

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by loss of elastic fibres from small airways and alveolar walls, with the decrease in elastin increasing with disease severity. It is unclear why there is a lack of repair of elastic fibres. We have examined fibroblasts cultured from lung tissue from subjects with or without COPD to determine if the secretory profile explains lack of tissue repair. In this study, fibroblasts were cultured from lung parenchyma of patients with mild COPD [Global initiative for chronic Obstructive Lung Disease (GOLD) 1, n= 5], moderate to severe COPD (GOLD 2–3, n= 12) and controls (non-COPD, n= 5). Measurements were made of proliferation, senescence-associated β-galactosidase-1, mRNA expression of IL-6, IL-8, MMP-1, tropoelastin and versican, and protein levels for IL-6, IL-8, PGE2, tropoelastin, insoluble elastin, and versican. GOLD 2–3 fibroblasts proliferated more slowly (P < 0.01), had higher levels of senescence-associated β-galactosidase-1 (P < 0.001) than controls and showed significant increases in mRNA and/or protein for IL-6 (P < 0.05), IL-8 (P < 0.01), MMP-1 (P < 0.05), PGE2 (P < 0.05), versican (P < 0.05) and tropoelastin (P < 0.05). mRNA expression and/or protein levels of tropoelastin (P < 0.01), versican (P < 0.05), IL-6 (P < 0.05) and IL-8 (P < 0.05) were negatively correlated with FEV1% of predicted. Insoluble elastin was not increased. In summary, fibroblasts from moderate to severe COPD subjects display a secretory phenotype with up-regulation of inflammatory molecules including the matrix proteoglycan versican, and increased soluble, but not insoluble, elastin. Versican inhibits assembly of tropoelastin into insoluble elastin and we conclude that the pro-inflammatory phenotype of COPD fibroblasts is not compatible with repair of elastic fibres.
Keywords: chronic obstructive pulmonary disease, pulmonary fibroblasts, pro-inflammatory phenotype, elastin, versican
Introduction
Chronic obstructive pulmonary disease (COPD), characterized by irreversible airflow obstruction in the small airways [1, 2], involves both small airway remodelling and emphysematous changes. In moderate and severe COPD elastic fibres are lost from the alveolar walls, a change thought to lead to weakening and loss of alveolar attachments to small airways and to their subsequent collapse in expiration [3]. The loss of elastic fibres in emphysema has been explained by the protease-antiprotease hypothesis [4], elaborated following the description in 1963 of emphysema due to α1-antitrypsin deficiency [5] and associated loss of inhibition of neutrophil elastase. There are reasons, however, for questioning whether neutrophil elastase and α1-antitrypsin are of central importance in COPD. Inherited deficiency of α1-antitrypsin is uncommon and only accounts for a small proportion of cases of COPD, and reduction of α1-antitrypsin levels in the lung has not been consistently demonstrated in individuals with COPD without α1-antitrypsin deficiency [6].
The majority of research on COPD has focussed on the idea that emphysema can be explained by progressive destruction of the alveolar walls but there are several lines of evidence to suggest that inhibition of repair may be important in the pathogenesis of COPD [7, 8]. Thus when damage occurs as a result of smoking there may be differences in the ability to repair this damage, and individuals with impaired repair mechanisms may be more likely to develop COPD. Impaired repair, however, does not appear to be due to the inability to synthesize structural matrix proteins such as elastin and collagen.
Elastin is a long-lived molecule with most elastin deposition occurring early in development with little turnover in adult life [9]. Several experimental studies, however, have reported that matrix proteins, including elastin and collagen, are re-synthesized in emphysematous lungs in animal models [10, 11]. Synthesis of elastin, however, does not result in restoration of physiological function, presumably because functional fibres fail to form [12]. Similarly, investigations on human emphysematous lungs report re-synthesis of matrix proteins, but with changed distribution patterns of collagen and elastin, including clumping of elastin deposits in the free edges of alveolar walls [13, 14].
More recently it has been reported that elastin mRNA expression is significantly increased in lungs with very severe COPD (GOLD 4) compared with non-COPD controls and mild or moderate COPD [15]. In situ hybridization localized this increased elastin expression to the alveolar walls, but neither the elastic fibre content nor desmosine levels were increased per lung volume.
Our recent research also provides support for the idea that loss of repair mechanisms may be important in the development of COPD. The impaired repair appears to be due to the inability to form functional insoluble elastic fibres rather than the lack of synthesis of the elastin precursor tropoelastin. We examined lobectomy lung tissue from patients with bronchial carcinoma and found that whereas elastic fibre content was significantly decreased in patients with mild to moderate COPD [16], versican, a matrix proteoglycan that mediates inflammatory changes, was increased [17]. This increase may be particularly relevant because versican has been shown to inhibit the assembly of tropoelastin into elastic fibres [18-20], which may explain the reduced capacity of COPD lung to form new elastic fibres despite the ability to synthesize the elastin precursor tropoelastin.
In order to examine more closely this inhibition-of-repair hypothesis, we have cultured pulmonary fibroblasts from the lungs of patients with variable degrees of COPD, and from lungs of individuals with a comparable smoking history but normal lung function, to determine if the synthetic profile of fibroblasts, with respect to elastin synthesis and deposition, and synthesis of versican and inflammatory cytokines, changes with disease progression and explains the inhibition of repair seen in lungs with advanced COPD.
Methods
Subjects
Primary fibroblasts were obtained from excised lung tissue collected during surgery for bronchial carcinoma at Auckland Hospital, Auckland, New Zealand. All patients had lung function measured preoperatively and those with a clearly documented history of asthma or bronchodilator reversibility [increase in forced expiratory volume in one second (FEV1) > 10% of predicted normal values] were excluded from the study. Patients with other lung diseases, such as bronchiectasis and interstitial lung disease, were also excluded. All enrolled subjects were divided into three groups according to the GOLD 2007 criteria, i.e. control (non-COPD), mild COPD (GOLD stage 1) and moderate to severe COPD (GOLD stage 2–3). The study was approved by the Northern X Regional Ethics Committee, Ministry of Health. Written informed consent for permission to use the lung tissue for research was obtained preoperatively.
Cultures of human lung fibroblasts
Fibroblasts were cultured from peripheral pleura-free parenchymal specimens of the resected lobe remote from the tumour. The tissues were immediately transferred into explant culture medium [DMEM, 10% foetal calf serum, penicillin (100 ng/ml) and streptomycin (100 ng/ml); Invitrogen Life Technologies, Grand Island, NY, USA], and then minced with a scalpel (1–2 mm2 pieces) and transferred into 25 cm2 culture flasks for primary culture at 37°C in 5% CO2. Cells were trypsinized when confluent and passaged. Cells were evaluated in passage 2 after primary culture to confirm that more than 99% of the cells were fibroblasts with typical fibroblast morphology, positive staining for anti-vimentin antibody (Cell Signaling, Danvers, MA, USA), negative staining for anti-pan cytokeratin antibody (Covance, Princeton, NJ, USA), and mostly negative staining for anti-α Smooth Muscle Action (SMA) antibody (Sigma-Aldrich, St. Louis, MO, USA). Cell-free supernanants and RNA were obtained from 48-hr subconfluent passage 3 cultures for further ELISA and PCR analysis.
Proliferation
Proliferation was measured on passage 3 fibroblasts using the AlarmaBlue (AB) assay (Invitrogen Life Technologies) as described previously [21]. Briefly, fibroblasts were seeded at 5000/well into 96-well plates in serum-free culture medium and AB added at a final concentration of 10%. After 6-hr incubation in a humidified atmosphere containing 5% CO2, the %AB reduction, which proved linear to cell number, was measured and recorded as the baseline reading. After additional culture for 24 hrs in complete culture medium (5% CO2 at 37°C), the %AB reduction was measured again. A proliferation index was calculated as the 24 hrs reading divided by the baseline reading.
Staining for senescence-associated β-galactosidase (SA-β-Gal)
A total of 5000 fibroblasts (passage 3) were transferred onto glass cover chamber slides (LAB-TEK, Nunc, 0.8 cm2). After culture for 24 hrs under standardized conditions (37°C, 5% CO2), staining for SA-β-Gal activity at pH 6.0 was performed using a SA-β-Gal staining kit (Cell Signaling Technologies, Beverly, MA, USA). Cells positive for the blue stain were counted under visible light. Total cell number was determined by counting DAPI stained cells under UV light.
Real-time PCR
Total RNA extraction was performed using Trizol Reagent (Invitrogen Life Technologies) and 1 μg of total RNA transcribed using SuperScript® ViLO™ cDNA synthesis kit (Invitrogen Life Technologies) according to the manufacturer's instructions. Real-time PCR was performed using Express SyBR® GreenERTM SuperMix with Premixed ROX (Invitrogen Life Technologies) following the manufacturer's instructions. PCR assays were performed in duplicate on a 7900HT real-time PCR machine (Applied Biosystems, Life Technologies) with cycler conditions as follows: incubation for 2 min. at 50°C followed by another incubation step at 95°C for 10 min., afterwards 15 sec. at 95°C and 1 min. at 60°C for 40 cycles. Reaction specificity was evaluated by melting curve analysis, performed by heating the plate from 55 to 95°C and measuring SYBR Green dissociation from the amplicons. The calculation of threshold cycles (Ct values) and further analysis of these data were performed by Sequence Detector (Applied Biosystems, Life Technologies) software. The relative mRNA expression of target genes in each sample was quantified and normalized to the GAPDH mRNA levels by the 2−ddCt method [22]. The sequences of each primer pair used for each mRNA analysed are listed in Table 1.
Table 1.
Details of each primer pair used for each mRNA
| Forward primer (5′−3′) | Reverse primer (5′−3′) | |
|---|---|---|
| Elastin | TCTGAGGTTCCCATAGGTTAGGG | CTA AGCCTGCAGCAGCTCCT |
| Collagen-1 | GGGCAAGACAGTGATTGAATA | ACGTCGAAGCCGAATTCCT |
| MMP-1 | CCCCAAAAGCGTGTGACAGTA | GGTAGAAGGGATTTGTGCG |
| Biglycan | TGTGTGTGTGTCTTGTGCTT | AGTGAAAGGGACAGGCGAAG |
| Decorin | TGGCAACAAAATCAGCAGAG | GCCATTGTCAACAGCAGAGA |
| Versican 1 | CCCAGTGTGGAGGTGGTCTAC | CGCTCAAATCACTCATTCGACGTT |
| EBP | CCATCCAGACATTACCTGGC | TTGATGGGCCCAGAGGGACA |
| IL-6 | GACAGCCACTCACCTCTTCA | TTCACCAGGCAAGTCTCCTC |
| IL-8 | CTGCGCCAACACAGAAATTATTGTA | TTCACTGGCATCTTCACTGATTCTT |
MMP-1: matrix metalloproteinase-1; EBP: elastin binding protein; IL: interleukin.
ELISA
For ELISA analyses the following commercial kits were used: matrix metalloproteinase-1 (MMP-1) (Calbiochem, Merck Chemicals, NJ, USA), prostaglandin E2 (PGE2) (R&D Systems, Minneapolis, MN, USA), Interleukin-6 (IL-6) and IL-8 (Becton and Dickinson, NJ, USA) and versican (CUSABio, Wuhan, China). The level of protein of interest in cell-free culture supernatants was determined according to the manufacturer's instructions.
Quantification for elastin
Passage 3 cells were seeded at 50,000cells/well in 6-well plates and cultured for 2 weeks. Cell-free supernatant was collected for measuring soluble elastin. For insoluble elastin, cell layers were scraped in 0.1N NaOH, sedimented by centrifugation, and boiled in 0.5 ml of 0.1N NaOH for 45 min. to solubilize all matrix components except elastin. The resulting pellets containing the insoluble elastin were solubilized by boiling in 5.7N HCl for 1 hr for further analysis. Fastin™ elastin assay kit from Biocolor (County Antrim, UK) was used to measure the protein concentration according to the manufacturer's instruction.
Statistics
All statistical analyses were performed using GraphPadPrism 5.0 for Windows. D'Agostino and Pearson omnibus normality test was performed on all data. Data normally distributed were expressed as mean ± S.D., and one-way analysis of variance followed by Bonferroni post-test was performed to determine whether the differences between the groups were statistically significant. Otherwise, data were expressed as median (minimum, maximum), and comparisons were made using the Kruskal–Wallis analysis followed by Dunn's multiple comparison test. Correlation analyses were carried out using Pearson or Spearman methods depending on the normality of the data distribution. Differences were considered significant at the level of P < 0.05.
Results
Clinical and demographic features of the subjects
Twenty-two patients met the criteria for the study and were divided into three groups according to the results of spirometry: control (non-COPD, n= 5), mild COPD (GOLD stage 1, n= 5) and moderate to severe COPD (GOLD stage 2–3, n= 12) (Table 2). The three groups were similar in age, gender and smoking status but differed significantly in lung function. As expected, the subjects in the moderate to severe COPD group (GOLD stage 2–3) had significantly lower FEV1% predicted and FEV1/forced vital capacity (FVC)% values compared with non-COPD group and mild COPD group (Table 2).
Table 2.
Clinical and demographic features of the subjects
| Control (non-COPD)* | Mild COPD (stage 1)† | Moderate to severe COPD (stage 2–3)§ | P value (one-way analysis of variance) | |
|---|---|---|---|---|
| Gender (M/F) | 3/2 | 3/2 | 7/5 | NS |
| Age (yrs) | 69.5 ± 3.1 | 62.3 ± 13.1 | 66.4 ± 9.2 | NS |
| Smoking pack-yrs | 30.2 ± 10.1 | 33.3 ± 15.3 | 47.1 ± 18.0 | NS |
| FEV1 (L) | 2.25 ± 0.39 | 1.84 ± 0.34 | 1.47 ± 0.36 | 0.002 |
| FEV1% pred | 90.2 ± 5.4 | 91.3 ± 10.3 | 59.8 ± 9.5 | <0.001 |
| FVC (L) | 2.971 ± 0.452 | 2.765 ± 0.621 | 2.747 ± 0.820 | NS |
| FVC% pred | 89.3 ± 10.2 | 104.3 ± 9.6 | 78.8 ± 17.3 | 0.046 |
| FEV1/FVC% | 79.3 ± 9.4 | 65.1 ± 6.2 | 54.9 ± 8.3 | <0.001 |
Data are presented as mean ± SD, and the comparisons were made by one-way analysis of variance followed by Bonferroni post-test. COPD: chronic obstructive pulmonary disease; M: male; F: female; FEV1: forced expiratory volume in one second; % pred: % predicted; FVC: forced vital capacity; NS: non-significant. *Subjects had an FEV1 ≥ 80% pred and FEV1/FVC > 70%. †Subjects had an FEV1 ≥ 80% pred and FEV1/FVC < 70%. §Subjects had an FEV1 < 80% pred and FEV1/FVC < 70%.
Cell morphology
The primary cultures of lung fibroblasts displayed morphological features typical of fibroblasts and immunostaining demonstrated that >99% of cells were vimentin-positive (Fig. 1A), anti-pan cytokeratin negative and mostly anti-αSMA negative (data not shown). Notably, the slower growing fibroblasts from GOLD stage 2–3 COPD patients were often less spindle-shaped and more flattened and spread than cells from control patients (non-COPD) (Fig. 1B and C), features shown previously to be associated with slower growth and an elastogenic phenotype [23, 24].
Fig 1.

Cultured fibroblasts from representative non-COPD subject and COPD subject. (A) Vimentin-stained (red) subconfluent fibroblasts from human lung tissue from non-COPD subject. All cells had morphological features typical of fibroblasts. Cell nuclei are stained with Hoechst (blue). (B) Methylene blue stained sparse fibroblasts from non-COPD subject showing typical spindle-shaped morphology predominant in cultures of fibroblasts cultured from the lungs of non-COPD subjects. (C) Methylene blue stained fibroblasts from COPD (GOLD stage 3) subject showing flattened spread morphology frequently seen in cultures of fibroblasts from COPD subjects.
Cell proliferation and cell senescence
Proliferation, determined by the AB assay, was significantly slower in fibroblasts from GOLD stage 2–3 COPD subjects (1.21±0.22) than from non-COPD subjects (1.65 ± 0.23) (P < 0.01) (Fig. 2A). Cell senescence, determined by β-galactosidase (SA-β-Gal) staining, was significantly increased in the GOLD stage 2–3 COPD fibroblasts (32.5%± 6.5%) compared to non-COPD controls (19.5%± 2.3%) (P < 0.001) and GOLD stage 1 fibroblasts (19.3%± 1.8%) (P < 0.01) (Fig. 2B).
Fig 2.
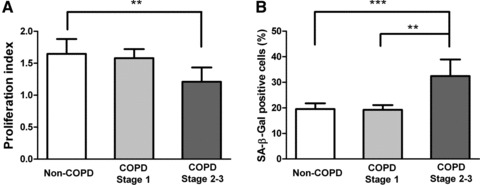
Reduced proliferation and increased senescence in fibroblasts from moderate and severe COPD subjects. (A) Proliferation index (Alarma Blue assay) showing significantly reduced proliferative capacity of fibroblasts cultured from the COPD GOLD stage 2–3 group compared with the control and GOLD stage 1 groups. (B) Significantly increased percentage of SA-β-Gal positive fibroblasts from COPD GOLD stage 2–3 subjects compared to control and GOLD stage 1 subjects. SA-β-Gal: senescence-associated β-galactosidase. **P < 0.01; ***P < 0.001.
mRNA and protein levels
In previous studies MMP-1 has been measured as an important indicator of airway inflammation and of developing COPD [25]. We found that MMP-1 mRNA was significantly increased (P < 0.05) in GOLD stage 2–3 compared with non-COPD and GOLD 1 cultures (Fig. 3). IL-6 and IL-8 mRNA expression levels were similarly significantly (P < 0.01) raised in the GOLD stage 2–3 cultures (Fig. 4A and B) compared to the other two groups, as were the respective protein levels (P < 0.05 and P < 0.001) (Fig. 4C and D). mRNA expression levels of IL-6 and IL-8 were negatively correlated with FEV1% of predicted (Spearman r=–0.632, P= 0.004; and Spearman r=–0.626, P= 0.003, respectively). Secreted levels of IL-6 and IL-8 also negatively correlated with FEV1% of predicted (Pearson r=–0.660, P= 0.014; and Pearson r=–0.821, P < 0.001, respectively).
Fig 3.
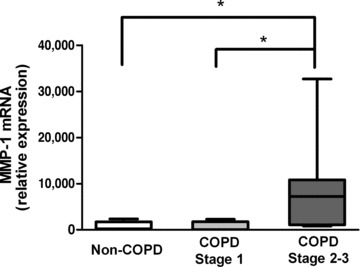
MMP-1 synthesis. MMP-1 mRNA expression, detected by real-time RT-PCR, relative to expression of GAPDH. Data are expressed as median, upper and lower quartiles (boxed area), and range. MMP-1: matrix metalloproteinase-1; *P < 0.05.
Fig 4.
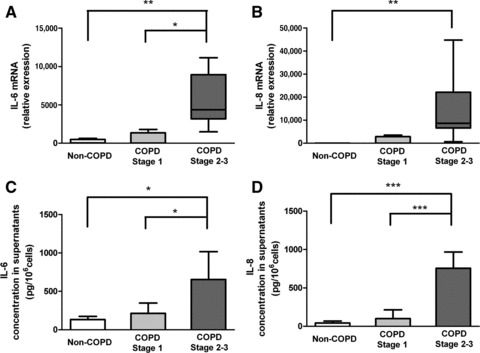
IL-6 and IL-8 synthesis and secretion. (A and B) mRNA expression for IL-6 and IL-8, detected by real-time RT-PCR, relative to expression of GAPDH. Data are expressed as median, upper and lower quartiles (boxed area), and range median (range). (C and D) IL-6 and IL-8 levels in cell-free supernatants. Data are expressed as mean ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001.
Levels of PGE2 in supernatants were significantly higher in the GOLD stage 2–3 COPD fibroblast cultures than in non-COPD cultures (P < 0.05) (Fig. 5). There was a weak but significant relationship between PGE2 and FEV1% of predicted (Pearson r=–0.502, P= 0.034).
Fig 5.

PGE2 secretion. Levels of PGE2 in supernatant were significant higher in GOLD stage 2–3 COPD group than in non-COPD and GOLD stage 1 groups. Data are expressed as mean ± S.D. PGE2: prostaglandin E2. *P < 0.05.
Elastin precursor mRNA (hnRNA) was significantly (P < 0.05) increased in fibroblasts from the GOLD stage 2–3 subjects compared to the non-COPD controls (Fig. 6A). Patients with a lower FEV1% of predicted had higher mRNA relative expression than subjects with higher FEV1% of predicted values (Spearman r=–0.751, P= 0.001) (Fig. 6B). Similarly, soluble elastin released into the media was significantly higher in the GOLD stage 2–3 cultures compared with non-COPD (P < 0.05) and GOLD stage 1 cultures (P < 0.05) (Fig. 6C). Soluble elastin level was also negatively related to the FEV1% of predicted value of patients (Spearman r=–0.768, P= 0.002) (Fig. 6D). There was no difference between groups, however, in the amount of insoluble elastin deposited into the cell layers (Fig. 6E), indicating that the increased amount of soluble tropoelastin in the GOLD stage 2–3 cultures was not incorporated into insoluble elastin. Variation in levels of insoluble elastin between subjects was not correlated with FEV1% of predicted (P= 0.5). Elastin binding protein (EBP) mRNA was also significantly increased (P= 0.032) in GOLD stage 2–3 cultures (Table 3).
Fig 6.

Synthesis and secretion of elastin. (A) Elastin precursor mRNA expression, detected by real-time RT-PCR, relative to expression of GAPDH. Data are expressed as median, upper and lower quartiles (boxed area), and range. (B) Correlation between FEV1% predicted and elastin mRNA relative expression. (C) Levels of soluble elastin in cell-free supernatants. Data are expressed as mean ± S.D. (D) Correlation between FEV1% of predicted and tropoelastin levels in the supernatant. (E) Insoluble elastin levels in cell layers. Data are expressed as mean ± S.D. *P < 0.05.
Table 3.
Comparison of relative mRNA expression index in primary human lung fibroblasts among the mild COPD, moderate to severe COPD and non-COPD group
| Control (non-COPD)* | Mild COPD (stage 1)† | Moderate to severe COPD (stage 2–3)§ | P value (Kruskal–Wallis test) | |
|---|---|---|---|---|
| Tropoelastin | 2.24 (0.92, 5.74) | 2.00 (1.00, 7.06) | 8.46 (3.12, 33.00) | 0.009 |
| Collagen-1 | 5374 (1523, 27,608) | 517.3 (445.6, 2160) | 12,610 (1349, 77,351) | 0.024 |
| MMP-1 | 203.5 (8.26, 2352) | 59.32 (11.9, 2287) | 7202 (822.6, 32,684) | 0.011 |
| Biglycan | 239.1 (109.8, 4294) | 79.8 (53.4, 350.6) | 1186 (130.2, 3582) | 0.028 |
| Decorin | 1131 (257.9, 1888) | 216.5 (53.4, 7757) | 3446 (0.03, 29,053) | NS |
| EBP | 0.25 (0.05, 1.30) | 0.10 (0.02, 0.73) | 2.40 (0.02, 11.39) | 0.032 |
| Versican 1 | 907.0 (134.7, 1445) | 177.1 (51.5, 2683) | 5196 (1965, 17,648) | 0.002 |
| IL-6 | 49.7 (3.13, 630) | 19.2 (9.8, 1805) | 4365 (1498, 11,147) | 0.002 |
| IL-8 | 0.79 (0.11, 3.4) | 28.54 (0.71, 3535) | 8675 (634, 44,780) | 0.002 |
Data are expressed as median (minimum, maximum), and comparisons were made by the Kruskal–Wallis test followed by Dunn's post-test. NS: non-significant; COPD: chronic obstructive pulmonary disease; MMP-1: matrix metalloproteinase-1; EBP: elastin binding protein; IL: interleukin; *Subjects had an FEV1 ≥ 80% predicted and FEV1/FVC > 70%. †Subjects had an FEV1 ≥ 80% predicted and FEV1/FVC < 70%. §Subjects had an FEV1 < 80% predicted and FEV1/FVC < 70%.
Versican mRNA (Fig. 7A) and versican levels measured by ELISA (Fig. 7C) were significantly (P < 0.05) increased in GOLD stage 2–3 cultures compared to the non-COPD controls. Moreover, versican mRNA expression was strongly negatively correlated with FEV1% predicted (Spearman r=–0.823, P= 0.001) (Fig. 7B). A similar correlation was detected for secreted versican (Pearson r=–0.709, P= 0.02) (Fig. 7D). Versican production by GOLD stage 1 fibroblasts was not significantly different from non-COPD fibroblasts, although versican mRNA was slightly non-significantly elevated (Fig. 7A). Biglycan mRNA was also increased (P= 0.028) in GOLD stage 2–3 cultures while the collagen-associated proteoglycan decorin showed a non-significant increase (Table 3). Collagen-1 mRNA was significantly increased (P= 0.024) in GOLD stage 2–3 cultures (Table 3).
Fig 7.
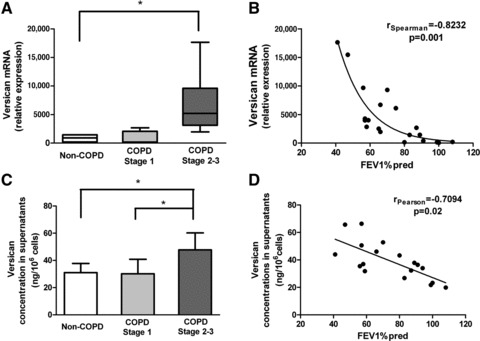
Synthesis and secretion of versican. (A) Versican (variant V1) mRNA expression, detected by real-time RT-PCR, relative to expression of GAPDH. Fibroblasts were cultured for 2 weeks. Data are expressed as median, upper and lower quartiles (boxed area), and range. (B) Correlation between FEV1% predicted and versican mRNA relative expression. (C) Versican levels in cell-free supernatants. Data are expressed as mean ± S.D. Correlation between FEV1% predicted and versican levels in supernatants. *P < 0.05.
Discussion
This study demonstrates that fibroblasts derived from the peripheral parenchyma of lungs of patients with moderate to severe COPD (GOLD stages 2–3) have a distinctly different phenotype from fibroblasts cultured from patients with mild COPD (GOLD stage 1) or without COPD. Fibroblasts from the GOLD stage 2–3 COPD group had slower proliferation rates and increased levels of SA β-Gal compared to fibroblasts from the control and mild COPD groups, but were significantly more metabolically active, synthesizing increased amounts of inflammatory cytokines IL-6 and IL-8 and PGE2, and the pro-inflammatory matrix proteoglycan versican. Synthesis of soluble tropoelastin was also increased. The increases in both versican and tropoelastin were negatively correlated with FEV1% of predicted values across all patients. Collectively, these data point to fibroblasts in the alveolar walls from lungs with COPD having an active pro-inflammatory secretory profile that is enhanced with deterioration of lung function.
The data are consistent with previous studies demonstrating an inflammatory and secretory phenotype in vivo for COPD lung, and with results on in vitro studies on cells from severe COPD, but the findings presented here additionally establish that the in vivo phenotype persists in vitro and that change in phenotype is closely related to disease progression with marked changes in levels of inflammatory and matrix molecules occurring from GOLD 1 to GOLD 2–3 stages of the disease.
The increased synthesis of soluble elastin indicates that COPD fibroblasts are capable of producing this extracellular component of major importance to alveolar structure and function; however, the increase in elastin message and in soluble tropoelastin was not matched by an increase in insoluble elastin, necessary for fibre formation. In previous immunohistochemical studies of COPD lung parenchyma we found that elastin content, measured by morphometry, progressively decreases as FEV1 decreases [16] and is associated with a reciprocal and progressive increase in versican [16, 17]. A more recent study has found that fibroblasts cultured from peripheral lung samples from COPD patients with severe COPD (GOLD 4) show elevated production of versican [26], as found in this current study for GOLD 2–3 patients. The increase in versican in COPD may be particularly relevant as versican inhibits the assembly of tropoelastin into insoluble cross-linked elastic fibres, both in cell culture and in vivo[18-20]. Thus the elevated production of versican in COPD is consistent with the lack of formation of new mature elastic fibres in COPD lung, despite fibroblasts synthesizing increased amounts of tropoeolastin.
Versican belongs to the family of large aggregating chondroitin sulphate (CS) proteoglycans located primarily within the extracellular matrix. CS chains, constituent to three isoforms of versican (V0, V1, V2), inhibit the assembly of tropoelastin into elastic fibres by displacing EBP from the cell surface. EPB is the receptor that chaperones tropoelastin through the golgi to the external cell membrane [19] and its displacement from the cell surface prevents the transfer of tropoelastin to the microfibrillar scaffold [19]. As a result, fibre formation is disrupted. The increase in EBP mRNA in GOLD stage 2–3 fibroblasts, along with the increase in soluble elastin, also indicates that these cells have elastogenic potential that is not fully realised. Notably, mRNA for biglycan, which also contains CS chains, was also increased and, similar to versican, this matrix proteoglycan also inhibits elastic fibre formation [27]. We postulate that the mismatch between levels of soluble and insoluble elastin, seen in this current study, can be explained by the elevated production of CS-containing matrix proteoglycans preventing fibre assembly.
Experimental evidence has shown that impaired elastic fibre assembly, caused by pericellular accumulation of CS proteoglycans, is reversible. Treatment of skin fibroblasts that overproduce versican, as occurs in Costello syndrome, with chondroitinase reverses the elastic fibre deficiency [19]. Knockdown of versican production by vascular smooth muscle cells (SMC) through overexpression of versican antisense similarly enhances elastic fibre deposition [20], as does overexpression of the glycosaminoglycan-deficient variant of versican, V3 [13]. Cells overexpressing V3 have a significantly lower level of the larger versican variant V1 on the cell surface, where it is attached to hyaluronan, which reduces CS in the pericellular region [23] allowing a longer residence time for EBP [18]. V3, versican antisense and chain-less mutant biglycan are also effective in vivo, and have been used to create elastin-rich, versican-depleted neointima [20, 24, 27]. Overexpression of V3, leading to decreased cell-associated CS, has also been used successfully to restore elastic fibre formation in cultures of skin fibroblasts from Costello syndrome patients [28].
Thus, findings from the present and previous studies [16, 17] raise a testable hypothesis, namely that repair of the elastin network in lung parenchyma, and possibly restoration or improvement of physiological function, might be achievable by reducing the CS proteoglycan content of alveolar walls. Lowering of versican in vivo might be particularly effective as the mechanical movements of ventilation likely have a role in determining the pattern and placement of elastic fibres during repair. Moreover, increased assembly of the secreted tropoelastin may lead to a decrease in the level of free tropoelastin-derived peptides that are capable of inducing production of elastolytic MMPs [29].
On the other hand, our study also showed that the inflammatory cytokines IL-6 and IL-8 mRNA and protein levels are elevated in fibroblasts from patients with moderate to severe COPD compared to control fibroblasts. IL-6 and IL-8 are regarded to play an important role in the inflammation of COPD. IL-6 acts as a proinflammatory cytokine and is also associated with epithelial apoptosis and injury in COPD [30]. IL-8 is a potent attractant for neutrophils and is partly responsible for the acute exacerbation and disease progression of COPD [31, 32]. The increased levels of IL-6 or IL-8 had been reported in sputum [31], exhaled breath condensate [32] and blood [33]. In this study, an increase in the mRNA levels of both IL-6 and IL-8 was found in primary human fibroblasts from GOLD 2–3 COPD patients compared to non-COPD controls. In addition, the levels were significantly and negatively correlated with the value of FEV1% of predicted.
These cytokines are also frequently associated with elevated levels of versican [34-36], which is also pro-inflammatory [37-41] and, acting through Toll-like receptor 2 (TLR2), a facilitator of metastasis in lung [37]. It is likely that persistent elevation of these cytokines, along with elevated MMP-1, inhibits reversion of COPD fibroblasts to the non-inflammatory phenotype that would be necessary for effective long-term remodelling and repair to take place. Elevation of PGE2 could also reduce repair responses as it inhibits several fibroblast repair responses, including chemotaxis [42] and proliferation [43, 44]; on the other hand it increases expression and activity of MMP-2 [44]. A further difficulty in activation of effective repair processes may be the expression by COPD fibroblasts of SA-β-Gal, also noted by others for emphysema [45], and thought to indicate premature aging. Telomere length, however, is not changed and any aging appears to be by a telomere-independent mechanism [45].
The findings of the present study suggest that a reduction in inflammation and in particular a reduction in CS-containing versican in lungs of patients with mild to moderate COPD may be an effective strategy for decreasing the rate of elastin loss, and possibly for restoration of elastin and repair of emphysematous changes. Increases in the elastin content of neointimal repair tissue in blood vessels subjected to angioplasty has been achieved through overexpression of versican antisense [20] and more recently it has been demonstrated that vascular SMC in vivo expressing the CS-depleted versican variant V3 result in increased intimal elastin content, decreased production of CS-containing versican and reduced macrophage ingress associated with cholesterol-induced inflammatory changes [46]. It remains to be determined if similar treatments, perhaps in combination with agents that reduce MMP levels, such as the anticholinergic bronchodilator tiotropium bromide [47, 48], might be effective in reducing the inflammatory changes associated with COPD and in creating a matrix environment permissive for the assembly of tropoelastin into insoluble fibres. Regardless, the persistence of an inflammatory phenotype in COPD would appear to be a major inhibitor of repair.
Authors’ contribution
Dr. Zhang contributed to collecting clinical tissue samples, performing cell biology experiments and writing up the methods and results. Dr. Wu's major contributions included experimental design, collecting clinical tissue samples, performing cell and molecular experiments, writing up the main structure of manuscript and editing manuscript. Professor Qu and Professor Bai contributed to project design and applying for research grants. Professor Merrilees contributed to original project conception and design, provided critical technical support in experimental design, and critical enhancement of the manuscript. Professor Black was instrumental in the project conception and design, in applying for project grants and in oversight of project direction.
Dedication
This article is dedicated to our close friend and mentor Professor Peter Black who died suddenly in January 2010. Peter was instrumental in the conception and design of this study. His death is a huge loss to us personally and to the COPD research community.
Acknowledgments
This work was supported by Auckland Medical Research Foundation, University of Auckland Faculty of Medical and Health Sciences Research Development Fund, the National Natural Science Foundation of China (No. 81000013) and the Shanghai Leading Academic Discipline Project (No. B115).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airways obstruction in chronic obstructive lung disease. N Engl J Med. 1968;278:1355–60. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 2.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–59. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 3.Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164:S28–38. doi: 10.1164/ajrccm.164.supplement_2.2106061. [DOI] [PubMed] [Google Scholar]
- 4.Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. 2008;5:475–7. doi: 10.1513/pats.200708-126ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurell CB, Eriksson S. The electrophoretic alpha-1-globulin pattern in alpha-1-antitrypsin deficiency. Scand J Clin Lab Invest. 1963;15:132–40. [Google Scholar]
- 6.Stone PJ, Calore JD, McGowan SE, et al. Smokers do not have less functional alpha 1-protease inhibitor in their lower respiratory tracts than nonsmokers. Chest. 1983;83:65S–6S. [PubMed] [Google Scholar]
- 7.Holz O, Zühlke I, Jaksztat E, et al. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J. 2004;24:575–9. doi: 10.1183/09031936.04.00143703. [DOI] [PubMed] [Google Scholar]
- 8.Togo S, Holz O, Liu X, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med. 2008;178:248–60. doi: 10.1164/rccm.200706-929OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro SD, Endicott SK, Province MA, et al. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons related radiocarbon. J Clin Invest. 1991;87:1828–34. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snider GL, Lucey EC, Stone PJ. State of the art: animal models of emphysema. Am Rev Resp Dis. 1986;133:149–69. doi: 10.1164/arrd.1986.133.1.149. [DOI] [PubMed] [Google Scholar]
- 11.Chambers RC, Laurent GJ. The lung. In: Comper WD, editor. Extracellular matrix, Volume 1: Tissue Function. The Netherlands: Harwood Academic Publishers GmbH; 1996. pp. 378–409. [Google Scholar]
- 12.Morris SM, Stone PJ, Snider GL, et al. Ultrastructural changes in hamster lung four hours to twenty-four days after exposure to elastase. Anat Rec. 1981;201:523–35. doi: 10.1002/ar.1092010309. [DOI] [PubMed] [Google Scholar]
- 13.Osman M, Cantor JO, Roffman S, et al. Cigarette smoke impairs elastin resynthesis in lungs of hampsters with elastase-induced emphysema. Am Rev Resp Dis. 1985;132:640–43. doi: 10.1164/arrd.1985.132.3.640. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda Y, Masuda Y, Ishizaki M. Morphogenesis of abnormal elastic fibers in panacinar and centriacinar emphysema. Hum Pathol. 1989;20:652–9. doi: 10.1016/0046-8177(89)90152-4. [DOI] [PubMed] [Google Scholar]
- 15.Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J. 2009;34:324–31. doi: 10.1183/09031936.00123008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merrilees MJ, Ching PS, Beaumont B, et al. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res. 2008;9:41. doi: 10.1186/1465-9921-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Black PN, Ching PS, Beaumont B, et al. Changes in elastic fibers in the small airways and alveoli in COPD. Eur Respir J. 2008;31:998–1004. doi: 10.1183/09031936.00017207. [DOI] [PubMed] [Google Scholar]
- 18.Hinek A, Mecham RP, Keeley F, et al. Impaired elastin fiber assembly related to reduced 67-kD elastin-binding protein in foetal lamb ductus arteriosus and in cultured aortic smooth muscle cells treated with chondroitin sulfate. J Clin Invest. 1991;88:2083–94. doi: 10.1172/JCI115538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinek A, Smith AC, Cutiongco EM, et al. Decreased elastin deposition and high proliferation of fibroblasts from Costello syndrome are related to functional deficiency in the 67-kD elastin-binding protein. Am J Hum Genet. 2000;66:859–72. doi: 10.1086/302829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang R, Merrilees MJ, Braun K, et al. Inhibition of versican synthesis by antisense alters smooth muscle cell phenotype and induces elastic fiber formation in vitro and in neointima after vessel injury. Circ Res. 2006;98:370–7. doi: 10.1161/01.RES.0000202051.28319.c8. [DOI] [PubMed] [Google Scholar]
- 21.Al-Nasiry S, Geusens N, Hanssens M, et al. The use of Alamar Blue assay for quantitative analysis of viability, migration and invasion of choriocarcinoma cells. Hum Reprod. 2007;22:1304–9. doi: 10.1093/humrep/dem011. [DOI] [PubMed] [Google Scholar]
- 22.Fink L, Seeger W, Ermert L, et al. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med. 1998;4:1329–33. doi: 10.1038/3327. [DOI] [PubMed] [Google Scholar]
- 23.Lemire JM, Merrilees MJ, Braun KR, et al. Over-expression of the V3 variant of versican alters arterial smooth muscle cell adhesion, migration, and proliferation in vitro. J Cell Physiol. 2002;190:38–45. doi: 10.1002/jcp.10043. [DOI] [PubMed] [Google Scholar]
- 24.Merrilees MJ, Lemire JM, Fischer JW, et al. Retrovirally mediated over-expression of versican V3 by arterial smooth muscle cells induces tropoelastin synthesis and elastic fiber formation in vitro and in neointima after vascular injury. Circ Res. 2002;90:481–7. doi: 10.1161/hh0402.105791. [DOI] [PubMed] [Google Scholar]
- 25.Joos L, He JQ, Shepherdson MB, et al. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet. 2002;11:569–76. doi: 10.1093/hmg/11.5.569. .Erratum in: Hum Mol Genet. 2003; 12: 803-4. [DOI] [PubMed] [Google Scholar]
- 26.Hallgren O, Nihlberg K, Dahlbäck M, et al. Altered fibroblast proteoglycan production in COPD. Respir Res. 2010;11:55. doi: 10.1186/1465-9921-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang JY, Johnson PY, Braun KR, et al. Retrovirally mediated overexpression of glycosaminoglycan-deficient biglycan in arterial smooth muscle cells induces tropoelastin synthesis and elastic fiber formation in vitro and in neointima after vascular injury. Am J Pathol. 2008;173:1919–28. doi: 10.2353/ajpath.2008.070875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinek A, Braun KR, Liu K, et al. Retrovirally mediated overexpression of versican V3 reverses impaired elastogenesis and heightened proliferation exhibited by fibroblasts from Costello syndrome and Hurler disease patients. Am J Pathol. 2004;164:119–31. doi: 10.1016/S0002-9440(10)63103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maquart F-X, Pasco S, Ramont L, et al. An introduction to matrikines: extracellular matrix-derived peptides which regulate cell activity. Implication in tumour invasion. Crit Rev Oncol/Hemat. 2004;49:199–202. doi: 10.1016/j.critrevonc.2003.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Eddahibi S, Chaouat A, Tu L, et al. Interleukin-6 gene polymorphism confers susceptibility to pulmonary hypertension in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:475–6. doi: 10.1513/pats.200603-038MS. [DOI] [PubMed] [Google Scholar]
- 31.Keatings VM, Collins PD, Scott DM, et al. Differences in interleukin-8 and tumour necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153:530–4. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- 32.Ko FW, Leung TF, Wong GW, et al. Measurement of tumour necrosis factor-alpha, leukotriene B4, and interleukin 8 in the exhaled breath condensate in patients with acute exacerbations of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2009;4:79–86. [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TM, Lin MS, Chang NC. Usefulness of C-reactive protein and interleukin-6 as predictors of outcomes in patients with chronic obstructive pulmonary disease receiving pravastatin. Am J Cardiol. 2008;101:530–5. doi: 10.1016/j.amjcard.2007.09.102. [DOI] [PubMed] [Google Scholar]
- 34.Le Bellego F, Perera H, Plante S, et al. Mechanical strain increases cytokine and chemokine production in bronchial fibroblasts from asthmatic patients. Allergy. 2009;64:32–9. doi: 10.1111/j.1398-9995.2008.01814.x. [DOI] [PubMed] [Google Scholar]
- 35.Beikler T, Peters U, Prior K, et al. Gene expression in periodontal tissues following treatment. BMC Med Genomics. 2008;1:30. doi: 10.1186/1755-8794-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klapperich CM, Bertozzi CR. Global gene expression of cells attached to a tissue engineering scaffold. Biomaterials. 2004;25:5631–41. doi: 10.1016/j.biomaterials.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Kim S, Takahashi H, Lin WW, et al. Carcinoma-produced factors activate myeloid cells through TRL2 to stimulate metastasis. Nature. 2009;547:102–6. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seidelmann SB, Kuo C, Pleskac N, et al. Athsq1 is an atherosclerosis modifier locus with dramatic effects on lesion area and prominent accumulation of versican. Arterioscler Thromb Vasc Biol. 2008;28:2180–6. doi: 10.1161/ATVBAHA.108.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng PS, Vais D, Lapierre D, et al. PG-M/versican binds to P-selectin glycoprotein ligand-1 and mediates leukocyte aggregation. J Cell Sci. 2004;117:5887–95. doi: 10.1242/jcs.01516. [DOI] [PubMed] [Google Scholar]
- 40.Potter-Perigo S, Johnson PY, Evanko SP, et al. Polyinosine-polycytidylic acid stimulates versican accumulation in the extracellular matrix promoting monocyte adhesion. Am J Respir Cell Mol Biol. 2010;43:109–20. doi: 10.1165/rcmb.2009-0081OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evanko SP, Potter-Perigo S, Johnson PY, et al. Organization of hyaluronan and versican in the extracellular matrix of human fibroblasts treated with the viral mimetic Poly I:C. J Histochem Cytochem. 2009;57:1041–60. doi: 10.1369/jhc.2009.953802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White ES, Atrasz RG, Dickie EG, et al. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol. 2005;32:135–41. doi: 10.1165/rcmb.2004-0126OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lama V, Moore BB, Christensen P, et al. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol. 2002;27:752–8. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol. 2004;286:C1089–99. doi: 10.1152/ajpcell.00461.2003. [DOI] [PubMed] [Google Scholar]
- 45.Müller KC, Welker L, Paasch K, et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res. 2006;7:32. doi: 10.1186/1465-9921-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merrilees MJ, Beaumont BW, Braun KR, et al. Neointima formed by arterial smooth muscle cells expressing versican variant V3 is resistant to lipid and macrophage accumulation. Arterioscler Thromb Vasc Biol. 2011;31:1309–16. doi: 10.1161/ATVBAHA.111.225573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asano K, Shikama Y, Shibuya Y, et al. Suppressive activity of tiotropium bromide on matrix metalloproteinase production from lung fibroblasts in vitro. Int J Chron Obstruct Pulmon Dis. 2008;3:781–9. doi: 10.2147/copd.s3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asano K, Shikama Y, Shoji N, et al. Tiotropium bromide inhibits TGF-β-induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways in vitro. Int J Chron Obstruct Pulmon Dis. 2010;5:277–86. doi: 10.2147/copd.s11737. [DOI] [PMC free article] [PubMed] [Google Scholar]