

Abstract
Phosphoprotein enriched in diabetes/phosphoprotein enriched in astrocytes-15 kD (PED/PEA-15) is an anti-apoptotic protein whose expression is increased in several human cancers. In addition to apoptosis, PED/PEA-15 is involved in the regulation of other major cellular functions, including cell adhesion, migration, proliferation and glucose metabolism. To further understand the functions of this protein, we performed a yeast two-hybrid screening using PED/PEA-15 as a bait and identified the 67 kD high-affinity laminin receptor (67LR) as an interacting partner. 67 kD laminin receptor is a non-integrin cell-surface receptor for the extracellular matrix (ECM), derived from the dimerization of a 37 kD cytosolic precursor (37LRP). The 67LR is highly expressed in human cancers and widely recognized as a molecular marker of metastatic aggressiveness. The molecular interaction of PED/PEA-15 with 67LR was confirmed by pull-down experiments with recombinant His-tagged 37LRP on lysates of PED/PEA-15 transfected HEK-293 cells. Further, overexpressed or endogenous PED/PEA-15 was co-immunoprecipitated with 67LR in PED/PEA-15-transfected HEK-293 cells and in U-373 glioblastoma cells, respectively. PED/PEA-15 overexpression significantly increased 67LR-mediated HEK-293 cell adhesion and migration to laminin that, in turn, determined PED/PEA-15 phosphorylation both in Ser-104 and Ser-116, thus enabling cell proliferation and resistance to apoptosis. PED/PEA-15 ability to induce cell responses to ECM-derived signals through interaction with 67LR may be of crucial importance for tumour cell survival in a poor microenvironment, thus favouring the metastatic spread and colonization.
Keywords: PED/PEA-15, 67 laminin receptor, adhesion, apoptosis
Introduction
PED/PEA-15 is a 15-kD ubiquitously expressed protein involved in the regulation of fundamental cellular functions, including apoptosis, proliferation and glucose metabolism [1]. PED/PEA-15 consists of an N-terminal nuclear export sequence, a death effector domain (DED), an extracellular-regulated kinase (ERK) binding site and two phosphorylation sites (Ser-104 and Ser-116) at the C terminus. PED/PEA-15 lacks enzymatic function and serves mainly as a molecular adaptor. Because it contains a DED, PED/PEA-15 regulates apoptosis by competitively inhibiting the binding of DED-containing proteins to initiator caspases [2, 3]. Apart from its apoptosis-related effects, PED/PEA-15 is a potent modulator of mitogen-activated protein kinase (MAPK) signalling cascades [4]. Unphosphorylated PED/PEA-15 binds ERK1/2 and prevents its translocation into the nucleus, thereby reducing the ERK1/2-mediated transcriptional activity and inhibiting cell proliferation [5, 6]. Moreover, PED/PEA-15 is an endogenous substrate for protein kinase C (PKC), calcium/calmodulin-dependent protein kinase II (CaM kinase II), and Akt. PKC phosphorylates PED/PEA-15 at Ser-104 [7] and CaM kinase II or Akt at Ser-116 [8, 9]. Phosphorylation of PED/PEA-15 at Serine-104 prevents ERK1/2-binding and phosphorylation at Serine-116 enhances the binding to Fas-associated protein with death domain (FADD) and caspase 8, resulting in the inhibition of apoptosis [7, 8].
Because of its functional role in ERK signalling and apoptosis, increased PED/PEA-15 levels may affect tumourigenesis and cancer progression as well as sensitivity to anti-cancer agents. We demonstrated that overexpression of PED/PEA-15 in a transgenic mouse model increases the susceptibility to chemically induced skin cancer [9]. Increased PEA-15 levels inhibit apoptosis in non–small cell lung cancer (NSCLC) [10], B-cell chronic lymphocytic leukaemia [11] and thyroid cancer [12]. In astrocytic tumours, PED/PEA-15 suppresses apoptosis [13] and prevents glucose deprivation-induced cell death via the ERK pathway [14], suggesting that PED/PEA-15 promotes tumour cell survival in a poor microenvironment.
PED/PEA-15 also plays a role in the regulation of cell adhesion and migration; indeed, its binding to ERK1/2 regulates the affinity for fibronectin (FN) of integrin adhesion receptors [15]. In astrocytes, PEA-15 prevents cell migration through a PKC delta-dependent pathway [16]. It has been recently reported that PED interacts with Rac1 and regulates cell migration/invasion processes in human NSCLC cells [17].
To further understand the functions of PED/PEA-15 in cancer, we performed a yeast two-hybrid screening using PED/PEA-15 as a bait and identified the 67LR as an interacting partner.
67 kD laminin (LM) receptor was originally identified as a non-integrin cell surface receptor for LM, an extracellular matrix molecule [18]. Laminins, other glycoproteins, collagen IV and proteoglycans constitute a tight network to form the basement membrane. Laminin-1, a 900-kD glycoprotein, is the major component of basement membranes and contains many bioactive domains involved in binding both integrin and non-integrin receptors [19]. Interactions between the non-integrin 67LR and LM play a major role in mediating changes in the cellular environment that affect cell adhesion [20], neurite outgrowth [19] and tumour growth and metastasis [21].
67 kD LM receptor derives from homo- or hetero-dimerization of a 37LRP, by fatty acid acylation [22, 23]. 67 kD LM receptor binds LM through different binding domains [24, 25]. Laminin conformation changes upon binding 67LR, thus interacting more efficiently with integrins [26] and becoming more sensitive to the action of proteolytic enzymes [27], with the release of motility fragments [28]. 67 kD LM receptor is co-expressed and can physically interact with the α6-integrin chain [29].
67 kD LM receptor expression is increased in neoplastic cells as compared to their normal counterparts and directly correlates with an enhanced invasive and metastatic potential [30], mediated by high-affinity interactions between 67LR and LM [31]. Thus, 67LR overexpression is considered a molecular marker of metastatic aggressiveness in cancers of many tissues, including breast, lung, ovary, prostate and also in leukaemia and lymphomas [32-34]. For these reasons, the specific targeting of 67LR with small-interfering RNAs (siRNAs), blocking antibodies and Sindbis viral vectors confers anti-tumour effects [35, 36].
Herein, we show 67LR interaction with both overexpressed and endogenous PED/PEA-15 and investigate the functional consequences of this interaction in the regulation of cell adhesion, migration, proliferation and apoptosis.
Materials and methods
Materials
Media, sera and antibiotics for cell culture and the Lipofectamine reagent were purchased from Invitrogen (Paisley, UK). Mouse monoclonal anti-p-Akt and p-PKC antibodies and the polyclonal anti-Akt antibody were from Cell Signaling Technology (Danvers, MA, USA). Mouse monoclonal anti-p-Erk and PKC antibodies, rabbit polyclonal anti-Erk2 and CamKII antibodies, anti-α6-integrin chain antibody (G0H3) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal anti-p-CamKII antibody was from Upstate (Billerica, MA, USA). Rabbit anti-67LR anti-serum Ab711, directed against residues 263–283 of the receptor (24), was from Abcam (Cambridge, UK); it does not contain sodium azide and is not toxic for the cells, as determined by measuring cells viability after 1 and 6 hrs of incubation. Anti-α3 and -β1 integrin chain antibodies were from Chemicon (Temecula, CA, USA). PED/PEA-15 antibodies have been previously reported [37]. Antisera against phospho-Serine104 and phospho-Serine116 PED/PEA-15 were prepared in rabbits by PRIMM (Milan, Italy) and have been previously reported [8]. Rac inhibitor NSC23766 and ERK2 inhibitor PD98059 were from Calbiochem (San Diego, CA, USA). Laminin-1 was from Engelbreth-Holm-Swarm (EHS) mouse tumour (BD Biosciences, Bedford, MA, USA), vitronectin was from human plasma (Promega, Madison, WI, USA), FN was from human plasma (Roche, Mannheim, Germany), collagen was from rat tail (Sigma-Aldrich, St. Louis, MO, USA), YIGSR-amide peptide was from Polypeptide Group (Strasbourg, France). SDS-PAGE reagents were purchased from Bio-Rad (Hercules, CA, USA). Western blotting and ECL reagents were from Amersham (Little Chalfont, UK). Cell proliferation was evaluated by a MTS [3-(4,5-dimethylthiazole-2yl)-5-(3-carboxymethoxyphenyl)-2-(4sulfophenyl)-2H-tetrazolium, inner salt] and PES (phenazine ethosulfate) assay (CellTiter 96 AQueous One Solution Reagent) provided by Promega (Madison, WI, USA). Ninety-six-well microtitre plates were from Costar (Edison, NJ, USA). Apoptosis was analysed using the ApoAlert Caspase-3 Colorimetric Assay Kit (Clontech, CA, USA). All other reagents were from Sigma-Aldrich.
Transformation of yeast strains and β-galactosidase assay
Plasmid DNA transformations were performed with high-efficiency lithium acetate procedure [38]. Co-transformants were propagated on Trp+, Leu− plates and potential interacting clones selected in Trp−, Leu−, His−, Ade− media. After 4 days of incubation at 30°C, positive clones were further tested for β-galactosidase activity by liquid culture assays using the substrate o-nitrophenyl-β-d-galactopyranoside as described by Miller et al. [39]. Clones of interest were analysed by DNA sequencing and BLAST analysis.
Cell culture
Two ninety-three kidney embryonic cells, featuring very low levels of the anti-apoptotic protein PED/PEA-15 [40], and human U-373 glioma cells were grown in DMEM medium supplemented with 10% heat-inactivated FCS. Transfected cells were grown in DMEM supplemented with 10% FBS and antibiotics.
Transfection
PED/PEA-15 cDNA was cloned in a pcDNA3 vector with resistance to Geneticin, and the resulting plasmid was named PED/PEA-15-pcDNA3. A total of 5 χ 106 293 cells were transfected with 10 μg of PED/PEA-15-pcDNA3 or with the empty vector pcDNA3 by 60 μl of Lipofectamine, for 5 hrs at 37°C (5% CO2). Transfected cells, named PED-293 and V-293 respectively, were selected by Geneticin at 1.5 mg/ml for 15 days, pooled and cultured in the presence of 0.5 mg/ml Geneticin.
Flow cytometric analysis of surface molecules
Flow cytometric analysis of cell surface molecules was performed as previously described [33]. Briefly, cells were incubated for 2 hrs at 4°C with 20 μg/ml anti-67LR or isotype control antibodies. This step was followed by a second incubation for 1 hr at 4°C with an anti-rabbit fluorescein-conjugated antibody. Finally, cells were washed and analysed with a FACSCalibur Cytofluorometer using Cell Quest software (Becton & Dickinson, San Fernando, CA, USA). A total of 104 events for each sample were acquired in all cytofluorometric analyses.
Western blot
V-293 and PED-293 cells were lysed in lysis buffer [50 mM HEPES (pH 7.5), 150 mM NaCl, 4 mM EDTA, 10 mM Na4PO7, 2 mM Na3VO4, 100 mM NaF, 10% glycerol, 1% Triton X-100] containing protease and phosphatase inhibitors, for 120 min. at 4°C. Cell lysates were clarified at 5000 χ g for 15 min. and the protein content was measured by a colorimetric assay. Fifty micrograms of protein was electrophoresed on a 15% SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% non-fat dry milk, and probed with the primary and secondary antibodies; immunoreactive bands were detected by ECL according to the manufacturer’s instructions.
For the detection of phosphorylated PED/PEA-15, ERK1/2, Akt, CaMKII and PKC, 5χ105 V-293 and PED-293 cells were plated onto 35 mm plates previously coated for 24 hrs with LM (20 μg/ml in PBS). After the indicated times, cells were washed with PBS, lysed and subjected to Western blot, as described earlier.
PED/PEA-15 interaction with 67LR
To investigate the interaction of PED/PEA-15 with 67LR, 37LRP-His-tag fusion protein was generated. To this end, wild-type 37LRP cDNA (33) was cloned into the pTrcHis B expression vector (Invitrogen, San Diego, CA, USA) and expressed in TOP-10 bacteria (Invitrogen). According to the procedures specified by Invitrogen, transformed bacteria were lysed in a denaturing lysis buffer (20 mM sodium phosphate, 500 mM sodium chloride, pH 7.8) containing 6M guanidium and His-tagged 37LRP (His-37LRP) was bound to nickel-NTA agarose beads, through its His-tagged N-terminus, in the same denaturing buffer containing 8M urea. Beads were washed several times at pH 6.0 and 5.3, to dissociate contaminating proteins and finally, His-37LRP bound to agarose beads was more than 90% pure, as assessed by SDS-PAGE and Coomassie stain. Thus, His-37LRP conjugated beads were washed in 50 mmol/l Tris (pH 7.5)-0.1% Triton X-100, to remove urea, and resuspended in the same buffer.
Lysates from PED/PEA-15-transfected 293 cells (500 μg) were incubated in the presence of 50 μl of agarose-bound His-37LRP (approximately 2 μg) for 2 hrs at 4°C. Beads were washed four times with 50 mmol/l Tris (pH 7.5)-0.1% Triton X-100, and then resuspended in Laemmli buffer followed by boiling for 6 min. and centrifugation at 25,000 χ g for 3 min. Supernatants were analysed by SDS-PAGE and blotted with anti-PED-PEA/15 antibodies.
Co-immunoprecipitation
Cells were harvested into 1 ml of lysis buffer, containing protease and phosphatase inhibitors. Total cell lysates were pre-cleared with 20 μl protein A-Sepharose beads (∼50% slurry; Amersham Biosciences) for 30 min. at room temperature (RT). Five micrograms of anti-67LR, anti PED/PEA-15 or non-immune antibodies was added to 500 μg of cleared lysate, and incubated for 2 hrs at 4°C. Then, 20 μl of protein A-Sepharose beads was added and the lysates were incubated 30 min. at RT. Immunoprecipitates were washed six times in lysis buffer and Western blotting with anti-67LR antibodies was used to detect 67LR co-immunoprecipitated with PED/PEA-15 in V-293 and PED-293 cells. Viceversa, Western blotting with anti-PED/PEA-15 antibodies was used to detect PED/PEA-15 co-immunoprecipitated with 67LR in U-373 cells. Separately, 25 μg of total cell lysate was immunoblotted for 67LR and PED/PEA-15, as a loading control.
Rac1 pull-down assay
V-293 and PED-293 cells were starved for 24 hrs and then plated on LM-coated wells (10 μg/ml) or BSA-coated wells, as a negative control, for the indicated times. After a quick wash with ice-cold PBS, cells were lysed with GST-Fish buffer (50 mM Tris-HCl pH 7.4, 2 mM MgCl2, 1% NP-40, 10% glycerol, 100 mM NaCl, 1 mg/ml leupeptin, 1 mg/ml pepstatin, 1 mg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride and 2 mM dithiothreitol). After 10 min. at 4°C under agitation, cells were scraped and lysates were cleared by centrifugation in a pre-cooled rotor. Five hundred micrograms of total protein extract was mixed with 10 μg of GST-PAK-CRIB domain coupled to glutathione-sepharose beads (Upstate Biotechnology, Danvers, MA, USA) and incubated 30 min. at 4°C under agitation. Beads were then rinsed three times rapidly with 1 ml of ice-cold GST-Fish buffer. The amounts of total Rac and Rac-GTP were estimated by immunoblot against Rac1 (Upstate Biotechnology).
Adhesion assay
Ninety-six-well flat-bottom microtitre plates were coated with 10 μg/ml of LM, vitronectin, FN, YIGSR peptide or 1% heat-denatured BSA-PBS as a negative control, and incubated overnight at 4°C. The plates were then blocked 1 hr at room temperature with 1% heat-denatured BSA-PBS. Cells were harvested by 2 mM EDTA-PBS and resuspended in Ca2+/Mg2+-containing PBS.
A total of 105 cells were plated in each coated well and incubated for 1 hr at 37°C. Attached cells were fixed with 3% paraformaldehyde in PBS for 10 min. and then incubated with 2% methanol for 10 min. Cells were finally stained for 10 min. with 0.5% crystal violet in 20% methanol. Stain was eluted by 0.1M sodium citrate in 50% ethanol, pH 4.2 and the absorbance at 540 nm was measured by a spectrophotometer.
Cell proliferation assay
V-293 and PED-293 cells were serum-starved overnight using DMEM 0.1% BSA, plated at 104 cells/well in 96-well plates coated with 10 μg/ml LM in the same medium and incubated for 1, 24, 48 and 72 hrs or 7 days at 37°C, 5% CO2. The growth medium was then removed and 20 μl/well of CellTiter 96 AQueous One Solution Reagent was added. After incubation at 37°C for 4 hrs, the absorbance was determined by an ELISA reader (Bio-Rad) at a wavelength of 490 nm.
Cell apoptosis assay
V-293 and PED-293 cells were serum-starved overnight using DMEM 0.1% BSA. A total of 106 cells were harvested by 2 mM EDTA-PBS and plated in 100 mm dishes pre-coated with 10 μg/ml LM. After 24, 48 and 72 hrs at 37°C, 5% CO2, apoptosis was analysed using a colorimetric assay that measures the proteolytic cleavage of the chromophore p-nitroanilide (pNA) by caspase-3. Some experiments were performed in the presence of the anti-67LR antibody or non-immune control Ig (10 μg/ml).
Cell migration and invasion assays
Cell migration assays were performed in Boyden chambers using 8 μm pore size PVPF polycarbonate filters coated with 50 μg/ml collagen, as an adhesion substrate. V-293 and PED-293 cells (2 χ 105) were plated in the upper chamber in DMEM 0.1% BSA. DMEM 0.1% BSA alone or containing 50 μg/ml LM was added in the lower chamber. Cells were allowed to migrate for 4 hrs at 37°C, 5% CO2.
For the invasion assay, filters were coated with 70 μg/ml Matrigel™ (BD Biosciences, San Jose, CA, USA) and incubated for 3 hrs for gelling. A total of 2 χ 105 cells were allowed to migrate towards DMEM medium supplemented with 10% FCS or 0.1% BSA, as a control, for 18 hrs at 37°C, 5% CO2.
At the end of both experiments, cells on the lower surface of the filter were fixed in ethanol, stained with haematoxylin, and counted at 200χ magnification (10 random fields/filter).
In a separate set of experiments, transfected cells were pre-incubated for 1 hr at 37°C with a polyclonal antibody directed to 67LR (10 μg/ml) or non-immune antibodies as a negative control, or with different pharmacological Rac1 and ERK1/2 inhibitors (10 μM NSC23766, 25 μM PD98059) or diluents as a control.
Cell migration and invasion were expressed as percent increase over control.
Statistical analysis
Differences between groups were evaluated by the Student’s t-test using PRISM software (GraphPad, San Diego, CA, USA). P ≤ 0.05 was considered statistically significant.
Results
Isolation and identification of the 37 kD LM receptor precursor (37LRP) as a novel PED/PEA-15 interacting protein
To search for proteins specifically interacting with PED/PEA-15, a yeast two-hybrid system was established using the full-length PED/PEA-15 gene (pG-BKT7-ped/pea-15) as a bait to screen a human HeLa library (Clontech). Upon HIS3 selection, 45 β-galactosidase positive cDNA clones were detected. Based upon sequence analysis and BLAST searching, three of these clones were shown to match the 37LRP sequence [22]. Moreover, two clones coding for phospholipase A2 and three clones coding for FADD protein, both well-known as PED/PEA-15 interactors [1, 7, 8], were also found.
To verify the interaction of 67LR with PED/PEA-15, HEK-293 cells, considered virtually PED/PEA-15 negative [40], were stably transfected with the PED/PEA-15 cDNA (PED-293) and PED/PEA-15 expression was assessed by Western blot with specific antibodies (Fig. 1A). Then, pull-down assays were performed with His-tag-fused recombinant 37LRP coupled to Ni-NTA agarose (Invitrogen) on PED-293 cell lysates (Fig. 1B). Recombinant 37LRP bound PED/PEA-15 in PED-293 cell extracts. No PED/PEA-15 was detectable using His-tag bound agarose, indicating that purified 37LRP specifically binds PED/PEA-15.
Fig 1.
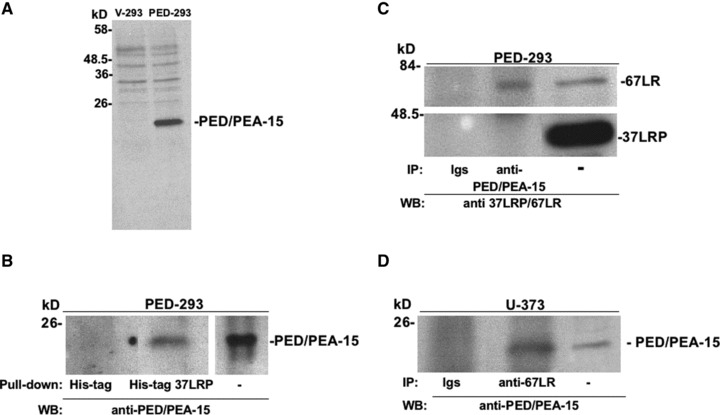
PED/PEA-15 interacts with 67LR. (A) HEK-293 cells were transfected with PED/PEA-15 cDNA (PED-293) or with the empty vector (V-293). Transfected cells were lysed and 50 μg of proteins was analysed by Western blot with PED/PEA-15-specific antibodies. (B) Lysates from PED-293 cells were incubated with agarose-bound recombinant His-tagged 37 kD laminin receptor precursor (His-tag 37LRP) or with agarose bound His-tag (His-tag), as a negative control. His-tag 37LRP conjugated beads were washed, resuspended in Laemmli sample buffer, boiled and supernatants were analysed by 15% SDS-PAGE and blotting with anti-PED/PEA-15 antibodies. Separately, 50 μg of total PED-293 lysate was immunoblotted. (C) PED-293 cell lysates were incubated with 5 μg of a polyclonal anti-PED/PEA-15 antibody or with non-immune immunoglobulins (Igs). The lysates were then immunoprecipitated with protein A Sepharose beads, washed and solubilized in Laemmli sample buffer. Western blotting with a polyclonal antibody, able to recognize both the 37LRP and the mature 67LR (anti-37LRP/67LR), was used to detect co-immunoprecipitated 67LR. Separately, 50 μg of total cell lysate was immunoblotted for 37LRP/67LR. (D) U-373 cell lysates were incubated with 5 μg of a polyclonal anti-67LR antibody. The lysates were then immunoprecipitated with protein A Sepharose beads, washed and solubilized in Laemmli sample buffer. Western blotting with a polyclonal anti-PED/PEA-15 antibody was used to detect co-immunoprecipitated PED/PEA-15. Separately, 25 μg of total cell lysate was immunoblotted for PED/PEA-15.
Dimerization of 37LRP [22,23] generates the mature form of the receptor, the 67LR. 37LRP is localized mostly in the cytoplasm acting as a precursor for 67LR, associated to ribosomes and in the nucleus, where it interacts with histones [30]. 67 kD LM receptor is localized in the plasma membrane, where it binds LM with high affinity, thus playing a crucial role in tumour invasion and metastasis [22–25, 30]. We sought to define by co-immunoprecipitation experiments the form of the receptor interacting with PED/PEA-15 in PED-293 cells, which express endogenous 37LRP and 67LR (Fig. 1C). Precipitation of lysates from PED-293 cells with anti-PED/PEA-15 antibodies, followed by blotting with antibodies able to recognize both 37LRP and 67LR, revealed that the mature membrane-bound form of the 67LR and not the 37LRP co-precipitates with PED/PEA-15, thus indicating a PED/PEA-15 interaction with 67LR. These results suggest that the PED/PEA-15 binding domain is exposed in purified recombinant 37LRP and in mature cellular 67LR whereas it is not available in cellular 37LR (Fig. 1B and C), as it occurs for LM, which binds bacterial recombinant 37LRP (our unpublished data and 41) while interacts only with cellular 67LR [18, 23, 42, 43].
Because PED/PEA-15 interaction with 67LR was observed in transfected cells, we assessed whether it occurred also in U-373 human glioblastoma cells, which endogenously express high levels of PED/PEA-15 [44] and 67LR. In fact, precipitation of lysates from U-373 cells with anti-67LR antibodies, followed by blotting with antibodies anti-PED/PEA-15, showed PED/PEA-15 co-precipitation (Fig. 1D).
Therefore, endogenous or overexpressed PED/PEA-15 specifically interacts with cellular 67LR.
PED/PEA-15 overexpression increases 67LR-mediated cell adhesion and migration to LM and extracellular matrix invasion
PED/PEA-15 is overexpressed in different human tumours, including breast and lung cancer, in which it determines refractoriness to anticancer therapy and resistance to apoptosis [14-17]. Therefore, we investigated the functional effects of PED/PEA-15 interaction with 67LR in PED/PEA-15 overexpressing cells.
Firstly, we sought to investigate whether PED/PEA-15 overexpression could influence 67LR-mediated cell functions, namely, cell adhesion and migration to LM. Thus, 293 cells transfected with the empty pcDNA3 vector (V-293) and 293 cells transfected with the PED/PEA-15-pcDNA3 plasmid (PED-293) were allowed to adhere to LM, FN and vitronectin (VN). PED-293 cell adhesion to LM was significantly increased, as compared to V-293; conversely, PED-293 cell adhesion to VN and FN was not affected (Fig. 2A), indicating that PED/PEA-15 overexpression in 293 cells specifically regulates the adhesion to LM. To investigate whether PED/PEA-15 overexpression could increase cell adhesion to LM through 67LR activation, V-293 and PED-293 cells were plated on wells coated with a peptide derived from the B1 chain of LM which specifically binds 67LR, the Tyr-Ile-Gly-Ser-Arg (YIGSR) pentapeptide [45]. Both V-293 and PED-293 cells were able to adhere to YIGSR coated wells; however, PED-293 cells showed a significantly increased binding to the YIGSR peptide, similar to that observed to full length LM (Fig. 2B).
Fig 2.
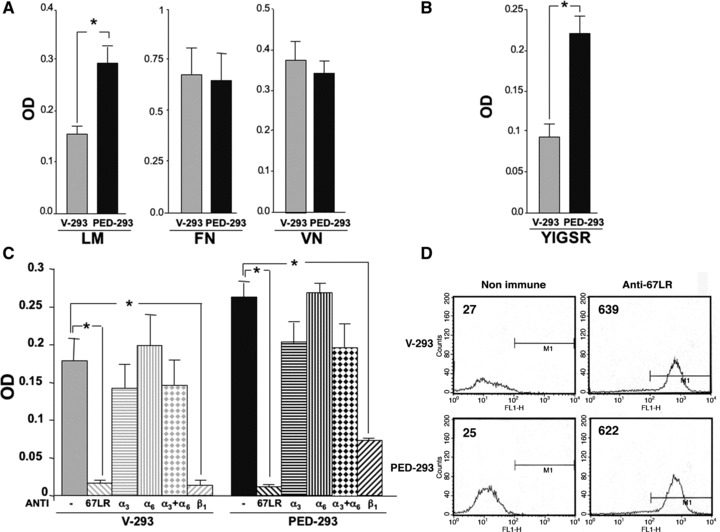
PED/PEA-15 overexpression increases 67LR-mediated cell adhesion to laminin. (A) V-293 () and PED-293 () cells were incubated for 1 hr on laminin (LM), vitronectin (VN), fibronectin (FN) or 1% heat-denatured BSA coated wells. The attached cells were fixed, permeabilized and stained with crystal violet. The stain was eluted and the absorbance at 540 nm (OD) was measured by a spectrophotometer. Cell adherence to BSA was subtracted from the reported values, representing the mean ± S.D. of six experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (B) V-293 () and PED-293 () cells were incubated for 1 hr on wells coated with an LM-derived peptide specifically binding to 67LR, the Tyr-Ile-Gly-Ser-Arg pentapeptide (YIGSR) or 1% heat-denatured BSA coated wells. The values represent the mean ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (C) V-293 () and PED-293 () cells were plated on LM-coated wells in the presence of 20 μg/ml of non-immune immunoglobulins (-), anti-67LR (,) and anti-α3 (,), anti-α6 (,), a mixture of anti-α3 and anti-α6 (,) and anti-β1 (,) and integrin polyclonal antibodies. The values represent the means ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (D) Flow cytometric analysis of cell surface 67LR expression was evaluated by incubating V-293 and PED-293 cells with a polyclonal anti-67LR antibody or an isotype control. Fluorescence intensity values are reported.
Then, to further discriminate 67LR and integrin contribution to the increased adhesion to LM of PED-293 cells, adhesion assays were performed in the presence of specific antibodies. V-293 and PED-293 cell treatment with anti-67LR antibodies showed that endogenous 67LR strongly contributes to V-293 cell adhesion to LM, even in the absence of PED/PEA-15, and confirmed that increased PED-293 cell adhesion to LM is mainly mediated by the 67LR (Fig. 2C). Accordingly, antibodies directed to the LM-binding site of α3 and α6 chains, the major LM-binding integrin subunits, did not affect significantly V-293 and PED-293 cell adhesion. Surprisingly, antibodies directed against the signal transducing β1-integrin chain, almost completely abolished the adhesion to LM of both V-293 and PED-293 cells, similarly to anti-67LR antibodies. These results suggest that, even integrins are not directly involved in the increased binding of PED-293 cells to LM, anyhow they contribute to generate a downstream signal, likely because 67LR is associated to the α6 β1-integrin on the cell membrane [29]. We thus hypothesize that 67LR mediates 293 cell binding to LM, but adhesion signals are mediated by its lateral association to β1-integrins.
We finally assessed whether PED/PEA-15 overexpression regulated 293 cell adhesion to LM by increasing 67LR expression. Cytofluorimetric analysis showed that V-293 and 293-PED cells expressed on their surface comparable levels of 67LR (Fig. 2D).
Then, the effect PED/PEA-15 overexpression on 67LR-dependent cell migration to LM was examined. V-293 and PED-293 cells were allowed to migrate towards LM in Boyden chambers.
PED/PEA-15 overexpression increased cell migration towards LM. Anti-67LR antibodies completely impaired PED-293 cell migration to LM; anti-67LR antibodies also impaired V-293 cell migration to LM, likely by blocking the endogenous 67LR, as in adhesion assays (Fig. 3A).
Fig 3.
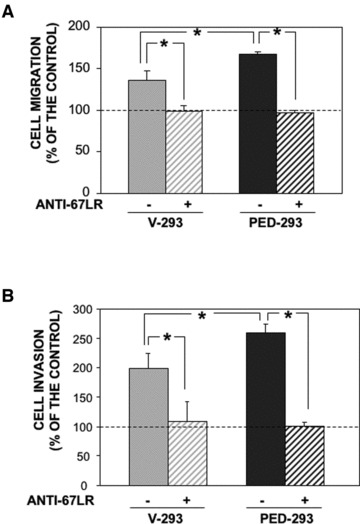
PED/PEA-15 overexpression increases 67LR-mediated migration to LM and matrigel invasion. (A) V-293 () and PED-293 () cells were pre-incubated with non-immune Ig (-) or a polyclonal anti-67LR antibody (+), plated in Boyden chambers and allowed to migrate towards 50 μg/ml LM on filters coated with 50 μg/ml collagen. Hundred per cent values represent cell migration in the absence of chemoattractants. The values are the mean ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (B) V-293 () and PED-293 () cells were pre-incubated with non-immune Ig (-) or a polyclonal anti-67LR antibody, plated in Boyden chambers and allowed to invade matrigel™. The values are the mean ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test.
Further, V-293 and PED-293 cells were plated on Matrigel, whose main component is LM, and allowed to migrate towards a generic chemoattractant, such as foetal bovine serum. Also in this case, PED/PEA-15 overexpression increased cell ability to invade Matrigel and anti-67LR antibodies abolished the observed increase (Fig. 3B).
These results suggest that PED/PEA-15 overexpression increases cell adhesion to LM by interacting with the 67LR and that 67LR requires β1-integrin partnership for an efficient intracellular signalling. Moreover, PED/PEA-15 overexpression, increases migration to LM and extracellular matrix invasion specifically via 67LR.
Adhesion to LM affects cell signalling in PED/PEA-15 overexpressing cells
Therefore, we investigated LM effects on the cell signalling in PED/PEA-15 overexpressing cells. PED/PEA-15 activates ERK/MAP Kinases through a Ras-dependent pathway [46]. It has been recently reported that, in an NSCLC cell line, PED/PEA-15 overexpression promotes the activation of Rac1, a member of the mammalian Rho GTPase protein family, in response to growth factor stimulation; in turn, active Rac1 increases ERK1/2 phosphorylation [17, 47].
Control V-293 and PED-293 cells were plated on LM; then, ERK1/2 and Rac1 activation were evaluated by Western blot analysis and by pull-down assay, respectively. PED/PEA-15 overexpressing cells, following LM stimulation, showed increased ERK1/2 activation, as compared to control V-293 cells (Fig. 4A, upper panel). Cell adhesion to LM also promoted Rac1 activation in PED/PEA-15 overexpressing cells, as compared to vector transfected cells (Fig. 4B, upper panel). Both ERK1/2 and Rac1 activation were specifically induced by the LM stimulus, as shown by plating PED-293 cells on uncoated or LM coated wells (Fig. 4A and B, lower panels).
Fig 4.
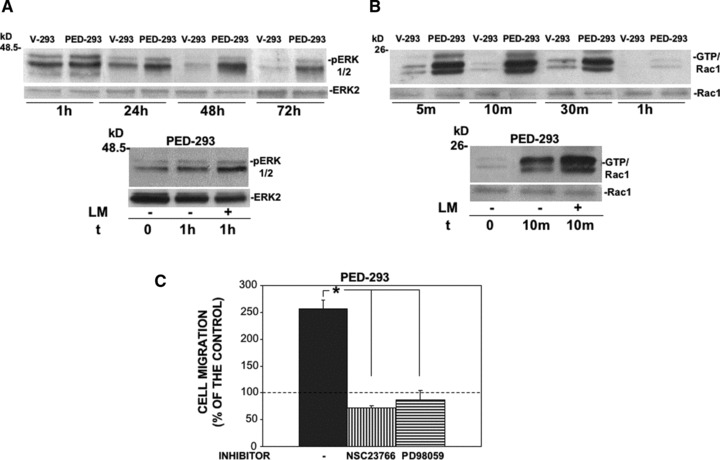
Cell adhesion to LM in PED/PEA-15 overexpressing cells activates ERK1/2 and Rac-1. (A) V-293 and PED-293 cells were serum-starved and plated on LM (upper panel); PED-293 cells were serum-starved and plated on BSA (-) or LM (+) coated wells (lower panel). The cells were harvested at the indicated times and lysed for Western blot analysis with anti-phospho-ERKs and anti-ERK 2 (as a loading control) antibodies. (B) V-293 and PED-293 cells were serum-starved and plated as in (A). The cells were harvested at the indicated times and lysed for Rac1-GTP pull down assay. The amounts of total Rac1 and Rac1-GTP were estimated by immunoblotting with Rac1. (C) PED-293 cells were pre-incubated with buffer (-), Rac1 (NSC23766) or ERK1/2 (PD98059) inhibitors, plated in Boyden chambers and allowed to migrate towards LM. Hundred per cent values represent cell migration in the absence of chemoattractants. The values are the mean ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test.
These results show that LM exerts its downstream effects in PED/PEA-15 overexpressing cells by activating Rac1 and ERK1/2. Because both Rac1 and ERK1/2 activation can mediate cell migration [47, 48], their involvement in the migration of PED/PEA-15 overexpressing cells towards LM was evaluated. The pharmacological inhibition of Rac1 and ERK1/2 by NSC23766 and PD98059, respectively, impaired PED-293 cell migration towards LM (Fig. 4B), demonstrating their involvement in LM-induced PED-293 cell migration.
67LR-mediated cell adhesion to LM regulates proliferation and apoptosis in PED/PEA-15 overexpressing cells by modulating PED/PEA-15 phosphorylation status
We then investigated whether 67LR can modulate PED/PEA-15 effects on cell proliferation and apoptosis. Firstly, we analysed V-293 and PED-293 cell proliferation after adhesion to LM by a colorimetric assay (Fig. 5A). As expected, LM induced a strong proliferative response in V-293 cells, whereas PED-293 cell proliferation was inhibited. However, after 24 hrs, PED-293 cells recovered their proliferative capacity, reaching control cells at 7 days. V-293 and PED-293 cell number, after adhesion to LM, was assessed at the indicated times and confirmed the results of the colorimetric assay (Fig. 5B).
Fig 5.
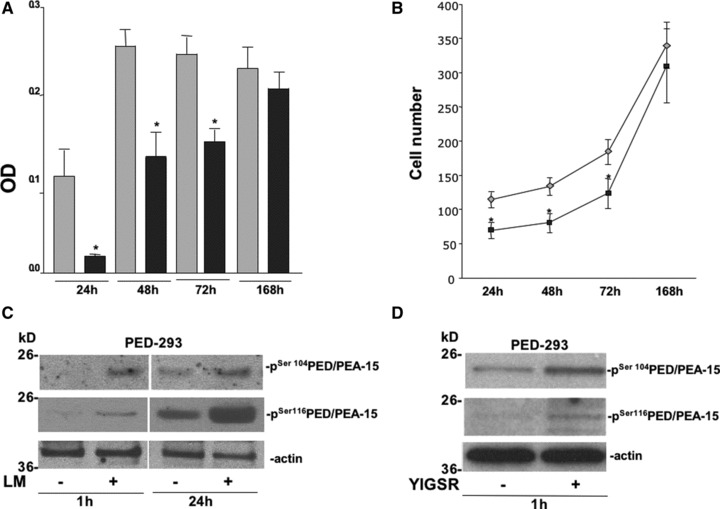
67LR interaction with PED/PEA-15 modulates LM-mediated cell proliferation and PED/PEA-15 phosphorylation status. (A) V-293 () and PED-293 () cell proliferation was evaluated by an MTS assay after adhesion to LM-coated wells. The growth medium was removed at the indicated times and 20 μl/well of reagent was added. The absorbance (OD) was determined at a wavelength of 490 nm. The values are the mean ± SD of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (B) V-293 () and PED-293 () cells were counted after adhesion to LM, at the indicated times. The values are the mean ± S.D. of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (C) PED-293 cells were serum-starved and plated on uncoated and LM-coated wells. The cells were harvested at the indicated times and lysed for Western blot analysis with anti-phospho-Ser104 PED/PEA-15 and anti-phospho-Ser116 PED/PEA-15 antibodies; anti-actin antibodies were used as a loading control. (D) PED-293 cells were serum-starved and plated on uncoated and YIGSR-coated wells. The cells were harvested after 1 hr and lysed for Western blot analysis with anti-phospho-Ser104 PED/PEA-15 and anti-phospho-Ser116 PED/PEA-15 antibodies; anti-actin antibodies were used as a loading control.
PED/PEA-15 can bind and retain into cytosol ERK1/2, independently of their phosphorylation status, thus impairing cell proliferation in PED/PEA-15 overexpressing cells (17, 49). PED/PEA-15 phosphorylation on Serine-104 inhibits its interaction with ERK1/2, thus allowing cell proliferation even in cells expressing high levels of PED/PEA-15 [49]. Therefore, we investigated whether 67LR-mediated cell adhesion to LM could alter PED/PEA-15 phosphorylation status, thus allowing the observed recovery of PED-293 cell proliferation. PED/PEA-15 phosphorylation after PED-293 cell adhesion to LM or BSA, as a negative control, was analysed. After 1 hr of cell adhesion to LM, PED/PEA-15 phosphorylation occurred both in Ser-104 and in Ser-116 and further increased at 24 hrs (Fig. 5C). Similar results were obtained after PED-293 cell adhesion to YIGSR, the LM peptide which specifically binds 67LR (Fig. 5D), demonstrating that PED/PEA-15 phosphorylation, likely responsible for the proliferation recovery of PED-293 cells on LM, can be specifically induced by 67LR-mediated binding to LM.
Then, we investigated whether increased 67LR-mediated binding to LM in PED/PEA-15 overexpressing cells could generate the signal leading to PED/PEA-15 phosphorylation. PED/PEA-15 is an endogenous substrate for PKC, calcium/calmodulin-dependent protein kinase II (CAM kinase II), and Akt. Therefore, PKC, CAMKII and Akt phosphorylation, after V-293 and PED-293 cells adhesion to LM, was evaluated by Western blot analysis. Akt was not activated whereas CAMKII was phosphorylated after 1 hr and still at 24 hrs; PKC was transiently phosphorylated at 1 hr (Fig. 6A).
Fig 6.
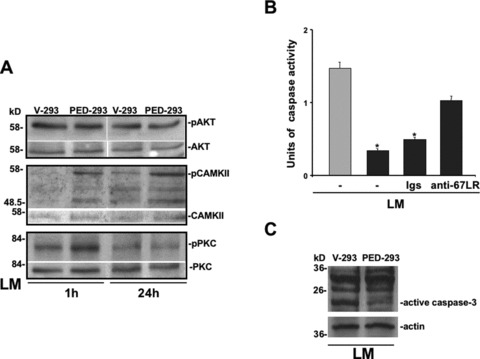
67LR-mediated cell adhesion to LM generates an intracellular signalling that inhibits apoptosis in PED/PEA-15 overexpressing cells. (A) V-293 and PED-293 cells were serum-starved and plated on LM coated wells. The cells were harvested at the indicated times and lysed for Western blot analysis with anti-phospho-Ser104 PED/PEA-15, anti-phospho-Ser116 PED/PEA-15, anti-phospho-CAMKII, anti-phospho-Akt and anti-phospho-PKC antibodies; anti-PED/PEA-15, CAMKII, Akt and PKC antibodies were used as a loading control. (B) V-293 () and PED-293 () cells were plated on LM in the absence of serum and apoptosis was evaluated at different times by ELISA assay in the presence of medium alone (-), 20 μg/ml of non-immune immunoglobulins (Igs) or anti-67LR polyclonal antibodies. The values are the mean ± SD of three experiments performed in triplicate. *P ≤ 0.05, as determined by the Student’s t-test. (C) V-293 and PED-293 cells were plated on LM for 48 hrs in the absence of serum and apoptosis was evaluated by Western blotting with anti-caspase 3 antibodies.
Therefore, 67LR binding to LM stimulates PED/PEA-15 phoshorylation in Ser-104, through PKC activation, thus enabling cell proliferation in response to LM in PED/PEA-15 overexpressing cells.
Because PED/PEA-15 phosphorylation in Ser-116, through CAMKII activation, induces its recruitment to the death-inducing signalling complex (DISC) causing the inhibition of the apoptotic process [6], we investigated the apoptotic response to serum deprivation in PED-293 cells, following adhesion to LM, which induces PED/PEA-15 phosphorylation. V-293 and PED-293 cells were plated on LM in the absence of serum and apoptosis was evaluated by ELISA assay. No apoptosis could be detected after 24 hrs of serum deprivation (not shown); at 48 hrs, V-293 cells underwent apoptosis whereas PED-293 cells showed a significant resistance (Fig. 6B). Noteworthy, cell-treatment with anti-67LR antibodies restored, at least in part, PED-293 cell sensitivity to serum deprivation-induced apoptosis. Resistance to apoptosis after 48 hrs of PED-293 cell adhesion to LM was also documented by the strong reduction of caspase 3 cleavage, as compared to V-293 cells (Fig. 6C).
Therefore, 67LR-dependent binding to LM, through PED/PEA15 phosphorylation, besides restoring cell proliferation, is also able to inhibit the apoptotic process in PED/PEA-15 overexpressing cells.
Discussion
PED/PEA-15 is an anti-apoptotic protein whose expression is increased in several human cancers. PED/PEA-15, being a member of the DED protein family, induces resistance to TRAIL-, FasL- and TNFα-mediated death in cancer, making therapy often ineffective. PED/PEA-15 is also a potent modulator of MAPK-signalling cascades. It binds ERK1/2 in the cytosol, avoiding their nuclear translocation and thereby reducing the ERK1/2-mediated transcriptional activity and cell proliferation.
To give new insights in its role in cancer, we aimed to find new PED/PEA-15 interactors. We performed a yeast two hybrid screening and identified several new candidate partners of PED/PEA-15, one of which was the 67LR.
67LR is a non-integrin cell surface receptor for the extracellular matrix whose expression is highly increased in human cancers and widely recognized as a molecular marker of metastatic aggressiveness [30].
We confirmed the interaction between PED/PEA-15 and 67LR through pull down and immunoprecipitation experiments, using both exogenous and endogenous proteins.
Then, we investigated the effects of PED/PEA-15 overexpression, occurring in different human tumours, such as breast and lung cancer, on 67LR-mediated cell functions. To this end, we evaluated cell adhesion and migration to LM in PED/PEA-15 transfected HEK 293 cells. PED/PEA-15 overexpression promoted cell adhesion and migration to LM, the major component of basement membranes [19], via the 67LR. The specific involvement of 67LR was unequivocally demonstrated by cell adhesion assays to an LM-derived pentapeptide, YIGSR, which specifically binds 67LR and by inhibition experiments with anti-67LR antibodies. Antibodies directed to LM-specific α6 and α3 integrin chains failed to abolish PED-293 cell adhesion to LM, confirming that increased binding to LM was mostly mediated by 67LR.
However, we also propose the involvement of β1-integrins in the generation of the downstream signal, because we observed that anti-β1-integrin antibodies exerted an effect similar to 67LR inhibition. In fact, 67LR is co-expressed and physically interacts with α6-containing integrins on the cell membrane [29] and PED/PEA-15 has been largely shown to functionally regulate integrin activation and integrin-dependent functions [6, 15, 50]. Therefore, PED/PEA-15 overexpression increases cell adhesion to LM through its interaction with 67LR, which is both able to bind LM and to recruit integrins, thus leading to an increased intracellular signalling. This mechanism regulates also the activity of other non-integrin cell surface receptors for the extracellular matrix, for instance the urokinase receptor, which is also a vitronectin receptor and requires integrins as signalling partners in cell adhesion and migration [51].
Then, we investigated the effects of cell adhesion to LM on the cell signalling. In NSCLC cells, PED/PEA-15 is associated with Rac1 and promotes its activation in response to growth factor stimulation [17]; in turn, Rac1 increases ERK1/2 phosphorylation [47]. We showed that LM stimulation of PED/PEA-15 overexpressing cells activates Rac1 and ERK1/2. Therefore, a PED/PEA-15-dependent signal transduction pathway, usually activated in response to growth factor stimulation, can be also activated after 67LR-mediated cell adhesion to basement membranes, a prerequisite for tumour invasion and metastasis.
Accordingly with our results, it has been recently reported that 67LR is overexpressed in astrocytoma and its down-regulation reduces the migratory activity of human glioma cells [52]. Findings on PED/PEA-15 are more controversial; it has been described a PED/PEA-15-mediated inhibition of cell migration/invasion in astrocytoma [16] whereas an increase has been shown in NSCLC [17]. Our study is in agreement with the last report; it is conceivable that this apparent discrepancy could be due to different regulatory mechanisms selectively controlling motility in a cell type-specific manner.
We then investigated whether the 67LR could also be involved in an LM-dependent modulation of PED/PEA-15 cellular functions, such as cell proliferation and apoptosis.
It has been reported that PED/PEA-15 overexpression inhibits ERK1/2 nuclear localization, thus blocking cell proliferation; PED/PEA-15 phosphorylation on Ser-104, induces ERK release to the nucleus and enables cell proliferation, even in the presence of high levels of PED/PEA-15 expression [49]. We demonstrated that 67LR-mediated cell adhesion to LM induces an intracellular signal that, through PCK and CaMKinase II activation, promotes PED/PEA-15 phosphorylation on Ser-104, thus enabling cell proliferation even in PED/PEA-15 overexpressing cells.
Interestingly, 67LR, such as PED/PEA-15, is involved in the regulation of cell proliferation and survival. Indeed, reduction of 67LR expression results in apoptosis [53, 54]; on the contrary, 67LR-dependent cell signalling pathways are important for cell survival [55, 56]. In agreement with these reports, we showed that 67LR-mediated cell adhesion to LM also determines PED/PEA-15 phosphorylation on Ser-116, which promotes resistance to apoptosis induced by growth factor deprivation [44].
In summary, when overexpressed, PED/PEA-15 increases cell adhesion and migration to LM via the 67LR, thus promoting LM-derived motility signals, which are crucial in intra-vasation and extra-vasation of cancer cells, during invasion and metastasis. Moreover, in PED/PEA-15 overexpressing cells, increased 67LR-mediated cell adhesion to LM activates a signal transduction pathway that restores cell proliferation and promotes resistance to apoptosis, thus also contributing to cancer cell survival in a poor microenvironment, as it occurs during metastatic spread and colonization.
Conflict of interest
The authors confirm that there are no conflict of interest.
References
- 1.Fiory F, Formisano P, Perruolo G, et al. Frontiers: PED/PEA-15, a multifunctional protein controlling cell survival and glucose metabolism. Am J Physiol Endocrinol Metab. 2009;297:E592–601. doi: 10.1152/ajpendo.00228.2009. [DOI] [PubMed] [Google Scholar]
- 2.Condorelli G, Vigliotta G, Cafieri A, et al. PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999;18:4409–15. doi: 10.1038/sj.onc.1202831. [DOI] [PubMed] [Google Scholar]
- 3.Kitsberg D, Formstecher E, Fauquet M, et al. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFalpha-induced apoptosis. J Neurosci. 1999;19:8244–51. doi: 10.1523/JNEUROSCI.19-19-08244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Formstecher E, Ramos JW, Fauquet M, et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell. 2001;1:239–50. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- 5.Callaway K, Abramczyk O, Martin L, et al. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry. 2007;46:9187–98. doi: 10.1021/bi700206u. [DOI] [PubMed] [Google Scholar]
- 6.Renganathan H, Vaidyanathan H, Knapinska A, et al. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. Biochem J. 2005;390:729–35. doi: 10.1042/BJ20050378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubes M, Cordier J, Glowinski J, et al. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes: identification of Ser104 and Ser116 phosphorylated, respectively, by protein kinase C and calcium/calmodulin kinase II in vitro. J Neurochem. 1998;71:1307–14. doi: 10.1046/j.1471-4159.1998.71031307.x. [DOI] [PubMed] [Google Scholar]
- 8.Trencia A, Perfetti A, Cassese A, et al. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol Cell Biol. 2003;23:4511–21. doi: 10.1128/MCB.23.13.4511-4521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Formisano P, Perruolo G, Libertini S, et al. Raised expression of the antiapoptotic protein ped/pea-15 increases susceptibility to chemically induced skin tumour development. Oncogene. 2005;24:7012–21. doi: 10.1038/sj.onc.1208871. [DOI] [PubMed] [Google Scholar]
- 10.Zanca C, Garofalo M, Quintavalle C, et al. PED is overexpressed and mediates TRAIL resistance in human non-small cell lung cancer. J Cell Mol Med. 2008;12:2416–26. doi: 10.1111/j.1582-4934.2008.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garofalo M, Romano G, Quintavalle C, et al. Selective inhibition of PED protein expression sensitizes B-cell chronic lymphocytic leukaemia cells to TRAIL-induced apoptosis. Int J Cancer. 2007;120:1215–22. doi: 10.1002/ijc.22495. [DOI] [PubMed] [Google Scholar]
- 12.Todaro M, Zerilli M, Ricci-Vitiani L, et al. Autocrine production of interleukin-4 and interleukin-10 is required for survival and growth of thyroid cancer cells. Cancer Res. 2006;66:1491–9. doi: 10.1158/0008-5472.CAN-05-2514. [DOI] [PubMed] [Google Scholar]
- 13.Hao C, Beguinot F, Condorelli G, et al. Induction and intracellular regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–70. [PubMed] [Google Scholar]
- 14.Eckert A, Böck BC, Tagscherer KE, et al. The PEA-15/PED protein protects glioblastoma cells from glucose deprivation-induced apoptosis via the ERK/MAP kinase pathway. Oncogene. 2008;27:1155–66. doi: 10.1038/sj.onc.1210732. [DOI] [PubMed] [Google Scholar]
- 15.Chou FL, Hill JM, Hsieh JC, et al. PEA-15 binding to ERK1/2 MAPKs is required for its modulation of integrin activation. J Biol Chem. 2003;278:52587–97. doi: 10.1074/jbc.M309322200. [DOI] [PubMed] [Google Scholar]
- 16.Renault-Mihara F, Beuvon F, Iturrioz X, et al. Phosphoprotein enriched in astrocytes-15 kDa expression inhibits astrocyte migration by a protein kinase C delta-dependent mechanism. Mol Biol Cell. 2006;17:5141–52. doi: 10.1091/mbc.E05-11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanca C, Cozzolino F, Quintavalle C, et al. PED interacts with Rac1 and regulates cell migration/invasion processes in human non-small cell lung cancer cells. J Cell Physiol. 2010;225:63–72. doi: 10.1002/jcp.22197. [DOI] [PubMed] [Google Scholar]
- 18.Rao NC, Barsky SH, Terranova VP, et al. Isolation of a tumour cell laminin receptor. Biochem Biophys Res Commun. 1983;111:804–8. doi: 10.1016/0006-291x(83)91370-0. [DOI] [PubMed] [Google Scholar]
- 19.Mecham RP. Receptors for laminin on mammalian cells. FASEB J. 1991;5:2538–46. doi: 10.1096/fasebj.5.11.1651264. [DOI] [PubMed] [Google Scholar]
- 20.Graf J, Ogle RC, Robey FA, et al. A pentapeptide from the laminin B1 chain mediates cell adhesion and binds the 67,000 laminin receptor. Biochemistry. 1987;26:6896–900. doi: 10.1021/bi00396a004. [DOI] [PubMed] [Google Scholar]
- 21.Taraboletti G, Belotti D, Giavazzi R, et al. Enhancement of metastatic potential of murine and human melanoma cells by laminin receptor peptide G: attachment of cancer cells to subendothelial matrix as a pathway for hematogenous metastasis. J Natl Cancer Inst. 1993;85:235–40. doi: 10.1093/jnci/85.3.235. [DOI] [PubMed] [Google Scholar]
- 22.Rao CN, Castronovo V, Schmitt MC, et al. Evidence for a precursor of the high-affinity metastasis-associated murine laminin receptor. Biochemistry. 1989;28:7476–86. doi: 10.1021/bi00444a047. [DOI] [PubMed] [Google Scholar]
- 23.Landowski TH, Dratz EA, Starkey JR. Studies of the structure of the metastasis-associated 67 kDa laminin binding protein: fatty acid acylation and evidence supporting dimerization of the 32 kD gene product to form the mature protein. Biochemistry. 1995;34:11276–87. doi: 10.1021/bi00035a037. [DOI] [PubMed] [Google Scholar]
- 24.Castronovo V, Taraboletti G, Sobel ME. Functional domains of the 67 kDa laminin receptor precursor. J Biol Chem. 1991;266:20440–6. [PubMed] [Google Scholar]
- 25.Kazmin DA, Hoyt TR, Taubner L, et al. Phage display mapping for peptide 11 sensitive sequences binding to laminin-1. J Mol Biol. 2000;298:431–45. doi: 10.1006/jmbi.2000.3680. [DOI] [PubMed] [Google Scholar]
- 26.Magnifico A, Tagliabue E, Butò S. Peptide G, containing the binding site of the 67 kDa laminin receptor, increases and stabilizes laminin binding to cancer cells. J Biol Chem. 1996;271:31179–84. doi: 10.1074/jbc.271.49.31179. [DOI] [PubMed] [Google Scholar]
- 27.Ardini E, Sporchia B, Pollegioni L, et al. Identification of a novel function for 67-kDa laminin receptor: increase in laminin degradation rate and release of motility fragments. Cancer Res. 2002;62:1321–5. [PubMed] [Google Scholar]
- 28.Berno V, Porrini D, Castiglioni F, et al. The 67 kDa laminin receptor increases tumour aggressiveness by remodeling laminin-1. Endocr Relat Cancer. 2005;12:393–406. doi: 10.1677/erc.1.00870. [DOI] [PubMed] [Google Scholar]
- 29.Ardini E, Tagliabue E, Magnifico A, et al. Co-regulation and physical association of the 67 kD monomeric laminin receptor and the α6β4 integrin. J Biol Chem. 1997;272:2342–5. doi: 10.1074/jbc.272.4.2342. [DOI] [PubMed] [Google Scholar]
- 30.Montuori N, Sobel ME. The 67-kDa laminin receptor and tumour progression. Curr Top Microbiol Immunol. 1996;213:205–14. doi: 10.1007/978-3-642-61107-0_13. [DOI] [PubMed] [Google Scholar]
- 31.Wewer UM, Taraboletti G, Sobel ME, et al. Role of laminin receptor in tumour cell migration. Cancer Res. 1987;47:5691–8. [PubMed] [Google Scholar]
- 32.Menard S, Tagliabue E, Colnaghi MI. The 67 kDa laminin receptor as a prognostic factor in human cancer. Breast Cancer Res Treat. 1998;52:137–45. doi: 10.1023/a:1006171403765. [DOI] [PubMed] [Google Scholar]
- 33.Montuori N, Selleri C, Risitano AM, et al. Expression of the 67-kDa laminin receptor in acute myeloid leukemia cells mediates adhesion to laminin and is frequently associated with monocytic differentiation. Clin Cancer Res. 1999;5:1465–72. [PubMed] [Google Scholar]
- 34.Chen A, Ganor Y, Rahimipour S, et al. The neuropeptides GnRH-II and GnRH-I are produced by human T cell and trigger laminin receptor gene expression, adhesion, chemotaxis and homing to specific organs. Nat Med. 2002;8:1421–6. doi: 10.1038/nm1202-801. [DOI] [PubMed] [Google Scholar]
- 35.Zuber C, Knackmuss S, Zemora G, et al. Invasion of tumourigenic HT1080 cells is impeded by blocking or downregulating the 37-kDa/67-kDa laminin receptor. J Mol Biol. 2008;378:530–9. doi: 10.1016/j.jmb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Scheiman J, Tseng JC, Zheng Y, et al. Multiple functions of the 37/67-kD laminin receptor make it a suitable target for novel cancer gene therapy. Mol Ther. 2010;18:63–74. doi: 10.1038/mt.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Condorelli G, Vigliotta G, Iavarone C, et al. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. EMBO J. 1998;17:3858–66. doi: 10.1093/emboj/17.14.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gietz D, St Jean A, Woods RA, et al. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JH. Experiments in molecular genetics, cold spring harbor laboratory. NY: Cold Spring Harbor; 1972. [Google Scholar]
- 40.Condorelli G, Trencia A, Vigliotta G, et al. Multiple members of the mitogen-activated protein kinase family are necessary for PED/PEA-15 anti-apoptotic function. J Biol Chem. 2002;277:11013–8. doi: 10.1074/jbc.M110934200. [DOI] [PubMed] [Google Scholar]
- 41.Jamieson KV, Hubbard SR, Meruelo D. Structure-guided identification of a laminin binding site on the laminin receptor precursor. J Mol Biol. 2011;405:24–32. doi: 10.1016/j.jmb.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douville PJ, Harvey WJ, Carbonetto S. Isolation and partial characterization of high affinity laminin receptors in neural cells. J Biol Chem. 1988;263:14964–9. [PubMed] [Google Scholar]
- 43.Stallmach A, Schuppan D, Dax J, et al. Identification of laminin binding proteins in cell membranes of a human colon adenocarcinoma cell line. Gut. 1990;31:70–6. doi: 10.1136/gut.31.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Botta G, Perruolo G, Libertini S, et al. PED/PEA-15 modulates coxsackievirus-adenovirus receptor expression and adenoviral infectivity via ERK-mediated signals in glioma cells. Hum Gene Ther. 2010;21:1067–76. doi: 10.1089/hum.2009.181. [DOI] [PubMed] [Google Scholar]
- 45.Massia SP, Rao SS, Hubbell JA. Covalently immobilized laminin peptide Tyr-Ile-Gly-Ser-Arg (YIGSR) supports cell spreading and co-localization of the 67-kilodalton laminin receptor with alpha-actinin and vinculin. J Biol Chem. 1993;268:8053–9. [PubMed] [Google Scholar]
- 46.Ramos JW, Hughes PE, Renshaw MW, et al. Death effector domain protein PEA-15 potentiates Ras activation of extracellular signal receptor-activated kinase by an adhesion-independent mechanism. Mol Biol Cell. 2000;11:2863–72. doi: 10.1091/mbc.11.9.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eblen ST, Slack JK, Weber MJ, et al. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002;22:6023–33. doi: 10.1128/MCB.22.17.6023-6033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumour progression and invasion. Cancer Metastasis Rev. 2003;22:395–403. doi: 10.1023/a:1023781114568. [DOI] [PubMed] [Google Scholar]
- 49.Krueger J, Chou FL, Glading A, et al. Phosphorylation of phosphoprotein enriched in astrocytes (PEA-15) regulates extracellular signal-regulated kinase-dependent transcription and cell proliferation. Mol Biol Cell. 2005;16:3552–61. doi: 10.1091/mbc.E04-11-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaidyanathan H, Opoku-Ansah J, Pastorino S, et al. ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc Natl Acad Sci USA. 2007;104:19837–42. doi: 10.1073/pnas.0704514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63:1028–37. doi: 10.1007/s00018-005-5428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen FX, Qian YR, Duanm YH, et al. Down-regulation of 67LR reduces the migratory activity of human glioma cells in vitro. Brain Res Bull. 2009;79:402–8. doi: 10.1016/j.brainresbull.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 53.Kaneda Y, Kinoshita K, Sato M, et al. The induction of apoptosis in HeLa cells by the loss of LBP-p40. Cell Death Differ. 1998;5:20–8. doi: 10.1038/sj.cdd.4400315. [DOI] [PubMed] [Google Scholar]
- 54.Susantad T, Smith DR. siRNA-mediated silencing of the 37/67-kDa high affinity laminin receptor in Hep3B cells induces apoptosis. Cell Mol Biol Lett. 2008;13:452–64. doi: 10.2478/s11658-008-0017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Givant-Horwitz V, Davidson B, Reich R. Laminin-induced signaling in tumour cells: the role of the M(r) 67,000 laminin receptor. Cancer Res. 2004;64:3572–9. doi: 10.1158/0008-5472.CAN-03-3424. [DOI] [PubMed] [Google Scholar]
- 56.Scheiman J, Tseng JC, Zheng Y, et al. Multiple functions of the 37/67-kD laminin receptor make it a suitable target for novel cancer gene therapy. Mol Ther. 2010;18:63–74. doi: 10.1038/mt.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]