced DNA damage activates Chk1 in wt cells, which prevents progression into mitosis via G2 arrest. Cells with non-repairable DNA damage go into apoptosis, whereas cells with repairable DNA damage slip through mitosis and arrest in G1 associated with senescence. (B) H2O2-induced DNA damage activates Chk1 in p53–/–/ p21–/– cells, preventing progression into mitosis via late G2 arrest. Because of a lack or low levels of p53/p21WAF1, respectively, cells undergo premature mitosis with their DNA largely unrepaired, which drives these cells into mitotic catastrophe. Importantly, Chk1 is required for recovery from G2 checkpoint arrest, leading to long-term senescence (A) and mitotic catastrophe (B).
Functional p53/p21WAF1
Firstly, Chk1 knockdown revealed that colorectal cancer cells with functional p53/p21WAF1 showed reduced establishment of G2 checkpoint arrest. Secondly, but most importantly, they failed to arrest in G1. Thus, in the presence of functional p53/p21WAF1, Chk1 directs re-entry of cells in G1, and they arrest there and go into senescence as long-term DNA damage response (Fig. 7A: HCT116 wt).
Absence of p53
In the absence of p53, Chk1 knockdown led to reduced establishment, but most notably to significant reduced override of G2 checkpoint arrest. Conceptually, in the absence of Chk1, cells may not re-enter the cell cycle in the M phase due to missing upstream Chk1-dependent G2 checkpoint override, but undergo cell cycle death from G2 phase. Thus, in p53–/– cells, Chk1 directs re-entry of cells in mitosis, which causes mitotic catastrophe as long-term DNA damage response (Fig. 7B: HCT116 p53–/–).
Absence of p21WAF1
In the absence of p21WAF1, G2 checkpoint arrest is realized by Chk1. Thus, Chk1-deficient cells fail to arrest cell cycle progression at first. Then, as a consequence, they go into premature mitosis. This drives DNA-damaged cells into significant mitotic catastrophe. Conceptually, as there is no upstream cell cycle arrest following Chk1 knockdown, cells cannot re-enter the cell cycle. Thus, in the absence of p21WAF1, Chk1 delays and reduces mitotic catastrophe as long-term DNA damage response by upstream G2 checkpoint arrest (Fig. 7B: HCT116 p21–/–).
Presence of mutated p53
To further strengthen our conclusion that Chk1 directs long-term DNA damage responses in colorectal cancer cells, we performed key experiments in the p53 mutant colorectal cancer cell line HT29 [43]. Our results show that G2 checkpoint arrest seems to be biphasic, whereas the first phase is not established via Chk1, but the second. Due to increased p21WAF1 level at 6 hrs, we propose that the establishment of the first G2 checkpoint arrest is p21WAF1 mediated. However, we observed an incomplete override of the first phase which led to G2 to M transit and cell death during mitosis in the process of mitotic catastrophe (Fig. 7B). Most importantly, both the incomplete G2 checkpoint override and the reduced, but prolonged, second G2 checkpoint arrest were Chk1 dependent. Thus, Chk1 protects HT29 cells from stronger mitotic catastrophe by means of downstream Chk1 dependent, prolonged G2 checkpoint arrest. In conclusion, Chk1 directs recovery from G2 checkpoint arrest also in HT29 cells, whereas (i) the establishment of the first G2 checkpoint arrest proceeds without involvement of Chk1, (ii) the override is not complete, but Chk1 dependent and (iii) the second, prolonged G2 checkpoint arrest is Chk1 dependent.
Acknowledgments
We thank Simone Staeck, Uta Schoenborn and Antje Schinlauer for their excellent technical assistance. We are grateful to Thomas Jonczyk-Weber and Bernd Wuesthoff for their crucial suggestions regarding manuscript preparation. This work was supported by a DAAD-fellowship to C.H.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Comet assay analysis of nuclear DNA in HCT116 wtand p53-/- cells at 48 and 72 hrs. Cells were exposed to30 mM H2O2 for 3 min. and further grown for 48 or 72 hrs.
Chk1 knockdown improves 5-FU treatment incolorectal cancer cell lines. (A) 5-FU (5 μM) inducesChk1-phosphorylation due to DNA damage as shown by increasedγ-H2AX expression in HCT116 wt, p53-/- andp21-/- cells, and HT29 cells. 5-FU also inducesapoptosis as indicated by the expression level of cleaved caspase3. Whole cell lysates were subjected to Western blot analysis. Foldexpression changes are given below the blots. β-actin wasimmunoblotted to control protein loading. (B) Twenty-fourhours after transfection with Chk1 siRNA, cells were treated with 5μM 5-FU for 48 or 72 hrs. The transfection medium alone (TR) anda control siRNA were used as negative controls for targeted siRNAtransfection. Whole cell lysates were subjected to Western blotanalysis. Fold expression changes are given below the blots.β-actin was immunoblotted to control protein loading.
References
- 1.Shapiro GI, Harper JW. Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest. 1999;104:1645–53. doi: 10.1172/JCI9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitt E, Paquet C, Beauchemin M, et al. DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J Zhejiang Univ Sci B. 2007;8:377–97. doi: 10.1631/jzus.2007.B0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci. 2007;96:2181–96. doi: 10.1002/jps.20874. [DOI] [PubMed] [Google Scholar]
- 4.Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6:877–82. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 5.Krek W, Nigg EA. Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 1991;10:3331–41. doi: 10.1002/j.1460-2075.1991.tb04897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 9.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 10.Peng CY, Graves PR, Thoma RS, et al. Mitotic and G2 checkpoint control: regulation of 14–3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–5. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 12.Latif C, den Elzen NR, O’Connell MJ. DNA damage checkpoint maintenance through sustained Chk1 activity. J Cell Sci. 2004;117:3489–98. doi: 10.1242/jcs.01204. [DOI] [PubMed] [Google Scholar]
- 13.den Elzen NR, O’Connell MJ. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 2004;23:908–18. doi: 10.1038/sj.emboj.7600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–45. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 16.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–13. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 17.Lleonart ME, Artero-Castro A, Kondoh H. Senescence induction; a possible cancer therapy. Mol Cancer. 2009 doi: 10.1186/1476-4598-8-3. ; in press: doi: 10.1186/1476-4598-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkisian CJ, Keister BA, Stairs DB, et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 19.Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–33. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 21.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–15. [PubMed] [Google Scholar]
- 22.Castedo M, Perfettini JL, Roumier T, et al. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 23.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–62. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 24.Kuntz K, O’Connell MJ. The G(2) DNA damage checkpoint: could this ancient regulator be the achilles heel of cancer. Cancer Biol Ther. 2009;8:1433–9. doi: 10.4161/cbt.8.15.9081. [DOI] [PubMed] [Google Scholar]
- 25.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habold C, Poehlmann A, Bajbouj K, et al. Trichostatin A causes p53 to switch oxidative-damaged colorectal cancer cells from cell cycle arrest into apoptosis. J Cell Mol Med. 2008;12:607–21. doi: 10.1111/j.1582-4934.2007.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbouti A, Doulias PT, Nousis L, et al. DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation of H(2)O(2) Free Radic Biol Med. 2002;33:691–702. doi: 10.1016/s0891-5849(02)00967-x. [DOI] [PubMed] [Google Scholar]
- 28.Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–6. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 29.Gali-Muhtasib H, Kuester D, Mawrin C, et al. Thymoquinone triggers inactivation of the stress response pathway sensor CHEK1 and contributes to apoptosis in colorectal cancer cells. Cancer Res. 2008;68:5609–18. doi: 10.1158/0008-5472.CAN-08-0884. [DOI] [PubMed] [Google Scholar]
- 30.Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol. 1998;141:875–85. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassing CH, Chua KF, Sekiguchi J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–10. doi: 10.1093/emboj/18.24.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang BD, Xuan Y, Broude EV, et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–18. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 35.Bhonde MR, Hanski ML, Notter M, et al. Equivalent effect of DNA damage-induced apoptotic cell death or long-term cell cycle arrest on colon carcinoma cell proliferation and tumour growth. Oncogene. 2006;25:165–75. doi: 10.1038/sj.onc.1209017. [DOI] [PubMed] [Google Scholar]
- 36.Macip S, Kosoy A, Lee SW, et al. Oxidative stress induces a prolonged but reversible arrest in p53-null cancer cells, involving a Chk1-dependent G2 checkpoint. Oncogene. 2006;25:6037–47. doi: 10.1038/sj.onc.1209629. [DOI] [PubMed] [Google Scholar]
- 37.Moran DM, Gawlak G, Jayaprakash MS, et al. Geldanamycin promotes premature mitotic entry and micronucleation in irradiated p53/p21 deficient colon carcinoma cells. Oncogene. 2008;27:5567–77. doi: 10.1038/onc.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabai VL, O’Callaghan-Sunol C, Meng L, et al. Triggering senescence programs suppresses Chk1 kinase and sensitizes cells to genotoxic stresses. Cancer Res. 2008;68:1834–42. doi: 10.1158/0008-5472.CAN-07-5656. [DOI] [PubMed] [Google Scholar]
- 39.Banáth JP, Macphail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res. 2004;64:7144–9. doi: 10.1158/0008-5472.CAN-04-1433. [DOI] [PubMed] [Google Scholar]
- 40.Gottifredi V, Karni-Schmidt O, Shieh SS, et al. P53 down-regulates CHK1 through p21 and the Retinoblastoma protein. Mol Cell Biol. 2001;21:1066–76. doi: 10.1128/MCB.21.4.1066-1076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol. 1998;141:875–85. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter LA, Cukier IH, Lee JM. Nuclear localization of cyclin B1 regulates DNA damage-induced apoptosis. Blood. 2003;101:1928–33. doi: 10.1182/blood-2002-04-1103. [DOI] [PubMed] [Google Scholar]
- 43.Rodrigues NR, Rowan A, Smith ME, et al. p53 mutations in colorectal cancer. Proc Natl Acad Sci USA. 1990;87:7555–9. doi: 10.1073/pnas.87.19.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comet assay analysis of nuclear DNA in HCT116 wtand p53-/- cells at 48 and 72 hrs. Cells were exposed to30 mM H2O2 for 3 min. and further grown for 48 or 72 hrs.
Chk1 knockdown improves 5-FU treatment incolorectal cancer cell lines. (A) 5-FU (5 μM) inducesChk1-phosphorylation due to DNA damage as shown by increasedγ-H2AX expression in HCT116 wt, p53-/- andp21-/- cells, and HT29 cells. 5-FU also inducesapoptosis as indicated by the expression level of cleaved caspase3. Whole cell lysates were subjected to Western blot analysis. Foldexpression changes are given below the blots. β-actin wasimmunoblotted to control protein loading. (B) Twenty-fourhours after transfection with Chk1 siRNA, cells were treated with 5μM 5-FU for 48 or 72 hrs. The transfection medium alone (TR) anda control siRNA were used as negative controls for targeted siRNAtransfection. Whole cell lysates were subjected to Western blotanalysis. Fold expression changes are given below the blots.β-actin was immunoblotted to control protein loading.