

Abstract
Vascular endothelium lines the entire cardiovascular system where performs a series of vital functions including the control of microvascular permeability, coagulation inflammation, vascular tone as well as the formation of new vessels via vasculogenesis and angiogenesis in normal and disease states. Normal endothelium consists of heterogeneous populations of cells differentiated according to the vascular bed and segment of the vascular tree where they occur. One of the cardinal features is the expression of specific subcellular structures such as plas-malemmal vesicles or caveolae, transendothelial channels, vesiculo-vacuolar organelles, endothelial pockets and fenestrae, whose presence define several endothelial morphological types. A less explored observation is the differential expression of such structures in diverse settings of angiogenesis. This review will focus on the latest developments on the components, structure and function of these specific endothelial structures in normal endothelium as well as in diverse settings of angiogenesis.
Keywords: PV1, PLVAP; caveolae; fenestrae; transendothelial channels; lipid rafts; vascular permeability
Introduction
Blood vessels have evolved to carry oxygen, nutrients and signaling molecules to distant organs in vertebrates higher than amphibians. The establishment of the primitive vascular plexus from angioblasts [1], known as vasculogenesis, followed by remodeling of this plexus into a hierarchical, mature vascular network, capable to accommodate the circulation of blood, are crucial for organ growth. The network expands with the growth of the organism via another process named angiogenesis, term defining the formation of neovessels from preexisting ones. In a more elaborate way, the term angiogenesis refers to the sprouting of new capillaries from the post-capillary venules that involves endothelial cell activation, extracellular matrix degradation, migration and proliferation, maturation and stabilization of new blood vessel by recruitment of pericytes and smooth muscle cells [2]. This contribution of angiogenesis to organ growth continues after birth until adulthood, when most blood vessels remain quiescent. In the adult, angiogenesis occurs during the menstrual cycles in the ovary and uterus as well as in the placenta during pregnancy. Endothelial cells maintain their ability to divide rapidly in response to physiological stimuli (i.e. hypoxia), angiogenesis being reactivated during wound healing in the adult. In many disorders, this stimulus becomes excessive, and the balance between stimulators and inhibitors is tilted, resulting in an angiogenic switch. The best known conditions in which angiogenesis is switched on are cancers, ocular and inflammatory disorders. However, many additional processes are affected, such as obesity, asthma, diabetes, cirrhosis, multiple sclerosis, endometriosis, AIDS, bacterial infections and autoimmune disease (for a comprehensive review see ref. [3]).
Endothelial phenotypic changes in diverse settings of angiogenesis are under intense scientific scrutiny in search for novel biomarkers with potential for therapy [3–5]. An interesting and less explored observation is the expression by endothelial cells of specific subcellular structures such as caveolae and their stomatal diaphragms, fenestrae, transendothelial channels (TEC) and vesiculo-vacuolar organelles (VVOs). This review will discuss the knowledge on the structure function and regulation of these structures in normal endothelia as well as in angiogenesis.
Endothelial phenotypes – morphology
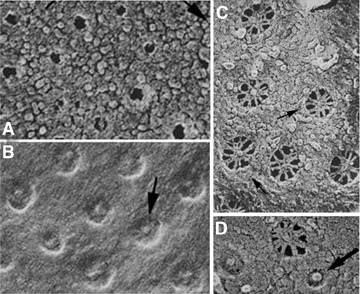
Vascular endothelium is a highly differentiated cellular monolayer with the organization of a simple squamous epithelium. It lines the entire cardiovascular system and thus constitutes a quasi-ubiquitous presence in organs and tissues throughout the body. From a morphological point of view, depending on their content of specific structures, endothelia have been classically defined into three main structural types:the continuous, fenestrated and discontinuous endothelium. The continuous endothelium (Fig. 1A) occurs in all large vessels (both arteries and veins) and microvessels of the body wall (skin, skeletal muscles) as well as those of the visceral muscles (myocardium included) and the lung. As reflected by its name, its main characteristic is the formation of a continuous, uninterrupted barrier between the blood and tissues. In a quiescent state it features a large population of caveolae or plasmalemmal vesicles [6, 7], extremely few TEC [8] and virtually no fenestrae. In select continuous endothelia (e.g. lung, tongue) caveolae can be provided with a stomatal diaphragm (SD). In addition to relatively fewer caveolae, the fenestrated endothelium (Fig. 1B) features specialized microdomains such as fenestrae, TEC and endothelial pockets [9–14]. This type of endothelium occurs in all endocrine glands, digestive tract mucosa and kidney (e.g. peritubullar capillaries). Caveolae and TEC are always provided with an SD and fenestrae are provided with a fenestral diaphragm (FD) in most cases within the fenestrated endothelium. Discontinuous or sinusoidal endothelium lines the sinusoids in the liver and bone marrow. It has extremely few caveolae always provided with an SD and sinusoidal gaps. The gaps are also called fenestrae in a large body of literature (reviewed in ref. [15]) but they differ from those of the fenestrated endothelium by being of a larger diameter, heterogeneous in size and by not being subtended by a diaphragm. Variations are found between these differentiated types of endothelium with respect to the surface density of each of the structures expressed [12, 14].
1.

Endothelium of continuous (A) and fenestrated (B) types as seen by freeze fracture. (A) The P face of the abluminal membrane of a heart endothelial cell shows the numerous caveolae. These are organized in linear arrays, better seen in the higher magnification images (insets). (B) Two endothelial cells of a jejunal capillary show the disposition of fenestrae in sieve plates. Higher magnification insets compare the linear disposition of caveolae (left) and fenestrae (right), suggesting attachment to cytoskeletal elements. Reproduced from reference [12], with permission.
Structure of endothelial stomatal and fenestral diaphragms in normal endothelium
Besides the common set of organelles featured by all mammalian cells, endothelia are provided with specific structures/microdomains that were discovered very early after the introduction of the electron microscope to biology in the 1950s. Microdomains such as fenestrae, caveolae, TEC and VVO were implicated as sites of transendothelial exchange between the blood plasma and the interstitial fluid in health and disease. These structures have continuously intrigued many researchers in the field especially as their components remained elusive for decades.
Caveolae and their stomatal diaphragms
Caveolae (or plasmalemmal vesicles) were first described in the endothelium of continuous type [6] but they occur in all types of endothelia as well as most mammalian cell types (for a review see ref. [16]). They are morphologically defined as spherical invaginations of plasma membrane of regular shape and size (∼70 nm average outer diameter). They can occur singly or in grape-like clusters attached to either front of the endothelium.
Only in select endothelia of continuous type (i.e. in lung, tongue, kidney vasa recta) and in all fenestrated and sinusoidal endothelia, caveolae are provided with an SD. The existence of caveolae with or without SDs has been clearly demonstrated by rapid-freeze deep-etch techniques ([17, 18] and Fig. 2A, B). By transmission electron microscopy, the SD is a thin (∼5–7 nm) protein barrier (Fig. 7A) that occurs in their necks or stoma or in between vesicles that are part of a cluster. The diameter of the SD is variable (<40 nm), most probably due to variable diameter of caveolar necks during caveolae internalization [19]. At the place of SD insertion, the plasma membrane forms a sharp angle (arrowheads in Figs 4C–D, 5B–C). Electron dense material is usually present on the intracellular aspect of the membrane within this angle, as shown in orthogonal transmission electron microscopy sections (Figs 4C–D, 5B–C arrowheads, Fig. 7A, left). Both orthogonal and en face views demonstrate a central density or knob on the SDs [7, 10]. Favorable en face views of the SDs (e.g. in the lung capillaries) show the central knob to be connected to the vesicular neck by very thin fibrils. The diaphragm is apparently lipid-free, as it does not have the trilaminar appearance of the plasma membrane bilayer. It is also destroyed by proteases [20, 21], which implies its protein nature (reviewed in ref. [16]).
2.
Caveolae with (B) and without (A) stomatal diaphragms, TEC (C) and Fenestrae (D) as seen in rapidly frozen deeply etched specimens. The stomatal diaphragms of caveolae and TEC look alike. Fenestrae have an octagonal symmetry, their fenestral diaphragms being constituted of radial fibrils starting at the rim and interweaving in a central mesh. Reproduced from [17] with permission.
7.
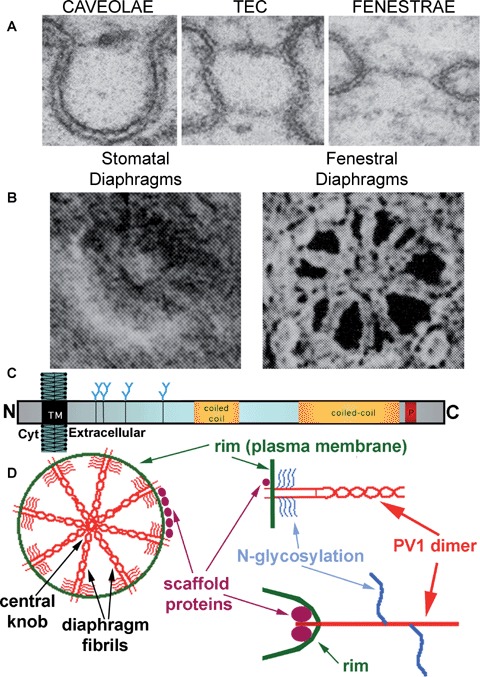
Proposed model for PV1 integration in the structure of the diaphragms.(A) Perpendicular sections of a caveola provided with stomatal diaphragm (left), TEC (middle) and fenestra (right).(B) En face views of stomatal diaphragms (left) and fenestral diaphragms (right), as shown by deep-etch rapid–freeze techniques, demonstrating the fibrils in the fenestral diaphragms and the hints of fibrils in their stomatal diaphragms counterparts. Reprinted from Bearer and Orci, JCB, 1985, with permission. (C) Schematic of the membrane insertion and features of the PV1 monomer. (D) Model of PV1 integration in the endothelial diaphragms:PV1 dimers participate in the formation of the fibrils inserted in the rim (via PV1 N-terminus) and interweaving in the central mesh (via PV1 C terminus). The glycan antennae (accounting for ∼15% of PV1 mass) are situated near the membrane, which would keep the protein ‘afloat’ by preventing collapse on the plasma membrane.
4.
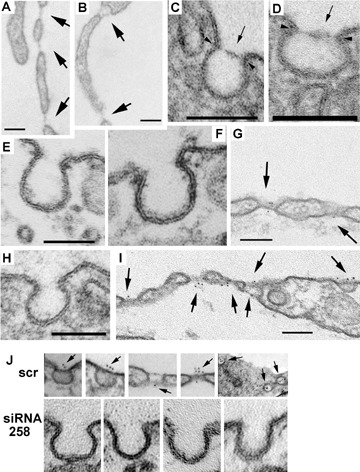
PMA induces TEC (B, I), fenestrae (A, B, G, I) as well as caveolar stomatal diaphragms (C, D, I) only in endothelial cells and not in non-endothelial cell types such as fibroblasts (F). Non-treated control endothelial cells (H) as well as fibroblasts (E) contain only caveolae devoid of stomatal diaphragms. PMA also upregulates PV1, which is found in the newly formed stomatal diaphragms and fenestral diaphragms as demonstrated by immunogold labeling (G, I).(J) PV1 mRNA silencing with siRNA prevents the formation of stomatal diaphragms of caveolae as well as the TEC and fenestrae altogether in HUVEC (lower panels) while the scrambled siRNA counterpart does not (scr, upper panels). Bars 100 nm.
5.
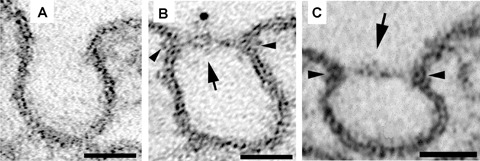
Transfection of PV1 in HUVEC leads to de novo formation of stomatal diaphragms of caveolae. No TEC or fenestrae are formed.(A) control HUVEC (B–C) HUVEC transfected with PV1-HA and stained with anti-HA gold (B).
Endothelial fenestrae
Endothelial fenestrae (Figs 2C, 7A, left), the landmarks of the endothelia of fenestrated type, are circular windows resembling ‘boat portholes’ that cut through the cell body. They are arranged in ordered linear arrays within large planar clusters called ‘sieve plates’. Individual fenestral pores have a remarkably constant diameter (∼62–68 nm) [10–12]. Their circumference is delineated by the fenestral rim that occurs where the luminal aspect of plasma membrane continues with the abluminal one under a sharp angle. As in the case of the SDs, electron dense material has been demonstrated in the cytoplasm within the sharp angle formed by the plasma membrane. The circumference of most fenestral pores is apparently round, but as demonstrated by Maul, 20–30% of them have an octagonal symmetry, which can be photographically enhanced by rotating the image several times around its center [22]. The octagonal symmetry has been clearly established by deep-etch of rapidly frozen specimens [17] and high resolution scanning electron microscopy [23].
The fenestrae are provided with a Fenestral Diaphragm (FD) [9] in all cases (i.e. in kidney peritubullar capillaries, all endocrine and exocrine glands, intestinal villi) except in capillaries of kidney glomerulus [24–27]. Although of a slightly larger diameter, the fenestral diaphragms are morphologically similar to the SDs by transmission electron microscopy (Fig. 7A, left). They appear as thin protein barriers anchored in the fenestral rim and provided with a central knob or density, as demonstrated in parallel and perpendicular sections. Moreover, in en face views obtained in oblique or grazing sections, the central density seems to be connected to the fenestral rim by thin fibrils [28]. Bearer and Orci exquisitely demonstrated the intimate organization of the fenestral diaphragm [17] (Fig. 2C). In rapidly frozen deep-etch specimens, they have shown the fenestral diaphragm to consist of radial fibrils, starting at the rim and interweaving in a central mesh, the equivalent of the central density/knob seen by transmission electron microscopy. Unfortunately, the SDs of cave-olae and TEC could not be resolved in the same detail due to their smaller size and experimental conditions (i.e. angle of shadowing) [17]. It was readily apparent that the caveolar diaphragms resembled those of the TEC both featuring a central particle [17] (Fig. 7A). Close examination also reveals hints of the same radial fibrils pattern as for fenestral diaphragms (Fig. 7B).
Finally, perfusion fixation of vascular beds provided with fenestrated endothelia where the fixatives have been dissolved in fluorocarbons (oxygen carriers used as blood plasma expanders) combined with tannic acid staining, revealed the presence of large (up to 400 nm) tufts or fascinae fenestrae on the luminal side of fenestral diaphragms (Fig. 2D) and their absence from the luminal side of fenestrae [29]. These tufts were absent from the caveolar SDs. The fibers forming these tufts have been interpreted as the morphologic equivalent of the heparan sulfate proteoglycans (HSPGs) shown to reside on the luminal surface of fenestrae by other means (see below).
Transendothelial channels (TEC)
TEC are patent pores spanning the endothelial cell body from lumen to ablumen (Fig. 7A, middle). TEC are rarely found in continuous endothelia where they seem to be formed by the fusion of either one caveola/plasmalemmal vesicle with both luminal and abluminal aspects of the plasmalemma or by chains of usually two to four caveolae [7, 8, 30]. In the fenestrated endothelia, TEC occur in the attenuated part of the endothelial cell and are provided with two SDs (one luminal and one abluminal) [11, 12, 30, 31]. As in the case of caveolar SDs, the fine structure of the SDs of TEC could not be clearly resolved by deep-etch due to technical limitations [17]. However, close inspections of the micrographs reveal the same pattern of radial fibrils and a central density (Figs 2B, 7B).
Vesiculo-vacuolar organelles (VVOs)
VVOs are morphologically defined as chains of interconnected vesicles of variable size that form intricate TEC spanning the cytoplasm of the endothelial cells from one front of endothelial cell to the other [32]. They are usually provided with SDs at the connection points between vesicles and vacuoles as well as at the level of their stoma or communication with the extracellular space [33]. The SDs of the VVOs closely resemble those of caveolae by transmission electron microscopy. A recent review discusses the biology of VVOs in great detail [34], therefore only data with relevance to the SDs will be considered here.
VVOs have been first described in the tumor vasculature [35] and are claimed to also occur in the normal endothelium of the post-capillary venules [32, 35, 36]. The definition of VVOs is purely morphological; therefore there is a difficulty in discerning VVOs from clusters of caveolae that occur in most endothelia [7, 12], which raised doubts as to their status as a bona fide, novel organelle [31]. This ‘novel organelle’ status might be bolstered by recent electron microscopy data showing that in caveolin 1 −/− mice (lacking caveolae in all non-muscle cell types, endothelia included), the venules were still provided with structures resembling VVOs [37, 38]. However, the relationship between these structures and VVOs remains to be clarified following biochemical evaluation.
Endothelial pockets
These are infrequent structures that by electron microscopy resemble a pocket or a large vacuole formed by cellular processes that contain fenestrae with the usual structure. The information on these is scarce and, so far, they seem to occur in very low numbers only in the fenestrated endothelia [13].
Structures present in the endothelium in neovascularization
Vasculogenesis and angiogenesis in the embryo
There are no systematic studies documenting the appearance of caveolae, fenestrations, TEC, pockets, VVO or diaphragms and their specific protein components during development. Most of the data available concern the formation of the fenestrae and their diaphragms.
In the blood island and vascular plexus stage of vascular development, examination of micrographs published in various papers shows a continuous endothelium devoid of fenestrae or TEC. Unfortunately, the magnification shown is usually too low for a qualitative assessment of the presence of caveolae or VVO and their respective diaphragms, most of the studies being concerned with the organization of the vasculature rather than the ultrastructural detail of endothelial cells.
The rat brain starts developing as an avascular mass or primordium surrounded by vascularized tissue called the perineurium, from where the developing brain will attract sinusoid-like vessels by sprouting angiogenesis between ∼ E12 and E16. At E12, the vessels in perineurium, have fenestrated endothelia [39] but it is not known when precisely fenestrae appear in these vessels, E12 being the earliest time point reported in this paper. Interestingly, diaphragmed fenestrae are present in the vessels that enter the nervous matter from E12 to E16 by sprouting angiogenesis but are lost upon the formation of the blood brain barrier beginning at E16 [39].
The capillaries of rat intestinal mucosa are initially continuous expressing caveolae and it is not clear whether the SDs are present. These continuous capillaries become fenestrated late in gestation at E15–E16, when they start to express fenestrae subtended by diaphragms [40, 41]. The reason for the delayed induction of the fenestrae in the intestine is not known. A possible explanation could be found by examining the situation in the adult where the vessels in the intestinal tunica muscularis are continuous and the endothelial cells have caveolae without SDs. As the vessels enter the mucosa (i.e. the villus), their endothelium remains continuous in the central bundle vessels but their caveolae have SDs. The capillary loops that come in contact with the epithelium are fenestrated and their fenestrae have SDs. The intestinal mucosa does not fully develop in the embryo but requires the postnatal establishment of the intestinal flora, which leads to the elongation of the villi and formation of fenestrated vessels by angiogenesis.
Interestingly, in the rat kidney development, the earliest fenestral pores appearing in the kidney glomerular capillaries are subtended by diaphragms. Moreover, these diaphragms bind cationic ferritin, a property common to all fenestrae in the adult embryo, conferred by heparan sulfate proteoglycans [26]. It is not known, however, at which precise stage fenestrae appear in the kidney. These diaphragms of glomerular fenestrae are gradually lost within a week or two after birth, resulting in the unique non-diaphragmed phenotype of the adult glomerulus [24–27].
In the liver at E10–E12, during the hematopoietic stage, the capillaries have a basement membrane and are fenestrated, but the fenestrae resemble those of the adult fenestrated endothelium: circular pores, regular in size (average 60–70 nm) and subtended by fenestral diaphragms. Starting at E17, these fenestrae are progressively replaced by larger, more heterogenous in size fenestrae that are typical of the adult liver and other sinusoidal endothelial cells [42, 43].
In summary, it is not known when fenestrae, TEC, cavolae, VVOs and their respective diaphragms first appear in different vascular beds. It is tempting to draw a parallel between the apparition of the glomerular fenestrae and those of the sinusoids is tempting to be drawn, as both types of non-diaphragmed fenestrae seem to be preceded by fenestrae subtended by diaphragms.
PV1 (PLVAP) protein is the only known marker (discussed below) of endothelial SDs and fenestral diaphragms. The mRNA is definitively expressed in both mouse and rat embryos by E7 [44], corresponding to the formation of blood islands. PV1 protein is detectable by E12 in most organs and is lost by E16-E18 in the brain at the time of blood-brain barrier formation [45]. Moreover, the loss of PV1 expression correlates with the loss of fenestrae upon BBB formation [46]. In mice at E11.5, PV1 is expressed in hind limb buds, but not forelimbs by DNA microarray, but the precise location is not clear [47]. PV1 is also expressed on bone marrow endothelial progenitor cells [48] as well as on the endothelial cells that spontaneously differentiate on embryoid bodies after 5 days outgrowth [49].
Arteriogenesis
Arteriogenesis is defined as development and growth of collateral arteries [2, 50]. There is little information regarding the ultrastructure of endothelium in this process. From the few ultrastructural studies done, there is no evidence of the occurrence of the endothelial specific structures on the endothelium during collateral growth [51, 52].
Angiogenesis in the adult
Angiogenesis in the adult occurs in the ovaries, uterus and during wound healing. The vasculature of the ovaries is fenestrated. One of the earliest observations on the induction of fenestrae is that during wound healing in regenerating muscle [53] the continuous endothelium of muscle capillaries becomes fenestrated, observation confirmed by different groups [54–56].
Pathologic angiogenesis
Electron microscopy studies documented pathological conditions where the continuous endothelium becomes fenestrated in cases such as the aberrant capillary loops that appear in the neovasculature of psoriatic lesions in skin [57] in the chronic inflammation of gingiva [58] during lung fibrosis [59], chronic allergic encephalomyelitis [60], delayed radiation necrosis [61], experimental lead encephalopathy [62], diabetic retinopathy [63], rheumatoid arthritis [64, 65] and especially the neovasculature of many solid tumours (reviewed in ref. [31]) [35, 62, 66–76].
Components of the stomatal and fenestral diaphragm
Plasmalemma vesicle associated protein (gp68, PV-1, PV1)
Recently, the stomatal and fenestral diaphragms were shown to share at least one biochemical marker, namely PV1 [77, 78]. PV1 protein was discovered as the antigen of a novel endothelial antibody (21D5 mAb) [79], which co-localized strictly on endothelial caveolae from rat lung immunoisolated on anti-caveolin 1 antibodies [80].
Gene features and expression of mRNA
PV1 gene product is encoded by Plasmalemmal Vesicle Associated Protein gene in humans (HUGO symbol PLVAP) and its presence has been documented in several other mammalian species including mouse, rat, bovine and chicken [16]. The gene and significant protein homologues are absent from the completed genomes of yeast (Saccharomyces cerevisiae) as well as worms (Caenorhabditis elegans) where the SDs and fenestral diaphragms are not present. The presence of the gene could not be confirmed so far in lower vertebrates such as amphibians (Xenopus laevis) or fish (Danio reiro and Fugu rubriprens), which might be due to the incomplete sequencing of these genomes or absence of the diaphragms in their vasculature. It is not clear whether the diaphragms are present in these species. In mammals, PV1 mRNA is expressed in most organs and tissues with the highest levels in lung, kidney, spleen, all endocrine glands and digestive tract [44, 77, 78].
Protein features and properties
PV1 is a single span, type II membrane glycoprotein that forms homodimers in situ[77, 79]. The size of the monomer lacking posttranslational modifications is 50 kD and 60 kD in its N-glycosylated, mature form. The N-linked glycans form ∼15% of the PV1 mass and contain terminal non-reducing Gal or GalNAc, GlcNAc as well as sialic acid in both α2–3 and α2–6 linkage [78]. The monomer has a very basic calculated pI (∼9.1) and the determined pI of the dimer in glycosylated form is still shifted to the basic (pI ∼7.8). PV1 binds avidly to heparin at physiological pH [81].
In humans PV1 has a short (27 aa) intracellular tail and a long (358 aa) extracellular C-terminal domain, topology that was confirmed with peptide antibodies directed against the C-terminus [44, 77, 78]. The intracellular domain does not contain any conserved known consensus site across mammalian species. It contains, however, two short identical stretches of amino acids. One is next to the transmembrane region (8 aa) and contains a putative caveolin 1 binding domain [16]. The other one at the extreme N-terminus (7 aa), and might play a role in the biology of PV1. The extracellular domain contains a regular spacing of nine cysteines, the odd number showing the possibility of dimer formation. It also contains four consensus N-glycosylation sites near the membrane, a proline-rich region near the C-terminus and two large coiled-coil domains. The secondary structure is predicted to be mostly alpha helical [44, 77]. This is sustained by the presence of two large consensus coiled-coil domains that are obligate alpha helix formers [82]. Every seventh amino acid of the alpha helix of the coiled-coil domain is hydrophobic, which results in a spiral hydrophobic interface through which one coiled-coil domain interacts with the hydrophobic interface on the cognate coiled-coil facilitating the formation of an intermolecular superhelix [82]. Moreover, seven of the cysteines are situated within the predicted coiled-coil domains. This would suggest that the coiled-coil mediated interaction between two monomers of a dimer would be ‘reinforced’ by several disulfide bonds. All these data suggest that PV1 might adopt a rod-like shape. The fact that the PV1 dimers can be solubilized in SDS suggests that the putative protein-protein interactions between adjacent dimers and other putative interacting proteins (e.g. other PV1 dimers or other protein(s)) are non-covalent in nature.
Localization
At protein level, PV1 was confirmed to be present in the organs where the mRNA is present [16, 78]. PV1 seems to be endothelium-specific, [78] and, moreover, to be restricted to a subset of endothelia such as the capillaries of the lung, choroid plexus, retina, adrenals, pancreas, intestinal villi, peritubular capillaries in the kidney, liver and spleen. PV1 is absent from the large vessels of the lung, aorta, vena cava and coronary artery as well as capillaries in the heart, skin, skeletal muscle, intestinal smooth muscle (where caveolae do not have SDs) and kidney glomerulus (where fenestrae do not have fenestral diaphragms). All in all, the pattern of expression of PV1 correlates well with the pattern of expression of fenestral and SDs ([78] and our unpublished data).
By immunocytochemistry (Fig. 3), PV1 was found to be specifically associated with the SDs of caveolae and TEC and the fenestral diaphragms, at both fronts of endothelial cells [77, 78], this being the first demonstration of a protein with such localization. The label was absent from other microdomains of the endothelial cells as well as from any other cell type in the organs investigated, bolstering the claims of endothelial specificity.
3.
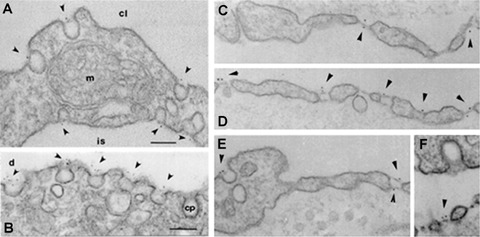
Immunolocalization of PV1 by immunodifussion to stomatal diaphragms of caveolae in lung (A–B, E), stomatal diaphragms of TEC (D–E) and fenestral diaphragms (C–D, F), as detected with anti-PV1 antibodies directed against its C-terminus.(A–B) lung, C–E) kidney, (F) intestine. The label was found specifically associated with the stomatals and fenestral diaphragms at both fronts of the cell. No label was found on the plasmalemma proper, clathrin-coated pits and vesicles, intercellular junctions as well as non-endothelial cellular types. Bars 100 nm.
There is some controversy as to the endothelial specificity of PV1 protein [81, 83–85], these reports claiming that PV1 is expressed in scores of different cell types. However, the data in these reports were obtained using novel antibodies that have not been validated by other groups. The data showing endothelial cell specificity are quite vast by comparison. Our data supporting endothelial specificity of PV1 have been independently confirmed by other groups [79, 86–91]. Moreover during the last few years it became apparent that PV1 protein is the antigen of two antibodies (i.e. MECA-32 and PAL-E) that were used for more than two decades as classical endothelial specific markers. An in depth discussion of this matter is found in ref. [38].
PV1 is necessary and sufficient to form fenestral and stomatal diaphragms
Besides being morphologically similar, the SDs and the fenestral diaphragms are biochemically related by sharing at least one protein namely PV1. Apart from PV1, there are no data on other specific components of the diaphragms. PV1 could function either as a structural component, function modulator component, or both, within these diaphragms. Recent data from our laboratory strongly suggest that PV1 is a key structural component of both the SDs and fenestral diaphragms, necessary and sufficient for diaphragm expression [92]. This is based on several lines of evidence: (i) PV1 forms homodimers in situ, [93]; (ii) several PV1 homodimers reside in close proximity within the same diaphragm as shown by cross-linking experiments of PV1 carried out in situ in rat lung and kidneys [93]; (iii) de novo formation of fenestral and SDs, correlates with de novo PV1 expression. As expected, PV1 could be found in the newly formed fenestral and SDs (Fig. 4A-E);(iv) PV1 knockdown by siRNA prevents the formation of both fenestral and SDs (Fig. 4F). Actually, this last approach prevented the formation of fenestrae and TEC as a whole. This finding suggested that both TEC and fenestrae require PV1 for their biogenesis, a point which was recently and independently confirmed [91];(v) Overexpression of tagged PV1 in cell types lacking PV1 expression and diaphragms, led to the formation of caveolar SDs. This result shows that either PV1 forms the diaphragms by itself or the other components of the diaphragms are ubiquitously expressed, PV1 being the limiting factor [92].
Other components
The chemical components of the SDs of caveolae and TEC and those of the fenestral diaphragms have been investigated with ‘general’, nonspecific probes such as lectins [94–97] and cationic molecules [21, 98–101]. It has been shown that SDs and fenestral diaphragms behave differently: the former bind lectins avidly and lack anionic sites, while the latter do not [94] or poorly [95] bind lectins and have multiple anionic sites conferred by HSPGs [20]. The presence of the lectin binding sites on SDs and fenestral diaphragms, combined with their protein nature (sensitive to protease degradation) [20] suggest the presence of glycoproteins in their structure. The HSPGs are present only on the luminal side of fenestral diaphragms [99], which correlates with the data obtained by fixation in the presence of fluorocarbons [29].
Working model of PV1 integration in the structure of the diaphragms
Based on the data in the literature a working hypothesis has been formulated (depicted in Fig. 7D) in which both SDs and fenestral diaphragms consist of a common framework of radial fibrils inserted in the rim of a pore (i.e. caveolar or TEC introit or fenestral pore) and interweave in the center of the diaphragm (Fig. 7D). The fibrils would consist of PV1 dimers whose C termini would form the central density of the diaphragm [44]. The dimers could be ‘kept afloat’ by the heavy (∼15% of the PV1 mass) glycosylation that occurs nearby the membrane (see Fig. 7C and ref. [44]). The diaphragm structure could be stabilized via interactions between PV1 C termini themselves or with another putative diaphragm stabilizing extracellular protein. Alternatively or in addition, the diaphragm could be stabilized by the existence of a rigid structure on the cytoplasmic face of the rim presumably connected to the cytoskeleton. This latter assumption seems to be sustained by data obtained with affinity chromatography and yeast two-hybrid screening using the intracellular domain of PV1, by which assays cytoskeletal linker molecules interact with PV1 (R. Stan, unpublished observations).
The HSPGs could interfere with the binding of the lectins in fenestrae, to explain the lack of fenestral binding [94] or poor binding [102] of lectins. PV1 binds most of the lectins that were shown to bind to the SDs [94] (e.g. WGA, GS I, RCA, Concanavalin A etc) [77] and unpublished data). Regarding the lack of anionic sites on SDs, PV1 protein backbone has a highly basic pI (∼9.0). Even with glycosylation, the pI of the native protein is still slightly basic (∼7.8) [77], which explains the absence of binding of cationized ferritin [20, 21] by the SDs of caveolae and TEC. Since caveolae are dynamic structures, it is conceivable that the protein lifetime is too short for binding circulating HSPGs.
The presence of the HSPGs on the luminal side of fenestrae could be explained by the following possibilities: (i) the caveolae from the opposite fronts of the endothelial cells fuse and form TEC that collapse to the minimal path length and form fenestrae. The HSPGs on fenestrae are a filtration residue [31, 103]. Or (ii) a different biogenetic pathway between fenestrae and caveolae with the HSPGs as a filtration residue or (iii) a different biogenetic pathway between fenestrae and caveolae by which HSPGs are specifically targeted to the fenestral pore.
The first hypothesis was proposed in an attempt to provide an explanation for the morphological similarity between the SDs and fenestral diaphragms and their apparent chemical differences (i.e. lectin binding and anionic sites). And this within the following paradigm of fenestrae biogenesis: TEC would arise by fusion of caveolae/plasmalemmal vesicles from both fronts of endothelial cells and fenestrae would be formed through the collapse of TEC to minimal path length [31]. As fenestrae were considered relatively stable structures spatially and temporally, the HSPGs could be added to their structure as a filtration residue, in view of their function as a permeability site [104]. The endothelial cells are know to shed HSPGs [105] although the extremely low levels of GAG chains or HSPGs circulating in blood plasma [105, 106], could be inconsistent with this hypothesis. However, if the fenestrae exist for a long enough time, PV1 could bind the necessary HSPGs.
Keeping the above caveat in mind, the second hypothesis seems more likely. This is supported by the fact that in caveolin 1 null mice, which completely lack endothelial caveolae, the TEC and fenestrae (both subtended by a fenestral diaphragm or not) are present at surface densities similar to those in the wild-type [[37, 107] and R.V. Stan, manuscript submitted]. Incidentally, the newly induced fenestrae in vivo by topical application of VEGF as well as those in the capillaries of VEGF-producing tumours seem to have either decreased or not to have anionic sites [108, 109]. Although cationized ferritin was shown to bind to the fenestrae imaged by rapid-freeze deep-etch [17], the tufts seen by fixation in presence of fluorocarbons [29] were lost. The reason for this discrepancy is not clear.
The third hypothesis is very attractive as it would be in agreement with the data from caveolin 1 −/− mice and it would lend some order to the control of permeability through fenestrae. It might also explain the ordered patterns seen in grazing sections in tissues fixed in presence of fluorocarbons [29]. The only observation here is that the targeting of HSPGs to fenestrae would occur only after the collapse of TEC. The HSPGs involved will most likely need to be expressed in polarized manner only on the luminal side of the endothelium.
Undoubtedly, most of this paradigm awaits further experimental confirmation. What the data have proven so far is that PV1 is present in both stomatal and fenestral diaphragms and that it is necessary for diaphragm formation.
PV1 and the endothelial diaphragms in ontogeny
Most of the data available concern the formation of the fenestrae in ontogeny, while little attention paid to TEC, caveolar SDs and VVOs. Milici and Bankston, showed that the fenestrated capillaries of intestine mucosa express fenestrae subtended by fenestral diaphragms only late in the gestation at E15–E16 [40, 41]. However, in the developing rat brain, the vessels present in the perineurium at E12 had endothelia that were all fenestrated [39]. Moreover, the fenestrae (provided with fenestral diaphragms) were present in the sinusoid-like vessels that entered the nervous matter by sprouting angiogenesis and were lost upon the formation of the blood brain beginning with E16. This is in keeping with the data obtained with the MECA32 mAb in mouse [45], as already noted. The reason for the delayed induction of fenestrae in the intestine is not known.
Interestingly, the first glomerular fenestrae that appear in ontogeny are subtended by fenestral diaphragms that bind cationized ferritin [26]. These fenestral diaphragms are lost after birth this resulting in the phenotype found in adult [24–27]. The liver fenestrae are also subtended by fenestral diaphragms at E10–E12 during the hematopoietic stage. They are progressively replaced by larger, more irregular fenestrae of the adult liver starting with E17 [42, 43]. [It should be noted that the lack of fenestral diaphragms in the glomerular fenestrae is contested by few reports in the literature (Rhodin 1962, [28]) the reason for this not being clear.]
By Northern blotting of RNA obtained from whole mouse embryos, PV1 mRNA is definitively expressed in both mouse and rat embryos by E7 [44], corresponding to the formation of blood islands. The only survey of the presence of PV1 protein reports that PV1 is detectable by E12 in most organs and is lost by E16-E18 in the brain when the blood-brain barrier develops [45]. Although no direct PV1-diaphragm correlation has been made (i.e., immunocytochemistry at the electron microscopy level with MECA-32 mAb at different developmental stages), the loss of PV1 expression correlates with the loss of fenestrae upon BBB formation [46]. Interestingly, in a comparison of the genes expressed in the mouse forelimb and hind limb buds at E11.5 by SAGE, PV1 is expressed only in the latter [47].
PV1 is also expressed on bone marrow endothelial progenitor cells [48] as well as on the endothelial cells that spontaneously differentiate on embryoid bodies after 5 days outgrowth [49]. It would be interesting to investigate embryos earlier than E12 for PV1 expression to see which of the diaphragms are present and when.
Regulation of endothelial specific structures
PV1 and diaphragms are lost upon endothelial cell dedifferentiation in cell culture
The phenotypic drift suffered by primary endothelial cells in culture is well established [110, 111]. This is shown by the differences in the surface density of endothelial cell specific structures as well as expression of gene products. Endothelial cells from bovine adrenals (BAEC) lose their fenestrae and TEC once in culture [112, 113]. Likewise, the number of caveolae and their SDs is drastically reduced in lung microvascular endothelial cells from both human and rat (HLMVEC and RLMVEC, respectively), in which case PV1 expression is also lost [92]. Also VVOs are lost in culture and need special treatment to be reinduced [114].
The diaphragms are inducible structures
Matrix components
The first demonstration of fenestrae and TEC induction in vitro was done by Milici et al. [14] who showed regeneration of bovine adrenal endothelial cell fenestrae and TEC when cultured onto a matrix deposited by MDCK cells. This suggested a role in fenestrae and TEC induction for a matrix component and/or a soluble factor that was deposited by the MDCK cells. This correlates well with the tightly controlled spatial distribution of fenestral sieve plates and TEC in situ where they are expressed on the attenuated part of endothelium, always toward the neighboring epithelium. It looks like the epithelium secretes either a matrix component(s) or soluble factors (or both) that dictate the spatial distribution of these structures. As discussed under the function heading of this review, this is in keeping with the presumed role of fenestrae in reabsorbing of water and solutes from the interstitium into the blood stream [115].
Phorbol esters
In a series of papers during the 1980s, Lombardi et al.[113, 116, 117] showed that bovine adrenal endothelial cells in culture could be induced to form fenestrae and TEC de novo upon treatment with nanomolar concentrations of phorbol myristate acetate (PMA). The formation of fenestrae was accompanied by dramatic changes in the morphology of endothelial cells from cobblestone to fibroblast-like shape with extreme thinning of the endothelial cell periphery. This presumed extensive cytoskeletal rearrangements of which lamellipodia formation was one of the most prominent features.
However, the formation of fenestrae took approximately 3 days of PMA treatment, which is a rather long time. The fenestrae formed by PMA had all the structural characteristics of the fenestrae in situ (clusters arranged in sieve plates, radial fibrils interweaving in a central mesh) as shown by deep-etch rapid freeze [113]. Most importantly, fenestral and TEC induction by PMA also occurred in endothelial cells that did not have SDs or fenestral diaphragms in situ, such as HUVEC or bovine pulmonary artery endothelial cells [117]. Moreover, the induction of fenestrae was endothelial cell specific and could not be achieved in non-endothelial cell types [117]. This modulation of fenestral and TEC numbers by PMA was enhanced by trans-retinoic acid and dimethylformamide and was decreased by TGFβ[116]. These results have been confirmed in situ where the induction of fenestrae and TEC by either retinoic acid or PMA has been shown also in the capillaries of the cerebral cortex that were infused for 4 weeks with the use of an osmotic pump (117).
Although demonstrating the possibility of fenestrae and TEC induction, PMA did not give clear indications as to the extracellular physiological factors involved in their formation. PMA, an analog of the second messenger diacylglycerol (DAG), has been shown to activate several classes of proteins includingPKC, PKD, Munc, Ras-GRP (an activator of Ras) as well as chimaerin (activator of Rac1) (reviewed in ref. [118]). This occurs by PMA binding to the C1 domains of these proteins that targets them to the membranes where they carry out their biological activity. In an attempt to understand the pathways involved in the induction of the diaphragms by PMA, in this system, it was found that besides TEC and fenestrae, PMA also induced SDs of caveolae in a variety of human endothelial cells, such as dermal microvascular and lung microvascular endothelial cells, as well as HUVEC [92]. The induction of SDs occurred much faster (12–24 hrs after treatment) than the induction of fenestrae or TEC (∼72 hrs) and was paralleled by induction of PV1 mRNA and protein in a dose and time-dependent manner. Moreover, the upregulation of PV1 is not PKC dependent but depends on the activation of a C1 domain containing protein and on the function of the Erk1/2 MAPK pathway. The upregulation is blocked by TGFβ 1, which inhibits the induction of fenestrae and TEC by PMA [116]. Surprisingly, these effects occurred only in human and bovine cells, as several rat and mouse endothelial cell types do not respond even to micro-molar doses of PMA (Stan RV, unpublished observations). Likewise, treatment of human endothelial cells with retinoic acid alone did not induce either stomatal or fenestral diaphragms and did not upregulate PV1 (Stan RV, unpublished observations) in our hands.
Growth factors—the role of VEGF
The most extensive literature related to the induction of fenestrae in vivo deals with their occurrence in the neovessels of tumours and wound healing. In 1992, W. Risau's group [119] made the observation that in adult tissues vascular endothelial growth factor (VEGF) was expressed in cell types adjacent to fenestrated endothelium (e.g. epithelial cells of the choroids plexus). As VEGF was also described as a permeability increasing agent [65, 120], they hypothesized that it would be responsible for maintaining fenestrations. Several years later, the same group showed that bovine adrenal endothelial cells could be induced to form fenestrae, TEC and VVOs in co-culture with EpH4 cells (mammary epithelial) stably expressing distinct isoforms of VEGF [121]. The presence or absence of caveolar SDs was not assessed. It also suggested that other components in the system such as matrix and/or the state of differentiation of endothelial cells are equally important.
In a separate effort, Roberts et al. demonstrated that topical application of VEGF to the rat cremaster muscle as well as by subcutaneous injection in the skin induced fenestrae, plasmalemma vesicles (caveolae) clusters (VVOs) and transendothelial gaps in situ after only 10-min exposure [108] as compared to heat inactivated VEGF, saline and histamine controls. This showed for the first time that:(i) the fenestrae could be induced by a physiological relevant factor in situ; (ii) components for forming the diaphragms were already present in the endothelial cells, the exposure time being too short for de novo protein synthesis. Moreover, the fenestrae induced in tumor vessels as well as those induced by chinese hamster ovary cells (CHO) stably expressing VEGF (VEGF:CHO) had a decreased or no binding of cationized ferritin, showing decreased occurrence of HSPGs on their luminal side. Based on purely morphological data, it was concluded that the VVOs were actually clusters of fused caveolae and the endothelial gaps were interpreted as openings of the intercellular junctions. The induction of both structures was considered to provide the structural basis of increased microvascular permeability caused by VEGF [35, 65].
Subsequent studies [103] showed that several tumours grown in nude mice had a significant number of the neovessels provided with fenestrated endothelium. Moreover, tumours formed by VEGF:CHO cells had fenestrae. Finally, slow releasing pellet implants placed on top of the cremaster, induced fenestrae at VEGF doses too low to induce angiogenesis [122]. The same tumours induced the formation of fenestrae also when implanted in the brain although the occurrence of VVOs and endothelial gaps were drastically reduced compared to vessels of tumours implanted in the flank [109], showing the importance of the host environment. Very strong evidence of the importance of VEGF in fenestrae induction came from another paper demonstrating that the vasculature of the tumours that were null for the VEGF locus, had continuous endothelia completely lacking fenestrae [123].
The induction of fenestrae by VEGF is also sustained by data showing that in a corneal angiogenesis assay only the endothelium of the neovasculature induced by VEGF is fenestrated as compared to that induced by basic FGF (FGF-2), another potent angiogenic factor [124] that does not induce fenestrae nor increase leakage of tracers. Interestingly, only fenestrae were induced in this assay by VEGF, the authors reporting a complete lack of transendothelial gaps. As a following, the increased permeability caused by VEGF (as demonstrated by tracer leakage into the interstitium) was linked to the formation of fenestrae. Dominant negative Rac1 GTPase rapidly and dramatically blocked both fenestral induction and the increase in tracer leakage when delivered as a transducible TAT-fusion protein (TAT-Rac1DN) concomitantly with VEGF. TAT peptides enable protein to cross the cell membrane-also called transduction-from the extracellular medium into the cytoplasm of cells via a yet unknown mechanism (for a review of the technology see ref. [125]). More importantly, TAT-Rac1DN did not block the angiogenic effects of VEGF, confirming that the permeability effects of VEGF could be separated at least in part from its angiogenic effect (for a review see ref. [126]). The role of Rac1 in forming fenestrae is not at all unexpected as these structures occur in very attenuated parts of the endothelial cells and their induction in culture is accompanied by cell thinning due to lamelipodia formation [113, 116, 117] where Rac1 is known to have a major role (for review see ref. [127] and references therein). In this system, PMA/DAG could activate Rac1 via chimaerin [118]. Finally, VEGF seems to activate Rac1 via PLCγ and PI3-kinase as determined using endothelial cells in culture stably expressing VEGF-R2. The requirement for both DAG and PLCγ is also supported by experiments in isolated frog capillaries, showing an inhibition of VEGF-induced permeability by PLCγ inhibitors as well as an increase of permeability by DAG analogs such as OAG [128].
Another paper [129] has shown an increase of both fenestrae and possibly VVO formation in endothelial cells (e.g. HUVEC and immortalized renal arteriolar endothelial cells) upon VEGF treatment when these cells are cultured on a matrix laid down by glomerular epithelial cells. This further sustains the requirement of both matrix/growth factor components.
With the availability of VEGF loxP mice [130] clear genetic evidence was obtained in support of VEGF requirement for fenestrae maintenance in normal tissues in mice both from pancreas islets capillaries [131] as well as kidney glomerular capillaries [132]. These papers both demonstrated that conditional deletion of VEGF switched the fenestrated phenotype to a continuous type.
In a series of very elegant papers [133–137], D. McDonald's group has demonstrated the critical role of VEGF in maintaining the endothelial cell phenotype. Transgenic overexpression of VEGF 165 iso-form in the airway epithelium leads to increased vascular density and fenestrae formation [137]. Inhibition of VEGF signaling via different means such as small molecule inhibitors or VEGFR2 or VEGF-Trap, a soluble form of VEGFR1, demonstrated that VEGF signaling is required for maintenance of a fenestrated phenotype of normal and tumour vessels [133–136].
Recently, Madden et al., showed by SAGE that PV1, the protein component of the diaphragms, was one of the most upregulated endothelial cell genes in glioblastoma vessels and that PV1 mRNA was induced by seeding human dermal endothelial cells onto Matrigel [138]. Next, it was shown that PV1 is upregulated by VEGF in HUVEC in culture via its VEGFR2 and that PV1 was expressed on endothelial cells from a multitude of tumours [139]. These findings were subsequently confirmed by data showing that PV1 is upregulated in endothelial cells by tumour cell conditioned medium, VEGF as well as HGF [140]. Moreover, PV1 was shown to facilitate endothelial cell migration, due to which it was proposed as a novel anti-angiogenic target [140]. However, there are other opinions on this [141]
Moreover, PV1 was also identified [142] as the antigen of PAL-E, a widely used human endothelial specific monoclonal antibody [143] shown to be expressed on endothelium in wound healing [144, 145], tumours [146–148] such as angiosarcoma [149, 150], Kaposi sarcoma [150–153], Wilm's tumour [154], brain tumours [155, 156], colorectal adenocarcinoma [144], melanoma [157, 158], hepatocellular carcinoma [159], breast cancer [160] and hemangiomas [161]. The presence of PV1 in tumour endothelium is illustrated in Figure 6.
6.
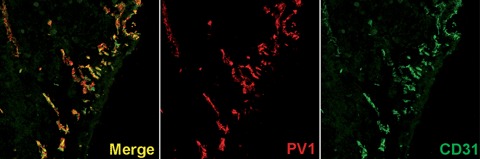
PV1 (red) is expressed in tumor vessels where it colocalizes with CD31 (green)
The induction of fenestrae by VEGF is not a matter of general agreement, however. The neovasculature of some VEGF-producing tumours was found devoid of fenestrae while they are present in other tumours [34, 35, 162]. Moreover, the numbers of fenestrated vessels in transgenic mice overexpressing VEGF in skin is not increased as compared to controls as reported in a review [34], citing unpublished observations. Also, the injection of VEGF in monkey eyes promotes formation of neovessels that are not fenestrated. The increased leakiness of these latter vessels was suggested to occur by increased vesi cular transport [163].
A model for VVO induction in cells in culture was also reported [114]. In this model, bovine adrenal endothelial cells were cultured on floating Matrigel/ Type I collagen matrices in presence of 500 ng/ml of VEGF, which concurred to the formation of VVOs. In contradistinction to the model of Esser et al., the VVO clusters stained with caveolin 1. However, the authors also report an increased expression of caveolae. This suggests that the difference between VVOs and caveolar clusters might be not be easy to make by morphology only, in absence of specific VVO biochemical markers to be used for double label immunocytochemistry. Another interesting finding was that VEGF did not increase the number of fenestrae over control adrenal endothelial cells (n.b. a small number of fenestrae were present in both control and VEGF treated samples) [114]. This is different from the data of Esser et al., noted above. The endothelial cells used by both groups were from bovine adrenal, a vascular bed provided with fenestrated endothelium. Therefore, the difference in VEGF effects reported by the two groups, must be explained by the factors secreted by the epithelial cells (i.e. soluble factors or matrix components) that enable induction of fenestrae by VEGF.
In a series of papers [83–85], Hnasko, et al, claim that PV1 is actually downregulated by VEGFR2 signaling in immortalized endothelial cells (MS-1 cells) or in the lungs of caveolin1 KO mice, whereas no effect was seen on the caveolin-2 KO or WT lungs. These data are in sharp contrast with the data obtained in cells in culture [138–140]. It is not clear whether this is specific to the lung, whether it is due to the side effects of the VEGF inhibitors used or, as discussed above, that the antibodies used by these investigators are not fully validated.
Other angiogenic factors that induce diaphragms
Leptin, a hormone with angiogenic activity secreted by the adipocytes, also causes fenestration of endothelia [164]in situ. In normal ovaries, human choriogonadotropin (hCG) was linked to the formation of fenestrae whereas interleukin 8 does not seem to have this function [165].
Function of endothelial diaphragms in normal vessels and angiogenesis
The precise function of the stomatal or fenestral diaphragms in normal vessels is not known. One of the hurdles in assessing their function is their occurrence in at least four different endothelial structures (caveolae, TEC, fenestrae and VVOs). However, by their localization at presumed sites of transendothelial exchange one could assume a sieving function for the diaphragms. The passage of select molecules from the blood plasma to the interstitial fluid could be either inhibited or facilitated (for reviews see refs. [16, 38, 166, 167]).
With respect to angiogenic vessels, the function of these structures becomes even more elusive as the newly formed capillaries, with very few exceptions, are usually immature, tortuous and leaky due to large gaps in the vessel wall [168–170].
Conclusions
From all these data a model emerges in which endothelial cells would be capable of forming highly ordered structures such as the SD or the fenestral diaphragm. An exciting development is that all of these structures are expressed by vessels in angio-genenesis as demonstrated by electron microscopy as well as by PV1 staining. Thus, PV1, the structural component of these differentiated microdomains of the endothelial cells, joins the widening ranks of novel tumour endothelium markers. Moreover, by its postulated function in endothelial cell migration PV1 might be a putative therapeutic target [140] although its presence in the normal endothelium from several organs would not recommend it [141].
We are far from having a clear understanding of how these endothelial structures come into being and what clear purpose(s) they serve. With a biochemical definition of the components of these entities all the striking images obtained by researchers in the past will truly come to life.
Acknowledgments
RVS is supported by NIH grants: HL65418 and HL83249.
References
- 1.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–45. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 2.Tirziu D, Simons M. Angiogenesis in the human heart: gene and cell therapy. Angiogenesis. 2005;8:241–51. doi: 10.1007/s10456-005-9011-z. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. Exs. 2005;94:209–31. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak HF. Angiogenesis: update 2005. J Thromb Haemost. 2005;3:1835–42. doi: 10.1111/j.1538-7836.2005.01361.x. [DOI] [PubMed] [Google Scholar]
- 6.Palade GE. Fine structure of blood capillaries. J Appl Phys. 1953;24:1424. [Google Scholar]
- 7.Palade GE, Bruns RR. Structural modulations of plasmalemmal vesicles. J Cell Biol. 1968;37:633–49. doi: 10.1083/jcb.37.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simionescu N, Siminoescu M, Palade GE. Permeability of muscle capillaries to small hemepeptides. Evidence for the existence of patent transendothelial channels. J Cell Biol. 1975;64:586–607. doi: 10.1083/jcb.64.3.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautier A, Bernhard W, Oberling C. Sur l'existence d'un appareil lacunaire pericapillaire du glomerule de Malpighi, revele par la microscopie electronique. Comptes rendus de la seances de la Societe de Biologie. 1950;144:1605–7. [PubMed] [Google Scholar]
- 10.Clementi F, Palade GE. Intestinal capillaries. II. Structural effects ofEDTA and histamine. J Cell Biol. 1969;42:706–14. doi: 10.1083/jcb.42.3.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clementi F, Palade GE. Intestinal capillaries. I. Permeability to peroxidase and ferritin. J Cell Biol. 1969;41:33–58. doi: 10.1083/jcb.41.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simionescu M, Simionescu N, Palade GE. Morphometric data on the endothelium of blood capillaries. J Cell Biol. 1974;60:128–52. doi: 10.1083/jcb.60.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milici AJ, Peters KR, Palade GE. The endothelial pocket. A new structure in fenestrated endothelia. Cell Tissue Res. 1986;244:493–9. doi: 10.1007/BF00212526. [DOI] [PubMed] [Google Scholar]
- 14.Milici AJ, Furie MB, Carley WW. The formation of fenestrations and channels by capillary endothelium in vitro. Proc Natl Acad Sci USA. 1985;82:6181–5. doi: 10.1073/pnas.82.18.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braet F, Wisse E, Bomans P, Frederik P, Geerts W, Koster A, Soon L, Ringer S. Contribution of high-resolution correlative imaging techniques in the study of the liver sieve in three-dimensions. Microsc Res Tech. 2007;70:230–42. doi: 10.1002/jemt.20408. [DOI] [PubMed] [Google Scholar]
- 16.Stan RV. Structure of caveolae. Biochim Biophys Acta. 2005;1746:334–48. doi: 10.1016/j.bbamcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Bearer EL, Orci L. Endothelial fenestral diaphragms: a quick-freeze, deep-etch study. J Cell Biol. 1985;100:418–28. doi: 10.1083/jcb.100.2.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noguchi Y, Shibata Y, Yamamoto T. Endothelial vesicular system in rapid-frozen muscle capillaries revealed by serial sectioning and deep etching. Anat Rec. 1987;217:355–60. doi: 10.1002/ar.1092170406. [DOI] [PubMed] [Google Scholar]
- 19.Pelkmans L, Helenius A. Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol. 2003;15:414–22. doi: 10.1016/s0955-0674(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 20.Simionescu M, Simionescu N, Silbert JE, Palade GE. Differentiated microdomains on the luminal surface of the capillary endothelium. II. Partial characterization of their anionic sites. J Cell Biol. 1981;90:614–21. doi: 10.1083/jcb.90.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simionescu N, Simionescu M, Palade GE. Differentiated microdomains on the luminal surface of the capillary endothelium. I. Preferential distribution of anionic sites. J Cell Biol. 1981;90:605–13. doi: 10.1083/jcb.90.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maul GG. Structure and formation of pores in fenestrated capillaries. J Ultrastruct Res. 1971;36:768–82. doi: 10.1016/s0022-5320(71)90030-x. [DOI] [PubMed] [Google Scholar]
- 23.Apkarian RP. The fine structure of fenestrated adrenocortical capillaries revealed by in-lens field-emission scanning electron microscopy and scanning transmission electron microscopy. Scanning. 1997;19:361–7. doi: 10.1002/sca.4950190503. [DOI] [PubMed] [Google Scholar]
- 24.Oberling C, Gautier A, Bernhardt W. La structure des capillaires glomerulairevue au microscope electronique. Presse medicale Parisiene. 1951;59:938–40. [PubMed] [Google Scholar]
- 25.Pease DC. Electron microscopy of the vascular bed of the kidney cortex. Anat Rec. 1955;121:701–12. doi: 10.1002/ar.1091210402. [DOI] [PubMed] [Google Scholar]
- 26.Reeves WH, Kanwar YS, Farquhar MG. Assembly of the glomerular filtration surface. Differentiation of anionic sites in glomerular capillaries of newborn rat kidney. J Cell Biol. 1980;85:735–53. doi: 10.1083/jcb.85.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamada E. The fine structure of the mouse kidney glomerulus. J Biophys Biochem Cytol. 1955;1:551–66. doi: 10.1083/jcb.1.6.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friederici HH. On the diaphragm across fenestrae of capillary endothelium. J Ultrastruct Res. 1969;27:373–5. doi: 10.1016/s0022-5320(69)80024-9. [DOI] [PubMed] [Google Scholar]
- 29.Rostgaard J, Qvortrup K. Electron microscopic demonstrations of filamentous molecular sieve plugs in capillary fenestrae. Microvasc Res. 1997;53:1–13. doi: 10.1006/mvre.1996.1987. [DOI] [PubMed] [Google Scholar]
- 30.Roberts WG, Palade GE. Endothelial fenestrae and fenestral diaphragms. In: Rissau W, Rubanyi GM, editors. Morphogenesis of endothelium. Amsterdam: Hardwood Academic Publishers; 2000. pp. 23–41. [Google Scholar]
- 31.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO):a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol. 1996;59:100–15. [PubMed] [Google Scholar]
- 32.Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 1999;6:23–44. [PubMed] [Google Scholar]
- 33.Feng D, Nagy JA, Dvorak HF, Dvorak AM. Ultrastructural studies define soluble macromolecular, particulate, and cellular transendothelial cell pathways in venules, lymphatic vessels, and tumour-associated microvessels in man and animals. Microsc Res Tech. 2002;57:289–326. doi: 10.1002/jemt.10087. [DOI] [PubMed] [Google Scholar]
- 34.Kohn S, Nagy JA, Dvorak HF, Dvorak AM. Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumour blood vessels. Lab Invest. 1992;67:596–607. [PubMed] [Google Scholar]
- 35.Vasile E, Dvorak AM, Stan RV, Dvorak HF. Isolation and characterization of caveolae and vesiculo-vacuolar organelles from endothelial cells cultured with VPF/VEGF and from human lung. Mol Biol Cell. 2000;11:121. [Google Scholar]
- 36.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chier KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–80. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stan RV. Channels across endothelial cells. In: Baluska F, Volkmann D, Barlowe PW, editors. Cell-cell channels. Georgetown, TX: Landes Biosciences; 2006. pp. 271–8. [Google Scholar]
- 38.Yoshida Y, Yamada M, Wakabayashi K, Ikuta F. Endothelial fenestrae in the rat fetal cerebrum. Brain Res Dev Brain Res. 1988;44:211–9. doi: 10.1016/0165-3806(88)90219-2. [DOI] [PubMed] [Google Scholar]
- 39.Milici AJ, Bankston PW. Fetal and neonatal rat intestinal capillaries: a TEM study of changes in the mural structure. Am J Anat. 1981;160:435–48. doi: 10.1002/aja.1001600407. [DOI] [PubMed] [Google Scholar]
- 40.Milici AJ, Bankston PW. Fetal and neonatal rat intestinal capillaries:permeability to carbon, ferritin, hemoglobin, and myoglobin. Am J Anat. 1982;165:165–86. doi: 10.1002/aja.1001650206. [DOI] [PubMed] [Google Scholar]
- 41.Naito M, Wisse E. Observations on the fine structure and cytochemistry of sinusoidal cells in fetal and neonatal liver. In: Wisse E, editor. Kupffer cells and other liver sinusoidal cells. Amsterdam: Elsevier; 1977. pp. 497–505. [Google Scholar]
- 42.Bankston PW, Pino RM. The development of the sinusoids of fetal rat liver:morphology of endothelial cells, Kupffer cells, and the transmural migration of blood cells into the sinusoids. Am J Anat. 1980;159:1–15. doi: 10.1002/aja.1001590102. [DOI] [PubMed] [Google Scholar]
- 43.Stan RV, Arden KC, Palade GE. cDNA and protein sequence, genomic organization, and analysis of cis regulatory elements of mouse and human PLVAP genes. Genomics. 2001;72:304–13. doi: 10.1006/geno.2000.6489. [DOI] [PubMed] [Google Scholar]
- 44.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202:325–32. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 45.Bradbury MW. The structure and function of the blood-brain barrier. Fed Proc. 1984;43:186–90. [PubMed] [Google Scholar]
- 46.Margulies EH, Kardia SL, Innis JW. A comparative molecular analysis of developing mouse forelimbs and hindlimbs using serial analysis of gene expression (SAGE) Genome Res. 2001;11:1686–98. doi: 10.1101/gr.192601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penn PE, Jiang DZ, Fei RG, Sitnicka E, Wolf NS. Dissecting the hematopoietic microenvironment. IX. Further characterization of murine bone marrow stromal cells. Blood. 1993;81:1205–13. [PubMed] [Google Scholar]
- 48.Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–31. [PubMed] [Google Scholar]
- 49.Helisch A, Schaper W. Arteriogenesis:the development and growth of collateral arteries. Microcirculation. 2003;10:83–97. doi: 10.1038/sj.mn.7800173. [DOI] [PubMed] [Google Scholar]
- 50.Scholz D, Ito W, Fleming I, Deindl E, Sauer A, Wiesnet M, Busse R, Schaper J, Schaper W. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis) Virchows Arch. 2000;436:257–70. doi: 10.1007/s004280050039. [DOI] [PubMed] [Google Scholar]
- 51.Wolf C, Cai WJ, Vosschulte R, Koltai S, Mousavipour D, Scholz D, Afsah-Hedjri A, Schaper W, Schaper J. Vascular remodeling and altered protein expression during growth of coronary collateral arteries. J Mol Cell Cardiol. 1998;30:2291–305. doi: 10.1006/jmcc.1998.0790. [DOI] [PubMed] [Google Scholar]
- 52.McKinney RV, Jr, Singh BB, Brewer PD. Fenestrations in regenerating skeletal muscle capillaries. Am J Anat. 1977;150:213–8. doi: 10.1002/aja.1001500117. [DOI] [PubMed] [Google Scholar]
- 53.Oki S, Desaki J, Matsuda Y, Shibata T, Okumura H. Capillaries with fenestrations around regenerating muscle fibers in the soleus muscle of the dystrophic (dy) mouse. J Orthop Sci. 1998;3:67–70. doi: 10.1007/s007760050023. [DOI] [PubMed] [Google Scholar]
- 54.Desaki J, Oki S, Matsuda Y, Sakanaka M. Morphological changes of capillaries in the rat soleus muscle following experimental tenotomy. J Electron Microsc. 2000;49:185–93. doi: 10.1093/oxfordjournals.jmicro.a023785. [DOI] [PubMed] [Google Scholar]
- 55.Ghatak NR, Nochlin D. Fenestrated blood vessels in human skeletal muscle. Acta Neuropathol. 1979;47:169–74. doi: 10.1007/BF00690543. [DOI] [PubMed] [Google Scholar]
- 56.Braverman IM, Yen A. Ultrastructure of the capillary loops in the dermal papillae of psoriasis. J Invest Dermatol. 1977;68:53–60. doi: 10.1111/1523-1747.ep12485169. [DOI] [PubMed] [Google Scholar]
- 57.De Almeida OP, Bohm GM. Vascular permeability in the rat gingiva. A model of vessel response in chronic inflammation. J Pathol. 1979;127:27–34. doi: 10.1002/path.1711270105. [DOI] [PubMed] [Google Scholar]
- 58.Kawanami O, Matsuda K, Yoneyama H, Ferrans VJ, Crystal RG. Endothelial fenestration of the alveolar capillaries in interstitial fibrotic lung diseases. Acta Pathol Japan. 1992;42:177–84. doi: 10.1111/j.1440-1827.1992.tb01669.x. [DOI] [PubMed] [Google Scholar]
- 59.Snyder DH, Valsamis MP, Stone SH, Raine CS. Progressive demyelination and reparative phenomena in chronic experimental allergic encephalomyelitis. J Neuropathol Exp Neurol. 1975;34:209–21. doi: 10.1097/00005072-197505000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Llena JF, Cespedes G, Hirano A, Zimmerman HM, Feiring EH, Fine D. Vascular alterations in delayed radiation necrosis of the brain. An electron microscopical study. Arch Pathol Lab Med. 1976;100:531–4. [PubMed] [Google Scholar]
- 61.Hirano A, Kawanami T, Llena JF. Electron microscopy of the blood-brain barrier in disease. Microsc Res Tech. 1994;27:543–56. doi: 10.1002/jemt.1070270609. [DOI] [PubMed] [Google Scholar]
- 62.Wallow IH, Geldner PS. Endothelial fenestrae in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1980;19:1176–83. [PubMed] [Google Scholar]
- 63.Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, Berse B, Dvorak HF. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. 1994;180:1141–6. doi: 10.1084/jem.180.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumour cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 65.Pappas GD, Sagen J. Fine structural correlates of vascular permeability of chromaffin cell transplants in CNS pain modulatory regions. Exp Neurol. 1988;102:280–9. doi: 10.1016/0014-4886(88)90221-x. [DOI] [PubMed] [Google Scholar]
- 66.Shibata S, Fukushima M, Mori K. Ultrastructure of capillary permeability in human brain tumours. 2: Mechanisms of contrast enhancement in gliomas. No Shinkei Geka. 1986;14:311–6. [PubMed] [Google Scholar]
- 67.Capo V, Ozzello L, Fenoglio CM, Lombardi L, Rilke F. Angiosarcomas arising in edematous extremities: immunostaining for factor VIII-related antigen and ultrastructural features. Hum Pathol. 1985;16:144–50. doi: 10.1016/s0046-8177(85)80063-0. [DOI] [PubMed] [Google Scholar]
- 68.Front D, Israel O, Kohn S, Nir I. The blood-tissue barrier of human brain tumours: correlation of scintigraphic and ultrastructural findings: concise communication. J Nucl Med. 1984;25:461–5. [PubMed] [Google Scholar]
- 69.Wagner RF, Jr, Gerard G, Sciubba JJ. Amelanotic melanoma of the lung and brain with fenestrated intrinsic tumour capillaries. Surg Neurol. 1981;15:384–8. doi: 10.1016/0090-3019(81)90177-4. [DOI] [PubMed] [Google Scholar]
- 70.Ludatscher RM, Gellei B, Barzilai D. Ultrastructural observations on the capillaries of human thyroid tumours. J Pathol. 1979;128:57–62. doi: 10.1002/path.1711280202. [DOI] [PubMed] [Google Scholar]
- 71.Kasantikul V, Glick AD, Netsky MG. Light and electron microscopic observations of blood vessels in neurilemoma. Arch Pathol Lab Med. 1979;103:683–7. [PubMed] [Google Scholar]
- 72.Wisse E, Braet F, Luo D, De Zanger R, Jans D, Crabbe E, Vermoesen A. Structure and function of sinusoidal lining cells in the liver. Toxicol Pathol. 1996;24:100–11. doi: 10.1177/019262339602400114. [DOI] [PubMed] [Google Scholar]
- 73.Madigan MC, Penfold PL. Human retinoblastoma:a morphological study of apoptotic, leukocytic, and vascular elements. Ultrastruct Pathol. 1997;21:95–107. doi: 10.3109/01913129709021310. [DOI] [PubMed] [Google Scholar]
- 74.Jin E, Fujiwara M, Nagashima M, Shimizu H, Ghazizadeh M, Pan X, Arai S, Ohaki Y, Gomibuchi M, Takemura T, Kawanami O. Aerogenous spread of primary lung adenocarcinoma induces ultrastructural remodeling of the alveolar capillary endothelium. Hum Pathol. 2001;32:1050–8. doi: 10.1053/hupa.2001.28243. [DOI] [PubMed] [Google Scholar]
- 75.Jin E, Ghazizadeh M, Fujiwara M, Nagashima M, Shaimizu H, Ohaki Y, Arai S, Gomibuchi M, Takerara T, Kawarami O. Angiogenesis and phenotypic alteration of alveolar capillary endothelium in areas of neoplastic cell spread in primary lung adenocarcinoma. Pathol Int. 2001;51:691–700. doi: 10.1046/j.1440-1827.2001.01264.x. [DOI] [PubMed] [Google Scholar]
- 76.Stan RV, Ghitescu L, Jacobson BS, Palade GE. Isolation, cloning, and localization of rat PV-1, a novel endothelial caveolar protein. J Cell Biol. 1999;145:1189–98. doi: 10.1083/jcb.145.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stan RV, Kubitza M, Palade GE. PV-1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc Natl Acad Sci USA. 1999;96:13203–7. doi: 10.1073/pnas.96.23.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghitescu LD, Crine P, Jacobson BS. Antibodies specific to the plasma membrane of rat lung microvascular endothelium. Exp Cell Res. 1997;232:47–55. doi: 10.1006/excr.1997.3490. [DOI] [PubMed] [Google Scholar]
- 79.Stan RV, Roberts WG, Predescu D, Ihida K, Saucan L, Ghitescu L, Palade GE. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae) Mol Biol Cell. 1997;8:595–605. doi: 10.1091/mbc.8.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hnasko R, McFarland M, Ben-Jonathan N. Distribution and characterization of plasmalemma vesicle protein-1 in rat endocrine glands. J Endocrinol. 2002;175:649–61. doi: 10.1677/joe.0.1750649. [DOI] [PubMed] [Google Scholar]
- 81.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–25. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 82.Hnasko R, Ben-Jonathan N. Developmental regulation of PV-1 in rat lung: association with the nuclear envelope and limited colocalization with Cav-1. Am J Physiol Lung Cell Mol Physiol. 2005;288:L275–84. doi: 10.1152/ajplung.00236.2004. [DOI] [PubMed] [Google Scholar]
- 83.Hnasko R, Carter JM, Medina F, Frank PG, Lisanti MP. PV-1 labels transcellular openings in mouse endothelial cells and is negatively regulated by VEGF. Cell Cycle. 2006;5:2021–8. doi: 10.4161/cc.5.17.3217. [DOI] [PubMed] [Google Scholar]
- 84.Hnasko R, Frank PG, Ben-Jonathan N, Lisanti MP. PV-1 is negatively regulated by VEGF in the lung of caveolin-1, but not caveolin-2, null mice. Cell Cycle. 2006;5:2012–20. doi: 10.4161/cc.5.17.3216. [DOI] [PubMed] [Google Scholar]
- 85.Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, Oh P, Schnitzer JE. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. 2004;22:985–92. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- 86.Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, Testa JE, Schnitzer JE. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–35. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- 87.Monfared A, Blevins NH, Cheung EL, Jung JC, Popelka G, Schnitzer MJ. In vivo imaging of mammalian cochlear blood flow using fluorescence microendoscopy. Otol Neurotol. 2006;27:144–52. doi: 10.1097/01.mao.0000190708.44067.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc Natl Acad Sci USA. 2006;103:407–12. doi: 10.1073/pnas.0506938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ioannidou S, Deinhardt K, Miotla J, Bradley J, Cheung E, Samuelsson S, Ng YS, Shina DT. An in vitro assay reveals a role for the diaphragm protein PV-1 in endothelial fenestra morphogenesis. Proc Natl Acad Sci USA. 2006;103:16770–5. doi: 10.1073/pnas.0603501103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stan RV, Tkachenko E, Niesman IR. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol Biol Cell. 2004;15:3615–30. doi: 10.1091/mbc.E03-08-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stan RV. Multiple PV1 dimers reside in the same stomatal or fenestral diaphragm. Am J Physiol Heart Circ Physiol. 2004;286:H1347–53. doi: 10.1152/ajpheart.00909.2003. [DOI] [PubMed] [Google Scholar]
- 93.Simionescu M, Simionescu N, Palade GE. Differentiated microdomains on the luminal surface of capillary endothelium: distribution of lectin receptors. J Cell Biol. 1982;94:406–13. doi: 10.1083/jcb.94.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pino RM. The cell surface of a restrictive fenestrated endothelium. II. Dynamics of cationic ferritin binding and the identification of heparin and heparan sulfate domains on the choriocapillaris. Cell Tissue Res. 1986;243:157–64. doi: 10.1007/BF00221864. [DOI] [PubMed] [Google Scholar]
- 95.Furuya S. Ultrastructure and formation of diaphragmed fenestrae in cultured endothelial cells of bovine adrenal medulla. Cell Tissue Res. 1990;261:97–105. doi: 10.1007/BF00329442. [DOI] [PubMed] [Google Scholar]
- 96.Bankston PW, Porter GA, Milici AJ, Palade GE. Differential and specific labeling of epithelial and vascular endothelial cells of the rat lung by Lycopersicon esculentum and Griffonia simplicifolia I lectins. Eur J Cell Biol. 1991;54:187–95. [PubMed] [Google Scholar]
- 97.Bankston PW, Milici AJ. A survey of the binding of polycationic ferritin in several fenestrated capillary beds:indication of heterogeneity in the luminal glycocalyx of fenestral diaphragms. Microvasc Res. 1983;26:36–48. doi: 10.1016/0026-2862(83)90053-5. [DOI] [PubMed] [Google Scholar]
- 98.Simionescu M, Simionescu N, Palade GE. Preferential distribution of anionic sites on the basement membrane and the abluminal aspect of the endothelium in fenestrated capillaries. J Cell Biol. 1982;95:425–34. doi: 10.1083/jcb.95.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Simionescu M, Simionescu N, Palade GE. Partial chemical characterization of the anionic sites in the basal lamina of fenestrated capillaries. Microvasc Res. 1984;28:352–67. doi: 10.1016/0026-2862(84)90006-2. [DOI] [PubMed] [Google Scholar]
- 100.Roberts WG, Palade GE. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:765–72. [PubMed] [Google Scholar]
- 101.Palade GF, Simionescu M, Simionescu N. Differentiated microdomains on the luminal surface of the capillary endothelium. Biorheology. 1981;18:563–8. doi: 10.3233/bir-1981-183-619. [DOI] [PubMed] [Google Scholar]
- 102.Rosenberg RD, Shworak NW, Liu J, Schwartz JJ, Zhang L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J Clin Invest. 1997;99:2062–70. doi: 10.1172/JCI119377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–73. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sorensson J, Fierlbeck W, Heider T, Schwarz K, Park DS, Mundel P, Lisanti M, Ballermann BJ. Glomerular endothelial fenestrae in vivo are not formed from caveolae. J Am Soc Nephrol. 2002;13:2639–47. doi: 10.1097/01.asn.0000033277.32822.23. [DOI] [PubMed] [Google Scholar]
- 105.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108:2369–79. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 106.Roberts WG, Delaat J, Nagane M, Huang S, Cavenee WK, Palade GE. Host microvasculature influence on tumour vascular morphology and endothelial gene expression. Am J Pathol. 1998;153:1239–48. doi: 10.1016/s0002-9440(10)65668-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rhodin JA. The diaphragm of capillary endothelial fenestrations. J Ultrastruct Res. 1962;6:171–85. doi: 10.1016/s0022-5320(62)90052-7. [DOI] [PubMed] [Google Scholar]
- 108.Schnitzer JE, Shen CP, Palade GE. Lectin analysis of common glycoproteins detected on the surface of continuous microvascular endothelium in situ and in culture:identification of sialoglycoproteins. Eur J Cell Biol. 1990;52:241–51. [PubMed] [Google Scholar]
- 109.Stolz DB, Jacobson BS. Macro- and microvascular endothelial cells in vitro: maintenance of biochemical heterogeneity despite loss of ultrastructural characteristics in vitro. Cell Dev Biol. 1991;27A:169–82. doi: 10.1007/BF02631005. [DOI] [PubMed] [Google Scholar]
- 110.Folkman J, Haudenschild C. Angiogenesis in vitro. Nature. 1980;288:551–6. doi: 10.1038/288551a0. [DOI] [PubMed] [Google Scholar]
- 111.Lombardi T, Montesano R, Furie MB, Silverstein SC, Orci L. Endothelial diaphragmed fenestrae: in vitro modulation by phorbol myristate acetate. J Cell Biol. 1986;102:1965–70. doi: 10.1083/jcb.102.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Madden SL, Cook BP, Nacht M, Weber WD, Callahan MR, Jiang Y, Dufault MR, Zhang X, Zhang W, Walter-Yohrling J, Rouleau C, Akmaev VR, Wang CJ, Cao X, St Martin TB, Roberts BL, Teicher BA, Klinger KW, Stan RV, Lucey B, Carson-Walter EB, Laterra J, Walter KA. Vascular gene expression in nonneoplastic and malignant brain. Am J Pathol. 2004;165:601–8. doi: 10.1016/s0002-9440(10)63324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vasile E, Qu H, Dvorak HF, Dvorak AM. Caveolae and vesiculo-vacuolar organelles in bovine capillary endothelial cells cultured with VPF/VEGF on floating matrigel-collagen gels. J Histochem Cytochem. 1999;47:159–67. doi: 10.1177/002215549904700205. [DOI] [PubMed] [Google Scholar]
- 114.Bendayan M. Pathway of insulin in pancreatic tissue on its release by the B-cell. Am J Physiol. 1993;264:G187–94. doi: 10.1152/ajpgi.1993.264.2.G187. [DOI] [PubMed] [Google Scholar]
- 115.Lombardi T, Montesano R, Furie MB, Silverstein SC, Orci L. In vitro modulation of endothelial fenestrae: opposing effects of retinoic acid and transforming growth factor beta. J Cell Sci. 1988;91(Pt 2):313–8. doi: 10.1242/jcs.91.2.313. [DOI] [PubMed] [Google Scholar]
- 116.Lombardi T, Montesano R, Orci L. Phorbol ester induces diaphragmed fenestrae in large vessel endothelium in vitro. Eur J Cell Biol. 1987;44:86–9. [PubMed] [Google Scholar]
- 117.Koya M, Chang L, Truong A, Brightman MW. Chemical induction of fenestrae in vessels of the blood-brain barrier. Exp Neurol. 1996;142:6–13. doi: 10.1006/exnr.1996.0174. [DOI] [PubMed] [Google Scholar]
- 118.Kazanietz MG. Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol Pharmacol. 2002;61:759–67. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- 119.Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–32. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- 120.Connolly DT, Olander JV, Heuvelman D, Nelson R, Monsell R, Siegel N, Haymore BL. Human vascular permeability factor. Isolation from U937 cells. J Clin Invest. 1989;264:20017–24. [PubMed] [Google Scholar]
- 121.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–59. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Böttinger EP, Letterio JJ, Roberts AB. Biology of TGF-beta in knockout and transgenic mouse models. Kidney Int. 1997;51:1355–60. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- 123.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumour-derived expression of vascular endothelial growth factor is a critical factor in tumour expansion and vascular function. Cancer Res. 1999;59:1592–8. [PubMed] [Google Scholar]
- 124.Post MJ, Laham R, Sellke FW, Simons M. Therapeutic angiogenesis in cardiology using protein formulations. Cardiovasc Res. 2001;49:522–31. doi: 10.1016/s0008-6363(00)00216-9. [DOI] [PubMed] [Google Scholar]
- 125.Wadia JS, Dowdy SF. Protein transduction technology. Curr Opin Biotechnol. 2002;13:52–6. doi: 10.1016/s0958-1669(02)00284-7. [DOI] [PubMed] [Google Scholar]
- 126.Bates DO, Hillman NJ, Williams B, Neal CR, Pocock TM. Regulation of microvascular permeability by vascular endothelial growth factors. J Anat. 2002;200:581–97. doi: 10.1046/j.1469-7580.2002.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 12. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 128.Chen J, Braet F, Brodsky S, Weinstein T, Romanov V, Noiri E, Goligorsky MS. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol. 2002;282:C1053–63. doi: 10.1152/ajpcell.00292.2001. [DOI] [PubMed] [Google Scholar]
- 129.Gerber HP, Hillan KJ, Ryan AM. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–59. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 130.Lammert E, Gu G, McLaughlin M, et al. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2003;13:1070–4. doi: 10.1016/s0960-9822(03)00378-6. [DOI] [PubMed] [Google Scholar]
- 131.Eremina V, Cui S, Gerber H. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–35. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 132.Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hn-Lowe DD, McDonald DM. Rapid vascular regrowth in tumours after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–21. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O'Brien SM, Davis RB, Gouen LC, Anderson KO, Thurston G, Joho S, Spinger ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–76. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 134.Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–59. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- 135.Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky OR, Thurston G, Yancopoulos GD, McDonald DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumour vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baluk P, Lee CG, Link H, Ator E, Haskell A, Elias JA, McDonald DM. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165:1071–85. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strickland LA, Jubb AM, Hongo JA, Zhong F, Burwick J, Fu L, Frantz GD, Koeppen H. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF) J Pathol. 2005;206:466–75. doi: 10.1002/path.1805. [DOI] [PubMed] [Google Scholar]
- 138.Carson-Walter EB, Hampton J, Shue E, Geynisman DM, Pillai PK, Sathanoori R, Madden SL, Hamilton RL, Walter KA. Plasmalemmal vesicle associated protein-1 is a novel marker implicated in brain tumour angiogenesis. Clin Cancer Res. 2005;11:7643–50. doi: 10.1158/1078-0432.CCR-05-1099. [DOI] [PubMed] [Google Scholar]
- 139.Sanders Strickland LA, Koeppen H. Comments on plasmalemmal vesicle associated protein-1 as a novel marker implicated in brain tumour angiogene-sis. Clin Cancer Res. 2006;12:2649–50. doi: 10.1158/1078-0432.CCR-05-2624. [DOI] [PubMed] [Google Scholar]
- 140.Niemela H, Elima K, Henttinen T, Irjala H, Salmi M, Jalkanen S. Molecular identification of PAL-E, a widely used endothelial-cell marker. Blood. 2005;106:3405–9. doi: 10.1182/blood-2005-01-0254. [DOI] [PubMed] [Google Scholar]
- 141.Schlingemann RO, Dingjan GM, Emeis JJ, Blok J, Warnaar SO, Ruiter DJ. Monoclonal antibody PAL-E specific for endothelium. Lab Invest. 1985;52:71–6. [PubMed] [Google Scholar]
- 142.Sundberg C, Ljungstrom M, Lindmark G, Gerdin B, Rubin K. Microvascular pericytes express platelet-derived growth factor-beta receptors in human healing wounds and colorectal adenocarcinoma. Am J Pathol. 1993;143:1377–88. [PMC free article] [PubMed] [Google Scholar]
- 143.Paavonen K, Puolakkainen P, Jussila L, Jahkola T, Alitalo K. Vascular endothelial growth factor receptor-3 in lymphangiogenesis in wound healing. Am J Pathol. 2000;156:1499–504. doi: 10.1016/S0002-9440(10)65021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schlingemann RO, Rietveld FJ, Kwaspen F, Van De Kerkhof PC, De Waal RM, Ruiter DJ. Differential expression of markers for endothelial cells, pericytes, and basal lamina in the microvasculature of tumours and granulation tissue. Am J Pathol. 1991;138:1335–47. [PMC free article] [PubMed] [Google Scholar]
- 145.Ruiter DJ, Schlingemann RO, Westphal JR, Denijn M, Rietveld FJ, De Waal RM. Angiogenesis in wound healing and tumour metastasis. Behring Inst Mitt. 1993;92:258–72. [PubMed] [Google Scholar]
- 146.Witmer AN, Van Blijswijk BC, Van Noorden CJ, Vrensen GF, Schlingemann RO. In vivo angiogenic phenotype of endothelial cells and pericytes induced by vascular endothelial growth factor-A. J Histochem Cytochem. 2004;52:39–52. doi: 10.1177/002215540405200105. [DOI] [PubMed] [Google Scholar]
- 147.Holden CA, Spaull J, Das AK, McKee PH, Jones EW. The histogenesis of angiosarcoma of the face and scalp: an immunohistochemical and ultrastructural study. Histopathology. 1987;11:37–51. doi: 10.1111/j.1365-2559.1987.tb02607.x. [DOI] [PubMed] [Google Scholar]
- 148.Kahn HJ, Bailey D, Marks A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi's sarcoma and a subset of angiosarcomas. Mod Pathol. 2002;15:434–40. doi: 10.1038/modpathol.3880543. [DOI] [PubMed] [Google Scholar]
- 149.Nadimi H, Saatee S, Armin A, Toto PD. Expression of endothelial cell markers PAL-E and EN-4 and Ia-antigens in Kaposi's sarcoma. J Oral Pathol. 1988;17:416–20. doi: 10.1111/j.1600-0714.1988.tb01307.x. [DOI] [PubMed] [Google Scholar]
- 150.Schulze HJ, Rutten A, Mahrle G, Steigleder GK. Initial lesions of HIV-related Kaposi's sarcoma–a histological, immunohistochemical, and ultrastructural study. Arch Dermatol Res. 1987;279:499–503. doi: 10.1007/BF00413279. [DOI] [PubMed] [Google Scholar]
- 151.Weninger W, Partanen TA, Breiteneder-Geleff S, Mager C, Kawalski H, Mildner M, Pammer J, Sturzl M, Kerjaschki D, Alitalo K, Tschachler E. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi's sarcoma tumour cells. Lab Invest. 1999;79:243–51. [PubMed] [Google Scholar]
- 152.Sarawar SR, Schlingemann RO, Kelsey A, Fleming S, Kumar S. A monoclonal antibody stains blastemal but not tubular components of Wilms' tumour. J Pathol. 1988;156:319–24. doi: 10.1002/path.1711560408. [DOI] [PubMed] [Google Scholar]
- 153.Leenstra S, Das PK, Troost D, Bosch DA, Claessen N, Becker AE. PAL-E, monoclonal antibody with immunoreactivity for endothelium specific to brain tumours. Lancet. 1990;335:671. doi: 10.1016/0140-6736(90)90465-h. [DOI] [PubMed] [Google Scholar]
- 154.Leenstra S, Troost D, Das PK, Claessen N, Becker AE, Bosch DA. Endothelial cell marker PAL-E reactivityin brain tumour, developing brain, and brain disease. Cancer. 1993;72:3061–7. doi: 10.1002/1097-0142(19931115)72:10<3061::aid-cncr2820721031>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 155.Renard N, Nooijen PT, Schalkwijk L, De Waal RM, Eggermont AM, Lienard D, Kroon BB, Lejeune FJ, Ruiter DJ. VWF release and platelet aggregation in human melanoma after perfusion with TNF alpha. J Pathol. 1995;176:279–87. doi: 10.1002/path.1711760310. [DOI] [PubMed] [Google Scholar]
- 156.Clarijs R, Schalkwijk L, Hofmann UB, Ruiter DJ, De Waal RM. Induction of vascular endothelial growth factor receptor-3 expression on tumour microvasculature as a new progression marker in human cutaneous melanoma. Cancer Res. 2002;62:7059–65. [PubMed] [Google Scholar]
- 157.Nakamura S, Muro H, Suzuki S, Sakaguchi T, Konno H, Baba S, Syed AS. Immunohistochemical studies on endothelial cell phenotype in hepatocellular carcinoma. Hepatology. 1997;26:407–15. doi: 10.1002/hep.510260222. [DOI] [PubMed] [Google Scholar]
- 158.Nielsen BS, Sehested M, Kjeldsen L, Borregaard N, Rygaard J, Dano K. Expression of matrix metalloprotease-9 in vascular pericytes in human breast cancer. Lab Invest. 1997;77:345–55. [PubMed] [Google Scholar]
- 159.Sauter B, Foedinger D, Sterniczky B, Wolff K, Rappersberger K. Immunoelectron microscopic characterization of human dermal lymphatic microvascular endothelial cells. Differential expression of CD31, CD34, and type IV collagen with lymphatic endothelial cells vs blood capillary endothelial cells in normal human skin, lymphangioma, and hemangioma in situ. J Histochem Cytochem. 1998;46:165–76. doi: 10.1177/002215549804600205. [DOI] [PubMed] [Google Scholar]
- 160.Feng D, Nagy JA, Brekken RA, Pettersson A, Manseau EJ, Pyne K, Mulligan R, Thorpe PE, Dvorak HF, Dvorak AM. Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressingtumours and adenoviral vectors. J Histochem Cytochem. 2000;48:545–56. doi: 10.1177/002215540004800412. [DOI] [PubMed] [Google Scholar]
- 161.Hofman P, Blaauwgeers HG, Tolentino MJ, Adamis AP, Nunes Cardozo BJ, Wensen GF, Schlingemann RO. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-A. Curr Eye Res. 2000;21:637–45. [PubMed] [Google Scholar]
- 162.Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc Natl Acad Sci USA. 2001;98:6390–5. doi: 10.1073/pnas.101564798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Goto J, Suganuma N, Takata K, Kitamura K, Asahina T, Kobayashi H, Muranaka Y, Furuhashi M, Kanayama N. Morphological analyses of inter-leukin-8 effects on rat ovarian follicles at ovulation and luteinization in vivo. Cytokine. 2002;20:168–73. doi: 10.1006/cyto.2002.1987. [DOI] [PubMed] [Google Scholar]
- 164.Stan RV. Endothelial diaphragms:nanogates of vascular permeability. Anat Rec. 2007 in press. [Google Scholar]
- 165.Stan RV. Endothelial structures involved in vascular permeability. Cambrige: Cambridge University Press; 2007. [Google Scholar]
- 166.Chang YS, Di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumours: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA. 2000;97:14608–13. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumour vessel leakiness. Am J Pathol. 2000;156:1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McDonald DM, Munn L, Jain RK. Vasculogenic mimicry:how convincing, how novel, and how significant? Am J Pathol. 2000;156:383–8. doi: 10.1016/S0002-9440(10)64740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]