

Abstract
Cyclic AMP (cAMP) is the archetypal smooth muscle relaxant, mediating the effects of many hormones and drugs. However, recently PGI2, acting via cAMP/PKA, was found to increase contraction-associated protein expression in myometrial cells and to promote oxytocin-driven myometrial contractility. Cyclo-oxygenase-2 (COX-2) is the rate-limiting enzyme in prostaglandin synthesis, which is critical to the onset and progression of human labour. We have investigated the impact of cAMP on myometrial COX-2 expression, synthesis and activity. Three cAMP agonists (8-bromo-cAMP, forskolin and rolipram) increased COX-2 mRNA expression and further studies confirmed that this was associated with COX-2 protein synthesis and activity (increased PGE2 and PGI2 in culture supernatant) in primary cultures of human myometrial cells. These effects were neither reproduced by specific agonists nor inhibited by specific inhibitors of known cAMP-effectors (PKA, EPAC and AMPK). We then used shRNA to knockdown the same effectors and another recently described cAMP-effector PDZ-GEF1-2, without changing the response to cAMP. We found that MAPK activation mediated the cAMP effects on COX-2 expression and that PGE2 acts through EP-2 to activate MAPK and increase COX-2. These data provide further evidence in support of a dual role for cAMP in the regulation of myometrial function.
Keywords: human myometrium, cyclic AMP, cycloxygenase-2, prostaglandins
Introduction
Preterm delivery is the prime cause of perinatal mortality and morbidity [1, 2]. However, despite intense laboratory and clinical investigations, the frequency of preterm birth has increased by over 30% in the last 20 years [3]. The early onset of labour is the most frequent cause of preterm birth and is most commonly related to intrauterine inflammation, uterine overdistension and placental abruption [4]. These factors shift the balance from myometrial quiescence to contractility and promote the onset of labour.
The second messenger, cAMP, influences a wide array of physiological and pathological events including smooth muscle contractility and inflammation. Indeed, both physiological (relaxin, CRH and CGRP) and pharmacological (β2-agonists) agents act via cAMP to induce myometrial relaxation. However, therapeutically in the management of preterm labour (PTL), β2-agonists are limited both by severe, potentially life threatening side effects and tachyphylaxis, the latter mediated by β2-agonist-induced down-regulation of myometrial β-adrenergic receptors [5, 6]. Consequently, other mechanisms to increase myometrial cAMP levels have been explored and a recent publication showed that using the phosphodiesterase type 4 inhibitor, rolipram, successfully reduced rates of PTL in a mouse model [7].
Prostaglandins (PGs) play a critical role in the onset of preterm and term labour, ripening the cervix [8] and promoting myometrial contractility [9]. These properties have been successfully exploited therapeutically on the one hand to induce labour and on the other by using inhibitors of PG synthesis to prevent PTL [9-11]. The key enzyme in PG synthesis is COX, and at least three isoforms are present in human myometrium, of which COX-2 is the most important. COX-2 is highly regulated by both transcriptional and post-transcriptional mechanisms [12, 13] and can be stimulated by growth factors, cytokines and endotoxins; its expression is increased in myometrium and amnion with the onset of labour [14, 15]. We have previously reported that the inflammatory cytokine, IL-1β, promotes COX-2 expression via NF-κB activation and that both IL-1β and mechanical stretch also act via MAPK to up-regulate COX-2 expression [16]. Previous studies have shown that cAMP is able to down-regulate both NF-κB and MAPK activity in a variety of tissues [17, 18] and could therefore be therapeutically useful in the prevention of PTL. However, interestingly, a recent study found that activation of the cAMP/PKA pathway by PGI2 leads to increased expression of the contraction associated proteins, connexin 43, α-SMA, h-caldesmon, calponin and SM2-MHC, suggesting that in response to certain signals, cAMP might promote the process of myometrial activation prior to the onset of labour [19]. Given that cAMP has the potential to be an effective tocolytic, but may also in some circumstances promote myometrial contractility, we have carried out this study to define whether cAMP increases myometrial COX-2 expression and studied the mechanisms responsible.
Materials and methods
Tissue collection
The local ethics committee approved the study and women donating tissue gave informed written consent. Human myometrial biopsies were taken at near term labour (approximately 39 weeks) from the upper margin of the uterine lower segment incision at the time of elective caesarean section prior to the onset of labour. The samples were processed for myocyte isolation and culture.
Isolation and culture of human myometrial cells
The myometrial tissue was carefully dissected and washed in ice-cold PBS several times. The tissue samples were digested for about 45–60 min. at 37°C in a collagenase solution 0.5 mg/ml collagenase 1A (Sigma-Aldrich Co. Ltd., Poole, Dorset, UK), 0.5 mg/ml collagenase XI (Sigma-Aldrich Co. Ltd.), 1 mg/ml bovine serum albumin in DMEM (Sigma-Aldrich Co. Ltd.). Digestion was stopped by addition of DMEM supplemented with 7.5% foetal calf serum (FCS; Sigma-Aldrich Co. Ltd.). The myometrial tissues suspension was agitated to further disperse the cells. The resulting suspension was then passed through a cell strainer (70 μm nylon cell strainer) and individual cells were collected by centrifugation at 3000 r.p.m. for 5 min. After washing, cells were grown in DMEM with supplementation of 7.5% FCS, 1% l-glutamine and 1% penicillin–streptomycin at 37°C and 5% CO2. The myometrial cells were used at either second or third passages. The culture medium was changed after 24 hrs and then every other day. Cells were exposed to different treatments as described for individual experiments. Cells were serum starved overnight prior to initiation of experiments.
Materials
In the following experiments, we use the different treatments and the final concentration of them was: forskolin 100 μM (Sigma-Aldrich Co. Ltd.), PGE2 10 μM, 8bromo-cAMP (Sigma-Aldrich Co. Ltd.), 250 μM, rolipram 10 μM (Sigma-Aldrich Co. Ltd.), Sp-6-phe-cAMP (PKA agonist 100nM; BIOLOG Life Science Institute, Bremen, Germany), 8pCPT-2’-O-Me-cAMP (Epac agonist, 50 μM; BIOLOG Life Science Institute), AICAr (AMPK agonist, 100μM; Merck Chemicals Ltd., Beeston, Nottingham, UK), KT5720 (PKA inhibitor 10μM; Sigma-Aldrich Co. Ltd.), brefeldin A (Epac inhibitor 100 μM; Sigma-Aldrich Co. Ltd.), compound C (AMPK inhibitor 100 μM; Merck Chemicals Ltd.), SB203580 (p38 inhibitor 10 μM; Tocris Cookson Ltd., Avonmouth, Bristol, UK), U0126 [ERK inhibitor 10 μM; New England Biolabs (UK) Ltd., Hitchin, Hertfordshire, UK], SP600125 (JNK inhibitor 20 μM; Tocris Cookson Ltd.), PKG inhibitor (Rp-8-Bromo-β-phenyl-1,N2-ethenoguanosine 3′:5′-cyclic monophosphorothioate sodium salt hydrate, 15 μM; (Sigma-Aldrich Co. Ltd.). Butaprost (EP-2 agonist 10 μM; Cayman Chemical Co., Ann Arbor, MI, USA), EP-4 agonist (10 μM, a gift from GlaxoSmithKline, Brentford, Middlesex, UK).
RT-PCR of COX-2 mRNA
Total RNA was extracted and purified from human myometrial cells grown in six-well plates using RNAeasy minikits purchased from Qiagen (Catalog No. 74106; Qiagen Ltd., Crawley, West Sussex, UK). After RNA quantification, 1.0 μg was reverse transcribed with oligo dT random primers using MuLV reverse transcriptase (Applied Biosystems Ltd., Warrington, Cheshire, UK). Primer sets for COX-2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed and obtained from Invitrogen Ltd. (Paisley, UK; Table 1). Assays were validated for all primer sets by confirming that single amplicons of appropriate size and sequence were generated according to predictions. Quantitative PCR was performed in the presence of SYBR Green (Applied Biosystems Ltd.), and amplicon yield was monitored during cycling in a RotorGene Sequence Detector (Corbett Research Ltd., Mortlake, Sydney, Australia) that continually measures fluorescence caused by the binding of the dye to double-stranded DNA. Pre-PCR cycle was 10 min. at 95°C followed by up to 45 cycles of 95°C for 20 sec., 58–60°C for 20 sec. and 72°C for 20 sec. followed by an extension at 72°C for 15 sec. The final procedure involves a melt over the temperature range of 72–99°C rising by 1° steps with a wait for 15 sec. on the first step followed by a wait of 5 sec. for each subsequent step. The cycle in which fluorescence reached a preset threshold (cycle threshold) was used or quantitative analyses. The cycle threshold in each assay was set at a level where the exponential increase in amplicon abundance was approximately parallel between all samples. All mRNA abundance data were expressed relative to the amount of constitutively expressed GAPDH.
Table 1.
Primer sequence, GenBank accession numbers and size of PCR products (bp)
Western blotting
Monolayers of human myometrial cells were lysed in cell lysis buffer obtained from New England BioLabs (UK) Ltd., scraped and collected in Eppendorf tubes. Samples were centrifuged at 13,000 χ g for 15 min. at 4°C. Supernatant were then transferred and stored at −80°C. Protein samples were denatured by heating 70°C for 10 min. and ran on a 10% SDS-PAGE for 30 min. at 80 V, and then 40–60 min. at 125 V, followed by a transfer to a Hybond ECL nitrocellulose membrane (GE Healthcare UK Ltd., Little Chalfont, Buckinghamshire, UK). The membrane was blocked with 5% milk protein solution overnight or for 1 hr, washed and hybridized with the primary antibody overnight at 4°C in a fresh blocking buffer (1χPBS, 1% milk protein and 0.1% Tween-20). Antibodies used included COX-2, EPAC (sc-1746, sc-8880, UK; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), p38, ERK, JNK, phospho-p38, phospho-ERK, phospho-JNK, PKA, AMPK [9212, 9102, 9252, 9212, 9101, 9251, 2532; New England BioLabs (UK) Ltd.], PDZ-GEF1,2 (WH0009693M1, WH0051735 M1; Sigma-Aldrich Corp., St. Louis, MO, USA) and β-actin (A1978; Sigma-Aldrich Chemie Gmbh, Munich, Germany). Subsequently, the membrane was incubated with the secondary antibody at room temperature for 2 hrs. For ECL detection of horse-radish peroxidise, ECL plus (GE Healthcare UK Ltd.) was used. Exposure for detection was at 25°C for 1–5 min.
PGE2, PGI2 and PGF2α Measurement
Following stimulation, the medium was collected and frozen at −80°C prior to analysis. Levels of PGE2, PGI2 and PGF2α were measured by ELISA using a Luminex Prostaglandin kit [R&D Systems, Inc., Minneapolis, MN, USA, Catalog. No. KGE004B; Enzo Life Sciences (UK) Ltd., Exeter, UK; Catalog No. 900-025; Cayman Chemical Co., Catalog No. 51601]. The ELISA was performed according to the manufacturer’s protocol. The concentrations of PGE2, PGI2 and PGF2α are expressed as pg/105 cells.
Transient gene transfection (knockdown PKA, Epac, AMPK, PDZ-GEF1,2 and COX-2 promoter-luciferase assay)
Cells were cultured in 24-well plates to about 80% confluence and then transfected by using Gene-Juice transfection reagent (Novogen Ltd. Windsor Berkshire, UK) according to the manufacturer’s protocol. A reporter construct containing 2419-bp of the promoter region of human COX-2 gene was used. The expression constructs and COX-2 reporter vector were co-transfected at concentrations of 300 ng/well and SV40-Renilla vector [pRL-SV40; Promega (UK) Ltd., Southampton, Hampshire, UK], which was used as a control for transfection efficiency at concentrations of 100 ng/well. The empty expression vector pSG5 and pGL4-Luc were included as filler constructs so that the total amount of transfected DNA per well was constant. Firstly, cells were treated for 24 hrs with transfection reagent and then with the specific stimulus for another 24 hrs. Finally, luciferase activity was measured by using a dual firefly/renilla luciferase assay [Luclite was obtained from PerkinElmer, Buckinghamshire, Seer Green, UK and Coelentrerazine was from CN Biosciences (UK) Ltd., Beeston, Nottingham, UK]. All transfections were performed in triplicates. Results of luciferase activity was first normalized to the level of Renilla luciferase activity and then calculated as fold induction relative to either the expression of vehicle-treated group, or the control empty expression vector. Based on preliminary transfection experiments, after transfection of relevant ShRNAs (OriGene Technologies, Rockville, MD, USA; Table 2) for the first 24 hrs, the medium was changed to one containing 7.5% serum, then after another 24 hrs, the medium was changed to one containing 0.1% puromycin (final concentration 1 ng/ml) medium. After 24–48 hrs, further transfections were carried out with the COX-2 promoter or treat with various stimuli for another 24 hrs.
Table 2.
ShRNA sequences used to knockdown cAMP effectors
| ShRNA DNA sequences Genes | |
|---|---|
| shRNA PKA | GGAACCACTATGCCATGAAGATCCTCGAC |
| GGAGATGTTCTCACACCTACGGCGGATCG | |
| GTCTCCATCAATGAGAAGTGTGGCAAGGA | |
| CTGGATTGCCATCTACCAGAGGAAGGTGG | |
| shRNA Epac | TGTTCTGCTCTTTGAACCACACAGCAAGG |
| CTGCGTGTGGACAAGCAGGACTTCAACCG | |
| GCTTCCTCCAGAAACTCTCAGACCTGGTG | |
| TACTCAACATGGTGTTGAGAAGGATGCAC | |
| shRNA AMPK | AGAAGATTCGGAGCCTTGATGTGGTAGGA |
| TTGGCAGTTGCCTACCATCTCATAATAGA | |
| GGAAGAATCCTGTGACAAGCACTTACTCC | |
| GAGTGATTCAGATGCTGAGGCTCAAGGAA | |
| shRNA PDZ-GEF1 | CAATGTCAGTGAGGCGAGAACTCTGTGCT |
| TGGTCAGTCTCAAGATGACAGCATAGTAG | |
| ATGGACGAGGAGAGTCTTCAGACATTATC | |
| GTCTGTGACTACGGAAGAAACCAAGCCTG | |
| shRNA PDZ-GEF2 | ACTCATCTTGCACTTACTGTGAAGACCAA |
| GGACTGAACAAGAGAAATCTGGTGTTCCT | |
| CTTCCAGAAGGACCTGTTGATTCTGAGGA | |
| AGCACGCTATGAGAGATACAGTGGCAATC |
Statistical analysis
All data were initially tested for normality using a Kolmogorov–Smirnoff test. Normally distributed data were analysed using a Student’s t-test for two groups and an ANOVA followed by a Dunnett’s or Bonferroni’s post-hoc test for three groups or more. Data that were not normally distributed were analysed using a Wilcoxon matched pair test for paired data and when comparing three groups or more a Friedman’s test, with a Dunn’s multiple comparisons post-hoc test. P < 0.05 was considered statistically significant.
Results
Induction of COX-2 mRNA expression, COX-2 protein and PG synthesis by cAMP
We initially investigated whether cAMP increased COX-2 mRNA expression by exposing human myometrial cells to forskolin (100 μM) and found increased COX-2 expression at 1 and 6 hrs (Fig. 1A). We confirmed these findings with two other cAMP agonists 8bromo-cAMP (250 μM) and rolipram (10 μM), a specific type 4 phosphodiesterase inhibitor, and found that COX-2 mRNA expression was significantly increased at 6 hrs of 8bromo-cAMP treatment (Fig. 1B) and at 1 and 6 hrs of rolipram treatment (Fig. 1C). We then performed a dose–response study and confirmed that forskolin increased in COX-2 mRNA at 6 hrs (100 μM, P < 0.05 only; Fig. 1D). This was associated with increased COX-2 protein synthesis at 6 and 24 hrs (Fig. 1F and G) and increased levels of PGE2, PGI2 and PGF2α at 24 and 48 hrs (Fig. 1H–J). In future experiments, we used the dose of 100 μM of forskolin and the 6-hr time point to assess both COX-2 mRNA expression and protein synthesis.
Fig 1.
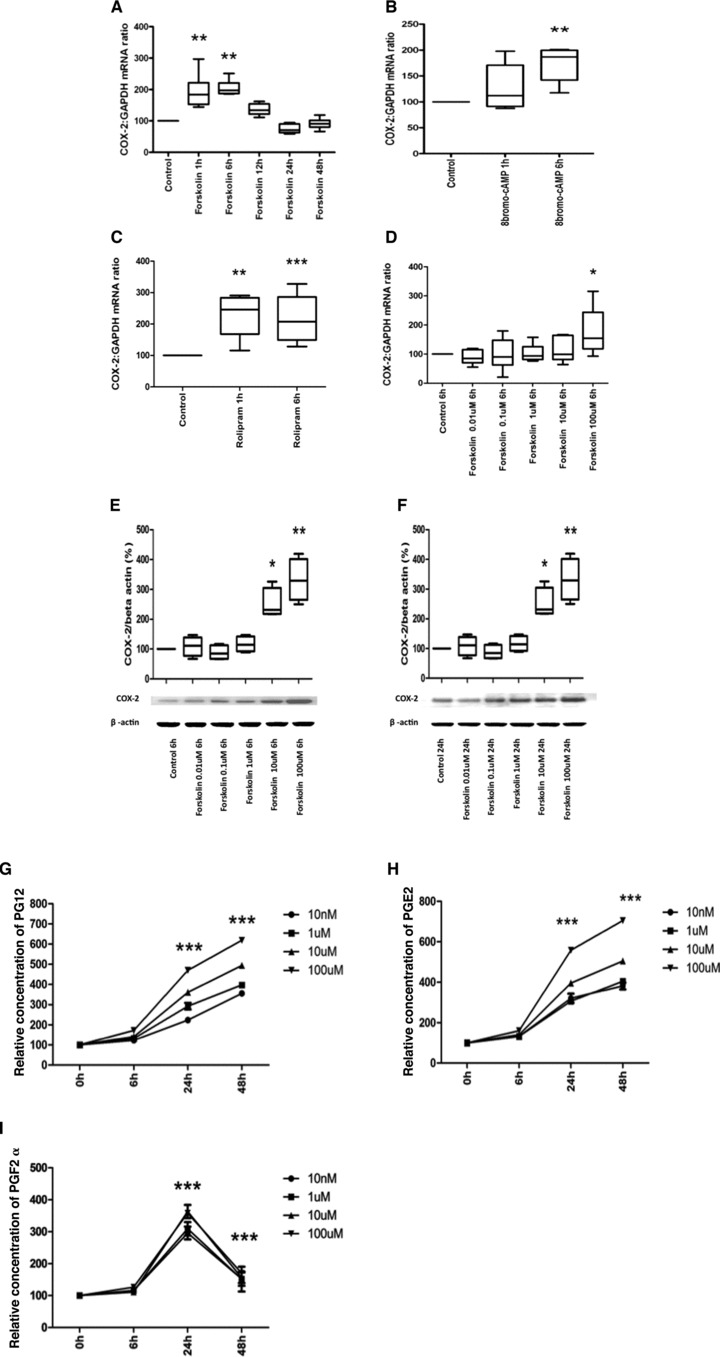
(A–E) Cyclic AMP exposure increases COX-2 mRNA expression, protein synthesis and activity. Human myometrial cells were treated with Forskolin (100 mM) for 0, 1, 6, 12, 24 and 48 hrs, and 8bromo-cAMP (250 μM) and rolipram (10 mM) for 0, 1 and 6 hrs (n = 6 in each study). mRNA was extracted, the levels of COX-2 mRNA measured using quantitative rtPCR. COX-2 mRNA expression was increased by forskolin (ANOVA P < 0.0001, Dunnett’s test P < 0.01 at 1 and 6 hrs; A), 8bromo-cAMP (ANOVA P = 0.008, Dunnett’s test P < 0.01 at 6 hrs; B) and rolipram (Friedman’s test = 0.0008, Dunnett’s test P < 0.01 at 1 hrs and P < 0.001 at 6 hrs; C). Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 and **P < 0.01 for control versus treatment. (D–F) In separate experiments, human myometrial cells were treated with forskolin 0.01, 0.1, 1.0, 10 and 100 mM for 6 hrs for COX-2 mRNA (measured by qPCR) and 6 and 24 hrs for COX-2 protein levels (measured by Western analysis). At 6 hrs, forskolin increased COX-2 mRNA expression (Friedman’s = 0.02, Dunnett’s test: 100 μM P < 0.05; D). COX-2 protein levels were increased by forskolin at 6 and 24 hrs (for 6 hrs, ANOVA P < 0.0001, Dunnett’s test: 100 and 10 μM P < 0.01; E, and for 24 hrs, ANOVA P < 0.0001, Dunnett’s test: 100 and 10 μM P < 0.01; F). Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 and **P < 0.01 for control versus forskolin. (G–I) In separate experiments, human myometrial cells were treated with forskolin 0.1, 1.0, 10 and 100 mM for 6, 24 and 48 hrs the medium was collected and the levels of PGE2, PGI2 and PGF2a measured with ELISA as described in the Materials and methods (G–I, n = 3). The data were analysed using ANOVA, with a Dunnett’s post-hoc test. Data are shown as the mean; *P < 0.05 and ***P < 0.001. The statistical significance is shown for the forskolin 100 μM concentration.
Intracellular mechanisms involved in cAMP-induced COX-2 synthesis
Role of PKA, EPAC, AMPK and PDZ-GEF1,2
Initially we attempted to identify which cAMP effector (PKA, EPAC or AMPK) was responsible for the cAMP-induced increase in COX-2 mRNA expression. Human myometrial cells were treated with forskolin (100 μM) and various cAMP effector activators: PKA (Sp-6-phe-cAMP, 100 nM), EPAC (8pCPT-2′-O-Me-cAMP, 50 μM) and AMPK (AICAR, 100 μM). Forskolin increased COX-2 mRNA expression (P < 0.05; Fig. 2A) and protein synthesis (P < 0.01; Fig. 2B), but none of the cAMP effector activators reproduced these results (Fig. 2A and B).
Fig 2.
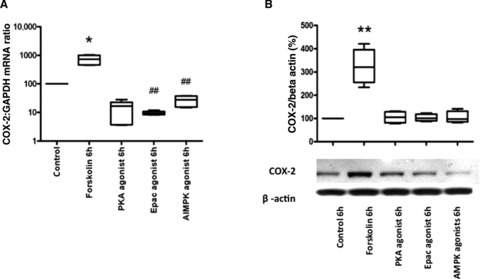
Cyclic AMP effector agonists do not reproduce the forskolin-induced increase in COX-2 mRNA expression and protein synthesis. Human uterine smooth muscle cells were treated with forskolin (100 μM) and different cAMP mediator agonists, Sp-6-phe-cAMP (PKA agonist) 100 nM, 8pCPT-2′-O-Me-cAMP (Epac agonist) 50 μM, AICAR (AMPK agonist) 100 μM for 6 hrs; mRNA and protein were extracted, the COX-2 mRNA levels measured using quantitative rtPCR (A) and the protein concentration assessed using western analysis (B). Control versus forskolin mRNA data were analysed using Wilcoxon matched pairs for paired data and Friedman’s test, with a Dunn’s multiple comparisons post-hoc test for analysis of three groups or more. The Western blot densitometry were analysed with an ANOVA followed by Bonferroni’s post-test. Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 and **P < 0.01 for control versus forskolin and ##P < 0.01 for forskolin versus cAMP agonists.
We then incubated human myometrial cells with forskolin (100 μM) alone or in the presence of cAMP effector antagonists: PKA (KT5720, 10 μM), EPAC (brefeldin A, 100 μM), AMPK (compound C, 100 μM). None of the inhibitors blocked the forskolin-induced increase in COX-2 mRNA expression, promoter activity or protein synthesis (Fig. 3A–C).
Fig 3.
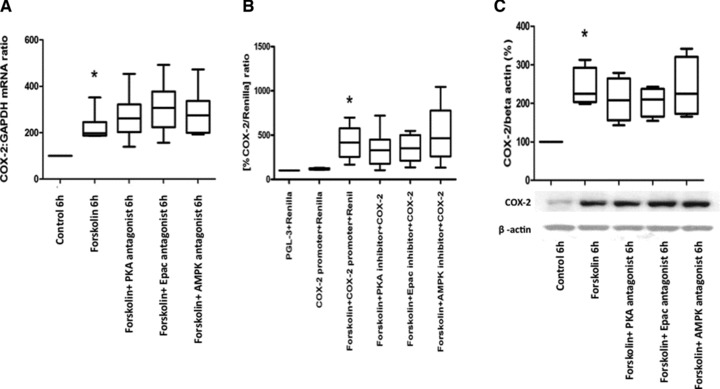
Cyclic AMP effector antagonists do not reduce forskolin-induced increase in COX-2 mRNA expression, promoter activity or protein synthesis. Human myometrial cells were treated with forskolin (100 μM) and different cAMP mediator antagonists, KT5720 10 μM (PKA inhibitor), brefeldin A 100 μM (Epac inhibitor) and compound C 100 μM (AMPK inhibitor) for 6 hrs, mRNA and protein were extracted, the COX-2 mRNA levels measured using quantitative rtPCR (A, n = 6) and the protein concentration assessed using Western analysis (C, n = 4). Human myometrial cells were transiently transfected with a COX-2 promoter construct and treated with forskolin in the presence and absence of cAMP-effector antagonists (as detailed earlier), after a 24 hrs the luciferase activity was measured (B, n = 6). The control versus forskolin mRNA data were analysed using Wilcoxon matched pairs for paired data and Friedman’s test, with a Dunn’s multiple comparisons post-hoc test for analysis of three groups or more. The Western blot densitometry and promoter studies were analysed with an ANOVA followed by Bonferroni’s post-test. Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 for control versus forskolin and ##P < 0.01 for forskolin versus cAMP antagonists.
To confirm the activator/antagonist data, we knocked down the expression of the cAMP effectors, using shRNA to PKA, EPAC and AMPK. In addition, we knocked down PDZ-GEF1,2, another potential cAMP effector without known chemical activators or antagonists. Western analysis for PKA, AMPK, EPAC and PDZ-GEF1,2 showed the efficiency of protein expression inhibition was approximately 80% (Fig. 4A–E). Knockdown of PKA, EPAC, AMPK and PDZ-GEF1,2 did not inhibit the cAMP-induced increase in COX-2 mRNA, promoter activity or COX-2 protein synthesis (Fig. 4F–H).
Fig 4.
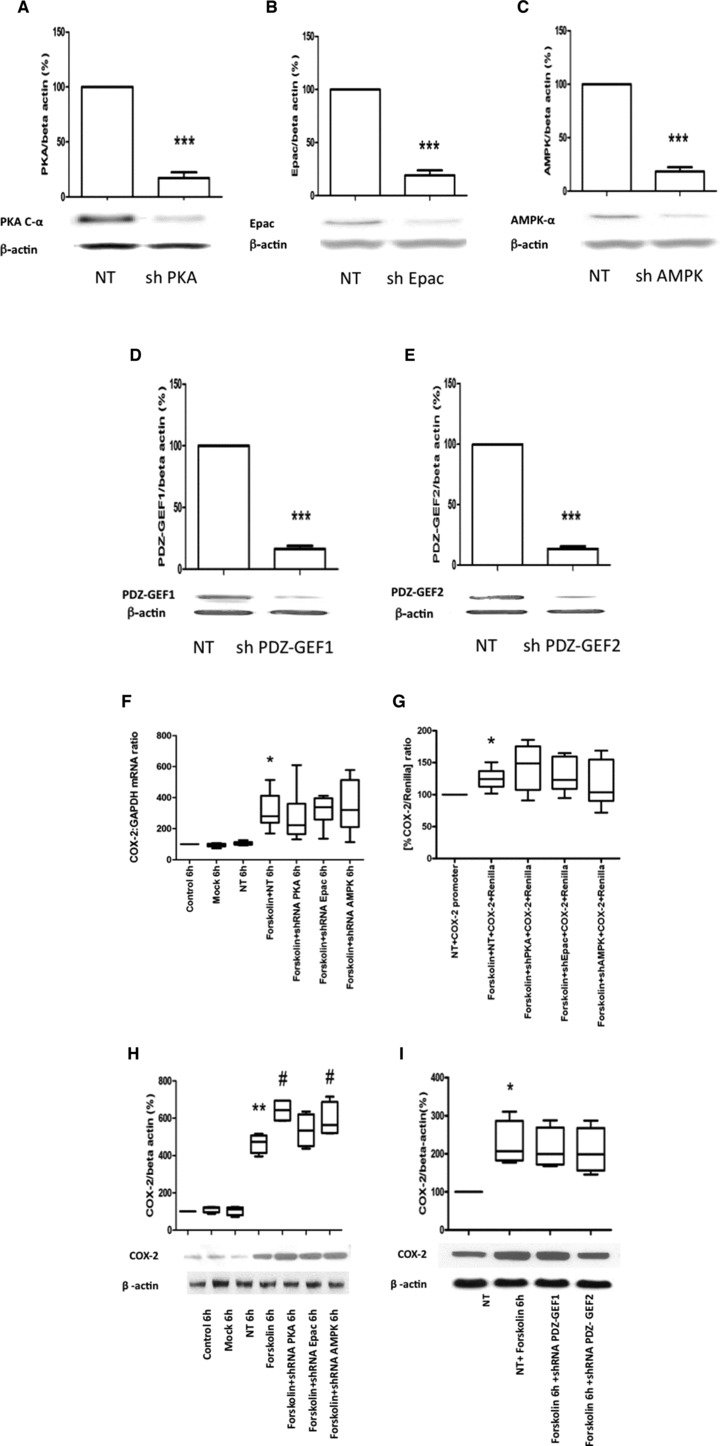
The effect of PKA, Epac and AMPK shRNA on cAMP-induced COX-2 mRNA expression and protein synthesis. Human myometrial cells were transfected with the specific shRNA for PKA, Epac, AMPK, and PDZ-GEF1 and PDZ-GEF2 and the respective protein concentrations assessed with Western blotting (data are expressed as mean ± S.E.M., paired t-test, A–E; ***P < 0.001). The cells were then treated with forskolin (100 μM) for 6 hrs, mRNA and protein were extracted and the COX-2 mRNA levels measured using quantitative rtPCR (F, n = 6) and the protein concentration assessed using Western analysis (H, n = 4). Human myometrial cells were also transiently transfected with a COX-2 promoter construct and treated with forskolin in the presence and absence of specific shRNA for PKA, EPAC, AMPK and PDZ-GEF1&2, after 24 hrs the luciferase activity was measured (G, n = 6). The control versus forskolin mRNA data were analysed using Wilcoxon matched pairs for paired data and Friedman’s test, with a Dunn’s multiple comparisons post-hoc test for analysis of three groups or more. The Western blot densitometry and promoter studies were analysed with an ANOVA followed by Bonferroni’s post-test. Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 and **P < 0.01 for control versus forskolin and #P < 0.05 for forskolin versus cAMP effector shRNA.
We confirmed that forskolin activated PKA and AMPK by using phospho-CREB and phospho-ACC (Fig. S1A and B). We used phospho-AKT as marker of EPAC activation, but found no increase of phosphorylation (Fig. S1C); to confirm that EPACs were present in human myometrial cells we assessed p38 phosphorylation with the EPAC agonist. In addition, we confirmed that the antagonists of PKA and AMPK were effective by co-incubating with KT5720 (PKA inhibitor, 10 μM) and compound C (AMPK inhibitor, 100 μM; Fig. S1A and B); we were unable to confirm that the EPAC antagonist, brefeldin A (EPAC inhibitor, 100 μM), was effective as we did not see any increase in AKT phosphorylation. We confirmed that the chemical stimulators of the cAMP effectors were active by assessing their respective down-stream substrates, for PKA (Sp-6-phe-cAMP, 100 nM) down-stream substrate phospho-CREB, for AMPK (AICAR, 100 μM), down-stream substrate phospho-ACC and for EPAC (8pCPT-2’-O-Me-cAMP, 50μM), down-stream substrate p38 (Fig. S2A–C).
We also investigated the role of PKG in mediating cAMP action because it has been recently recognized that cAMP can act via PKG to induce biological effects. Co-incubation of human myometrial cells with forskolin and a PKG inhibitor (Rp-8-Bromo-β-phenyl-1,N2-ethenoguanosine 3’:5’-cyclic monophosphorothioate sodium salt hydrate, 15 μM) had no effect on cAMP-driven COX-2 expression (Fig. S3A).
Role of MAPKs
It is well-established that myometrial COX-2 expression is regulated by MAPK-dependent pathways [16]. Consequently, we next assessed whether cAMP-increased COX-2 expression in human myometrial cells involves MAPK activation. We found that forskolin (100 mM) significantly increased the phosphorylation of p38 and ERK1/2 for up to 60 min. (Fig. 5A and B) and of JNK for 30 min. (Thr183 and Tyr185; Fig. 5C). Subsequently, we used specific inhibitors of MAPK activity and found that the forskolin-induced increases in COX-2 mRNA expression, COX-2 promoter activity and COX-2 protein synthesis were reduced (Fig. 5D–F).
Fig 5.
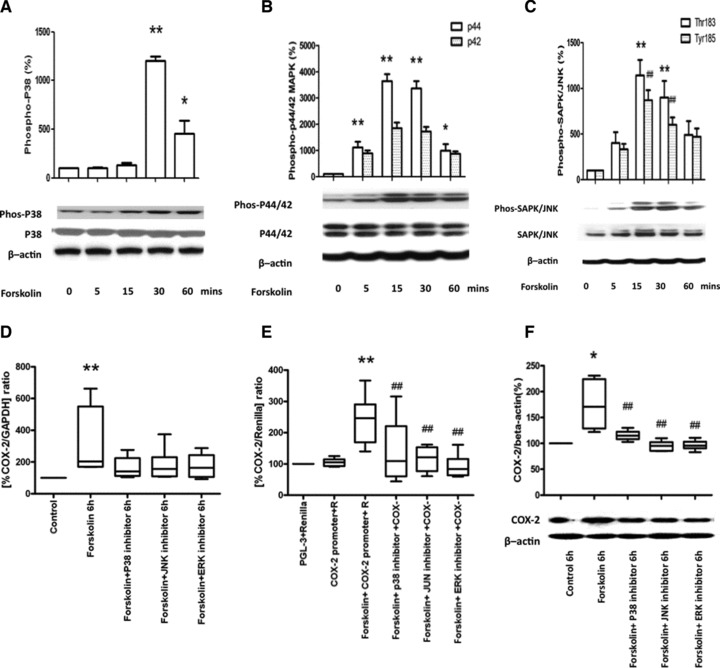
MAPK mediates the cAMP-induced increase in COX-2 mRNA expression, protein synthesis and promoter activity. (A–C) Human myometrial cells were treated with forskolin (100 μM) for 0, 5, 15, 30 and 60 min. Protein was extracted and Western analysis performed for the phosphoforms of p38, ERK1/2 and JNK, followed by densitometry. Forskolin increased p38 phosphorylation (ANOVA P < 0.0001, Dunnett’s test at 30 and 60 min. P < 0.01 and P < 0.05 respectively, A), ERK phosphorylation (for p44, ANOVA P < 0.0001, Dunnett’s test at 5, 15 and 30 min. P < 0.01 and at 60 min. P < 0.05, p42 ANOVA not significant, B) and JNK phosphorylation (for Thr183 ANOVA P = 0.0012, Dunnett’s test at 15 and 30 min. P < 0.01, for Tyr185 ANOVA P = 0.0003, Dunnett’s test at 15 and 30 min. P < 0.01, C). The data are expressed as mean ± S.E.M. *P < 0.05 and **P < 0.01 for p38, ERK-p44 and JNK-threonine 183 and ##P < 0.01 for JNK-tyrosine 185. For all n = 6. (D–G) Human myometrial cells were treated with forskolin (100 μM) in the presence and absence of specific MAPK inhibitors (p38 inhibitor SB203580 10 μM, ERK inhibitor U0126 10 μM, JNK inhibitor SP600125 20 μM) for 6 hrs. The increases in COX-2 mRNA expression (ANOVA P = 0.011, Bonferroni’s, control versus forskolin P < 0.01; D, n = 6), in COX-2 promoter activity (ANOVA P < 0.0001, Bonferroni’s, control versus forskolin P < 0.001 and forskolin versus forskolin and p38, ERK and JNK inhibitors P < 0.001; E, n = 6). and in COX-2 protein synthesis (ANOVA P = 0.0013, Bonferroni’s, control versus forskolin P < 0.05 and forskolin versus forskolin and p38, ERK and JNK inhibitors <0.01 at 6 hrs; F, n = 6) were inhibited. Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05 and **P < 0.01 between control and forskolin treatment and #P < 0.05, ##P < 0.01 between forskolin alone and in the presence of MAPK inhibitors.
The effect of PGE2 on COX-2 expression, protein synthesis and promoter activity
To confirm the physiological relevance of these data, we assessed the effect of PGE2 and found that it increased both COX-2 mRNA (Fig. 6A) and protein (Fig. 6B), acting via its EP-2 receptor (Fig. 6C–E). PGE2 also significantly increased the phosphorylation of p38 (Fig. 7A), ERK (Fig. 7B) and JNK (Fig. 7C). The PGE2-induced increases in COX-2 mRNA expression, promoter activity and protein synthesis were reduced in the presence of the MAPK inhibitors (Fig. 7D–F).
Fig 6.
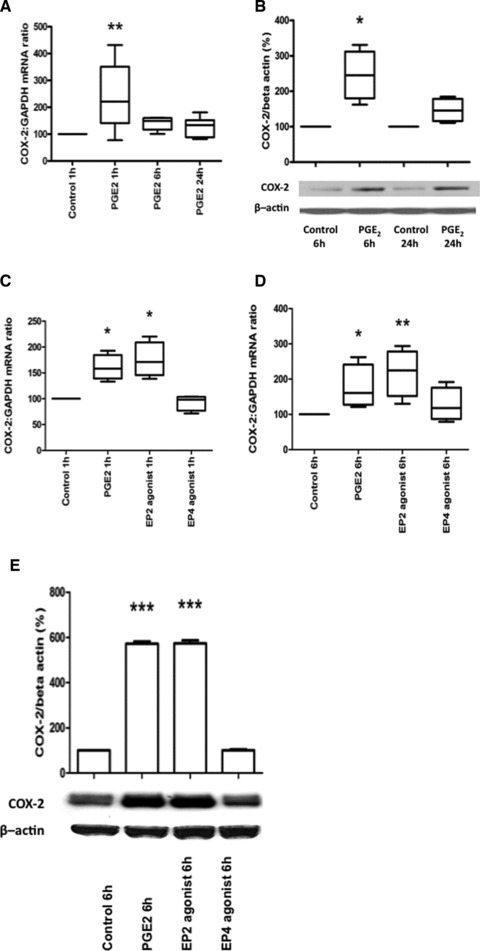
PGE2 increases COX-2 mRNA expression and protein synthesis via EP-2. Human myometrial cells were treated with PGE2 (10 μM) 1, 6 and 24 hrs, COX-2 mRNA increased (ANOVA P = 0.018, Dunnett’s test P < 0.01 at 1 hr, A; n = 6). In separate experiments, human myometrial cells were treated with PGE2 (10 μM) for 6 and 24 hrs COX-2 protein levels were increased at 6 hrs (paired t-test at 6 hrs, P = 0.023 and at 24 hrs, P = 0.056, B; n = 4). In a separate experiment, human myometrial cells were treated with PGE2 (10 μM), Butaprost (10 μM, EP-2 agonist) and an EP4 agonist (10 μM) for 1 and 6 hrs, COX-2 mRNA increased (at 1 hr, ANOVA P = 0.006, Dunnett’s test P < 0.05 for PGE2 and P < 0.01 for Butaprost, C; at 6 hrs, ANOVA P = 0.028, Dunnett’s test P < 0.05 for Butaprost, D; n = 3). In separate experiments, human myometrial cells were treated with PGE2 (10 μM), Butaprost (10 μM, EP-2 agonist) and EP4 agonists (10 μM) for 6 hrs COX-2 protein levels were increased (ANOVA P = 0.0005, Dunnett’s test P < 0.01 for PGE2 and Butaprost, E; n = 3). Data are shown as the median, the 25th and 75th percentiles and the range; *P < 0.05 and **P < 0.01.
Fig 7.
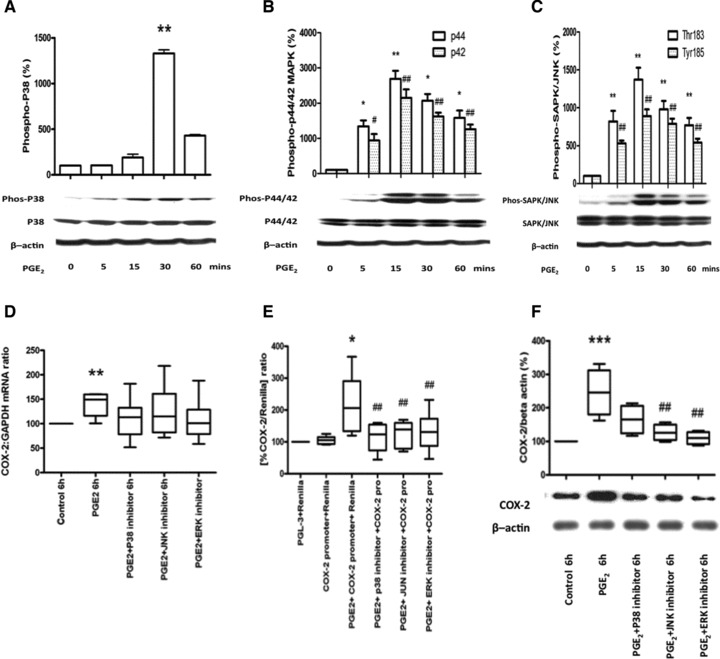
MAPK mediates the PGE2-induced increase in COX-2 mRNA expression, protein synthesis and promoter activity. Human myometrial cells were treated with PGE2 (10 μM), which increased the phosphorylation of p38 at 30 min. (ANOVA P < 0.0001, Dunnett’s test at 30 min. P < 0.01, A), ERK at 5, 15, 30 and 60 min. (p44, ANOVA P = 0.0028, Dunnett’s test at 5 min. P < 0.05, at 15 and 30 min. P < 0.01 and at 60 min. P < 0.05; p42 ANOVA P = 0.0003, Dunnett’s test at 5 min. P < 0.05 and at 15, 30 and 60 min. P < 0.01, B) and JNK at 5, 15, 30 and 60 min. (Thr183 ANOVA P = 0.0005, Dunnett’s test at 5, 15, 30 and 60 min. P < 0.01; Tyr185 ANOVA P < 0.0001, Dunnett’s test at 5, 15, 30 and 60 min. P < 0.01, C). The data are expressed as mean ± S.E.M. *P < 0.05 and **P < 0.01 between control and PGE2 treatment for p38, ERK-p44 and JNK-threonine 183; #P < 0.05 and ##P < 0.01 for ERK-p42 and JNK-tyrosine 185. In all cases n = 6. (D–F) In separate experiments, the studies were repeated in the presence and absence of specific MAPK inhibitors (p38 inhibitor SB203580 10 μM, ERK inhibitor U0126 10μM, JNK inhibitor SP600125 20 μM) for 6 hrs, mRNA and protein were extracted and the COX-2 mRNA levels measured using quantitative rtPCR (D, n = 6) and the protein concentration assessed using Western analysis (F, n = 6). Human myometrial cells were also transiently transfected with a COX-2 promoter construct and treated with forskolin in the presence and absence of specific MAPK inhibitors after 24 hrs the luciferase activity was measured (E, n = 6). The MAPK inhibitors reduced the PGE2-induced increase in COX-2 mRNA (D), in COX-2 promoter activity (ANOVA P = 0.0005, Bonferroni’s, control versus PGE2P < 0.001 and PGE2versus PGE2 with p38 P < 0.001, ERK and JNK inhibitors P < 0.01; E) and in COX-2 protein (6 hrs: ANOVA P = 0.0009, Bonferroni’s, control versus PGE2P < 0.001 and PGE2versus PGE2 with ERK and JNK inhibitors P < 0.01; F). Data are shown as the median, the 25th and 75th percentiles and the range. *P < 0.05, **P < 0.01 and ***P < 0.001 between control and PGE2 treatment and ##P < 0.01 and ###P < 0.001 between PGE2 alone versus in the presence of MAPK inhibitors.
Discussion
Our data show that cAMP increases the expression, synthesis and activity of COX-2, which is critical to the onset and progression of labour. These data provide more evidence that cAMP has two opposing effects on human myometrium, on the one hand inducing relaxation and on the other priming the uterus for contraction. These observations are consistent with previous reports that cAMP increases COX-2 mRNA expression in cultures of several human primary cells, including leucocyte, cardiomyocytes, endothelial cells and granulosa cells [20, 21]. Despite the evidence that cAMP increases COX-2 synthesis in other tissues, to prove that this was not an artefact of either the culture system or of forskolin administration, we used other cAMP agonists 8bromo-cAMP and rolipram and performed a dose/time–response study for forskolin.
The recent paper of Fetalvero et al. found that cAMP enhanced the synthesis of the contraction associated proteins connexin 43, α-SMA, h-caldesmon, calponin and SM2-MHC, suggesting that cAMP be important in myometrial activation prior to the onset of labour [19]. In this paper, the effects of cAMP were mediated via PKA as shown by the inhibitory effect of PKA knock-down [19]. In other tissues, cAMP has been shown to act via PKA (osteoblasts, Ref. 22) and AMPK expression (renal podocytes, Ref. 23) to increase COX-2 expression and to act via EPAC to suppress lipopolysaccharide (LPS)-induced interferon expression in macrophages [24]. However, using a combination of chemical inhibitors and activators, complimented by knock-down using shRNA, we showed that cAMP in our system was acting independent of known effectors. Given that we have previously shown that the MAPK system is essential for both IL-1β and stretch-induced COX-2 mRNA expression [25], we assessed whether cAMP could be acting via MAPK. We found that cAMP induced a robust activation of all MAPK isoforms and that MAPK inhibition blocked the cAMP-induced increase in COX synthesis. This suggested that the cAMP could be working through a guanine nucleotide exchange factor (GEF) to activate MAPK. cAMP-dependent Ras/MAPK activation has been reported in many cell types, but the Ras activator involved was not always identified [26]. In some reports, EPAC was suggested to have a role [27], but we had already excluded a role for EPAC in our studies. In melanoma cells, PDZ-GEF (also known as RAGEF, nRapGEP or CNrasGEF) was reported to mediate the cAMP activation of Ras/Erk pathway, leading to alterations in melanogenesis [28]. However, when we knocked down PDZ-GEF, we again found that the forskolin induced changes in COX-2 mRNA expression were unaltered. These data suggest that cAMP up-regulates COX-2 expression via MAPK, but that the exact pathway involved is as yet unidentified.
Most data suggest that cAMP stimulates the COX-2 promoter via the CRE via either CREB or AP-1 proteins [29-31], both of which can be activated by MAPK [32]. MAPK activation can also prolong COX-2 mRNA half-life [32]. These effects seem to be isoform specific, because transforming growth factor-α increased COX-2 mRNA in epidermal keratinocytes via ERK-induced increases in transcription and p38-induced increases in mRNA half-life [33]. We observed an increase in COX-2 mRNA associated with increased COX-2 promoter activity with a greater increase in COX-2 protein. In mouse cardiac smooth muscle cells, activation of MAPK-activated protein kinase-2 (MK2), a kinase downstream of p38, increased COX-2 protein levels with out altering COX-2 mRNA levels or protein stability [34], which is consistent with our observations. Interestingly, we found that COX-2 protein remained elevated after COX-2 mRNA had returned to baseline probably reflecting the stability of COX-2 protein.
Most studies suggest that cAMP increases COX-2 expression typically on the background of an inflammatory stimulus. A recent review concluded that cAMP should be considered as a positive modulator of COX-2 expression, rather than an inducer in it own right and suggested that the clinical use of β2-mimetics or PG receptor agonists would not impact on prostanoid synthesis [35]. However, our data show that cAMP and PGE2 (via the cAMP linked EP-2 receptors) have a robust effect on COX-2 expression and prostanoid synthesis in human myometrial cells in the absence of an inflammatory stimulus, suggesting that in the myometrium at least, cAMP agonists have a significant effect on prostanoid synthesis. We found that the effect of cAMP was most marked for PGE2 and least for PGF2α. The cAMP-induced increase in both PGE2 and PGI2 could establish a positive feedback loop culminating in greater COX-2 expression. Clearly, the effect of cAMP on PG synthesis does not occur in isolation and, as cAMP agonists reduce spontaneous myometrial contractility, at least acutely [36], and rolipram pre-treatment reduces PTL in the mouse LPS model [37], it seems likely the overall effect of an increase in cAMP is to reduce the risk of PTL. However, our data taken with those of Fetalvero et al. [19], suggest that cAMP can also activate the myometrium and it is possible that the balance of these opposing effects determines the overall contractile state of human myometrium and may therefore offer an explanation for the limited effect of β-agonists in the treatment of threatened PTL. Thus, although it is accepted that cAMP induces myometrial relaxation acutely, through non-genomic effects, future work must define what its effects are on myometrial contractility in the medium to long-term and the mechanisms involved. This would clarify whether therapies based on increasing intracellular cAMP could be of potential benefit in the management of PTL.
Acknowledgments
This work was supported by a grant from Action Medical Research (SP4573). The authors thank all the women and staff on the labour ward of Chelsea and Westminster Hospital, who assisted with the provision and collection of human myometrial tissue.
Conflict of interests
The authors confirm that there are no conflicts of interest.
References
- 1.MacDorman MF, Mathews TJ. Behind international rankings of infant mortality: how the United States compares with Europe. NCHS Data Brief. 2009:1–8. [PubMed] [Google Scholar]
- 2.Meloni A, Melis M, Alba E, et al. Medical therapy in the management of preterm birth. J Matern Foetal Neonatal Med. 2009;3:72–6. doi: 10.1080/14767050903198256. [DOI] [PubMed] [Google Scholar]
- 3.Martin JA, Hamilton BE, Sutton PD, et al. Births: final data for 2005. Natl Vital Stat Rep. 2007;56:1–103. [PubMed] [Google Scholar]
- 4.Goldenberg RL, Culhane JF, Iams JD, et al. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landau R, Morales MA, Antonarakis SE, et al. Arg16 homozygosity of the beta2-adrenergic receptor improves the outcome after beta2-agonist tocolysis for preterm labor. Clin Pharmacol Ther. 2005;78:656–63. doi: 10.1016/j.clpt.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Simhan HN, Caritis SN. Prevention of preterm delivery. N Engl J Med. 2007;357:477–87. doi: 10.1056/NEJMra050435. [DOI] [PubMed] [Google Scholar]
- 7.Mehats C, Schmitz T, Oger S, et al. PDE4 as a target in preterm labour. BMC Pregnancy Childbirth. 2007;1:S12. doi: 10.1186/1471-2393-7-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparey C, Robson SC, Bailey J, et al. The differential expression of myometrial connexin-43, cyclooxygenase-1 and -2, and Gs alpha proteins in the upper and lower segments of the human uterus during pregnancy and labor. J Clin Endocrinol Metab. 1999;84:1705–10. doi: 10.1210/jcem.84.5.5644. [DOI] [PubMed] [Google Scholar]
- 9.Olson DM, Ammann C. Role of the prostaglandins in labour and prostaglandin receptor inhibitors in the prevention of preterm labour. Front Biosci. 2007;12:1329–43. doi: 10.2741/2151. [DOI] [PubMed] [Google Scholar]
- 10.Blumenfeld YJ, Lyell DJ. Prematurity prevention: the role of acute tocolysis. Curr Opin Obstet Gynecol. 2009;21:136–41. doi: 10.1097/GCO.0b013e3283292455. [DOI] [PubMed] [Google Scholar]
- 11.Kaminski K, Rechberger T, Oleszczuk J, et al. Biochemical and clinical evaluation of the efficiency of intracervical extraamniotic prostaglandin F2 alpha and intravenous oxytocin infusion to induce labour at term. Aust N Z J Obstet Gynaecol. 1994;34:409–13. doi: 10.1111/j.1479-828x.1994.tb01258.x. [DOI] [PubMed] [Google Scholar]
- 12.Cao Z, Liu LZ, Dixon DA, et al. Insulin-like growth factor-I induces cyclooxygenase-2 expression via PI3K, MAPK and PKC signaling pathways in human ovarian cancer cells. Cell Signal. 2007;19:1542–53. doi: 10.1016/j.cellsig.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyesanya RA, Lee ZP, Wu J, et al. Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. Faseb J. 2008;22:2639–51. doi: 10.1096/fj.07-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slater DM, Berger LC, Newton R, et al. Expression of cyclooxygenase types 1 and 2 in human foetal membranes at term. Am J Obstet Gynecol. 1995;172:77–82. doi: 10.1016/0002-9378(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 15.Zakar T, Olson DM, Teixeira FJ, et al. Regulation of prostaglandin endoperoxide H2 synthase in term human gestational tissues. Acta Physiol Hung. 1996;84:109–18. [PubMed] [Google Scholar]
- 16.Mohan AR, Sooranna SR, Lindstrom TM, et al. The effect of mechanical stretch on cyclooxygenase type 2 expression and activator protein-1 and nuclear factor-kappaB activity in human amnion cells. Endocrinology. 2007;148:1850–7. doi: 10.1210/en.2006-1289. [DOI] [PubMed] [Google Scholar]
- 17.Jung WK, Lee DY, Park C, et al. Cilostazol is anti-inflammatory in BV2 microglial cells by inactivating nuclear factor-kappaB and inhibiting mitogen-activated protein kinases. Br J Pharmacol. 2010;159:1274–85. doi: 10.1111/j.1476-5381.2009.00615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minguet S, Huber M, Rosenkranz L, et al. Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur J Immunol. 2005;35:31–41. doi: 10.1002/eji.200425524. [DOI] [PubMed] [Google Scholar]
- 19.Fetalvero KM, Zhang P, Shyu M, et al. Prostacyclin primes pregnant human myometrium for an enhanced contractile response in parturition. J Clin Invest. 2008;118:3966–79. doi: 10.1172/JCI33800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H, Xu B, Inoue H, et al. P38 MAPK mediates COX-2 gene expression by corticosterone in cardiomyocytes. Cell Signal. 2008;20:1952–9. doi: 10.1016/j.cellsig.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Yaqub S, Tasken K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumour immune responses by regulatory T cells. Crit Rev Oncog. 2008;14:57–77. doi: 10.1615/critrevoncog.v14.i1.40. [DOI] [PubMed] [Google Scholar]
- 22.Wadhwa S, Choudhary S, Voznesensky M, et al. Fluid flow induces COX-2 expression in MC3T3-E1 osteoblasts via a PKA signaling pathway. Biochem Biophys Res Commun. 2002;297:46–51. doi: 10.1016/s0006-291x(02)02124-1. [DOI] [PubMed] [Google Scholar]
- 23.Faour WH, Gomi K, Kennedy CR. PGE(2) induces COX-2 expression in podocytes via the EP(4) receptor through a PKA-independent mechanism. Cell Signal. 2008;20:2156–64. doi: 10.1016/j.cellsig.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Xu XJ, Reichner JS, Mastrofrancesco B, et al. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol. 2008;180:2125–31. doi: 10.4049/jimmunol.180.4.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sooranna SR, Engineer N, Loudon JA, et al. The MAPK dependent expression of prostaglandin H synthase-2 and interleukin 8 mRNA by myometrial cells: the differential effect of stretch and interleukin-1{beta} J Clin Endocrinol Metab. 2005;90:3517–27. doi: 10.1210/jc.2004-1390. [DOI] [PubMed] [Google Scholar]
- 26.Yin F, Wang YY, Du JH, et al. Noncanonical cAMP pathway and p38 MAPK mediate beta2-adrenergic receptor-induced IL-6 production in neonatal mouse cardiac fibroblasts. J Mol Cell Cardiol. 2006;40:384–93. doi: 10.1016/j.yjmcc.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Ster J, De Bock F, Guerineau NC, et al. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci U S A. 2007;104:2519–24. doi: 10.1073/pnas.0611031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amsen EM, Pham N, Pak Y, et al. The guanine nucleotide exchange factor CNrasGEF regulates melanogenesis and cell survival in melanoma cells. J Biol Chem. 2006;281:121–8. doi: 10.1074/jbc.M507595200. [DOI] [PubMed] [Google Scholar]
- 29.Schroer K, Zhu Y, Saunders MA, et al. Obligatory role of cyclic adenosine monophosphate response element in cyclooxygenase-2 promoter induction and feedback regulation by inflammatory mediators. Circulation. 2002;105:2760–5. doi: 10.1161/01.cir.0000018127.10968.34. [DOI] [PubMed] [Google Scholar]
- 30.Subbaramaiah K, Chung WJ, Dannenberg AJ. Ceramide regulates the transcription of cyclooxygenase-2. Evidence for involvement of extracellular signal-regulated kinase/c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways. J Biol Chem. 1998;273:32943–9. doi: 10.1074/jbc.273.49.32943. [DOI] [PubMed] [Google Scholar]
- 31.Kirtikara K, Raghow R, Laulederkind SJ, et al. Transcriptional regulation of cyclooxygenase-2 in the human microvascular endothelial cell line, HMEC-1: control by the combinatorial actions of AP2, NF-IL-6 and CRE elements. Mol Cell Biochem. 2000;203:41–51. doi: 10.1023/a:1007045600664. [DOI] [PubMed] [Google Scholar]
- 32.Wu MH, Wang CA, Lin CC, et al. Distinct regulation of cyclooxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J Clin Endocrinol Metab. 2005;90:286–95. doi: 10.1210/jc.2004-1612. [DOI] [PubMed] [Google Scholar]
- 33.Matsuura H, Sakaue M, Subbaramaiah K, et al. Regulation of cyclooxygenase-2 by interferon gamma and transforming growth factor alpha in normal human epidermal keratinocytes and squamous carcinoma cells. Role of mitogen-activated protein kinases. J Biol Chem. 1999;274:29138–48. doi: 10.1074/jbc.274.41.29138. [DOI] [PubMed] [Google Scholar]
- 34.Streicher JM, Ren S, Herschman H, et al. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010;106:1434–43. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein T, Shephard P, Kleinert H. Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim Biophys Acta. 2007;1773:1605–18. doi: 10.1016/j.bbamcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Simpkin JC, Kermani F, Palmer AM, et al. Effects of corticotrophin releasing hormone on contractile activity of myometrium from pregnant women. Br J Obstet Gynaecol. 1999;106:439–45. doi: 10.1111/j.1471-0528.1999.tb08297.x. [DOI] [PubMed] [Google Scholar]
- 37.Schmitz T, Souil E, Herve R, et al. PDE4 inhibition prevents preterm delivery induced by an intrauterine inflammation. J Immunol. 2007;178:1115–21. doi: 10.4049/jimmunol.178.2.1115. [DOI] [PubMed] [Google Scholar]