

Abstract
Bicarbonate transport (BT) has been previously shown to participate in apoptosis induced by various stress factors. However, the precise role of BT in ischaemia-induced apoptosis is still unknown. To investigate this subject, rat coronary endothelial cells (EC) were exposed to simulated ischaemia (glucose free anoxia at Ph 6.4) for 2 hrs and cells undergoing apoptosis were visualized by nuclear staining or by determination of cas-pase- 3 activity. To inhibit BT, EC were either treated with the inhibitor of BT 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS, 300 μmol/l) or exposed to ischaemia in bicarbonate free, 4-(2-hydroxyethyl)-I-piperazi-neethanesulphonic acid (HEPES)-buffered medium. Simulated ischaemia in bicarbonate-buffered medium (Bic) increased caspase-3 activity and the number of apoptotic cell (23.7 + 1.4%versus 5.1 + 1.2% in control). Omission of bicarbonate during ischaemia further significantly increased caspase-3 activity and the number of apoptotic cells (36.7 1.7%). Similar proapoptotic effect was produced by DIDS treatment during ischaemia in Bic, whereas DIDS had no effect when applied in bicarbonate-free, HEPES-buffered medium (Hep). Inhibition of BT was without influence on cytosolic acidification during ischaemia and slightly reduced cytosolic Ca2+ accumulation. Initial characterization of the underlying mechanism leading to apoptosis induced by BT inhibition revealed activation of the mitochondrial pathway of apoptosis, i.e., increase of cytochrome C release, depolarization of mitochondria and translocation of Bax protein to mitochondria. In contrast, no activation of death receptor-dependent pathway (caspase-8 cleavage) and endoplasmic reticulum- dependent pathway (caspase-12 cleavage) was detected. In conclusion, BT plays an important role in ischaemia-induced apoptosis of coronary EC by suppression of mitochondria-dependent apoptotic pathway.
Keywords: apoptosis, endothelial cells, ischaemia, bicarbonate transport, cytochrome C, Bax
Introduction
Apoptosis of the coronary endothelial cells (EC) can lead to endothelial dysfunction [1] and may induce the apoptosis of cardiac myocytes [2]. Myocardial ischaemia has been shown to be an important trigger of apoptosis [3, 4]. Within numerous pro-apoptotic stress factors, alteration of ion homeostasis is an important cause of apoptosis under ischaemic environment. Particularly, extra- and intracellular accumulation of H+, which is one of the essential features of the ischaemic insult, has been shown to induce apoptosis in many cell types [5–7], including coronary EC [8]. Regulation of the intracellular H+ homeostasis in EC is dependent on the activity of two iontransporting systems: i.e., Na+/H+ -exchanger and Na+– or Cl−-coupled bicarbonate transport (BT) [9]. Although the involvement of Na+/H+ exchanger in regulation of apoptosis has been shown under various stress conditions, including ischaemia [3], the role of BT is still poorly understood. Under serum deprivation BT seems to play an anti-apoptotic role [10], while apoptosis induced by staurosporine can be ameliorated through BT inhibition [11]. Whether BT also modulates the apoptosis induced by ischaemia and which signalling mechanisms are involved is unknown. Therefore, the aim of the present study was to investigate the role of BT in endothelial apoptosis under ischaemic conditions.
As an experimental model, a primary culture of rat coronary EC was exposed to in vitro simulated ischaemia consisted of glucose-free anoxia in combination with extracellular acidosis. This model has been characterized in detail in our previous studies with respect to cytosolic Ca2+ and pH homeostasis and apoptotic signalling mechanisms [8, 12]. To analyse the role of BT, treatment with a stilbene derivative, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), the tool most frequently used for the inhibition of BT, was applied. This irreversible anion channel inhibitor is not selective for the BT and has also inhibitory effect on Cl− channels [13]. Because of this limitation the experiments with DIDS were compared with another set of experiments in which BT was blocked by omission of bicarbonate from media. Applying these two tools, we found that inhibition of BT significantly increased apoptosis of EC due to activation of mitochondrial pathway.
Materials and methods
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Cell culture
Coronary EC were isolated from 250 to 300 g male Wistar rats and maintained in Eagle's minimal essential medium 199 supplemented with 10% foetal calf serum and 10% newborn calf serum as previously described [14]. The purity of the cell culture (> 95% EC) was confirmed by immunochemical staining with antibodies against von Willebrand factor (vWF) and by uptake of DiI-ac-low-density lipoprotein (LDL) as previously described [15]. Experiments were performed with monolayers reaching 80–90% confluence and 18 hrs prior to experiments serum content in culture medium was reduced from 20% to 5%.
In vitro simulated ischaemia
To simulate ischaemic conditions, cells were exposed to anoxia in combination with glucose-deprivation and acidosis as previously described [8]. Dishes with cultured EC were incubated for 2 hrs at 37°C in a gastight chamber under continuous flush with humidified N2 or gas mixture (95% N2+ 5% CO2). Analysis of buffer pH after 2 hrs of simulated ischaemia did not reveal any significant alterations.
Capase-3 activity assay
Activity of caspase-3 in cell extracts was detected using a colorimetric caspase-3 cellular activity assay kit (Calbiochem) based on cleavage of the synthetic caspase substrate-1 linked to the chromophore p-nitroanilide (Ac-DEVD- pNA). Preparation of cell extracts and analysis of caspase-3 activity was performed according to the manufacturer's protocol. The amount of hydrolysed substrate was measured at an optical density of 405 nm. The activity of caspase-3 was expressed in arbitrary units defined as the maximal increase of optical density, derived by linear regression, per 0.5 × 106 cells during 30 min.
Hoechst-33342 and propidium iodide staining
To distinguish between apoptotic and necrotic cells, nuclear staining with Hoechst-33342 and propidium iodide was applied as described previously with slight modifications [16]. Briefly, cells were trypsinized, washed with phosphate buffered soline (PBS) and incubated for 10 min with 1 μg/ml Hoechst-33342 and 3 μg/ml propidium iodide. The stained nuclei were visualized with convert fluorescent microscope at a magnification of 700x using excitation light at 350 nm for Hoechst-33342 and 540 nm for propidium iodide.
For quantitative assay, a blind analysis of 200–300 nuclei from randomized 4–5 fields was performed. Cells were scored as apoptotic, when nuclei stained with Hoechst-33342 produced unequivocal bright blue fluorescence due to chromatin condensation/fragmentation [8]. Propidium iodide stained nuclei with normal nuclear morphology, i.e. without signs of chromatin condensation, were scored as necrotic. Cells exhibiting both chromatin alteration and propidium iodide stained nuclei (i.e. 'late-stage apoptotic cells') were included in the apoptotic population. The number of these cells did not exceed 5% of all cells.
Western blot
Western blot analyses were performed as described previously [5]. Primary antibodies were: Bax (Cell Signalling), cytochrome C (Sigma), cytochrome oxidase IV (Molecular Probes), β-actin (Chemicon International), caspase-8 (Bio-Vision), caspase-12 (recognizing the residues 100–116, Oncogene). Specific bands were visualized after incubation with peroxidase-linked horseradish peroxidase (HRP)-labelled secondary antibodies by chemiluminescence using ECL+ kit (Amersham Pharmacia). Equivalent sample loading was confirmed by stripping membranes with the Blot Restore Membrane Stripping buffer (Pierce) followed by treatment with antibodies against β-actin.
Subcellular fractionation
Subcellular fractionation was performed as described by Heiden et al.[17] with small modifications. Briefly, cells were permeabilized by resuspension in ice cold buffer containing 210 mM sucrose, 20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 0.1mM phenylmethylsulfonyl fluoride, 1:100 protease inhibitor cocktail (Sigma) and 0.04% digitonin. After initial centrifugation of cell suspension at 750 × g for 10 min, supernatant was further centrifuged at 10,000 × g for 25 min resulting in mitochondrial fraction in the pellet. Supernatant was used as a mitochondrial free fraction. The purity of this fraction was confirmed by the absence of Cox-IV.
Cytosolic Ca2+ and pH analysis
Measurement of cytosolic free Ca2+ and H+ concentration with fluorescent indicators Fura-2 and 2′, 7′-bis-(2-carboxyethyl)- 5-(and-6)-carboxyfluorescein (BCECF) and calibration of Fura-2 and BCECF ratios were performed as previously described [12].
Mitochondrial membrane potential analysis
Mitochondrial membrane potential was assessed with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolocar-bocyanin iodide (JC-1) as described previously [18]. Briefly, EC were loaded for 30 min with 5 μmol/l JC-1 in medium M199 at 30°C. Recordings were begun after the cells had incubated in dye-free medium at 30°C for an additional 20 min. Excitation of dye was performed at 496 nm. Emission was measured at 530 and 590 nm. Emission signal ratio (590/530) was normalized to the ratio obtained under normoxic condition, i.e. before simulated ischaemia.
Immunocytochemistry and confocal microscopy
Briefly, cells fixed with 4% paraformaldehyde were treated for 15 min with 0.5 mg/ml MitoTracker Red (Molecular Probes). After washing with PBS, the cells were additionally fixed for 5 min with 4% paraformaldehyde, permeabilized with 0.05% Triton X-100 and incubated with a rabbit polyclonal anti-Bax antibody (1:250, Cell Signaling) followed by treatment with fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (Dianova). The cells were examined by a laser scanning confocal microscope (Leica TCS SP2). Series of confocal optical sections were taken at 0.5 micrometer intervals using a Leica Planapo 63/1.4 objective lens. Each recorded image was taken using dual-channel scanning and consisted of 1024 × 1024 pixels.
Statistical analysis
Data are given as mean ± SEM.The comparison of means between the groups was performed by oneway analysis of variance (ANOVA) followed by Bonferroni post-hoc test. Statistical significance was accepted when P<0.05.
Results
Inhibition of BT enhanced the pro-apoptotic effect of simulated ischaemia
Exposure of EC to simulated ischaemia for 2 hrs in Bic led to a rise of caspase-3 activity and apoptotic cell number compared to control (Fig. 1A). Inhibition of BT by omission of bicarbonate during ischaemic incubation (Hep) significantly increased the apoptotic rate. Previously we reported that simulated ischaemia induced apoptosis in coronary EC in a caspase-dependent manner. To prove whether the enhancement of apoptosis induced by inhibition of BT is also caspase-dependent, a treatment under simulated ischaemia with pan-caspase inhibitor Z-Val- Ala-Asp(OMe)-fluoromethyl ketone (zVAD)-fmk was applied. Under this treatment, apoptosis rate did not differ from the normoxic control demonstrating that caspase-dependent apoptosis is the predominant apoptosis form induced by inhibition of BT during simulated ischaemia.
1.
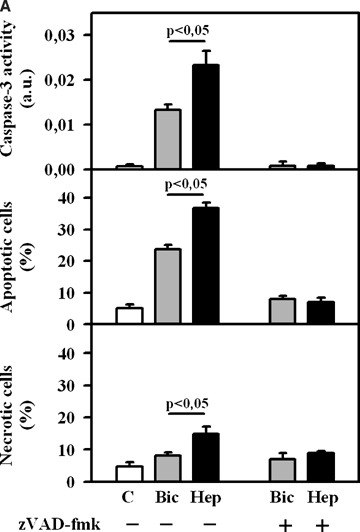
(A) Caspase-3 activity and the number of apoptotic and necrotic EC under control (C) condition (no treatment) or after 2 hrs of simulated ischaemia either in bicarbonate-buffered medium (Bic) or in HEPES-buffered medium (Hep) in the presence or absence of pan-caspases inhibitor zVAD-fmk (50 μmol/L). Values are mean ± SEM, n = 9–12. (B) Caspase-3 activity and the number of apoptotic and necrotic EC under control (C) condition or after 2 hrs of simulated ischaemia either in bicarbonate-buffered medium (Bic) or in HEPES-buffered medium (Hep) in the presence or absence of stilbene derivative 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS, 300 μmol/l). Values are mean ± SEM, n = 9–11.
In a second set of experiments, BT was inhibited by applying the widely used inhibitor of bicarbonate-coupled ion transport DIDS (300 μmol/l). This concentration was used on the basis of the results of our initial experiments with various DIDS concentrations (100–1000 μmol/l) demonstrating the marked pro-apoptotic and the minimal necrotic effect of DIDS at 300 μmol/l. Similar to bicarbonate withdrawal, treatment with DIDS led to a significant rise of caspase-3 activity and apoptotic cell number (Fig. 1B). In contrast, at the similar concentration DIDS had no pro-apoptotic action in normoxic, control EC.
Since DIDS is not specific inhibitor for BT and may also influence apoptosis rate due to side effects, an additional set of experiments was designed. Here, treatment with DIDS during simulated ischaemia was performed in bicarbonate-free, Hep. Under this condition, DIDS has no effect on apoptotic parameters (Fig. 1B). Therefore, the pro-apoptotic action of DIDS is due to its known inhibitory effect on BT.
Cytosolic acidosis and Ca2+ overload are not involved in apoptosis induced by BT inhibition
Previously, we reported that cytosolic acidosis leading to Ca2+ leak from the endoplasmic reticulum (ER) followed by the Ca2+ overload in EC are the upstream events in simulated ischaemia-induced apoptosis [8]. Therefore, we tested whether inhibition of BT, which is an important regulator of cytosolic pH homeostasis in EC [9], leads to apoptosis due to increased cytosolic H+ and/or Ca2+ accumulation. Exposure of EC to simulated ischaemia for 2 hrs in bicarbonate-buffer medium led to cytosolic acidosis (pH = 6.52 ± 0.05) and Ca2+ -overload (1073 ± 47 nmol/l) (Fig. 2). Both protocols of BT inhibition did not influence significantly cytosolic acidification. The accumulation of Ca2+ in the cytosol was slightly, but significantly, attenuated by both protocols of BT inhibition after 60 min exposure to simulated ischaemia (Fig. 2B). Thus, pro-apoptotic effect of BT inhibition is not due to increased cytosolic H+ and Ca2+ accumulation under simulated ischaemia.
2.
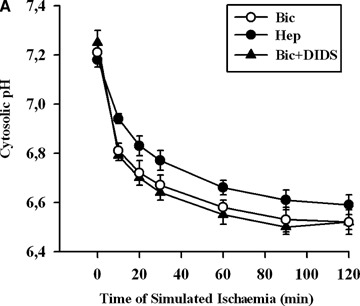
Time courses of cytosolic pH (A) and Ca2+ concentration (B) in EC exposed to simulated ischaemia in bicarbonate- buffered medium without (Bic, O-) or with DIDS (Bic + DIDS, 300 μmol/l, -Δ-), or in HEPES-buffered medium (Hep, -•-). Values are mean ± SEM, n = 5–6. *–P < 0.05 versus Hep and Bic + DIDS.
Inhibition of BT leads to apoptosis due to activation of cytochrome C release
To find out which apoptotic pathway is activated by inhibition of BT, first the cleavage of ER-bound caspase- 12 was investigated. This approach was based on our previous data demonstrating that caspase- 12 cleavage is a main intrinsic pathway of simulated ischaemia-induced apoptosis in coronary EC [8]. In line with this previous report, a significant cleavage of caspase-12 was found after simulated ischaemia in Bic (Fig. 3A). Inhibition of BT either by omission of bicarbonate or by treatment with DIDS reduced rather than increased cleavage of caspase- 12, thus excluding the involvement of the ER-dependent pathway in the pro-apoptotic action of BT inhibition.
3.
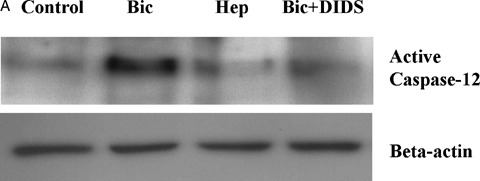
Western blots of cleaved caspase-12 (A) and cytosolic cytochrome C (B) prepared from extracts of control EC or EC exposed to simulated ischaemia in bicarbonate-buffered medium without (Bic) or with DIDS (Bic + DIDS, 300 μmol/l) or in HEPES-buffered medium (Hep). Data are representative from three independent experiments with similar results.
We further analysed two other main signaling pathways previously described for ischaemia-induced apoptosis in various cell type, i.e. the death receptor- and mitochondria-dependent pathways. To prove a contribution of the death receptor-mediated pathway, cleavage of caspase-8 was analysed. No cleavage of caspase-8 could be detected after exposure of EC to simulated ischaemia under all conditions (data not shown).
To prove the role of the mitochondrial pathway, a release of cytochrome C from mitochondria was investigated. We found that simulated ischaemia in Bic led to a slight elevation of cytosolic cytochrome C (Fig. 3B). This effect of simulated ischaemia was significantly augmented by both protocols of BT inhibition. Therefore, release of cytochrome C from mito-chondria seems to be an event responsible for the pro-apoptotic effect of BT inhibition.
Inhibition of BT during simulated ischaemia leads to mitochondrial depolarization
Alteration of the mitochondrial membrane potential (ΔΨm) has been shown to be an essential event associated with mitochondrial cytochrome C release under various stress conditions [19]. To test whether increased cytochrome C release induced by inhibition of BT is associated with changes in ΔΨm, mito-chondrial membrane potential was measured applying the specific fluorescent dye JC-1 (Fig. 4). Exposure of EC to simulated ischaemia for 2 hrs in Bic only slightly reduced ΔΨm, which did not differ from the initial, pre-ischaemic level. In contrast, ischaemic treatment of EC in Hep significantly depolarized the mitochondria. A similar effect was observed under DIDS treatment in Bic. Thus, it seems that bicarbonate-coupled ion transport is important for control of ΔΨm under simulated ischaemia.
4.
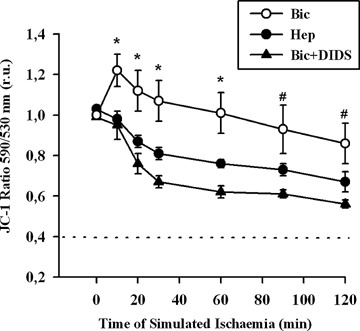
Time course of mitochondrial membrane potential measured with fluorescent dye JC-1 in EC exposed to simulated ischaemia in bicarbonate-buffered medium without (Bic, -O-) or with DIDS (Bic + DIDS, 300 μmol/l, -•-), or in HEPES-buffered medium (Hep, -•-). Values are mean ± SEM, n = 4–5. *–P< 0.05 versus Hep and Bic + DIDS. #–P<0.05 versus Bic + DIDS.
Inhibition of BT during simulated ischaemia leads to mitochondrial translocation of Bax
Translocation of pro-apoptotic members of the Bcl-2 family proteins, e.g. Bax, from cytosol to mitochon- dria is an important initial mechanism for the release of mitochondrial cytochrome C [20]. In the present study we tested, thus, whether BT inhibition leads to mitochondrial Bax translocation. For this purpose, two methods were applied. First, a western blot analysis of Bax protein in the mitochondrial fraction was performed at the end of simulated ischaemia. We found that simulated ischaemia in bicarbonate buffered medium slightly increased mitochondrial Bax localization (Fig. 5). This Bax binding to mitochondria was significantly augmented by both protocols of BT inhibition. Second, an immunostaining of Bax at the end of ischaemia was performed. With this method, only weak co-localization of Bax with mitochondria was detected in control cells or in cells after simulated ischaemia in bicarbonate buffer (Fig. 6) In contrast, a pronounced colocalization of Bax with mitochondria was detected under withdrawal of bicarbonate (HEPES-buffer, data not shown) or treatment with DIDS during simulated ischaemia. Thus, translocation of Bax to mitochondria under simulated ischaemia is dependent on BT activity.
5.
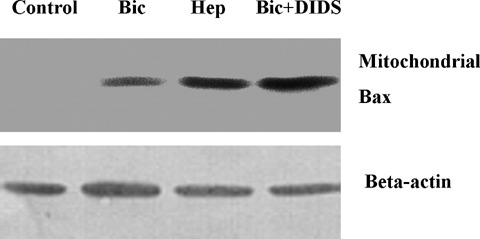
Western blot of Bax pre-pared from the mitochondrial fraction of EC (similar experimental conditions as in Fig. 3). Data are representative from three to four independent experiments with similar results.
6.
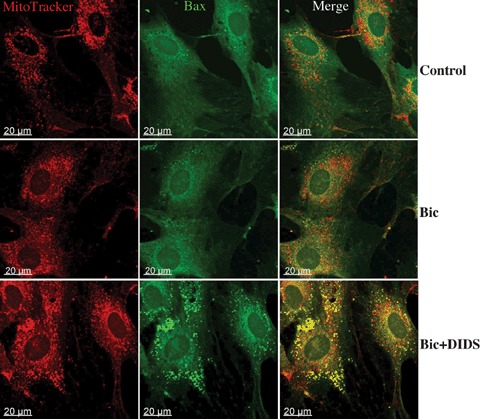
Immunostaining analysis of Bax location in EC (similar experimental conditions as in Fig.3). Mitochondria were labeled with Mitotracker (red) and Bax with specific antibodies (green). Yellow spots in merge images represent the colocalization of Bax with mitochondria. Data are representative from three independent experiments with similar results.
Discussion
The aim of the present study was to find out whether BT plays a role in simulated ischaemia-induced apoptosis of coronary EC and which signalling path-ways might be involved. The main findings are the following: (i) BT inhibition significantly enhanced apoptosis of ischaemic EC. (ii) The main mechanism responsible for pro-apoptotic action of BT inhibition is the activation of the mitochondrial pathway, i.e. cytochrome C release, mitochondrial depolarization and Bax translocation to mitochondria.
To simulate ischaemia, combination of glucose-free anoxia with extracellular acidosis (pHo6.4) was performed in the present study. The applied extracellular acidification seems to be relevant to in vivo myocardial ischaemia, where extracellular pH can decrease to as low as 6.0 [21]. This model of simulated ischaemia was characterized in our previous studies, which demonstrated the importance of cytosolic acidification as an initial trigger for Ca2+ -release from the ER [12] followed by the cleavage of the ER-bound caspase-12 [8]. Surprisingly, mitochondrial pathway of apoptosis plays, however, only a minor role in the simulated ischaemia-induced apoptosis. In the present study, we further demonstrated that the mitochondrial pathway of apoptosis is under control of BT. To analyse the role of BT, two treatment protocols were applied. First, bicarbonate was withdrawn from the incubation medium during simulated ischaemia by substitution of bicarbonate-buffer with HEPES-buffer. Second, BT was directly inhibited by treatment with the stilbene derivative DIDS. Both treatments significantly elevated apoptotis rate during exposure of EC to simulated ischaemia. Although DIDS, additionally to inhibition of BT, can also suppress Cl− channels [12], the pro-apoptotic action of DIDS is unlikely due to inhibition of Cl channels. First, specific inhibition of Cl− channels under various stress conditions has an anti-apoptotic effect [22]. Second, in the present study treatment with DIDS in bicarbonate-free, Hep did not influence apoptosis of ischaemic EC (Fig. 1B), indicating that DIDS jeopardizes EC only in the presence of bicarbonate. Thus, inhibition of BT rather than side effects seems to be responsible for pro-apoptotic action of DIDS.
In contrast to our data, several previous studies demonstrated an anti-apoptotic effect of DIDS treatment or bicarbonate withdrawal [11, 23]. This discrepancy may be, first of all, related to differences in the cell injury model. Indeed, in the report of Fujita and colleagues [11] staurosporine-induced apoptosis of bovine carotid artery EC was associated with cytosolic alkalosis due to transmembrane HCO3− influx. The causal role of cytosolic alkalinization in apoptosis induction has been previously demonstrated [24]. Therefore, the prevention of a cytosolic pH-rise by inhibition of BT in Fujita's study could be the underlying protective mechanism of DIDS treatment, whereas it unlikely may have a similar effect under profound cytosolic acidification (pHi∼6.5) in our ischaemic model. Furthermore, treatment with DIDS may have an anti-apoptotic effect due to a BT-independent action, e.g. due to inhibition of Cl− channels [23], which, as discussed above, is not relevant for our model. In line with the present data, a few recent reports have demonstrated that BT inhibition promotes cell death [10, 25]. The mechanism of this action was not analysed till now.
To find out which apoptotic pathways were activated by BT transport inhibition, cleavage of caspase-8, caspase-12 and release of cytochrome C were investigated. Activation of the death receptor pathway is unlikely responsible for the pro-apoptotic effect of the BT inhibition, since no caspase-8 cleavage could be detected under any experimental conditions. Cleavage of the ER-bound caspase-12 was previously reported to be a main apoptotic pathway in coronary EC under simulated ischaemia [8]. Similarly, a marked cleavage of caspase-12 was found in ischaemic EC in the present study. However, both protocols of BT inhibition rather suppressed than increased cleavage of caspase- 12. Thus, the pro-apoptotic effect of the BT inhibition is not due to activation of the caspase-12 dependent pathway. In contrast, cytochrome C release from mitochondria was dramatically augmented by both protocols of BT inhibition. Therefore, it seems likely that inhibition of BT leads to apoptosis through activation of a mitochondrial pathway.
The mechanisms leading to cytochrome C release under stress conditions are still debatable. Although several models have been proposed, two of them seem to play a major role in apoptosis induced by various stimuli. One of the earliest models postulated the link between cytochrome C release and mitochondrial swelling, e.g. due to opening of the mitochondrial permeability transition (MPT) pores, followed by dissipation of ΔΨm and rupture of the outer mitochondrial membrane. This allows the release of cytochrome C from the intermembrane space to the cytosol. In the other model, permeabilization of the outer mitochondrial membrane by pro-apoptotic Bcl-2 family proteins, e.g. Bax, was postulated as an initial mechanism for cytochrome C release [26, 27]. In this model, stress-induced activation and translocation of Bax to the outer mitochondrial membrane followed by its oligomerization [27] leads to formation of pores permeable for cytochrome C. In the present study, inhibition of BT led to a marked translocation of pro-apoptotic Bax to mitochondria and to significant mito-chondrial depolarization. These findings argue, therefore, for the applicability of both these models of cytochrome C release in our study. In agreement with our data, recent reports suggested the "combined model" for cytochrome C release, where binding of Bax to mitochondria and formation of MPT followed by mitochondrial depolarization mutually affect each other [28, 29]. Altogether, (i) release of cytochrome C, (ii) mitochondrial Bax translocation and (iii) mito-chondrial depolarization provide strong evidence for activation of mitochondrial pathway of apoptosis by BT inhibition in ischaemic EC.
The role of BT in the regulation of mitochondrial pathway of apoptosis is still obscure. Cytosolic Ca2+ overload can jeopardize mitochondrial function followed by cytochrome C release [30]. Our previous reports suggested that in ischaemic EC cytosolic Ca2+ overload is caused by cytosolic acidification [8, 12]. Plasmalemmal BT, e.g. Na+ -HCO3− symporter, together with Na+/H+ -exchanger plays an essential role in regulation of cytosolic pH in EC [9]. Therefore, it can be supposed that inhibition of BT in ischaemic EC may reduce the H+ extrusion from the cytosolic and enhance the cytosolic acidosis followed by a further rise of cytosolic Ca2+ overload, which may be a cause for mitochondrial dysfunction. However, analysis of cytosolic ion homeostasis in the present study revealed that the inhibition of BT under simulated ischaemia had no effect on cytosolic acidosis and rather slightly reduced Ca2+ overload. Thus, activation of mitochondrial apoptotic pathway under BT inhibition is not due to increased cytosolic Ca2+ and H+ accumulation.
Beside plasmalemmal BT, the involvement of mitochondrial- localized BT in the regulation of mitochondrial pathway of apoptosis may be hypothesized. Although a few previous reports provided evidence for bicarbonate flux across the mitochondrial membrane [31, 32], little is known about precise bicarbonate transporting systems in mitochondria. Several anion channels in mitochondrial inner and outer mitochondrial membranes were described, which may also be permeable for bicarbonate. It has been shown that permeability of two mitochondrial anion channels, i.e. voltage dependent anion channel and inner membrane anion channel, can be modulated by DIDS [33, 34]. Further work needs to be done in order to discover the precise bicarbonate transporting system modulating cytochrome C release under ischaemic stress, which was beyond the scope of the present study.
In conclusion, this study for the first time demonstrated the importance of BT for ischaemic apoptosis of the coronary EC. Inhibition of BT leads to activation of the mitochondrial apoptotic pathway, i.e. Bax translocation to mitochondria, mitochondrial depolarization and cytochrome C release. Further investigation of precise bicarbonate transporting systems modulating mitochondrial pathway may lead to discovery of new therapeutic approaches for treatment of diseases accompanied by apoptosis.
Acknowledgments
The technical help of G. Scheibel and K. Rezny is gratefully acknowledged. This study was supported by Grant STE 495/6–1 of the Deutsche Forschungsgemeinschaft and was part of the thesis of S. Kumar submitted in fulfillment of the requirements for the degree of Doctor of Philosophy at the Ruhr-Universität Bochum (Germany).
References
- 1.Werner N, Wassmann S, Ahlers P, Kosiol S, Nickenig G. Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:112–6. doi: 10.1161/01.ATV.0000191634.13057.15. [DOI] [PubMed] [Google Scholar]
- 2.Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, Ferrari R, Knight R, Latchman D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischaemia/reperfusion injury. Circulation. 2001;104:253–6. doi: 10.1161/01.cir.104.3.253. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti S, Hoque AN, Karmazyn M. A rapid ischaemia-induced apoptosis in isolated rat hearts and its attenuation by the sodium-hydrogen exchange inhibitor HOE 642 (cariporide) J Mol Cell Cardiol. 1997;29:3169–74. doi: 10.1006/jmcc.1997.0561. [DOI] [PubMed] [Google Scholar]
- 4.Hinescu ME. Cardiac apoptosis: from organ failure to allograft rejection. J Cell Mol Med. 2001;5:143–52. doi: 10.1111/j.1582-4934.2001.tb00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyama K, Burns DM, Suh SW, Garnier P, Matsumori Y, Shiina H, Swanson RA. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab. 2005;25:358–70. doi: 10.1038/sj.jcbfm.9600043. [DOI] [PubMed] [Google Scholar]
- 6.Webster KA, Discher DJ, Kaiser S, Hernandez O, Sato B, Bishopric NH. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J Clin Invest. 1999;104:239–52. doi: 10.1172/JCI5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gottlieb RA, Giesing HA, Zhu JY, Engler RL, Babior BM. Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H(+)-ATPase. Proc Natl Acad Sci USA. 1995;92:5965–8. doi: 10.1073/pnas.92.13.5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Kasseckert S, Kostin S, Abdallah Y, Schafer C, Kaminski A, Reusch HP, Piper HM, Steinhoff G, Ladilov Y. Ischaemic acidosis causes apoptosis in coronary endothelial cells through activation of caspase-12. Cardiovasc Res. 2007;73:172–80. doi: 10.1016/j.cardiores.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Taylor CJ, Nicola PA, Wang S, Barrand MA, Hladky SB. Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol. 2006;576:769–85. doi: 10.1113/jphysiol.2006.117374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Arcangelo D, Ambrosino V, Giannuzzo M, Gaetano C, Capogrossi MC. Axl receptor activation mediates laminar shear stress anti-apoptotic effects in human endothelial cells. Cardiovasc Res. 2006;71:754–63. doi: 10.1016/j.cardiores.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Fujita H, Yanagisawa A, Ishikawa K. Suppressive effect of a chloride bicarbonate exchanger inhibitor on staurosporine-induced apoptosis of endothelial cells. Heart Vessels. 1997;12:84–8. [PubMed] [Google Scholar]
- 12.Ladilov Y, Schafer C, Held A, Schafer M, Noll T, Piper HM. Mechanism of Ca(2+) overload in endothelial cells exposed to simulated ischaemia. Cardiovasc Res. 2000;47:394–403. doi: 10.1016/s0008-6363(00)00108-5. [DOI] [PubMed] [Google Scholar]
- 13.Kokubun S, Saigusa A, Tamura T. Blockade of Cl− channels by organic and inorganic blockers in vascular smooth muscle cells. Pflugers Arch. 1991;418:204–13. doi: 10.1007/BF00370515. [DOI] [PubMed] [Google Scholar]
- 14.Piper HM, Spahr R, Mertens S, Krützfeldt A, Watanabe H. Microvascular endothelial cells from heart. In: Piper HM, editor. Cell Culture Techniques in Hearth and Vessel Research. Heidelberg: Springer; 1990. pp. 158–177. [Google Scholar]
- 15.Ando H, Kubin T, Schaper W, Schaper J. Cardiac microvascular endothelial cells express alpha-smooth muscle actin and show low NOS III activity. Am J Physiol. 1999;276:H1755–68. doi: 10.1152/ajpheart.1999.276.5.H1755. [DOI] [PubMed] [Google Scholar]
- 16.Terminella C, Tollefson K, Kroczynski J, Pelli J, Cutaia M. Inhibition of apoptosis in pulmonary endothelial cells by altered pH, mitochondrial function, and ATP supply. Am J Physiol. 2002;283:L1291–302. doi: 10.1152/ajplung.00246.2001. [DOI] [PubMed] [Google Scholar]
- 17.Heiden MGV, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-XL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–37. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 18.White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–97. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halestrap AP, Doran E, Gillespie JP, O'Toole A. Mitochondria and cell death. Biochem Soc Trans. 2000;28:170–7. doi: 10.1042/bst0280170. [DOI] [PubMed] [Google Scholar]
- 20.Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2003;7:249–57. doi: 10.1111/j.1582-4934.2003.tb00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari R, Cargnoni A, Bernocchi P, Pasini E, Curello S, Ceconi C, Ruigrok TJ. Metabolic adaptation during a sequence of no-flow and low-flow ischaemia. A possible trigger for hibernation. Circulation. 1996;94:2587–96. doi: 10.1161/01.cir.94.10.2587. [DOI] [PubMed] [Google Scholar]
- 22.Jiao JD, Xu CQ, Yue P, Dong DL, Li Z, Du ZM,Yang BF. Volume-sensitive outwardly rectifying chloride channels are involved in oxidative stress-induced apoptosis of mesangial cells. Biochem Biophys Res Commun. 2006;340:277–85. doi: 10.1016/j.bbrc.2005.11.175. [DOI] [PubMed] [Google Scholar]
- 23.Tanabe S, Wang X, Takahashi N, Uramoto H, Okada Y. HCO(3)(-)-independent rescue from apoptosis by stilbene derivatives in rat cardiomyocytes. FEBS Lett. 2005;579:517–22. doi: 10.1016/j.febslet.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 24.Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 2004;11:953–61. doi: 10.1038/sj.cdd.4401466. [DOI] [PubMed] [Google Scholar]
- 25.Araki T, Hayashi M, Saruta T. Anion-exchange blocker enhances cytoplasmic vacuole formation and cell death in serum-deprived mouse kidney epithelial cells in mice. Cell Biol Int. 2006;30:93–100. doi: 10.1016/j.cellbi.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Putcha GV, Deshmukh M, Johnson EM. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–85. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–76. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Precht TA, Phelps RA, Linseman DA, Butts BD, Le SS, Laessig TA, Bouchard RJ, Heidenreich KA. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005;12:255–65. doi: 10.1038/sj.cdd.4401552. [DOI] [PubMed] [Google Scholar]
- 29.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–70. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 30.Smaili SS, Hsu YT, Youle RJ, Russell JT. Mitochondria in Ca 2+ signaling and apoptosis. J Bioenerg Biomembr. 2000;32:35–46. doi: 10.1023/a:1005508311495. [DOI] [PubMed] [Google Scholar]
- 31.Selwyn MJ, Walker HA. Permeability of the mito-chondrial membrane to bicarbonate ions. Biochem J. 1977;166:137–9. doi: 10.1042/bj1660137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simpson DP, Hager SR. Bicarbonate-carbon dioxide buffer system: a determinant of the mitochondrial pH gradient. Am J Physiol. 1984;247:F440–6. doi: 10.1152/ajprenal.1984.247.3.F440. [DOI] [PubMed] [Google Scholar]
- 33.Beavis AD, Davatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J Bioenerg Biomembr. 1996;28:207–14. doi: 10.1007/BF02110652. [DOI] [PubMed] [Google Scholar]
- 34.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–63. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]