

Abstract
Infiltration of bone marrow derived cells is part of the angiogenic switch required for uncontrolled tumour growth. However, the nature of the tumour-infiltrating cells from bone marrow has not been fully elucidated. To investigate the phenotype of bone marrow derived cells within a tumour, we employed the Lewis lung carcinoma (LLC) murine tumour model. We followed bone marrow derivation of tumour-infiltrating cells through transplantation of CD45.2 bone marrow cells into pre-irradiated CD45.1 mice. We found robust CD45.2 donor type chimerism in bone marrow and blood of CD45.1 recipient tumour-bearing mice. Flow cytometric analysis of LLC tumours showed, in addition to previously described pro-angiogenic CD45+VEGFR2+‘endothelial progenitor cells’ (EPC), or CD45+Tie2+‘Tie2-expressing monocytes’ (TEM), incorporation of donor type lineage marker negative (Lin−) and Lin−Sca1+ undifferentiated haematopoietic cell types. Immunohistochemical analysis confirmed the extravasal location of the primitive haematopoietic cells. Flow-cytometric sorting of bone marrow cells and subsequent analysis in haematopoietic colony-forming assays revealed that cells with a Lin−Sca1+ phenotype, which were initially negative for VEGFR2 and Tie2, gave rise to VEGFR2+ and/or Tie2+ cells. Moreover, Lin− bone marrow cells pre-labelled with the membrane dye PKH26 (a red fluorochrome) and transplanted i.v. into tumour-bearing mice were found to extravasate and incorporate into LLC tumours within 24 hrs. Thus, primitive haematopoietic precursors which are thought to be precursors of EPC and TEMs, constitute a part of the tumour microenvironment. This makes them an attractive target cell population for tumour-directed cellular therapies.
Keywords: haematopoietic progenitor cells, colony-forming cells, homing, tumour
Introduction
The relationship between tumour development and blood vessel formation has been intensively studied over the last years. In 1971, Folkman published the hypothesis that tumour growth is angiogenesis dependent [1]. Subsequent studies revealed that initiation of new vessel formation is a key step in tumour progression, and confirmed that the switch to the angiogenic phenotype resulted from a change in the balance between several positive and negative regulators of angiogenesis [2–4]. More recent studies have indicated that bone marrow derived endothelial cells and monocytic cells can contribute significantly to vessel growth in ischemic and malignant tissues [5–7]. This postnatal vasculogenesis seems to involve the recruitment and in situ differentiation of endothelial progenitor cells (EPC) to endothelial cells. In addition, bone marrow derived cells have been shown to contribute to angiogenic network formation by their capacity to give rise to endothelial or mural cells and/or by releasing pro-angiogenic factors like vascular endothelial growth factor (VEGF), angiopoietin-1, hepatocyte growth factor, epidermal growth factor, transforming growth factor β1 or thrombospondin-1 [7–10].
Primitive haematopoietic cells in the bone marrow have been characterized as a hierarchy of haematopoietic stem and progenitor cells, which can be enriched by using a combination of cell surface markers. These cells include the long-term bone marrow reconstituting haematopoietic stem cells (LT-HSC) which were described as Lin −/lo c-kithi Sca-1hi Thy 1.1low Flk2− and short-term repopulating haematopoietic stem cells (ST-HSC) described as Lin−/low c-kithi Sca-1hi Thy 1.1low Flk2+[11]. In addition to the above mentioned HSC types, multipotential Lin−/low c-kithi Sca-1hi Thy 1.1− Flk2+, common lymphoid or common myeloid progenitors as well as bi- or unilineage-determined progenitor cell types have been defined [12]. A further antigen which has been shown to be expressed on both HPC/HSC and in endothelial cells is Tie2, a receptor tyrosine kinase [13, 14]. Tie2 expression characterizes also pro-angiogenic CD45+ cells of haematopoietic origin, including ‘Tie2-expressing monocytes’ (TEM), and CD45− pericyte precursors of mesenchymal origin [15].
In this study we asked whether cells with haematopoietic progenitor cell phenotype may reside in tumours. We therefore established a Lewis lung carcinoma (LLC) model in mice and characterized the tumour incorporation of bone marrow derived cell populations. We report that Lin− and Lin− Sca-1+ progenitors are present at significant frequencies in the tumour in addition to TEM and EPC. We further show that cells with TEM and EPC phenotypes can directly be differentiated in methylcellulose culture from Lin− and Lin−Sca-1+ cells, and that fluorescence marked Lin− cells home directly to tumour. Thus, cells with primitive haematopoietic phenotype can contribute to the tumour microenvironment.
Materials and methods
Cells and reagents
Antibodies against Lineage markers CD11b/Mac-1 (clone M1/70), Gr-1 (clone RB6–8C5), CD3, B220 (clone RA3–6B2), TER119, against CD45 (clone 30-F11) or CD45 isoforms CD45.1 (clone A20) and CD45.2 (clone 104), Sca-1 (clone D7), c-kit (clone 2B8), CD31 (clone MEC 13.3), VEGFR2/flk1 (clone Avas 12α1), as well as IgG controls were obtained from BD Pharmingen (San Jose, CA, USA). Tie-2 (clone TEK4) was from eBioscience (San Diego, CA, USA), anti-pan-Laminin from Sigma (Munich, Germany) and anti fibronectin ab23750 from Abcam (Cambridge, UK). MethoCult medium was from Stem Cell Technologies (Vancouver, Canada) and was supplemented with 10 ng/ml of rmIL-3 (R&D Systems, Wiesbaden, Germany), rmIL-6 (TeBU-bio, Offenbach, Germany), rmSCF (Peprotech, Offenbach, Germany), rhTPO (TeBU-bio, Offenbach, Germany) and FLT3-L (TeBU-bio). For enrichment of Lin− cells, bone marrow cells were prepared from femurs and tibiae of C57Bl\6 45.2 donor mice (Charles River Laboratories, Sulzfeld, Germany) after cervical dislocation. Enrichment of stem and progenitor cells from bone marrow was performed by immunomagnetic depletion of lineage-committed cells using the Lineage Cell Depletion Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol.
Murine tumour model
The LLC cell line (provided from American Type Culture Collection [ATCC], Manassas, VA, USA) was maintained in RPMI 1640 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco, Karlsruhe, Germany) and 10% foetal bovine serum (PanBiotech, Passau, Germany). To induce tumour formation, single cell suspensions of 1 × 106 LLC cells suspended in 100 μl PBS were injected subcutaneously into the flanks of 8–10-week-old C57BL/6 mice. Tumour growth was followed by measurement of length and width diameter of tumours, and tumour volumes were calculated using the formula V= 4π (L1 ×L2)2/3, with V = volume (mm3), L1 = longest diameter (mm) and L2 = shortest diameter (mm). The tumours were processed for analysis 14–26 days after LLC cell inoculation. All procedures were approved by the Animal Care Committee at Frankfurt am Main University.
Immunohistochemistry
Frozen tumour tissue sections (6 μm) were prepared using Tissue Teck (Sakura Finetek Europe, Zoeterwoude, The Netherlands) and kept at –20°C. After thawing, slides were fixed at RT with 4% paraformaldehyde for 10 min., washed twice with PBS, permeabilized and blocked by incubation for 30 min. with PBS supplemented with 3% bovine serum albumin and 0.1% Triton X-100. The sections were immunostained with primary FITC conjugated antibody against CD45, CD11b, Sca-1, PE conjugated anti-c-kit antibody, or alexa-Fluor 488 or 555 anti-Rabbit antibody (Invitrogen, Carlsbad, CA, USA) at a final concentration of 5 μg/ml. After 1 hr incubation at room temperature, the slides were washed three times with PBS and mounted with fluorescence protective mounting medium (Vectashield; Vector Laboratories, Burlingame, CA, USA) containing DAPI for nuclear counterstaining and photo-documented using a CCD camera and Axio Vision 3.1 software.
Flow-cytometric sorting and Colony Forming Unit (CFU) assays
LLC tumour cells were isolated by flushing tumours through a 40 μm cell sieve. Subsequently, mononuclear cells were separated by density gradient centrifugation using Ficoll. Mononuclear cell suspensions were then incubated for 1 hr at 4°C with an antigen-presenting cell (APC) conjugated mixture containing optimized concentrations of antibodies to CD2, CD3, CD5, CD8, Mac-1, Gr-1, TER119, CD45R and CD19; PE conjugated anti-VEGFR2, and FITC conjugated anti-CD11b, and subsequently sorted flow cytometrically using a FACSAria (Becton Dickinson, San Jose, CA). The sorted subpopulations were pelleted and an aliquot of each was taken for reanalysis. The sorted cell populations were cultured in methylcellulose/ 10% FBS supplemented with 20 ng/ml of recombinant haematopoietic growth factors IL-3, IL-6, SCF, TPO, FLT3-L at a plating density of 1000 cells/well in 24-well plates. Culture dishes were incubated at 37°C and infused with 5% CO2. The number of colonies was counted using an inverted microscope after 7 days of culture. In addition, the colonies were analysed for cell surface staining by flow cytometry.
Homing assay in mice
PKH Fluorescent Cell Linkers provide fluorescent labelling of living cells over an extended period of time. To label the progenitor cells prior to transplantation experiments, Lin− cells were washed three times with PBS by centrifugation at 400 ×g for 5 min. The pellet was dissolved in Diluent C and PKH26 (red fluorochrome) was added to a final concentration of 1.7 × 10−5 M. After 5 min. incubation at RT under continuous gentle shaking on rocking platform, 100 μl of FCS was added for 1 min. to inactivate the reaction. Finally, the cells were washed once with PBS/10%FCS, then two times with PBS, and were resuspended in 500 μl PBS. Cell viability was assessed before injection using the Trypan blue method and PKH fluorescent cells were quantified by flow cytometric analysis.
Statistical analysis
Calculation of statistical significances was performed with a Student’s t-test. A P-value ≤ 0.05 was considered significant.
Results
Bone marrow derived cells have been proposed to be crucially involved in angiogenesis and tumour growth. One important function of these cells is to provide soluble factors required for tumour neovascularization. Furthermore, gene transduction experiments with HSV-Thymidin kinase (Tk) have shown that gene transfer of this enzyme to tumour infiltrating bone marrow derived cells and subsequent systemic activation of Tk by Gancyclovir may be a means to limit tumour growth and to support the animals’ survival [7, 15]. We have addressed whether in addition to the known population of bone marrow derived cells, such as TEM and EPC, cells with haematopoietic progenitor phenotype may be also recruited to tumours.
We first established a transplantation model using lethally irradiated CD45.1 (also termed Ly 5.1) recipient mice which were transplanted with bone marrow cells form CD45.2 (Ly 5.2) mice and were challenged with LLC cells 2–3 weeks later (Fig. 1A). Within a further 2–3 weeks, LLC tumours grew exponentially to a size up to approximately size of ≥1 cm (Fig. 1B, C). In addition to tumour size, we quantified the donor haematopoietic chimerism. As shown in Fig. 2, donor cells constituted on average 50–70% of all CD45+ cells, not only in bone marrow and blood, but also in the tumours of the transplanted mice albeit with a lower mean value, which was however statistically not significantly different from bone marrow or blood,. Thus, the tumours contained a comparable amount of donor type bone marrow derived cells as in bone marrow or blood.
Fig 1.
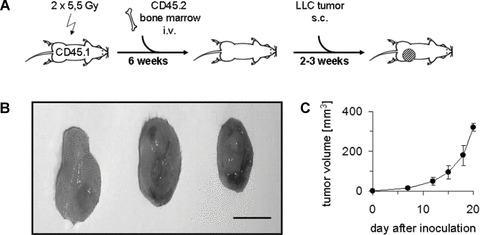
Establishment of donor haematopoiesis by bone marrow transplantation and growth of LLC tumours. (A) Experimental scheme. (B) Photographs of three representative tumours. Size bar, 1 cm. (C) Volumes of LLC tumours. Values are means ± S.D.; n= 6.
Fig 2.
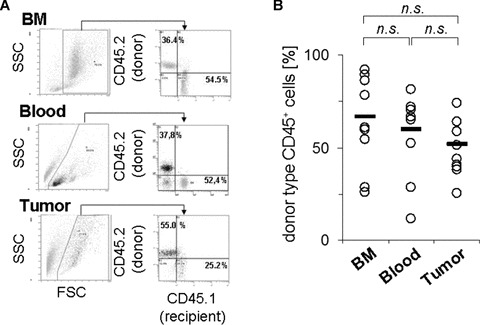
Engraftment of donor type haematopoietic cells in transplanted mice. Analysis was performed by tracing of CD45.2 donor type cells tumour-bearing mice which had been transplanted into CD45.1 mice prior to tumour inoculation. Bone marrow cells or mononuclear cell fractions from peripheral blood or tumour cell suspensions were isolated and stained with fluorescence-conjugated antibodies specific for the CD45.1 and CD45.2 isoforms, and analysed by flow cytometry. (A) Results from one animal, showing gating and analysis strategy. (B) Results form quantitative evaluation in 10 transplanted mice. Circles represent values from individual mice, bars give mean values. n.s.; no statistically significant difference was detected between the indicated groups.
To further investigate the phenotype of the donor cells recovered from LLC tumours, we performed flow cytometric analysis from tumour cell suspensions. These analyses included antibodies against lineage markers to exclude more differentiated haematopoietic cells, as well as antibodies against surface markers expressed on HPC and HSC such as c-kit and Sca-1 (Fig. 3A). We observed that donor type Lin− and Lin−Sca-1+ cells were detected at relatively comparable levels in bone marrow, blood and tumours (5–10% and, respectively, 1–2%; Fig. 3B). Still, most of the mononuclear cells found in tumours were differentiated cells, since 90.3 ± 3.2% of these cells were found to stain positive for haematopoietic lineage markers (Lin+; data not shown). We also determined the frequency of cells with the phenotype of TEM and EPC in all three compartments, and found increased levels of TEM and EPC in tumours compared with bone marrow or blood. Immunohistochemical analysis of tumour specimens revealed the presence of CD45+ cells, CD11b+ (lineage marker) cells and Sca 1+ cells, the latter in part co-expressing c-kit (Fig. 3C–F). Thus, cells with a primitive haematopoietic phenotype were detected not only in haematopoietic organs, but also in LLC tumours.
Fig 3.
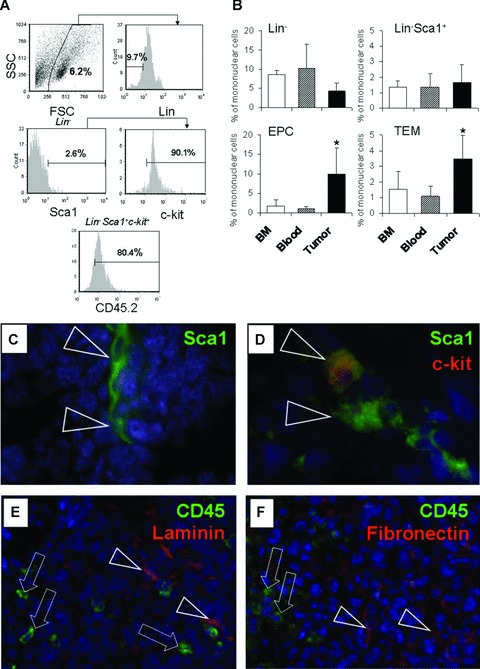
Analysis of bone marrow derived cells engrafted in bone marrow, blood and in tumours. (A) Representative flow-cytometric analysis from a tumour cell suspension of CD45.1 tumour-bearing mice previously transplanted with CD45.2 bone marrow cells. (B) Results form quantitative evaluation of (A), showing the percentage of cells with the indicated phenotypes. Values are means ± S.D.; n= 6 per group. *, statistically significant difference compared with bone marrow or blood (P < 0.05) (C-F) Immunohistochemical analysis of tumour cryosections prepared from LLC tumours and stained with fluorescence-marked antibodies against the indicated antigens. Nuclei were counterstained using DAPI. Triangles and arrows show examples of cells staining positive for the indicated antigens. Representative frames are shown. (C, D), 63-fold magnification, (E, F), 20-fold magnification.
To address the question whether the undifferentiated haematopoietic progenitors could be the origin of the more mature pro-angiogenic cell types typically found in tumours, such as TEMs and EPCs, we selected primitive HPCs via flow cytometric cell sorting and subsequently plated them in methylcellulose culture in the presence of haematopoietic growth factors. We found that haematopoietic colony forming cells were virtually absent both in the VEGFR2+ and Lin+ bone marrow cell compartments (Fig. 4A, B). In contrast, Lin− CD11b−, Lin− CD11b+ and Lin− Sca-1+ c-kit+ cells were highly clonogenic (30–50 CFU/1000 initially plated cells; Fig. 4B). Flow cytometric reanalysis of the cells from the grown colonies showed the presence of VEGFR2+ and Tie2+ cells (Fig. 4C, and data not shown). This indicates an intrinsic differentiation potential of the isolated primitive progenitors to cells with a pro-angiogenic phenotype.
Fig 4.
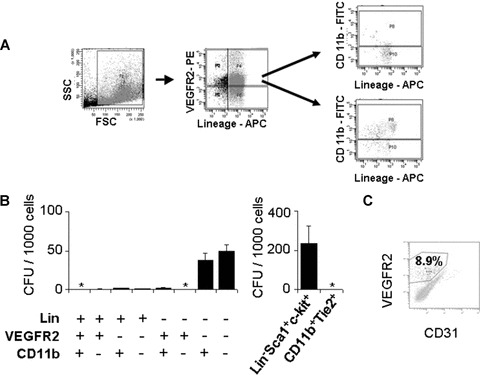
Lin− Populations have colony forming potential. Mononuclear cell fractions from single cell suspensions prepared from LLC tumours were subjected to flow cytometric cell sorting. (A) Flow cytometric isolation strategy of a representative sample after labelling with Lin−APC, VEGFR2-PE and Sca1-FITC antibodies. (B) Results from colony formation assays in methylcellulose supplemented with SCF, IL-1β, IL-3, IL-6, TPO after seeding of 1000 isolated progenitors of the indicated cell populations. Results are means ± S.D.; n= 3–5. *, zero values were obtained. (C) Flow cytometric analysis of cells from a colony grown from sorted Lin−Sca1+c-kit+ HPC after 7 days of culture in methylcellulose in the presence of haematopoietic growth factors as indicated in (B) and staining with anti-VEGFR2 and anti-CD31 antibodies.
Since this creates the hypothesis that primitive haematopoietic cells home to tumours, where they can form TEM and EPC, we investigated whether i.v. injected fluorescence marked Lin− HPCs can directly home to the tumours. We established a homing assay in tumour-bearing mice into which we i.v injected PKH26 fluorescence labelled Lin− bone marrow cells. As shown in Fig. 5, Lin− cells were detected at a frequency of approximately 200–300/100,000 investigated cells in bone marrow and spleen, and approximately 30–50/ 100,000 investigated cells in tumour by flow cytometric quantitation (Fig. 5A, B). In lungs, we detected fewer cells (mean, 17 ± 12/100,000 cells). Immunofluorescence analysis demonstrated the presence of PKH26+ homed cells in the tumour outside of blood vessels (Fig. 5C–F).
Fig 5.
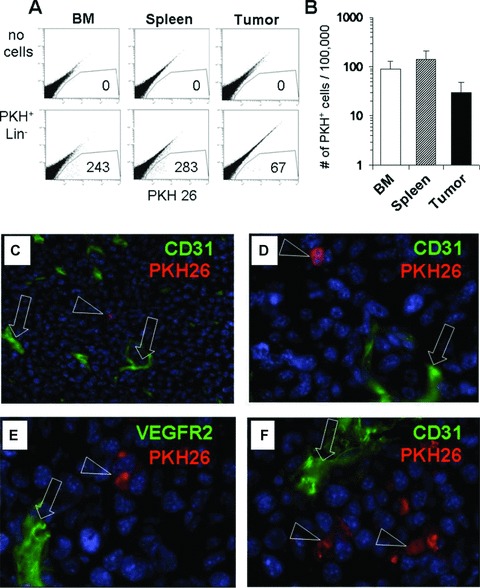
Lin− myeloid progenitors home to tumours. Lin− cells were isolated from bone marrow of normal mice, labelled with the vital fluorescent dye PKH26, and injected i.v. into mice with established LLC tumours. Twenty-four hours later, animals were killed, and cell suspensions or tissue samples were prepared for cryosectioning. (A) Flow cytometric analysis for the presence of PKH+ cells in a non-transplanted (‘no cells’) control mouse and a mouse that received 1.5 × 106 PKH+ Lin− cells. Numbers indicate number of cells within the gate after analysis of 100,000 cells (B) Quantitative analysis in three different tissues of PKH+ cell frequencies from seven mice with LLC tumours and transplanted with 1–2 × 106 PKH+ Lin− bone marrow cells. Values are means ± S.D. (C–F) Tissue sections were prepared from tumours of injected mice and stained for endothelial cells using CD31-FITC or VEGFR2-FITC (green), showing PKH+ cells in red. Triangles and arrows indicate cells stained positive for the indicated antigens. Blue counterstain with DAPI was performed to visualize nuclei. Magnification 63-fold.
Discussion
A number of recent studies in tumour biology have indicated that cells derived from adult BM like EPC and monocytic or haematopoietic progenitor cells contribute to neovessel formation in tumours [5, 7, 15, 17]. However, the phenotype of these cells as well as the mechanisms of their recruitment to the tumour microenvironment have not been fully elucidated.
In our study, we established a bone marrow transplantation model followed by LLC tumour induction to reveal the capacity of donor bone marrow derived cells to target the tumour. The presence of donor type haematopoietic CD45+cells, including cells with undifferentiated phenotype like Lin− and Lin− Sca+ cells, within tumours and also in the bone marrow and blood indicates that these cells are mobilized through the blood stream into the tumour. Additionally, we quantified cells with the phenotype of EPC and TEM in these three tissues, which have been described as cells with pro-angiogenic capacity, and found levels comparable to the data in the literature [15, 21]. Immunohistochemical analyses have demonstrated an extravascular lodgement of the primitive haematopoietic cells within the tumour microenvironment.
So far, several groups have suggested that bone marrow derived cells of different types can be mobilized through the blood to tumours and incorporated into sites of neovascularization [4–7, 15–20]. To elucidate whether cells with primitive phenotype may give rise to cells with pro-angiogenic phenotype such EPC and TEM, we performed in vitro colony forming assays from selected subpopulations of bone marrow HPC. We could demonstrate that the haematopoietic colony forming cells are enriched in a different population than EPC and TEM, whereas EPC and TEM lacked of in vitro colony proliferation potential. However, flow cytometric reanalysis of the colonies grown from Lin− CD11b−, Lin− CD11b+ and Lin− Sca1+ c-kit+ cells showed the development of VEGFR2+ and Tie2+ expressing cells. Our data show that primitive haematopoietic cells also circulate in blood of tumour-bearing mice indicate that, in addition to the previously shown mobilization of EPC and TEM [5–7, 15], more primitive bone marrow derived cells exist in tumours which may differentiate into known pro-angiogenic cell types.
We furthermore demonstrated that transplanted HPCs circulate in the blood stream and target the tumour. In in vivo transplantation experiments, we quantified the BM Lin− PKH26+ cells in bone marrow, blood and tumour and showed that these cells may be recruited to subcutaneously implanted LLCs. This complements the data of our flow cytometric and immunohistochemical analysis in tumour-bearing mice, where HPC were found in tumour in similar frequency as in bone marrow and blood, and reflects the findings of Jin et al.[10], who have demonstrated the direct homing of both, Lin+ and Lin− cells to murine tumours. Immunohistological stainings showed that the bone marrow derived cells are manly located at a distance from blood vessels inside the tumour tissue. The comparable incidence of the Lin- cell populations (around 5% of mononuclear cells, 1% of Lin−Sca1+ cells) in circulating blood and tumour tissue, and the immunohistological staining results both indicate that the transplanted BM Lin− PKH26+ cells are present in the tumour tissue and were not simply circulating through the tumour vasculature.
In conclusion, the present study demonstrates that bone marrow derived HPC are recruited to LLC tumours and may to differentiate into cells with angiogenic phenotype. Our data suggest that HPC, in addition to their capacity to give rise to pro-angiogenic cells in the bone marrow which can circulate as mature cells in blood and home to tumours, are present themselves in blood of tumour-bearing mice and can be directly recruited to the tumour microenvironment. This creates new hypotheses about the roles of primitive haematopoietic cells in tumour biology, and the questions whether this could be exploited to target tumour in a therapeutically desirable fashion, e.g. by ex vivo expanding haematopoietic stem cells transfected with suicide genes for repeated i.v. delivery as an efficacious anti-tumour cellular therapy.
Acknowledgments
We thank Monika Stein, Christine Ströbele, Victoria Lang and Sabrina Boehme for excellent technical support. The study was supported through the German Research Foundation (DFG) by SFB T/R23 project C3 (to J.G. and R.H.) and through the DFG Excellence Cluster ‘Cardiopulmonary Systems’ (to E.S. and R.H.)
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during angiogenesis. Cell. 1996;86:353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 3.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 5.Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 6.Rafii S, Lyden D, Benezra R, et al. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy. Nat Rev Cancer. 2002;2:826–35. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 7.De Palma M, Vennert MA, Galli R, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 9.Ruzinova MB, Schoer RA, Gerald W, et al. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell. 2003;4:277–89. doi: 10.1016/s1535-6108(03)00240-x. [DOI] [PubMed] [Google Scholar]
- 10.Jin H, Aiyer A, Su J, et al. A homing mechanism for bone marrow–derived progenitor cell recruitment to the neovasculature. J. Clin. Invest. 2006;116:652–62. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc Natl Acad Sci USA. 2001;98:14541–6. doi: 10.1073/pnas.261562798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Passegué E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics. Proc Natl Acad Sci USA. 2003;100:11842–9. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont DJ, Gradwohl GJ, Fong GH, et al. The endothelial-specific receptor tyrosine kinase, tek, is a member of a new subfamily of receptors. Oncogene. 1993;8:1293–301. [PubMed] [Google Scholar]
- 14.Arai F, Hirao A, Ohmura M, et al. Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 15.De Palma M, Venneri MA, Roca C, et al. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–95. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- 16.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 17.Davidoff AM, Ng CY, Brown P, et al. Bone marrow-derived cells contribute to tumor neovasculature and, when modified to express an angiogenesis inhibitor, can restrict tumor growth in mice. Clin Cancer Res. 2001;7:2870–9. [PubMed] [Google Scholar]
- 18.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–12. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 19.Reyes M, Dudek A, Jahagirdar B, et al. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002;109:337–46. doi: 10.1172/JCI14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nolan DJ, Ciarrocchi A, Mellick AS, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–58. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]