

Abstract
Radiotherapy is an important treatment modality against cancer resulting in apoptosis and inhibition of cell growth. Survivin is an important cancer biomarker conferring to tumour cells increased survival potential by inhibiting apoptosis. In the present study, we investigated the implication of breast cancer cells features, as hormone receptors and p53 status, in the radio-resistance of breast cancer cells and in the regulation of survivin’s expression by nuclear factor (NF)-κB and c-myc. Six breast cancer cell lines Michigan Cancer Foundation (MCF-7), MCF-7/Human Epidermal Growth Factor Receptor (HER)2, M. D. Anderson – Metastatic Breast (MDA-MB-231), SK-BR-3, BT-474 and Human Breast Lactating (HBL-100) were irradiated and cell viability as well as cell cycle distribution were evaluated by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and flow cytometry, respectively. Survivin mRNA and protein levels were evaluated by real time PCR and Western blot analysis. Survivin and HER2 gene knockdown was performed with siRNA technology and investigation of transcription factors binding to survivin and c-myc gene promoters was assessed by chromatin immunoprecipitation. Student’s t-test and F-statistics were used for statistical evaluation. Our results demonstrated that only HER2+ breast cancer cells up-regulated survivin upon irradiation, whereas HER2 knockdown in HER2+ cells led to survivin’s down-regulation. Survivin and especially HER2 knockdown abolished the observed G2/M cell cycle checkpoint and reduced the radio-resistance of HER2 overexpressing breast cancer cells. Additionally, HER2 was found to regulate survivin’s expression through NF-κB and c-myc transcription factors. This study revealed the significance of HER2 in the radio-resistance of HER2+ breast cancer cells through induction of transcription factors NF-κB and c-myc, leading to activation of survivin, a downstream target oncogene preventing apoptosis.
Keywords: survivin regulation, HER2/neu, NF-κβ, c-myc, irradiation, breast cancer
Introduction
Breast cancer is the most common malignancy among females in Western countries who have an overall lifetime risk of 10% of developing invasive breast cancer [1]. Radiation therapy is considered a common treatment modality for cancer patients, because of its ability to elicit complex cellular responses in various cancerous cells resulting in inhibition of cell growth or apoptosis [2–4]. However, the cellular and molecular mechanisms underlying the response of tumours to radiation therapy have not been thoroughly investigated.
Survivin, a member of the inhibitor of apoptosis (IAP) family, appears to be an important cancer therapeutic target and modulation of its expression and/or function may provide rational approaches for cancer therapeutics [5, 6]. IAPs aberrantly function as apoptosis regulators by directly binding and inhibiting caspases, prolonging thus cell viability and contributing possibly to cancer by facilitating mutations and promoting resistance to therapy [7]. Survivin has been found to be activated in most human cancers, although it is either undetectable or expressed at very low levels in differentiated adult tissues [8]. It is estimated that survivin is up-regulated in around 60–70% of breast cancers and its expression has been associated with significant reduction of apoptosis compared to survivin-negative tumours [8, 9]. Recently, it was suggested that in tumours with high endogenous survivin expression, failure to induce apoptosis may result in resistance to radiation therapy [10]. Moreover, down-regulation of survivin was shown to sensitize human tumour cells to ionizing radiation, as well as cytotoxic drugs, such as etoposide and cisplatin [11, 12].
Survivin’s aberrant expression in the majority of human tumours is unlikely to occur exclusively as a result of gene amplification, as amplicons of the survivin locus have not been observed. Most likely, regulation of survivin expression occurs at the level of transcription as a consequence of the de-regulation of upstream transcriptional regulators and transcription factors, as c-myc, E2F family members, b-catenin/TCF (T-cell factor), nuclear factor (NF)-κB and signal transducer and activator of transcription-3 (STAT3) [6, 13–16]. Additionally, different features of tumour cells, as HER2 and p53, have been shown to be responsible for survivin’s regulation, when cells are treated by irradiation or chemotherapy agents and furthermore survivin’s expression has been correlated with HER2/neu gene overexpression [17, 18]. HER receptors are involved in the control of diverse biological processes, such as proliferation, differentiation, migration and apoptosis and HER2 plays an important role in the progression of breast cancer tumorigenesis and metastasis [19]. Overexpression or gene amplification of HER2, which occurs in 20–30% of breast cancers, has been found to predict for a poor clinical outcome and resistance to therapy [20].
In the present study, we investigated the effect of irradiation on survivin’s expression in six different breast cancer cell lines, in association with their characteristics, such as HER2 and p53 status. We also investigated the role of NF-κB and c-myc transcription factors in regulating survivin’s expression upon exposure to ionizing radiation.
Materials and methods
Cell cultures and irradiation
The breast cancer cell lines that were used were : MCF-7 (p53 wild-type, ERα+, weakly positive for HER2), MCF-7/HER2 (p53 wild-type, ERα+, constitutively expressing HER2), MDA-MB-231 (p53 mutant, ERα–, weakly positive for HER2), HBL100 (p53 wild-type, ERα–, weakly positive for HER2), SK-BR-3 (p53 mutant, ERα–, HER2+) and BT-474 (p53 thermosensible mutant, acting as mutant when cells are grown at 37 °C, ERα+, HER2+). Cells in exponential growth were irradiated with 10 and 20 Gy doses of ionizing radiation, using a 6 MV photon linear accelerator with a dose rate of 4.90 Gy/min. at room temperature. Ten and 20 Gy doses were chosen based on our previous dose titration data [21]. Two separate experiments were carried out for each cell line studied. In order to ensure a uniform dose build-up and homogeneous irradiation cells were situated at the isocentre of two tangential opposed photon fields of 20 × 20 field size. For the accurate positioning and immobilization of the culture plates and flasks and to ensure reproducibility and accurate doses of irradiation, a homogeneous polymethyl methacrylate C5H8O2 phantom was constructed, as previously described by us [21].
Cell viability
Cell viability was determined with the MTT assay using the TACS MTT kit (R&D Systems, Minneapolis, MN, USA) according to manufacturer’s instructions. The MTT cell proliferation and viability assay is an in vitro assay for the measurement of cell proliferation or reduction of cell viability, when metabolic events lead to apoptosis or necrosis.
Quantification of survivin and HER2 mRNA expression
Total RNA was extracted using Trizol reagent according to manufacturer’s instructions (Gibco, Paisley, Scotland, UK). Preservation of 28S and 18S rRNA species was used to assess RNA integrity. Only samples with prominent 28S and 18S rRNA components were included in the study. Total RNA was reversed transcribed to cDNA using SuperScript First Strand synthesis (Invitrogen, Carlsbad, CA, USA) for RT-PCR using the oligo(dT) primer according to manufacturer’s instructions. Real-time RT-PCR for survivin, was performed with FastStart Universal Synergy Brands (SYBR) Green Master (ROX) (Roche, Mannheim, Germany) in a iCycler Optical Module (Bio-Rad, Hercules, CA, USA). Reactions were performed in triplicate using 2 μl of cDNA per reaction and the primers sequences used were for survivin: forward: 5′-CGAGGCTGGCTTCATCCA-3′; reverse: 5′-GCAACCGGACGAATGCTTT-3′, for HER2: forward: 5′-CTCGTTGGAAGAGGAACAGC-3′; reverse: 5′-CTGAATGGGTCGCTTTTG TT-3′ and for human porphobilinogen deaminase: forward: 5′-AGAGTGATTCGC GTGGGTACC-3′; reverse: 5′-GGCTCCGATGGTGAAGCC-3′.
Western blot analysis
Irradiated and non-irradiated cells were trypsinized, collected and centrifuged for 7 min. at 2000 rpm. Cell pellets were lysed using Nonidet P-40 lysis buffer containing 30 mM Tris (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Nonidet P-40 and a cocktail of protease inhibitors for 30 min. on ice, followed by centrifugation for 20 min. at 12,000 rpm. Supernatants were transferred in new tubes and stored at −80°C. Protein concentration was quantified using the Bio-Rad Bradford protein assay, with bovine serum albumin as standard. Equal amounts of protein were electrophoresed and separated by 10% SDS-PAGE (Bio-Rad) and transferred to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). The membrane was incubated with specific antibodies to survivin (sc-10811; Santa Cruz Biotechnology, Heidelberg, Germany) and HER2 (MS-441-S; Thermo Fisher Scientific, Loughborough, UK) (1:800) and signals were detected using anti-rabbit immunoglobulin IgG conjugated with horseradish peroxidase (1:5000). The chemiluminescence was resolved by an enhanced chemiluminescence ECL kit (Amersham, Milan, Italy). The results were normalized by anti-actin monoclonal antibody.
Chromatin immunoprecipitation (ChIP) assays for c-myc, mad1, max, p53, acetylated H3 and NF-κB
ChIP was performed with a ChIP assay kit (Upstate USA, Inc., Charlottesville, VA, USA) on irradiated and non-irradiated cells. Briefly, cells were cross-linked by incubating them in 1% (vol/vol) formaldehyde-containing medium for 10 min. at 37°C and then sonicated to make soluble chromatin with DNA fragments between 200 and 1000 bps. Samples of total chromatin were taken at this point to use as a positive control in the PCRs (input chromatin). The cell lysates were pre-cleared by incubation with G-Sepharose beads and then incubated with the polyclonal antibodies anti-max (sc-197), anti-c-myc (sc-764), anti- mad1 (sc-222), anti-p53 (sc-6243), anti NF-κB (sc-109) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-acetylated histone H3 (06–599) (Upstate Biotechnology, Lake Placid, NY, USA) overnight at 4°C. DNA–protein complexes were collected with G-Sepharose beads followed by several rounds of washing, eluted and reverse cross-linked. Following treatment with protease K (Sigma-Aldrich, Munich, Germany), the samples were extracted with phenol-chloroform and precipitated with ethanol. The recovered DNA was resuspended in Tris-HCl-EDTA (TE) buffer and used for the PCR amplification. The primer sequences used for survivin promoter were: forward: 5′-CTG CAC GCG TTC TTT GA-3′; reverse: 5′-GCG GTG GTC CTT GAG A-3′ and for c-myc promoter: forward: 5′-ACT TTG CAC TGG AAC TTA CAA CAC-3′; reverse: 5′-CGA AAA AAA TCC AGC GTC TAA G-3′. The PCR products were fractionated on 3% agarose gels and stained with ethidium bromide.
Knockdown of survivin and HER2 mRNA expression
Cells were transfected with dsRNA oligonucleotides for survivin (141244; Invitrogen) and HER2 (s65633; Ambion, Austin, TX, USA) using Lipofectamine 2000 reagent (Invitrogen) according to manufacturer’s instructions. Cells were cultured in Optimem medium (Invitrogen) in 96 well plates for 24 hrs in order to achieve a 50% confluence by the time of transfection.
Cell cycle distribution
The cell cycle distribution of irradiated and non-irradiated survivin knocked-down cells was determined by flow cytometry as previously described by us [21].
Statistical analysis
Statistical significance for mRNA expression analysis, was determined using Student’s t-test using a confidence level of 95% (P < 0.05), whereas the effect of siRNA against HER2 on HER2 and survivin mRNA expression, and of siRNA against survivin on survivin mRNA expression, was measured utilizing single factor anova. The metric used to quantify the investigated effects is the F-statistic, which shows the amount of overall variance that is induced due to the knockdown effect. In order to statistically evaluate the combined effects of HER2 or survivin knockdown, along with the irradiation process on cell viability, we employed 2-factor anova.
Results
Survivin mRNA and protein expression profiles after irradiation of breast cancer cells
We found that the HER2+ breast cancer cell lines (MCF-7/HER2, SK-BR-3 and BT-474) demonstrated significantly higher survivin mRNA expression levels compared to HER2– cell lines (MCF-7, MDA-MB-231 and HBL100) (data not shown). Protein levels were high in all non-irradiated cell lines, with the exception of MCF-7 cells (Fig. 1).
Fig 1.
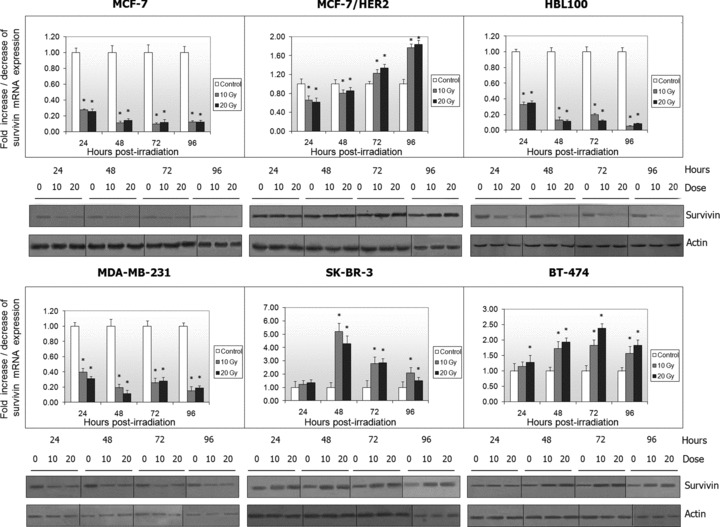
Survivin mRNA and protein expression was evaluated by real time PCR and Western blotting in non-irradiated and irradiated breast cancer cell lines respectively. Histograms represent the fold increase or decrease of survivin mRNA expression in irradiated cells, compared to non-irradiated cells. The normalized survivin mRNA expression of non-irradiated cells was set to 1 (*P < 0.05).
Cells were then irradiated with 10 and 20 Gy and survivin mRNA and protein levels were evaluated and compared with those of unirradiated cells. As shown in Figure 1, irradiated HER2– cells demonstrated significant down-regulation of survivin mRNA expression (P < 0.05), which was correlated with reduction in protein levels. On the contrary, irradiated HER2+ SK-BR-3 and BT-474 cells demonstrated an increase in both survivin mRNA and protein expression levels. MCF-7/HER2 cells were differentiated from the parental MCF-7 cell line exhibiting an initial down-regulation observed especially at the mRNA level, followed by up-regulation at 72 hrs after irradiation and afterwards (P < 0.05). Oestrogen and progesterone receptors, as well as p53 status were not correlated with survivin’s expression.
Implication of HER2 in survivin regulation
Because only HER2 seemed to be critical for survivin’s regulation upon irradiation, we evaluated HER2 protein expression levels in all irradiated breast cancer cell lines (Fig. 2A). Weakly expressing HER2 cells demonstrated low HER2 protein levels before and after irradiation, whereas HER2+ cells demonstrated high HER2 protein levels, which further increased after irradiation.
Fig 2.
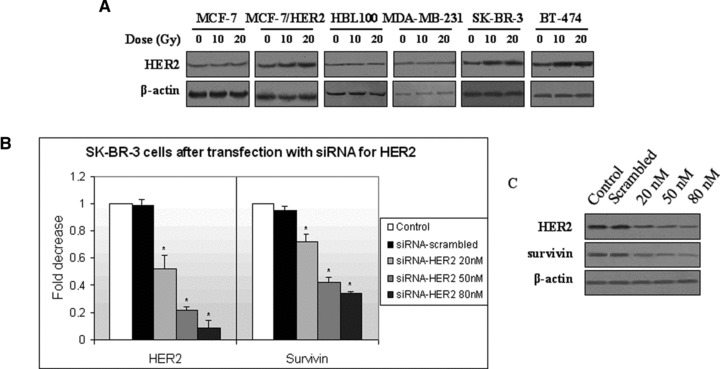
Effect of HER2 expression in the regulation of survivin mRNA expression levels. (A) A total of 40 μg of protein from non-irradiated, 10 and 20 Gy irradiated cells were subjected to Western blot analysis of HER2 protein. Analysis for β-actin was performed to show equal loading. For simplicity reasons, only 48 hr blots are represented here (72 hrs for MCF-7/HER2 cells). (B) Effect of HER2 knockdown on HER2 and survivin mRNA expression levels. (Bars: standard errors; *P < 0.05). (C) Effect of HER2 knockdown on HER2 and survivin proteins, as demonstrated by Western blot analysis.
We then proceeded by knocking down HER2 gene in HER2 overexpressing SK-BR-3 cells using siRNA against HER2 and transfection with liposomes. We observed a significant dose-dependent decrease in HER2 mRNA (Fig. 2B) (F= 205.4, P < 0.05) and protein (Fig. 2C) expression followed by an analogous decrease in survivin mRNA (Fig. 2B) (F= 105.5, P < 0.05) and protein levels (Fig. 2C) at 24 hrs after transfection.
HER2 and survivin knockdown render breast cancer cells less resistant to irradiation
We then silenced HER2 or survivin and subsequently irradiated HER2 overexpressing SK-BR-3 cells in order to study their contribution to cell viability and radio-resistance. HER2 knockdown caused a significant decrease (almost 60%, 24 hrs after transfection) in cell viability (F= 3620.3≫1, P < 0.05), which was higher than the decrease caused by irradiation (F= 222.7≫1, P < 0.05) (Fig. 3A). Univariate 2-factor anova indicated that both examined factors (silenced HER2 and irradiation) have a significant effect on cell viability, reducing it by 80% 24 hrs after irradiation (F= 3869.5≫1, P < 0.05). We then performed survivin knockdown using siRNA against survivin (Fig. 3B demonstrates the efficiency of the procedure) and found that it resulted in significant reduction in cell viability in non-irradiated and in 10 Gy irradiated SK-BR-3 cells, although significantly lower than the reduction observed after HER2 knockdown (Fig. 3C). anova yielded high F-statistics for both irradiation (F= 87.5) and survivin knockdown (F= 342.4), exhibiting a pronounced combined effect (F= 448.6) (P < 0.05).
Fig 3.
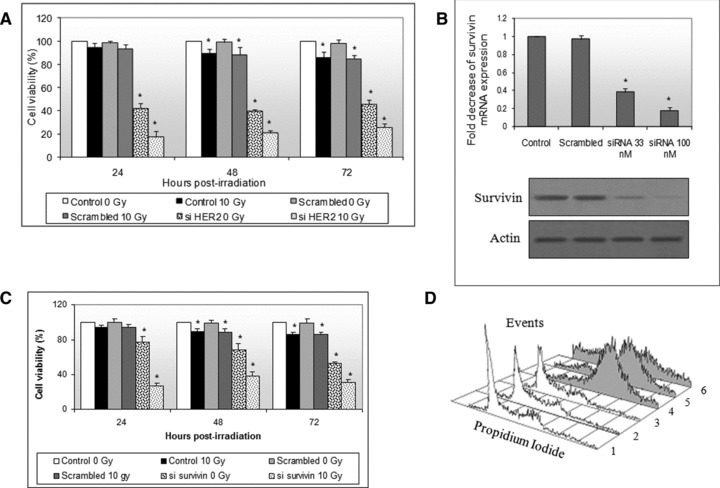
(A) SK-BR-3 cells were transfected with siRNA against HER2, irradiated twenty-four hours after transfection with a single 10 Gy dose and cell viability was assessed by MTT assay 24, 48 and 72 hrs after irradiation. Cell viability in non-transfected, non-irradiated cells was considered to be at 100% (Bars: standard errors; *P < 0.05). (B) SK-BR-3 cells were transfected with siRNA against survivin. Increasing doses of siRNA (33 and 100 nM) reduced survivin mRNA and protein levels. (C) SK-BR-3 cells were transfected with 33 nM of siRNA against survivin, irradiated with a single 10 Gy dose and measured for cell viability using MTT assay 24, 48 and 72 hrs after irradiation (Bars: standard errors; *P < 0.05). (D) SK-BR-3 cells were transfected with siRNA against survivin and subsequently irradiated with a single 10 Gy dose. Cell cycle distribution analysis was performed for non-irradiated/non-transfected (1), non-irradiated/scrambled (2), non-irradiated/survivin knocked down (3), irradiated/non-transfected (4), irradiated/scrambled (5) and irradiated/survivin knocked down (6) cells. For simplicity reasons, only 48 hrs after irradiation histograms are presented here.
It has been proposed that survivin knockdown alters cell cycle distribution. We evaluated SK-BR-3 cell cycle distribution modifications after survivin knockdown and subsequent irradiation. We found that survivin knockdown caused no cell cycle modifications in non-irradiated SK-BR-3 cells (Fig. 3D). However, 10 Gy irradiated, non-transfected and scrambled cells demonstrated a significant G2/M arrest (with a concomitant survivin’s up-regulation). After survivin’s knockdown and subsequent 10 Gy irradiation, cells demonstrated a smaller G2/M arrest and a great increase of the apoptotic subG0 fraction.
Study of the occupancy of survivin gene promoter by transcription factors
We then proceeded to investigate the regulation of survivin expression upon irradiation of breast cancer cells. The binding of transcription factors of the myc/mad/max complex, of p53 and the acetylation status of survivin gene promoter were assessed by ChIP assay. ChIP assay revealed no p53 binding (Fig. 4), whereas p53 was found to bind p21 promoter in all cell lines except BT-474 (data not shown).
Fig 4.
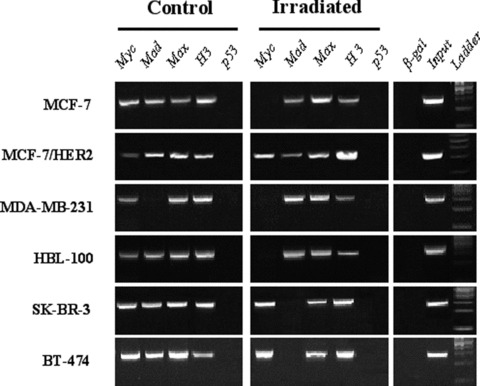
Occupancy of the survivin promoter in irradiated and non-irradiated breast cancer cell lines by ChIP analysis.
Regarding the myc/mad/max protein network, we found a strong binding of both c-myc and mad1 in non-irradiated cells. Max protein was found to bind to survivin promoter in all cells and under all conditions. Irradiated HER2– MCF-7, MDA-MB-231 and HBL100 cells (with down-regulated survivin) showed no c-myc binding, although mad1 binding was observed. In HER2 overexpressing SK-BR-3 and BT-474 irradiated cells (with up-regulated survivin), the mad1 binding became undetectable at 48 and 72 hrs, respectively. MCF-7/HER2 cells demonstrated signals for all precipitations for non-irradiated and irradiated cells up to 96 hrs, although a weaker signal was observed for c-myc in non-irradiated cells and for mad1 in irradiated cells (especially at 96 hrs after irradiation). Concerning histone acetylation status, we found that acetylated histone H3 signal was increased in irradiated HER2+ cell lines and was reduced in HER2– cells. As a control, chromatin was immunoprecipitated with β-galactosidase antibodies and no survivin promoter sequences could be detected, demonstrating the specificity of the procedure. Input chromatin was found positive for all samples tested.
Study of the occupancy of c-myc promoter by NF-κB, downstream target of HER2
The preferential binding of c-myc to survivin promoter leading to survivin’s up-regulation in irradiated HER2+ cells prompted us to study the possible molecular pathways connecting HER2 and survivin through myc. C-myc protein levels were found increased in irradiated HER2+ and decreased in irradiated HER2– cell lines (Fig. 5A). Similar results were obtained when c-myc mRNA levels were studied (data not shown). We, then, studied possible regulators of c-myc promoter, also constituting intracellular signalling molecules associated with HER2 activation. NF-κB protein which is activated downstream of HER2 and AKT and has been found to transcriptionally regulate c-myc, could be an attractive link between HER2 and survivin. The occupancy of c-myc promoter by NF-κB was therefore investigated by ChIP assay. We found that only HER2+ cells demonstrated strong signals, whereas NF-κB precipitates from non-irradiated breast cancer cells demonstrated low presence of c-myc promoter (Fig. 5B).
Fig 5.
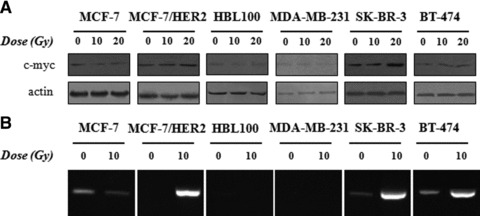
(A) Non-irradiated, 10 and 20 Gy irradiated cells were harvested at 24, 48, 72 and 96 hrs, lysed and 40 μg of total protein were subjected to Western blot analysis of c-myc protein. Analysis for β-actin was performed to show equal loading. For simplicity reasons, only 48 hr blots are represented here (72 hrs for MCF-7/HER2 cells). (B) Occupancy of the c-myc promoter in irradiated and non-irradiated breast cancer cells by NF-κB transcription factor by ChIP analysis.
Discussion
Because radiotherapy plays an important role in the management of breast cancer, understanding the molecular mechanisms employed by tumour cells to evade the inhibitory activity of radiotherapy is of major importance. In the present study, we investigated the response of six different irradiated breast cancer cell lines in survivin’s expression and regulation in relation to breast cancer cells characteristics, as oestrogen, progesterone, HER2/neu Receptors and p53 status. We found that HER2 overexpressing breast cancer cells demonstrated significantly higher survivin mRNA and protein expression levels compared to their HER2– counterparts, confirming previous reports proposing a significant relationship between survivin expression and HER2 overexpression in primary breast tumours [17, 18]. We also found that ionizing radiation caused further survivin and HER2 up-regulation in breast cancer cell lines overexpressing the receptor. Up-regulation of survivin has been associated with increased tumour aggressiveness and decreased patient survival rates, in addition to poor response to chemotherapy [18, 22]. It has also been shown that tetracycline-regulated short term induction of HER2 expression induced up-regulation of survivin in HER2– MCF-7 cells [23].
In order to further investigate HER2 implication in survivin’s regulation after irradiation of breast cancer cells, we proceeded by knocking down HER2 gene in the HER2+ SK-BR-3 cells and we observed significant down-regulation of survivin mRNA and protein levels suggesting a more extended role of HER2 in inhibition of apoptosis in breast cancer. Genetic and pharmacologic approaches to block HER2 expression have been previously assayed in HER2+ BT-474 cells and survivin was also found to be down-regulated [18].
We then proceeded by investigating the implication of HER2 and/or survivin in cell viability and radio-resistance of breast cancer cells, after knockdown of either HER2 or survivin and irradiation of the radio-resistant HER2+ SK-BR-3 cells. We found that survivin and especially HER2 knockdown reduced significantly the radio-resistance of SK-BR-3 cells. We have demonstrated in a previous report of ours the implication of HER2 in human Telomerase Reverse Transcriptase (hTERT) telomerase regulation and the effect of hTERT knockdown in the reduction of the radio-resistance of SK-BR-3 cells [24]. Taken together, the above findings suggest, for the first time to our knowledge, the implication of HER2 in the induction of at least two oncogenes, survivin and telomerase in breast cancer providing an explanation for the radio-resistance of HER2 overexpressing breast cancers.
We also observed that although irradiation caused a significant G2/M arrest (with a concomitant survivin up-regulation), survivin knockdown and subsequent irradiation of HER2 overexpressing SK-BR-3 cells, abolished the cells ability to arrest at the G2/M phase and increased the apoptotic subG0 fraction, suggesting that survivin’s induction upon irradiation induces a G2/M arrest necessary for SK-BR-3 cells to overcome the irradiation effects. It has been reported that survivin knockdown alters cell cycle distribution in colorectal and sarcoma cell lines, resulting in an increased G2/M fraction 24 hrs after transfection causing thus, at the time of irradiation, blockage in a more radio-sensitive stage of the cell cycle [25, 26].
In continuation, we were interested to investigate the activity of survivin’s promoter upon irradiation in breast cancer cells. Survivin’s promoter contains a response element for proteins of the myc/mad/max complex, whereas survivin’s overexpression in tumours has been linked to loss of wild-type p53 [6, 27]. It has been suggested that survivin may belong to a group of genes targeted for transcriptional repression by p53, providing thus a supplementary mechanism by which p53 exerts its important role in tumour suppression [27, 28]. We found that wild-type and also some mutant forms of p53 were able to up-regulate downstream targets, such as p21, but no direct binding was obvious on survivin promoter. p53 could be responsible for the observed survivin repression in HER2– MCF-7, HBL100 and MDA-MB-231 cells through an indirect mechanism, which could involve the p21/E2F pathway or another p53-induced transcriptional repressor of survivin [13]. This could also be applied to MCF-7/HER2 and SK-BR-3 cells, where, initially, survivin was not up-regulated. Transcriptional repression by p53 is complex and most repressed genes have no classical p53-binding sites in their promoter. The mechanism is generally ascribed to sequestration of components of the basal transcription machinery by p53 through protein–protein interactions in the absence of DNA binding [29, 30]. It has been reported that accumulation of wild-type p53 in human ovarian cancer cells induced survivin transcriptional repression which did not require direct sequence specific DNA binding to survivin promoter.
Regarding the activity of the proximal E-box of survivin promoter, we observed a switch of myc and mad proteins binding, at the time-points after irradiation when survivin expression was enhanced or reduced respectively. Max protein was found to bind to survivin promoter in all cells and under all conditions, which was expected, because max forms heterodimers with either myc or mad for the regulation of target genes. Here, we provide, for the first time to our knowledge, evidence of a more general involvement of c-myc and its network proteins in different breast cancer cells in association with their HER2 status and ionizing radiation. The implication of myc/mad/max protein network in the regulation of survivin’s expression had been previously demonstrated in SK-BR-3 cells [6]. Also, it has been proposed that transactivation by c-myc may involve the recruitment of histone acetyltransferases, which could result in increased histone acetylation of the promoter [30]. We found that acetylated histone H3 signal was increased in irradiated HER2+ cells and reduced in HER2– cells, proposing a supplementary mechanism by which c-myc may mediate transcriptional activation.
The up-regulation and preferential binding of c-myc to the survivin promoter leading to survivin’s up-regulation in irradiated HER2+ cells prompted us to investigate the possible molecular pathways connecting HER2 and survivin, through myc. HER2 is a receptor type tyrosine kinase with distinct intracellular signalling pathways, including mitogen-activated protein kinase (MAPK) and PI3K/AKT pathways [31]. The importance of the PI3K/AKT pathway in the cell survival signal of growth factor receptors has been previously demonstrated and because NF-κB can be activated downstream of AKT, it is likely that survivin’s expression could be regulated by this cascade. Additionally, c-myc has been found to be one of the molecular targets of NF-κB [32, 33]. In our study, we found that NF-κB was able to bind and activate c-myc only in irradiated HER2+ cells, whereas no activation was observed in HER2– cells. The importance of the HER2-PI3K/AKT pathway in the regulation of survivin’s expression has already been demonstrated in HER2+ SK-BR-3 cells treated with Herceptin [31]. This is the first time to our knowledge, that the importance of this pathway and of the downstream targets NF-κB and c-myc is demonstrated for different breast cancer cell lines upon irradiation.
In conclusion, our findings point towards the implication of HER2 and p53 in the regulation of survivin’s expression in irradiated breast cancer cells, with p53 being responsible for survivin’s repression through an indirect mechanism, although HER2 positivity enables breast cancer cells to overcome this repression by inducing survivin among other possible target genes. This study also supports the implication of the PI3K/AKT pathway downstream of HER2, through NF-κB and c-myc induction, leading to the activation of target oncogenes, as survivin. In order to verify our findings in vivo, we are already in the process of evaluating our findings in fresh cancer tissues derived from breast cancer patients.
Acknowledgments
The authors thank Dr. Ioannis Tsougos for his valuable assistance during irradiation of breast cancer cell lines.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Feuer EJ, Wun LM, Boring CC, et al. The lifetime risk of developing breast cancer. J Natl Cancer Inst. 1993;85:892–7. doi: 10.1093/jnci/85.11.892. [DOI] [PubMed] [Google Scholar]
- 2.Cuddihy AR, Bristow RG. The p53 protein family and radiation sensitivity: yes or no. Cancer Metastasis Rev. 2004;23:237–57. doi: 10.1023/B:CANC.0000031764.81141.e4. [DOI] [PubMed] [Google Scholar]
- 3.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–29. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 4.Liu W, Ip MM, Podgorsak MB, et al. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;115:43–50. doi: 10.1007/s10549-008-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 6.Cosgrave N, Hill AD, Young LS. Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J Mol Endocrinol. 2006;37:377–90. doi: 10.1677/jme.1.02118. [DOI] [PubMed] [Google Scholar]
- 7.Deveraux QL, Leo E, Stennicke HR, et al. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–51. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li QX, Zhao J, Liu JY, et al. Survivin stable knockdown by siRNA inhibits tumor cell growth and angiogenesis in breast and cervical cancers. Cancer Biol Ther. 2006;5:860–6. doi: 10.4161/cbt.5.7.2893. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka K, Iwamoto S, Gon G, et al. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin Cancer Res. 2000;6:127–34. [PubMed] [Google Scholar]
- 10.Jin XD, Gong L, Guo CL, et al. Survivin expressions in human hepatoma HepG2 cells exposed to ionizing radiation of different LET. Radiat Environ Biophys. 2008;47:399–404. doi: 10.1007/s00411-008-0165-0. [DOI] [PubMed] [Google Scholar]
- 11.Saito T, Hama S, Izumi H, et al. Centrosome amplification induced by survivin suppression enhances both chromosome instability and radiosensitivity in glioma cells. Br J Cancer. 2008;98:345–55. doi: 10.1038/sj.bjc.6604160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma H, Sen S, Lo Muzio L, et al. Antisense-mediated downregulation of anti-apoptotic proteins induces apoptosis and sensitizes head and neck squamous cell carcinoma cells to chemotherapy. Cancer Biol Ther. 2005;4:720–7. doi: 10.4161/cbt.4.7.1783. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, Saavedra HI, Holloway MP, et al. Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem. 2004;279:40511–20. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- 14.Kanda N, Seno H, Konda Y, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921–9. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 15.Kawakami H, Tomita M, Matsuda T, et al. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer. 2005;115:967–74. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]
- 16.Ma H, Nguyen C, Lee KS, et al. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–31. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 17.Chu JS, Shew JY, Huang CS. Immunohistochemical analysis of survivin expression in primary breast cancers. J Formos Med Assoc. 2004;103:925–31. [PubMed] [Google Scholar]
- 18.Xia W, Bisi J, Strum J, et al. Regulation of survivin by ErbB2 signaling: therapeutic implications for ErbB2-overexpressing breast cancers. Cancer Res. 2006;66:1640–7. doi: 10.1158/0008-5472.CAN-05-2000. [DOI] [PubMed] [Google Scholar]
- 19.Liang K, Lu Y, Jin W, et al. Sensitization of breast cancer cells to radiation by trastuzumab. Mol Cancer Ther. 2003;2:1113–20. [PubMed] [Google Scholar]
- 20.Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 21.Satra M, Tsougos I, Papanikolaou V, et al. Correlation between radiation-induced telomerase activity and human telomerase reverse transcriptase mRNA expression in HeLa cells. Int J Radiat Biol. 2006;82:401–9. doi: 10.1080/09553000600800090. [DOI] [PubMed] [Google Scholar]
- 22.Tamm I, Wang Y, Sausville E, et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–20. [PubMed] [Google Scholar]
- 23.Siddiqa A, Long LM, Li L, et al. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8:129. doi: 10.1186/1471-2407-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papanikolaou V, Iliopoulos D, Dimou I, et al. The involvement of HER2 and p53 status in the regulation of telomerase in irradiated breast cancer cells. Int J Oncol. 2009;35:1141–9. doi: 10.3892/ijo_00000430. [DOI] [PubMed] [Google Scholar]
- 25.Kappler M, Bache M, Bartel F, et al. Knockdown of survivin expression by small interfering RNA reduces the clonogenic survival of human sarcoma cell lines independently of p53. Cancer Gene Ther. 2004;11:186–93. doi: 10.1038/sj.cgt.7700677. [DOI] [PubMed] [Google Scholar]
- 26.Rodel F, Hoffmann J, Distel L, et al. Survivin as a radioresistance factor, and prognostic and therapeutic target for radiotherapy in rectal cancer. Cancer Res. 2005;65:4881–7. doi: 10.1158/0008-5472.CAN-04-3028. [DOI] [PubMed] [Google Scholar]
- 27.Mita AC, Mita MM, Nawrocki ST, et al. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 28.Johnson RA, Ince TA, Scotto KW. Transcriptional repression by p53 through direct binding to a novel DNA element. J Biol Chem. 2001;276:27716–20. doi: 10.1074/jbc.C100121200. [DOI] [PubMed] [Google Scholar]
- 29.Martin DW, Munoz RM, Subler MA, et al. p53 binds to the TATA-binding protein-TATA complex. J Biol Chem. 1993;268:13062–7. [PubMed] [Google Scholar]
- 30.Martinato F, Cesaroni M, Amati B, et al. Analysis of Myc-induced histone modifications on target chromatin. PLoS One. 2008;3:e3650. doi: 10.1371/journal.pone.0003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asanuma H, Torigoe T, Kamiguchi K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–25. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 32.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6:1059–70. doi: 10.1158/1541-7786.MCR-07-2088. [DOI] [PubMed] [Google Scholar]