

Abstract
Adhesion molecules of the integrin beta1 family are thought to be involved in the malignant progression renal cell carcinoma (RCC). Still, it is not clear how they contribute to this process. Since the hematogenous phase of tumour dissemination is the rate-limiting step in the metastatic process, we explored beta1 integrin alterations on several RCC cell lines (A498, Caki1, KTC26) before and after contacting vascular endothelium in a tumour-endothelium (HUVEC) co-culture assay. Notably, alpha2, alpha3 and alpha5 integrins became down-regulated immediately after the tumour cells attached to HUVEC, followed by re-expression shortly thereafter. Integrin down-regulation on RCC cells was caused by direct contact with endothelial cells, since the isolated endothelial membrane fragments but not the cell culture supernatant contributed to the observed effects. Integrin loss was accompanied by a reduced focal adhesion kinase (FAK) expression, FAK activity and diminished binding of tumour cells to matrix proteins. Furthermore, intracellular signalling proteins RCC cells were altered in the presence of HUVEC membrane fragments, in particular 14-3-3 epsilon, ERK2, PKCdelta, PKCepsilon and RACK1, which are involved in regulating tumour cell motility. We, therefore, speculate that contact of RCC cells with the vascular endothelium converts integrin-dependent adhesion to integrin-independent cell movement. The process of dynamic integrin regulation may be an important part in tumour cell migration strategy, switching the cells from being adhesive to becoming motile and invasive.
Keywords: adhesion, beta1 integrins, HUVEC, membrane fragments, renal cell carcinoma, signalling proteins
Introduction
Renal cell carcinoma (RCC) is the most common renal tumour and the third most common malignancy encountered in urological oncology. Metastasized RCC is generally fatal, despite conventional chemotherapy, radiotherapy and hormonal therapy. Immunotherapy, with administration of interferons or interleukin-2 has shown some promise, however, the total response rate is only about 20% and the treatment is occasionally associated with severe adverse effects.
Novel treatment strategies are therefore eagerly awaited. However, the complex mechanism of tumour dissemination is far from being completely understood. Before potential drug targets can be identified, it is obligatory to analyze those events, which are related to RCC metastasis. Several reports document that adhesion molecules of the integrin family may be responsible for regulating tumour cell proliferation, invasion and malignant progression. Integrins are heterodimeric molecules formed by a combination of an alpha integrin subunit with a member of the beta subunit. Classification occurs on the basis of their beta subunit, whereby the beta1 subtypes are considered to be the prototype key mediators of tumour dissemination. They not only provide the mechanical link between tumour cells, vascular endothelium and extracellular matrix, but also mediate intracellular signalling through integrin-associated adaptor molecules.
Publications, which have focussed on alterations of the integrin expression level during cancer progression, are contradictory. When sections of squamous-cell carcinomas were examined by immunohistochemistry, considerable variations in integrin expression were observed, ranging from normal expression of a particular integrin to a complete integrin loss or even integrin over-expression both within a single tumour and between tumours [1]. Studies with breast cancer cells have demonstrated that blocking of beta1-integrin receptors leads to the reversion of the malignant phenotype, but re-expression of beta1 induced similar results [2, 3]. Tawil and co-workers observed increased beta1 integrin expression in metastatic mammary carcinoma cells [4], whereas the inhibition of motility and invasion of the metastatic breast carcinoma cell lines was also accompanied by an increased integrin expression [5]. Finally, integrin beta1 upregulation is supposed to correlate with progression of prostate carcinoma, although mRNA levels of beta1 were reduced in neoplastic compared to normal prostate tissues [6].
The unclear function of integrins is reflected in the lack of data dealing with beta1 integrins in RCC. Since the hematogenous phase of tumour dissemination is the ratelimiting step in the metastatic process, we established a tumourendothelium co-culture assay and explored beta1 integrin alterations on several RCC cell lines before and after endothelial cell contact. The paper presents evidence that the interaction of RCC cells with human vascular endothelial cells is coupled with a rapid and reversible loss of beta1 integrin subtypes, notably, alpha2, alpha3 and alpha5 integrins. We, furthermore, demonstrate that this process is accompanied by a reduced focal adhesion kinase (FAK) expression, FAK activity and diminished binding of tumour cells to matrix proteins. Further intracellular signalling components are altered in RCC cells after contacting HUVEC membrane fragments, which altogether may represent the dynamics of the tumour cell migration strategy.
Materials and methods
Antibodies
Monoclonal antibodies were used, directed against proteins indicated subsequently: Cell cycle proteins: Cdk1/Cdc2 (IgG1, clone 1), Cdk2 (IgG2a, clone 55), Cdk4 (IgG1, clone 97), Cyclin A (IgG1, clone 25), Cyclin B (IgG1, clone 18), Cyclin D3 (IgG2b, clone 1), Rb (IgG2a, clone 2), Rb2 (IgG2a, clone 10), RBBP (IgG1, clone 12), p70s6kinase (IgG1, clone 16) were purchased from BD Biosciences (Heidelberg, Germany). Signal transduction proteins: MEK1 (IgG2a, clone 25), MEK2 (IgG2a, clone 96), MEK5 (IgG1, clone 21), ERK1 (IgG1, clone MK12), ERK2 (IgG2b, clone 33), panERK (IgG2a, clone 16), p90Rsk (IgG2a, clone 78), 14-3-3 epsilon (IgM, clone 12), JNK (IgG1, clone 37), phospho-specific JNK (pT183/pY185; IgG1, clone 41), p38 (IgG1, clone 27), phospho-specific p38 (pT180/pY182;IgG1, clone 30) were obtained from BD Biosciences. PKC proteins: PKCalpha (IgG2b, clone 3), PKCbeta (IgG2b, clone 36), PKCdelta (IgG2b, clone 14), PKCepsilon (IgG2a, clone21), PKCiota (IgG2b, clone23), PKClambda (IgG1, clone41), PKCtheta (IgG1, clone 27), DGKtheta (IgG1, clone 24), RACK1 (IgM, clone20) were from BD Biosciences. Integrin activation: anti integrin-linked kinase (ILK; clone 3), anti focal adhesion kinase (Fak; clone 77) and anti phospho-specific Fak (pY397;clone 18) were derived from BD Biosciences. Anti-beta-actin monoclonal antibody was obtained from Sigma (Taufenkirchen, Germany).
Cell cultures
Kidney carcinoma Caki-I and KTC-26 cells were purchased from LGC Promochem (Wesel, Germany). A498 were derived from CLS (Heidelberg, Germany). Tumour cells were grown and subcultured in RPMI 1640 medium (Seromed, Berlin, Germany) supplemented with 10% FCS, 100 IU/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified, 5% CO2 incubator.
Endothelial cells (HUVEC) were isolated from human umbilical veins and harvested by enzymatic treatment with chymotrypsin. HUVEC were grown in Medium 199 (Biozol, Munich, Germany), 10% fetal calf serum (FCS; Gibco, Karlsruhe, Germany), 10% pooled human serum (Blood Bank of The German Red Cross, Frankfurt am Main, Germany), 20 μg/ml endothelial cell growth factor (Boehringer, Mannheim, Germany), 0.1% heparin (Roche, Basel, Switzerland), 100 ng/ml gentamycin (Gibco) and 20 mM HEPES-buffer (Seromed, Berlin, Germany). Subcultures from passages 2–4 were selected for experimental use.
Isolation of HUVEC plasma membranes
HUVEC were pooled and washed in cold STM buffer (0.25 M sucrose, 5 mM Tris, pH 8.0, 0.5 mM MgCl2), minced and homogenized using a Dounce homogenizer. Nuclei and cell debris were removed by centrifugation at 280 g for 5 min. The supernatant was saved and the pellet resuspended in 0.25 M STM using a Dounce homogenizer. The suspension was again centrifuged as above. First and second supernatants were combined and centrifuged at 1500 x g for 10 min. The resulting pellets, containing the plasma membrane fraction, were resuspended in 0.25 M STM and adjusted to 1.18 g/cm3 sucrose density using 2 M sucrose in 5 mM Tris, pH 8.0, and 0.5 mM MgCl2. Sucrose density was determined at room temperature with an Abbe refractometer. Samples were transferred to ultracentrifuge tubes and overlaid with 0.25 M sucrose. After centrifugation for 60 min at 82, 000 x g in a Beckman L5-65 centrifuge (SW 28 rotor), the pellicle at the interface was collected and resuspended in sufficient 0.25 M sucrose to obtain a density of 1.05 g/cm3. Protein concentration was determined by the method of Lowry using BSA as a standard.100 μg protein/ml was used to activate tumour cells.
For western blot experiments, which required high membrane yield, cell membranes were additionally prepared by a method based on that of Hill et al. [7]. HUVEC were harvested by mechanical scraping, pooled, and lysed by repeated freeze (147°C) -thawing (+37°C). The lysed cell membranes were harvested by centrifugation at 10, 000 g for 30min and stored in aliquots at −80°C prior to use. To exclude cytosolic or nucleic contamination, beta-actin content and GAPDH mRNA was evaluated by western blot or reverse transcriptase-polymerase chain reaction (RT-PCR), respectively, and compared to intact HUVEC as the positive control.
Tumour cell attachment to extracellular matrix components
24-well plates were coated with collagen (Seromed;diluted to 100 μg/ml in PBS), laminin (BD Biosciences; diluted to 50 μg/ml in PBS), or fibronectin (BD Biosciences;diluted to 50 μg/ml in PBS) overnight. Plastic dishes served as the background control. Plates were washed with 1% BSA (bovine serum albumin) in PBS to block non-specific cell adhesion. Thereafter, 1 × 105 tumour cells/well (stimulated with plasma membrane fragments for 4hrs versus non-stimulated) were added for 60 min. Subsequently, non-adherent tumour cells were washed off, the remaining adherent cells were fixed with 1% glutaraldehyde and counted microscopically. The mean cellular adhesion rate, defined by adherent cellscoated well adherent cellsbackground, was calculated from five different observation fields.
Evaluation of integrin surface expression
Tumour cell monocultures were washed in blocking solution (PBS, 0.5% BSA) and then incubated for 60 min at 4°C with FITC-or PE-conjugated monoclonal antibodies anti-alpha1beta1 (Becton Dickinson; clone SR84), anti-alpha2beta1 (Becton Dickinson; clone AK-7), anti-alpha3beta1 (Becton Dickinson; clone C3II1), anti-alpha4beta1 (Cymbus Biotechnology, Hofheim, Germany; clone HP2I1), anti-alpha5beta1 (Cymbus Biotechnology; clone SAM-1), or anti-alpha6beta1 (Becton Dickinson; clone GOH3). Integrin expression of tumour cells was then measured using a FACscan (Becton Dickinson; FL-1H or FL-2H (log) channel histogram analysis;1 104 cells/scan) and expressed as mean fluorescence units (MFU). A mouse IgG1-FITC or a mouse IgG-PE was used as an iso-type control (Cymbus Biotechnology).
Alpha2, alpha3 and alpha5 expression dynamics was evaluated in an RCC-HUVEC co-culture model. The 0.5 106 tumour cells/well were added to a HUVEC monolayer grown in a 6-well plate. After different time periods, tumour cells were harvested by accutase treatment and washed in blocking solution (PBS, 0.5% BSA). Cells were fixed with 100 μl fixation medium (Fix & Perm; Biozol-An der Grub Bioresearch, Eching, Germany) and washed twice in blocking solution (PBS, 0.5% BSA). Subsequently, they were incubated for 60 min at 4°C with 100 μl permeabilization medium (Fix & Perm) together with the monoclonal antibody anti Factor VIII-associated antigen (Von Willebrand factor; clone F8/86; Dako, Hamburg, Germany). Finally, tumour cells were marked with monoclonal antibodies anti alpha2, anti alpha3, or anti alpha5, and FACS analysis was carried out.
Confocal microscopy
Integrin alpha3 localization were analyzed on A498 tumour cells. In the monoculture assay, tumour cells were labelled with PE marked anti alpha3 monoclonal antibodies. In the co-culture assay, tumour cells were added to HUVEC for 60 min or 24 hrs. Non-adherent cells were washed off thereafter. The remaining cells were stained with FITC labelled anti factor VIII-associated antigen monoclonal antibodies and with PE labelled anti-integrin alpha3 monoclonal antibodies as described above. To prevent photobleaching of the fluorescent dye, specimen were embedded in an antifade reagent/mounting medium mixture (ProLong™ Antifade Kit, MoBiTec, Göttingen, Germany) and mounted on slides. The slides were viewed using a confocal laser scanning microscope (LSM 10; Zeiss, Jena, Germany) with a plan-neofluar x100/1.3 oil immersion objective.
mRNA expression of beta1 integrins
mRNA expression of beta1 integrins was evaluated by reverse RT-PCR. Tumour cells were seeded in 50 ml culture flasks (25 cm2 growth area; Falcon Primaria, Becton Dickinson) and cultured with or without HUVEC membrane fragments. Total RNA was extracted by using RNeasy kit (Qiagen, Hilden, Germany) and RNA samples were then treated with 80 U/ml of Rnase-free Dnase I (Boehringer Mannheim, Mannheim, Germany) for 60 min at 37°C, to eliminate amplifiable contaminating genomic DNA. Subsequently, samples were incubated for 10 min at 65°C to inactivate Dnase. Complementary DNA was synthesized from 1 μg of total RNA per sample with a 60 min incubation at 42°C, using the Moloney murine leukaemia virus reverse transcriptase (Invitrogen, Karlsruhe, Germany) and oligo-(dT) priming (Boehringer Mannheim). Amplification was carried out using gene specific primers and Platinum-Taq polymerase (Invitrogen) in a Mastercycler Gradient thermocycler (Eppendorf, Hamburg, Germany). Reactions were performed in the presence of 0.5 μl cDNA, with an initial incubation step at 95°C for 2 min. Cycling conditions consisted of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and extension at 72°C for 30 sec over a total of 30 cycles. The reaction was completed by another 10 min incubation step at 72°C. The specific sequences for sense and anti-sense primers are as follows: alpha2: 5′-GCATCTCAGAAGTCTGTTGCC-3′ and 5′-CCTGTTGT-TACCTTCAGGGAG-3′; (335 bp); alpha3: 5′-TACGTGC-GAGGCAATGACCTA-3′ and 5′-TTTGGGGGTGCAGGAT-GAAGCT-3′; (306 bp); alpha5: 5′-CA GACTTTCTT-GCAGCGG-3′ and 5′-GAATGGGAGTCTGAAATTGG-3′; 312 bp). Internal controls for the polymerase chain reaction (PCR) reaction were performed byrunning parallel reaction mixtures with the housekeeping gene GAPDH:5′-ATC TTC CAG GAG CGA GAT CC-3′ and 5′-ACC ACT GAC ACG TTG GCA GT-3′ (509 bp). The PCR products were subject-ed to electrophoresis in 1.5% agarose gel and visualized by ethidium bromide.
Western blotting
Cell cycle, cell signalling and integrin activation proteins were evaluated by Western blot analysis: A498 cell lysates, prepared from unstimulated cells or after stimulation with HUVEC plasma membrane fragments, were applied to a 7% polyacrylamide gel and electrophoresed for 90 min at 100 V. The protein was then transferred to nitrocellulose membranes. After blocking with non-fat dry milk for 1 hr, the membranes were incubated overnight with the respective antibodies listed above. HRP-conjugated goat-anti-mouse IgG or IgM (Upstate Biotechnology, Lake Placid, New York; dilution 1:5000) served as the secondary antibodies. The membranes were briefly incubated with ECL detection reagent (ECL™, Amersham) to visualize the proteins and exposed to an x-ray-film (Hyperfilm™ EC™, Amersham).
Statistics
All experiments were performed 3–6 times. Statistical significance was investigated by the Wilcoxon–Mann-Whitney-U-test. Differences were considered statistically significant at a P-value less than 0.05.
Results
Integrin beta1 expression pattern
Beta1 integrin subtype expression was evaluated on three kidney carcinoma cell lines. Alpha1, alpha3 and alpha6 subtypes were labelled with PE, and background fluorescence controlled by mouse IgG-PE. Alpha2, alpha4 and alpha5 subtypes were labelled with FITC and background fluorescence controlled by using mouse IgG-FITC. Flow cytometry, depicted as FL-1H or FL-2H (log) channel histogram analysis, demonstrated the same integrin surface pattern on A498, Caki-I and KTC-26 mono-cultures (Fig. 1). Alpha3 subtypes were detected most extensively on all cell lines and were, therefore, analyzed in detail in further studies. Alpha1, alpha2 and alpha5 were expressed to a lower extent, whereas alpha4 and alpha6 subtypes were not significantly elevated over the background values. Out of the low expressed integrins, alpha2 and alpha5 subtypes were included in subsequent experiments.
1.
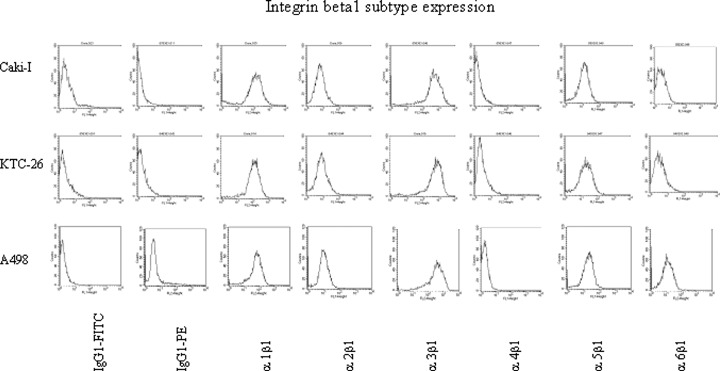
FACS analysis of integrin beta1 subtype expression on Caki-I, KTC-26 and A498 tumour cells. Cells were washed in blocking solution and then stained with specific monoclonal antibodies as listed in Materials and methods. A mouse IgG1-FITC was used as an isotype control for FITC conjugated antibodies. To evaluate background staining of PE conjugated antibodies, goat, anti-mouse IgG1-PE was used. Fluorescence was analyzed using a FACScan flow cytometer, and a histogram plot was generated to show FITC/PE-fluorescence. One from 6 independent experiments.
Interaction of tumour cells with HUVEC induces integrin down-regulation
To explore if integrins become altered in the course of tumour cell attachment to HUVEC, we analyzed integrin receptor expression on tumour cells in a co-culture model. A498, KTC26 or Caki1 were added to a HUVEC monolayer, and integrin expression on the tumour cells was determined after different time periods. Cell cultures were double stained using a monoclonal antibody directed against the HUVEC specific Factor VIII-associated antigen and mono-clonal antibodies directed against the alpha integrin of question. Dot plot quadrant analyses have then been carried out to show Factor VIII-positive cell populations (VIII+/alpha integrin+; VIII+/alpha inte-grin-), which reflect HUVEC, and Factor VIII-negative cell populations (VIII-/alpha integrin+; VIII-/alpha integrin-) which reflect tumour cells (Fig. 2A; representative for A498). Time-dependent FACscan (FL-1H/FL-2H [log] channel) histogram analysis of alpha2, alpha3 and alpha5 expression during the process of tumour cell adhesion to HUVEC revealed a strong down-regulation of integrin subtypes on A498, KTC26 or Caki1 cells immediately after attachment to HUVEC. This phenomenon was of transient nature with a maximum 30–60 min after tumour cell addition to HUVEC (Fig. 2B).
2.
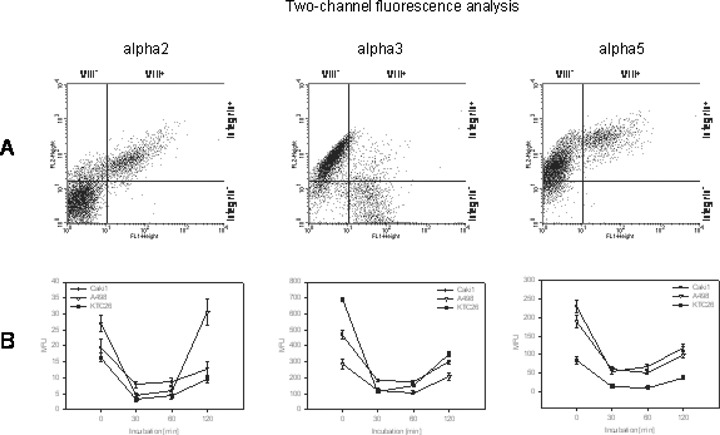
Down-regulation of alpha integrins on RCC cell lines co-cultivated with HUVEC. Cell cultures were double stained using the HUVEC specific Factor VIII-associated antigen monoclonal antibody (clone F8/86) and monoclonal antibodies directed against alpha2, alpha3 or alpha5 integrins. Both cell populations were defined in the FL-1 versus FL-2 dot blot diagram (Fig. 2A, representative for A498) and tumour cells (VIII-) analyzed separately. Dynamics of integrin expression is depicted in Fig. 2B as MFU values.
Expression and localization of alpha3 integrins were visualized by con-focal laser-scanning microscopy. In this context, alpha3 was distributed homogenously along the cell membrane of A498 cells in the monoculture system (Fig. 3A, arrows). However, alpha3 became undetectable in tumour cells in the co-culture model 60 min after they had attached firmly to the HUVEC monolayer (Fig. 3B, arrows). To discriminate HUVEC from A498, endothe-lial cells were coloured green (VIII-FITC), whereas A498 were coloured red (alpha3-PE). Twenty-four hours after tumour cell attachment to HUVEC, A498 cell aggregates were visualized which started to destruct the HUVEC integrity. According to the FACS data, alpha3 integrins were re-expressed in the late phase of tumour cell adhesion and detected again along A498 cell membranes (Fig. 3C, arrows).
3.
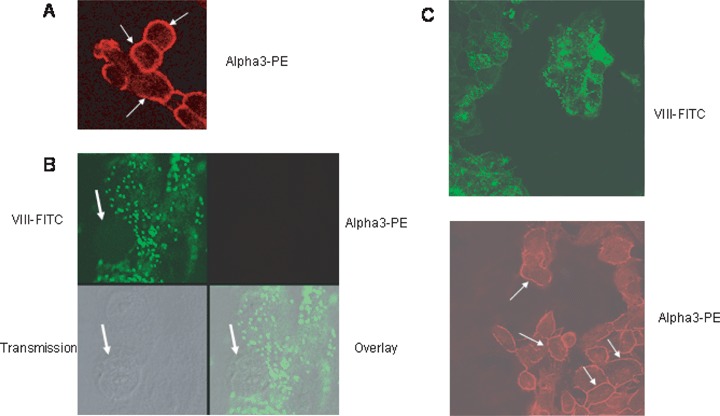
Confocal images of the distribution pattern of alpha3 integrin receptor molecules on A498 tumour cells.(A) shows distinct alpha3 localization along the tumour cell surface in the monoculture model (arrows).(B) and (C) discriminate HUVEC from A498 in the co-culture model, specimen were stained against Factor VIII-associated antigen, labelled with FITC (green fluorescence), and alpha 3 integrin, labelled with PE (red fluorescence). Overlay images of the red and green fluorescence channels are shown in the right hand corner. Transmission pictures were taken to localize adherent tumour cells (arrow). Note that integrin alpha3 is lost on adherent A498 cells after 60 min (B) but restored after 24 hrs (C).
Contact to HUVEC membranes is responsible for integrin down-regulation
Reduced integrin expression seen in the tumour cell-HUVEC co-culture system pointed to two options:A) Interaction of tumour cells with endothelium may evoke membrane triggered alterations of integrin expression level, or B) Interaction of tumour cells with endothelium may evoke the release of soluble mediators which then modify integrin expression. To address this question, A498 cells were treated with isolated HUVEC membrane fragments for 60 min and integrin expression was analyzed thereafter. Data were compared to non-treated cells and to tumour cells activated with supernatant taken from HUVEC tumour cell co-cultures. Figure 4 demonstrates distinct and significant integrin alpha2, alpha3 and alpha5 mRNA down-regulation when A498 were exposed to isolated endothelial membranes for 60–120 min. This effect occurred transiently. Twenty-four hours after contact induction, mRNA activity increased again and was then similar to the activity seen under control conditions. Fluorometry data revealed that down-regulation of alpha3 and alpha5 subtypes on the A498 cell surface was caused by the contact with endothelial membranes but not by the cell culture supernatant.
4.
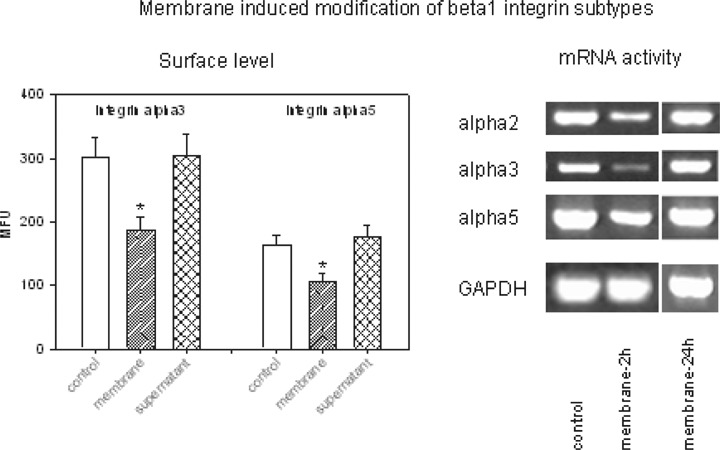
Endothelial membrane fragments but not cell culture supernatant reduce alpha3 and alpha5 surface expression and alpha2, alpha3 and alpha5 mRNA of A498 tumour cells. Surface level was analyzed by flow-cytometry and expressed as MFU values.*indicates significant difference to controls (n = 6). RNA was extracted, reverse-transcribed and submitted to semi-quantitative reverse transcription-PCR using gene-specific primers as described in Materials and methods. The internal control for the RT-PCR reaction was performed by running parallel reaction mixtures with the housekeeping gene GAPDH. One representative of three separate experiments is shown.
Quantitative loss of integrins may not necessarily be coupled with reduced receptor activity. We therefore explored ILK, FAK and phosphorylation of FAK (pFAK), which are involved in the regulation of inte-grin function. The experiments were carried out before and after treatment of A498 cells with HUVEC membrane fragments. Figure 5 demonstrates that both FAK and pFAK was down-regulated in A498 cells in the presence of HUVEC membranes.
5.
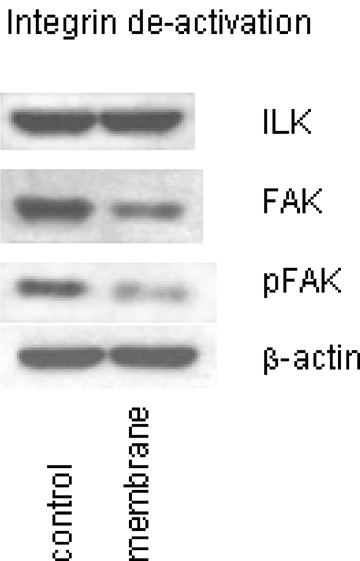
Endothelial membrane fragments modulate focal adhesion kinase (FAK) and FAK phosphorylation (pFAK) but not integrin-linked kinase (ILK). Analysis was carried out by Western blot technique. Cell lysates were subjected to SDS-PAGE and blotted on the membrane incubated with specific monoclonal antibodies (1:1000 dilution), listed in materials and methods. βactin served as the internal control. The figure shows one representative from three separate experiments.
Integrin loss is attributed to diminished adhesion capacity of tumour cells
Our data indicated that the adhesion of kidney carcinoma cells to endothelium alters integrin alpha2, alpha3 and alpha5 expression. Integrin beta1 subtypes are predominantly involved in cell adhesion to the extracellular matrix. We consequently explored whether heterogenous contact of tumour cells with endothelial cell membranes leads to modified binding to the matrix proteins collagen, fibronectin or laminin. In fact, pre-treatment of A498 cells with endothelial membrane proteins was accompanied by a reduced binding activity in the order laminin > fibronectin > collagen (Fig. 6).
6.
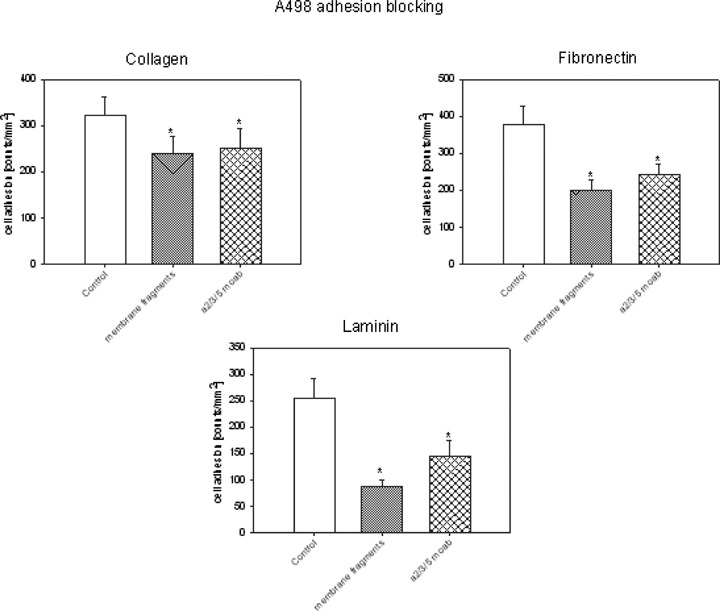
Contact inhibition of A498. A498 cells were treated with endothelial membrane fragments or with monoclonal antibodies directed against alpha2, alpha3 and alpha5 integrins (20μg/ml each). After preincubation, tumour cells were added to immobilized collagen, laminin, or fibronectin. Adherent cells were counted after 60 min and mean values calculated (cells/mm2). *indicates significant difference to controls One representative of 6 experiments is shown.
The relevance of alpha2, alpha3 and alpha5 inte-grins for adhesion events was explored by blocking studies using monoclonal antibodies. In this context, adhesion of A498 cells to fibronectin, laminin, or collagen was drastically inhibited by integrin blocking antibodies (but not by the corresponding IgG isotype control), which speaks for a link between integrin down-regulation and blocking tumour cell-matrix interaction (Fig. 6).
Intracellular signalling of tumour cells is modified by HUVEC
Integrins serve as important downstream elements. We therefore examined modifications of the intracellular signalling cascade in A498 tumour cells before and after exposure to isolated HUVEC membrane fragments. The experiments concentrated on PKC isoforms, the Mitogen-Activated Protein Kinase signalling pathway (MEKs, ERK, JNK, p38), and proteins, which are involved in cell-cycle regulation (cyclins, CDKs, Rb, p70s6kinase). Compared to the controls, the addition of membrane proteins to tumour cell cultures evoked down-regulation of PKCdelta, PKCepsilon and PKClambda, whereas PKCalpha, PKCbeta, PKCiota, PKCtheta remained unchanged (Fig. 7). MEK1 and ERK1 became down-regulated, whereas ERK2 was enhanced in the presence of membrane proteins. JNK, p38 (total and activated) and RACK1 were reduced, however, 14-3-3 epsilon was strongly elevated.
7.
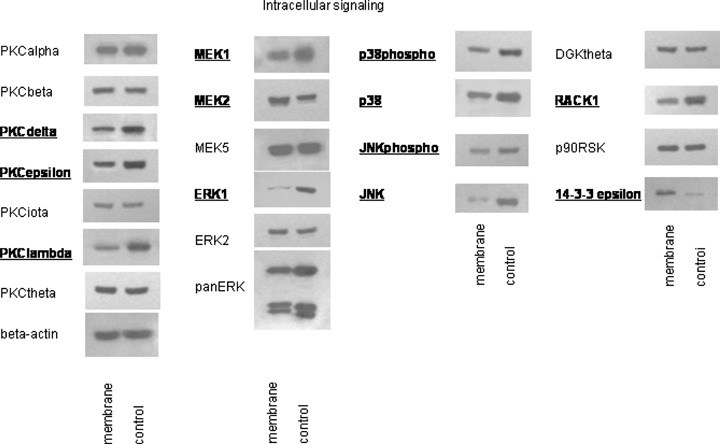
Western blot analysis of intracellular signalling proteins, listed in materials and methods. A498 cells were treated with endothelial membrane fragments for 60 min (control:untreated). Cell lysates were subjected to SDS-PAGE and blotted on the membrane incubated with the respective monoclonal antibodies. Concentrations were as follows: 1:250: JNK (total and activated), DGKtheta, PKCbeta, PKCtheta, PKCiota, PKClambda; 1:500: PKCdelta; 1:1000: 14-3-3 epsilon, MEK1, MEK5, p90RSK, PKCalpha, PKCepsilon;1:2500: MEK2, p38 (activated), RACK1;1:5000:ERK1, ERK2, panERK, p38 (total). Beta-actin served as the internal control. Bold types indicate changes to controls. The figure shows one representative from three separate experiments.
Expression level of cell cycle regulating proteins was quantified additionally. Cyclin B and p70s6kinase were both enhanced, RB and RB2 diminished in membrane treated A498 cells, compared to the untreated controls. Cyclin A, cyclin D3 and CDK subtypes were not altered in treated versus non-treated cell cultures (Fig. 8).
8.
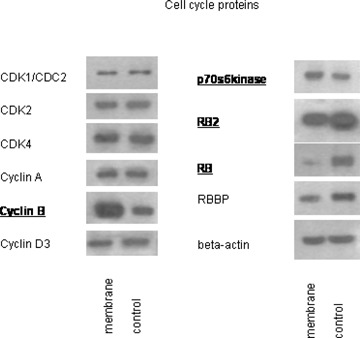
Western blot analysis of cell cycle proteins, listed in materials and methods. A498 cells were treated with endothelial membrane fragments for 60 min (control: untreated). Cell lysates were subjected to SDS-PAGE and blotted on the membrane incubated with the respective monoclonal antibodies. Concentrations were as follows: 1:250: CDK4, cyclin A, RB; 1:1000: p70s6kinase, cyclin B, cyclin D3, RB2, RBBP; 1:2500: CDK1, CDK2. Beta-actin served as the internal control. Bold types indicate changes to controls. The figure shows one representative from three separate experiments.
Discussion
Beta1 integrin family members govern the adhesive interactions between cancer cells and vascular endothelium. Still, it is not clear which subtypes are involved in RCC transmigration and how they contribute to this process. Our fluorescence analysis of A498, Caki1 and KTC26 tumour cells revealed distinct expression of alpha1, alpha2, alpha3 and alpha5 subtypes, whereas alpha4 and alpha6 subtypes were not detected. Accordingly, RCC specimens taken from metastatic tissue were all found to be alpha2, alpha3 and alpha5 positive and alpha4 negative by immunohistochemistry, with a positive correlation between integrin expression and tumour grading [8]. Different results have been presented in another immunohistochemical study, where all inte-grin alpha subtypes were present in RCC tissue specimens, including alpha4, and where tumours of higher grade showed decreased alpha3, alpha5 and alpha6 expression [9].
Considering that only snapshots of expression are provided in tissue analyses and that the same tech-nique was applied in the above mentioned experiments, dynamic and reciprocal alterations of the integrin expression level at discrete stages of tumour metastasis may be assumed which account for the observed differences. We propose that not simply the presence or absence of integrins may determine cancer progression and grading, but transient receptor modifications occur while RCC cells cross the endothelial barrier. In fact, our in vitro experiments present strong evidence that alpha2, alpha3 and alpha5 integrins became down-regulated immediately after the tumour cells attached to the endothelial cell monolayer followed by re-expression shortly there-after. The rapid integrin turnover seems not to be a unique phenomenon as the alphaVbeta5 receptor down- and up-regulates in a short time span during tissue morphogenesis [10, 11].
Integrin down-regulation on RCC cells was caused by direct contact with endothelial cells, as isolated endothelial membrane fragments but not cell culture supernatant contributed to the observed effects. We conclude that mechanical interaction is the necessary precondition to allow integrin down-regulation. However, it is not clear if integrins became reduced after binding to their respective ligands on endothelial cells or after binding to matrix proteins exposed on endothelial cells. Because blocking beta1 integrins did not influence attachment of epithelial cancer cell lines to vascular endothelium [12] but stopped tumour attachment to laminin secreted by endothelial cells [13], it seems likely that RCC integrins interact with extracellular matrix proteins deposited on the endothelial cell monolayer, rather than interacting with HUVEC.
The loss of alpha integrin subunits was accompanied by a reduced FAK expression, FAK activity and diminished binding of tumour cells to matrix proteins. It is generally accepted that FAK promotes cell adhesion, and in fact, integrin-stimulated cell adhesion requires FAK and FAK activity [14]. However, cells lacking FAK become refractory to motility signals independent of integrin receptors [15]. Achiwa and co-workers have speculated that continuous FAK phosphorylation following tumour-matrix attachment reflects decreased focal adhesion turnover and, consequently, loss or down-regulation of FAK may be necessary to maintain the malignant phenotype and initiate tumour motility [16]. In line with this assumption, dephosphorylation of FAK resulted in the loss of adhesion signals and promoted enhanced tumour migration in a breast carcinoma model [17]. It, therefore, seems plausible that the dynamics of tumour transmigration are not always controlled by the FAK-integrin pathway, but further regulatory proteins are involved at a particular time.
Indeed, the conversion from beta1-integrin-dependent movement to beta1-integrin-independent single-cell motility has been observed recently on primary melanoma explants [18]. Accordingly, blocking of beta1 integrins on RCC cells impaired binding to extracellular matrix proteins but not polarized cell movement [19, 20]. We propose that dynamic and reversible alterations of the integrin profile play an important part in tumour cell migration strategy, switching the cells from being adhesive to becoming motile and invasive [21]. No experiments have been carried out dealing with this issue. However, MCF-7 breast cancer cell lines adhered less efficiently, migrated more efficiently and became invasive when FAK was dephosphorylated [22]. Transition from adhesive interaction toward amoeboid locomotion was initiated in melanoma and neural crest cells after blocking beta1 integrins [18, 23].
In context with our experiments, the loss of inte-grins may facilitate detachment of RCC cells from the vasculature and promote dynamic movement and spread into the target organ. We further conclude that changes of the protein expression pattern may be associated with this process. In fact, 14-3-3 epsilon proteins that were amplified in our in vitro model were recently demonstrated to force metastatic progression of breast or lung cancer [24, 25]. There is also evidence that ERK2 contributes to enhanced tumour motility and spread. Transendothelial migration of monocytes as well as enhanced chemotactic activity of neutrophils has been shown to be accompanied by elevated ERK2 but not ERK1 levels [26, 27]. This also occurred in our co-culture assay, which points to the specific involvement of the ERK2 isoform in RCC dissemination. PKCepsilon is linked to integrin beta chains and to integrin-mediated cell adhesion in several types of cancer [28, 29]. Down-regulation of PKCepsilon in context with reduced beta1 subtype expression may therefore underline the concept of switching from integrin-dependent adhesion to inte-grin-independent cell movement. We also found reduced PKCdelta in RCC cells after treatment with HUVEC membranes, process of which drives tumour cells to become highly motile [30]. In accordance with our observations down-regulation of RACK1 has been recorded in a multicellular spheroid coculture model of breast tumour cells and fibroblasts [31]. This is of particular importance as RACK1 has been demonstrated to convert tumour cells from being adhesive to becoming motile [17].
Nevertheless, although changes of the intracellular signalling cascade support our speculation of switching the tumour cell migration strategy, we are aware that the interpretation of the protein data is difficult. Numerous functions are ascribed to each single protein and alterations of a particular protein may not adequately reflect the dynamic interactions between tumour and endothelium. Changes of the protein profile as a total should be considered, which, furthermore, may not exclusively be linked to adhesion events. Although purely speculative integrin down-regulation may also reflect tumour escape mechanism by reducing immunoreactivity. Alterations of cell cycle regulating proteins, which became evident in our model, refer to modulations of the tumour cell proliferation program. In fact, recent findings focus on distinct alterations of tumour behaviour during vascular transmigration by diminishing their adhesive forces and simultaneously elevating their proliferative capacity [32, 33].
To sum up, we present evidence that the interaction of RCC cells with human vascular endothelial cells is coupled with a rapid and reversible loss of beta1 integrin subtypes and integrin-dependent sig-nalling. Hypothetically, RCC may alter the adhesive strength of tumour-HUVEC and/or tumour-matrix interactions, to undergo transition from one migration program to another. Because cell migration may comprise diverse cellular and molecular mechanisms, future therapeutic targeting of the invasion cascade will require taking such migratory plasticity into consideration.
Acknowledgments
We would like to thank Heinz Schewe for technical assistance and Karen Nelson for critically reading the manu-script. This work was supported by the “Horst Müggenburg-Stiftung”, “Matthias Lackas-Stiftung”, “Jung-Stiftung”, “Ebert-Stiftung”and “Held-Hecker-Stiftung”.
References
- 1.Janes SM, Watt FM. New roles for integrins in squamous-cell carcinoma. Nat Rev Cancer. 2006;6:175–83. doi: 10.1038/nrc1817. [DOI] [PubMed] [Google Scholar]
- 2.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zutter MM, Santoro SA, Staatz WD, Tsung YL. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci USA. 1995;92:7411–5. doi: 10.1073/pnas.92.16.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tawil NJ, Gowri V, Djoneidi M, Nip J, Carbonetto S, Brodt P. Integrin alpha3beta1 can promote adhesion and spreading of metastatic breast carcinoma cells on the lymph node stroma. Int J Cancer. 1996;66:703–10. doi: 10.1002/(SICI)1097-0215(19960529)66:5<703::AID-IJC20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Seftor RE, Seftor EA, Sheng S, Pemberton PA, Sager R, Hendrix MJ. Maspin suppresses the invasive phenotype of human breast carcinoma. Cancer Res. 1998;58:5681–5. [PubMed] [Google Scholar]
- 6.Perlino E, Lovecchio M, Vacca RA, Fornaro M, Moro L, Ditonno P, Battaglia M, Selvaggi FP, Mastropasqua MG, Bufo P, Languino LR. Regulation of mRNA and protein levels of beta1 integrin variants in human prostate carcinoma. Am J Pathol. 2000;157:1727–34. doi: 10.1016/s0002-9440(10)64809-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill MB, Phipps JL, Hughes P, Greaves M. Anti-endothelial cell antibodies in primary antiphospholipid syndrome and SLE: patterns of reactivity with membrane antigens on microvascular and umbilical venous cell membranes. Br J Haematol. 1998;103:416–21. doi: 10.1046/j.1365-2141.1998.00979.x. [DOI] [PubMed] [Google Scholar]
- 8.Anastassiou G, Duensing S, Steinhoff G, Kirchner H, Ganser A, Atzpodien J. In vivo distribution of integrins in renal cell carcinoma:integrin-phenotype alteration in different degrees of tumor differentiation and VLA-2 involvement in tumor metastasis. Cancer Biother. 1995;10:287–92. doi: 10.1089/cbr.1995.10.287. [DOI] [PubMed] [Google Scholar]
- 9.Markovic-Lipkovski J, Brasanac D, Muller GA, Muller CA. Cadherins and integrins in renal cell carcinoma:an immunohistochemical study. Tumori. 2001;87:173–8. doi: 10.1177/030089160108700312. [DOI] [PubMed] [Google Scholar]
- 10.Yamada S, Yamada KM, Brown KE. Integrin regulatory switching in development: oscillation of beta 5 inte-grin mRNA expression during epithelial-mesenchymal interactions in tooth development. Int J Dev Biol. 1994;38:553–6. [PubMed] [Google Scholar]
- 11.Salmivirta K, Gullberg D, Hirsch E, Altruda F, Ekblom P. Integrin subunit expression associated with epithe-lial-mesenchymal interactions during murine tooth development. Dev Dyn. 1996;205:104–13. doi: 10.1002/(SICI)1097-0177(199602)205:2<104::AID-AJA2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 12.Yu LG, Andrews N, Zhao Q, McKean D, Williams JF, Connor LJ, Gerasimenko OV, Hilkens J, Hirabayashi J, Kasai K, Rhodes JM. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J Biol Chem. 2007;282:773–81. doi: 10.1074/jbc.M606862200. [DOI] [PubMed] [Google Scholar]
- 13.Andrews EJ, Wang JH, Winter DC, Laug WE, Redmond HP. Tumor cell adhesion to endothelial cells is increased by endotoxin via an upregulation of beta-1 integrin expression. J Surg Res. 2001;97:14–9. doi: 10.1006/jsre.2001.6090. [DOI] [PubMed] [Google Scholar]
- 14.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 15.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 16.Achiwa H, Lazo JS. PRL-1 tyrosine phosphatase regulates c-Src levels, adherence, and invasion in human lung cancer cells. Cancer Res. 2007;67:643–50. doi: 10.1158/0008-5472.CAN-06-2436. [DOI] [PubMed] [Google Scholar]
- 17.Kiely PA, Leahy M, O'Gorman D, O'Connor R. RACK1-mediated integration of adhesion and insulin-like growth factor I (IGF-I) signaling and cell migration are defective in cells expressing an IGF-I receptor mutated at tyrosines 1250 and 1251. J Biol Chem. 2005;280:7624–33. doi: 10.1074/jbc.M412889200. [DOI] [PubMed] [Google Scholar]
- 18.Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-inte-grin function, and migration strategies. Cancer Res. 2002;62:2125–30. [PubMed] [Google Scholar]
- 19.Brenner W, Gross S, Steinbach F, Horn S, Hohenfellner R, Thuroff JW. Differential inhibition of renal cancer cell invasion mediated by fibronectin, collagen IV and laminin. Cancer Lett. 2000;155:199–205. doi: 10.1016/s0304-3835(00)00429-8. [DOI] [PubMed] [Google Scholar]
- 20.Brenner W, Benzing F, Gudejko-Thiel J, Fischer R, Farber G, Hengstler JG, Seliger B, Thuroff JW. Regulation of beta1 integrin expression by PKCepsilon in renal cancer cells. Int J Oncol. 2004;25:1157–63. [PubMed] [Google Scholar]
- 21.Furuya M, Kato H, Nishimura N, Ishiwata I, Ikeda H, Ito R, Yoshiki T, Ishikura H. Down-regulation of CD9 in human ovarian carcinoma cell might contribute to peritoneal dissemination: morphologic alteration and reduced expression of beta1 integrin subsets. Cancer Res. 2005;65:2617–25. doi: 10.1158/0008-5472.CAN-04-3123. [DOI] [PubMed] [Google Scholar]
- 22.Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1beta in vivo and in vitro. Breast Cancer Res Treat. 2005;89:5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- 23.Dufour S, Duband JL, Humphries MJ, Obara M, Yamada KM, Thiery JP. Attachment, spreading and locomotion of avian neural crest cells are mediated by multiple adhesion sites on fibronectin molecules. EMBO J. 1988;7:2661–71. doi: 10.1002/j.1460-2075.1988.tb03119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li DQ, Wang L, Fei F, Hou YF, Luo JM, Wei-Chen, Zeng R, Wu J, Lu JS, Di GH, Ou ZL, Xia QC, Shen ZZ, Shao ZM. Identification of breast cancer metastasis-associated proteins in an isogenic tumor metastasis model using two-dimensional gel electrophoresis and liquid chromatography-ion trap-mass spectrome-try. Proteomics. 2006;6:3352–68. doi: 10.1002/pmic.200500617. [DOI] [PubMed] [Google Scholar]
- 25.Qi W, Liu X, Qiao D, Martinez JD. Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int J Cancer. 2005;113:359–63. doi: 10.1002/ijc.20492. [DOI] [PubMed] [Google Scholar]
- 26.Sultana C, Shen Y, Johnson C, Kalra VK. Cobalt chloride-induced signaling in endothelium leading to the augmented adherence of sickle red blood cells and transendothelial migration of monocyte-like HL-60 cells is blocked by PAF-receptor antagonist. J Cell Physiol. 1999;179:67–78. doi: 10.1002/(SICI)1097-4652(199904)179:1<67::AID-JCP9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Hii CS, Stacey K, Moghaddami N, Murray AW, Ferrante A. Role of the extracellular signal-regulated protein kinase cascade in human neutrophil killing of Staphylococcus aureus and Candida albicans and in migration. Infect Immun. 1999;67:1297–302. doi: 10.1128/iai.67.3.1297-1302.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Besson A, Wilson TL, Yong VW. The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem. 2002;277:22073–84. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- 29.Wu D, Thakore CU, Wescott GG, McCubrey JA, Terrian DM. Integrin signaling links protein kinase Cepsilon to the protein kinase B/Akt survival pathway in recurrent prostate cancer cells. Oncogene. 2004;23:8659–72. doi: 10.1038/sj.onc.1207900. [DOI] [PubMed] [Google Scholar]
- 30.Renault-Mihara F, Beuvon F, Iturrioz X, Canton B, De Bouard S, Leonard N, Mouhamad S, Sharif A, Ramos JW, Junier MP, Chneiweiss H. Phosphoprotein enriched in astrocytes-15 kDa expression inhibits astro-cyte migration by a protein kinase C delta-dependent mechanism. Mol Biol Cell. 2006;17:5141–52. doi: 10.1091/mbc.E05-11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seidl P, Huettinger R, Knuechel R, Kunz-Schughart LA. Three-dimensional fibroblast-tumor cell interaction causes downregulation of RACK1 mRNA expression in breast cancer cells in vitro. Int J Cancer. 2002;102:129–36. doi: 10.1002/ijc.10675. [DOI] [PubMed] [Google Scholar]
- 32.Margulis A, Zhang W, Alt-Holland A, Pawagi S, Prabhu P, Cao J, Zucker S, Pfeiffer L, Garfield J, Fusenig NE, Garlick JA. Loss of intercellular adhesion activates a transition from low- to high-grade human squamous cell carcinoma. Int J Cancer. 2006;118:821–31. doi: 10.1002/ijc.21409. [DOI] [PubMed] [Google Scholar]
- 33.Blattner SM, Kretzler M. Integrin-linked kinase in renal disease:connecting cell-matrix interaction to the cytoskeleton. Curr Opin Nephrol Hypertens. 2005;14:404–10. doi: 10.1097/01.mnh.0000172730.67746.5b. [DOI] [PubMed] [Google Scholar]