

Abstract
Rare cells not normally present in the peripheral bloodstream, such as circulating tumour cells, have potential applications for development of non-invasive methods for diagnostics or follow up. Obtaining these cells however require some means of discrimination, achievable by cell type specific antibodies. Here we have generated a microselection method allowing antibody selection, by phage display, targeting a single cell in a heterogeneous population. One K562 cell (female origin) was positioned on glass slide among millions of lymphocytes from male donor, identifying the K562 cell by FISH (XX). Several single cell selections were performed on such individual slides. The phage particles bound to the target cell is protected by a minute disc, while inactivating all remaining phage by UV-irradiation; leaving only the phage bound to the target cell viable. We hereby retrieved up to eight antibodies per single cell selection, including three highly K562 cell type specific.
Keywords: microselection, single cell selection, phage display, antibody selection, K562, UV irradiation, shadow-stick, rare cell detection, FISH
Introduction
Technological achievements within cell biology and imaging systems have during the last decades revealed many novel cell types that people have previously been unaware of. The majority of these are very rare cell types such as certain stem cell populations and endocrine cells. However, also cells commonly present at one location but rare at an unfamiliar location, such as circulating non-blood cells have been documented. Examples here are circulating tumour cells (CTCs) originating from primary tumours and foetal cells in the peripheral blood of pregnant women. Both types have drawn a great deal of attention because the ability to identify and isolate such rare cells in the peripheral blood may provide a way for improved diagnostic procedures.
Today the knowledge of these rare cells in the peripheral blood is limited. The estimated number of CTCs is one in millions of peripheral blood mononuclear cells (PBMC), depending on disease progression and tumour class [1]. Likewise, foetal cells crossing the placenta barrier into the maternal peripheral blood are extremely rare – estimated to be around one per five million PBMC [2].
Further knowledge, especially identification of novel biomarkers, is therefore essential before efficient enrichment and identification of these cell types can be applied for routine diagnostics. Today the identification processes for characterizing newly discovered very rare cells are quite laborious. After the initial characterization the conventional way to recognize these rare cells is by means of cell specific antibodies, if such are available.
Currently a number of suitable systems for in vitro antibody generation are available, of which phage display is the most thoroughly tested. Since the first publication introducing the method in 1985 [3], phage display has evolved into a widely used technique for obtaining antibodies [4]. Nonetheless, in the case of very rare cells it has proven to be quite a challenging task. Due to lack of cell specific markers, it is difficult to enrich pure population in high number, thus preventing application of standard methods for antibody generation. One way to circumvent the problems of insufficient accessibility to target cells is to reduce the requirement to only one identified target cell.
Attempts along these lines have been performed, namely by utilizing retrieval of a target cell with adherent phage by laser capture microdissection (LCM) [5]. However, this approach has so far not proven successful in achieving single cell selections [6]. Analogous attempts have been done using FACS (fluorescence activated cell sorting), performed on a more common cell sub-population [7], but so far without success when applied for a single cell.
Here we present a novel method by which we have managed to isolate specific scFv (single chain variable region fragment) antibodies against one single cell spiked onto a slide with a heterogeneous cell population consisting of lymphocytes. The method is adapted from the selective ultraviolet radiation fractionation (SURF) method, developed for single cell PCR (Shibata et al.). In our design we allow binding of phage to all the cells on the slide containing the spiked target cell. Subsequently we inactivate the phage on all non-target cells by UV irradiation. During this UV irradiation, the phage bound to the target cell are protected by a minute gold-disc, positioned over the target cell by micromanipulation equipment, shielding it from the UV irradiation. After UV irradiation viable phage is retrieved containing the DNA sequences encoding the antibodies bound to the UV-shielded target cell. Subsequently the antibodies gained by the selection can be produced monoclonally and tested by various methods.
Using this microselection method we have demonstrated the generation of specific antibodies from single cell selections, and successfully applied these antibodies for the identification of this rare cell type in a heterogeneous cell population.
Materials and methods
Cells
K562 cell culture was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and grown according to protocol in RPMI 1640 (Sigma-Aldrich, St Louis, MO, USA) medium supplemented with 10% foetal bovine serum, 50 units/ml of penicillin, and 50 mg/ml of streptomycin. Partially purified lymphocytes were obtained by lymphoprep enrichment or sedimentation. Blood sample were obtained from male donors giving informed consent.
Slide preparation was performed by applying a monolayer of male lymphocytes onto a poly-lysine coated glass slide. Slides were then dried, after which one to five K562 cells were spiked onto marked locations on the glass slides. Slides were subsequently allowed to air dry. Following fixation using methanol and paraformaldehyde, fluorescence in situ hybridization (FISH) with colour swapping was performed, with probes specific for the human X and Y chromosomes, according to protocol [8]. Using this FISH protocol single K562 cells can be recognized by the presence of two X chromosome signals, in contrast to the lymphocytes, which were male cells.
Single chain variable region fragment antibodies
Tomlinson I and Tomlinson J repertoire were used as the source of scFv antibodies. http://www.geneservice.co.uk/products/proteomic/datasheets/tomlinsonIJ.pdf
Phage rescue was performed with the KM13 helper phage [9]. Soluble scFv were expressed from the pKBJ3 vector, according to protocol [10].
Shadow stick
Custom made glass pipette with a flat gold disc at the end were manufactured by Unisence, Aarhus, Denmark (Fig. 2). An angle of either 135° or 165° was introduced between gold disc and stick in order to allow the positioning of the disc on top of a cell using micromanipulation equipment. The diameters of the gold discs varied, ranging from 35 to 120 μm. For the handling of the shadow stick we used micromanipulation equipment from Narishige (Model MM-188, Nikon, Tokyo, Japan).
Fig 2.
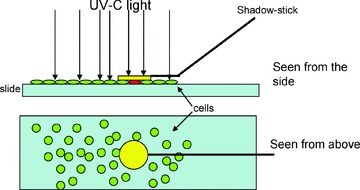
Cartoon illustrating the shadow stick and the UV-shielding of the target cell. Top shows slide and shadow stick viewed from the side, bottom shows slide and shadow stick viewed from the top. UV light is illustrated with arrows. The target cells (red) found underneath the gold disc (yellow) is protected from UV irradiation in contrast to non-target cells (green).
Selection
Initially a single K562 cell was located and a mark was made underneath the slide at the position of the cell in order to simplify the subsequent relocation. Slides were soaked in 1× PBS for 10 min. to loosen cover slide before removal. Slides were subsequently washed three times in 1× PBS, and then blocked in 2% weight/volume low fat milk powder in PBS (MPBS), for 1 hr. Slides were then washed three times in 1× PBS and incubated with phage scFv antibody library in 2% MPBS, 4°C with mild shaking over night. Input titre was approximately 1013; representing around 100 copies of each antibody displayed monovalent on the phage particles containing their antibody coding sequence.
The next day slides were washed three times with 1× PBS, followed by 5 × 3 min. with 1× PBS, with shaking. The final wash was made with 10% glycerol in 1× PBS. Slides were then allowed to dry, except for the area of interest which was kept soaked by application of a drop of final washing buffer. Individual XX cells (K562) were then relocated and the shadow stick placed over such a XX cell. The whole slide was then irradiated with UV light, wavelength 254 nm, for 5 min. with a hand held UV lamp, model UVSL-14P, UVP, Upland, CA, USA. The shadow stick was then removed and phage from the region of interest eluted with 50 μl (1 mg/ml) trypsin. Released phage were subsequently washed off with 4 × 200 μl 1× PBS. Twenty-five microlitres foetal calf serum was added to the eluted phage and the mixture was stored on ice for 30 min. before freezing at –18°C over night.
Phage infection
TG-1 culture OD600 0.5–0.7 was infected with half of the eluted phage for 30 min. at 37°C and another culture with the other half of the phage. The two cultures were grown from different colonies of the minimal plate, and grown independently. When performing selection on rare cells it is preferred to perform the two infections with half the eluate in different experiments to avoid losing all selected antibodies if the procedure failed. The infected TG-1 suspension was spun down and resuspended in an appropriate volume of medium and plated on TYE plates with ampicillin and glucose. Incubation at 30°C was performed over night in order to obtain colonies.
ELISA
ELISA was performed with monoclonal phage displaying the antibodies that had been selected by the microselection method. Single phage antibody clones were screened for binding to antigen in ELISA as described [11], except that phage rescuing was performed with KM13 helper phage. ELISA was carried out in ELISA plates (MAXI-sorp™, NUNC, Roskilde, Denmark). Coatings of the ELISA wells were performed with whole K562 cells or lymphocytes. Cells were added in 1× PBS, approximately 10,000 per well. Plates were centrifuged, after which the liquid was poured out by a rapid invert movement of the plates. Subsequently the plates were allowed to dry before usage or frozen at –18°C for later applications.
Pre-cleared culture supernatant from phage antibody clones grown and rescued monoclonally, were added together with MPBS, to a final concentration of 2% MPBS and a volume of 100 μl, and incubated for 2 hrs. The ELISA plates were washed and phage were detected with a 1:5000 dilution of anti-M13-HRP conjugated antibody (Pharmacia Biotech, Uppsala, Sweden). The ELISA was developed with OPD (o-phenylenediamine) tablets (DAKO, Denmark) according to the manufacturer’s instructions. In the case of ELISA using pKBJ derivatives of scFv antibodies these were detected with the anti-Myc tag antibody (9E10) and a HRP-conjugated rabbit antimouse antibody (DAKO, Glostrup, Denmark).
Western blot
K562 cells and lymphocytes were lysed in loading buffer, respectively, followed by SDS-PAGE. Membrane was stained with ponceau after blotting. Blot was washed and blocked for 1 hr with 2% MPBS, followed by washing with PBS twice. Western blot performed with phage, 5 × 1010 colony forming units (c.f.u.)/ml in 2% MPBS, 1 hr incubation with rotation. Blot was washed three times 5 min. in PBS supplemented with 0.05% tween and three times 5 min. in PBS with rotation. Phage were detected with a 1:5000 dilution of anti-M13-HRP conjugated antibody (Pharmacia). Blots were developed with ECL Plus Western Blotting Detection System (Amersham, GE Healthcare Bio-Sciences, Piscataway, NJ, USA).
Expression and purification of soluble scFv antibodies
Subcloning of antibody coding sequences was performed by double digest of the phagemid DNA with the restriction endonucleases NcoI and NotI (New England Biolabs, Beverly, MA, USA), according to the manufacturer’s protocol, followed by ligation of the insert into the pKBJ3 vector.
Soluble scFv antibodies were expressed from the pKJB3 vector and purified on Ni-NTA beads by His-Tag according to His-Tag protocol (by Qiagen, Valencia, CA, USA), with the exception that the washing buffer contained 30 mM and elution buffer 300 mM Imidazol, respectively.
Imidazol was removed by dialysis and biotinylation performed according to the manufacturer’s protocol, ECL™ Protein Biotinylation Module (Amersham). Direct coupling of fluorochromes were performed with ‘FluoroLink™-Ab Cy5 labelling kit’ (Amersham).
Cell staining
Initially the cells were applied onto the slide and allowed to dry. Permeabilization was performed by treatment with ice cold pure methanol, 10 min. at –20°C. Methanol was removed and slide washed two times with 1× PBS. Subsequently cells were fixed in 2% paraformaldehyde, at room temperature for 10 min. Finally slides were washed three times with PBS and used immediately or allowed to dry and then stored at –20°C until use.
Prior to antibody staining the slides were blocked 1 hr with 2% MPBS, and subsequently washed two times with 1× PBS. Purified conjugated soluble scFv were diluted to appropriate concentration in 2% MPBS and incubated on target cells for 1 hr with mild shaking. Slides were washed five times of 3 min. with 1× PBS. Mounting medium (Vectashield, Vector Laboratories, Burlingame, CA, USA) with DAPI (4′,6-diamidino-2-phenylindole) was applied and a cover glass mounted. Slides were subsequently analysed under microscope (Zeiss LSM510 Meta Confocal microscope and Zeiss fluorescence microscope, Oberkochen, Germany).
Results
Designing the method
For the development of the method the myelogenous leukaemia cell line K562 [12] were chosen. Initially attempts of isolating K562 cells were performed with laser microdissection pressure catapulting, but this method proved unsatisfactory, mainly due to drying of the slide, a step which is required in LCM, but which prevents efficient recovery of phage by host infection. Analysis of the drying effect was performed by selections on four slides treated different after washing and counting the c.f.u. thereby estimating the phage output titre (Fig. 1). It is seen that there is a severe loss of phage, when the slides are allowed to dry. Because there will be a very limited number of phage bound to a single cell, high recovery of phage is critical for successful selection; it is therefore important that the area containing the cell of interest is not allowed to dry. During the optimization it was furthermore found that the target cell was best kept in a buffer consisting of 10% glycerol in PBS.
Fig 1.
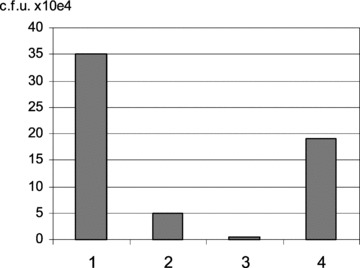
Drying effect of phage. Effect on phage particle recovery when drying the slide, measured as output titre. Mock selection performed with mild washing and elution from whole slide (approximately 106 cells) with input phage titre 1011 c.f.u. Output titre show as c.f.u. ×104 (i) Elution just before slide is dry (relative output of 100) (ii) Dried 10 min. at room temperature. (iii) Dried 20 min. at 37°C. (iv) Same as treatment (iii); however, last wash was performed with glycerol added to the PBS (10% glycerol in PBS). Output titre estimated from a 250-fold dilution of the eluate. A large decrease in output titre is observed as the time of drying is increased. The effect is however reduced when last wash is in buffer supplemented with glycerol, as an approximate 55-fold difference is observed between treatment 3 (4000 c.f.u.) and 4 (190,000 c.f.u.).
Because laser microdissection pressure catapulting proved unsatisfactory, we developed another approach, based on SURF. This technique was developed in connection with single cell PCR to ensure specific amplification of DNA from limited number of cells in a heterogeneous population. In SURF a small dot of UV impermeable ink was placed on top of cells of interest, preventing cross-linking of DNA in the target cells during UV irradiation. The UV irradiation will prevent replication of DNA from all cells exposed to the irradiation but spare the cells that have been shielded by the ink [13].
For the phage particle to be able to replication, the phage DNA must be capable to replicate in the host bacteria, as previously been shown by phage inactivation by UV irradiation [14]. Instead of removal of the target cell with bound phage, we therefore chose to use UV irradiation to in-activate the phage bound to cells that are not of interest in a manner similar to SURF. Initially we tested the ability of UV-irradiation to inactivate the phage particles, as their ability to replicate in the host bacteria. Irradiation of phage particles, 1011 c.f.u., for 30 sec. or less revealed little phage inactivation, whereas 60 sec. showed a phage inactivation of 65%, 90 sec. showed 97.5% and 120 sec. showed 99.7%. Multiple tests revealed that 300 sec. consistently resulted in 100% inactivation.
In the SURF method shielding of a single cell were obtained by placing ink on top. This, however, would be problematic in a phage selection method, because viable phage has to be retrieved and must not dry out. In addition the physical contact between the Ink and the phage might interfere with phage recovery. To solve this we used micromanipulation equipment to place different loose particles over the area to be shielded. However, due to difficulties in retaining the particle in position, this turned out not to be a reliable method. Subsequent attempts were performed coupling the shielding particle to the end of an injection pipette (35° bend, 7 μm tip). The resulting device was composed of a minute gold disc as the shielding particle, coupled to a custom made glass pipette for movement and positioning of the disc. The device was named ‘shadow stick’ (Fig. 2).
Initial attempt of shielding a target cell with the shadow stick was performed with discs of a diameter of 35 μm, which should be sufficient to cover a large cell. However, no active phage were obtained from these tests. We consider that lack of control of reflection from the UV source and light entering the protected area from the sides can be the cause of this unwanted inactivation, and we therefore increased the diameter of the disc to reduce this effect. The final working diameter of the disk was 100–120 μm, which ensured that the phage particles bound to the cell of interest would not be irradiated. Flowchart of the whole microselection process is illustrated in Fig. S1, and in the cartoon in Fig.S2.
Selections
Based on initial logistic problems, the whole procedure took place over several additional hours and locations, resulting in a problem maintaining the area of interest wet without introducing a background of non-specific phage. To keep the area of interest wet during transportation, different approaches were attempted, such as using EasiSeal (Thermo Scientific, Franklin Lakes, MA, USA). Consequently variable outputs of eluted phage particles were obtained (ranging from 0 to 400). Subsequent optimization of the method and improvements of the logistics in the lab yielded a steady outcome of 0–8 phage per single cell selection.
In total 37 individual selections were performed during the optimization procedure. From these selections a total 1536 phage antibodies were produced and tested monoclonally in ELISA against both K562 cells and lymphocytes. A problem in high throughput screenings of phage antibodies is a general variation and lower phage production in 96-well microtitre plates, compared to production in conical flasks. This most probably results from reduced oxygenation of the small volume in the 96-well microtitre plate, resulting in titres on the order of 1010 c.f.u./ml compared to 1012 c.f.u./ml when cultured in conical flasks. In the initial screen for positive clones this often results in far from optimal phage antibody concentration added to each ELISA well. At low phage antibody concentration the difference in signal strength between positive and negative wells can be marginal, thereby obscuring putative candidates. For a second screen only candidate’s recognizing K562 better than lymphocytes by a factor of more than 2.5 were chosen. The clones chosen for the second screen were cultivated in conical flask, thereby giving the possibility to add higher concentration of phage antibodies in the ELISA, resulting in a larger signal to noise ratio.
One of the K562 cells used for selection (selection #3.2) is shown in Fig. 3. From two of the single cell selections analysed in detail, three clones showed an increased recognition of the K562 cells compared to lymphocytes (Fig. 4). The three clones showed variability in the specificity level, so that clone S2.1A8 recognized K562 18.6 times higher than lymphocytes. Clones S3.1A9 45 times and clone S3.1B2 7.8 times, suggesting that different biomarkers are recognized.
Fig 3.
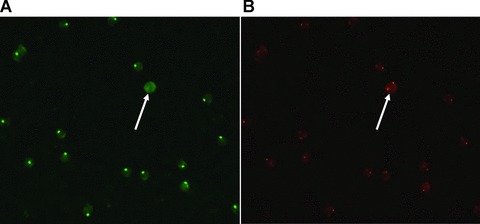
K562 cell (female origin) spiked onto slide containing male lymphocytes. Show Target cell of selection #3.2. (A) Shows the result of FISH using a Y-chromosome specific probe (green) and (B) shows the result of FISH using an X-chromosome specific probe (red). Hybridization was performed twice, reversing the colours of the chromosomes; the figure showing the last FISH. After the first hybridization Y- and X-chromosomes were red and green, respectively. Colours were reversed in a second hybridization (shown in the figure) to eliminate false positive cells.
Fig 4.
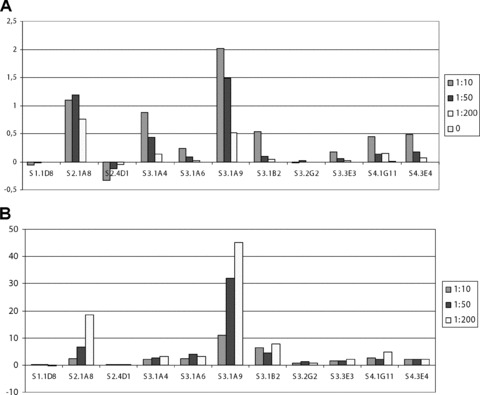
ELISA results. (A) OD signal from ELISA against K562 – OD signal from ELISA against Lymphocytes. (B) (K562-background)/(Lymp-background). Phage diluted from stock preparation, approximately 1013 phage titre. Background is ELISA signal against blank control (no cells coated). Zero is no phage added in the ELISA. Wells were coated with approximately 10,000 cells per well and treated with methanol and paraformaldehyde prior to blocking. ELISA made in duplicates. S1.1D8, S2.4D1 and S3.2G2 were chosen based on stronger or similar binding to lymphocytes compared to K562 in initial screening. S2.1A8, S3.1A9 and S3.1B2 were found to bind stronger to K562 compared to lymphocytes in a concentration dependent manner.
Immunostaining of cells
To further analyse the specificity of the selected antibodies and characterize the localization of cognate biomarkers, we tested the K652-specific antibodies by immunostaining on fixed cells. Purified scFv, from the pKBJ3 vector, were directly conjugated with biotin or fluorochrome and used for immunostaining of K562 and lymphocytes. Prior to immunostaining the scFv format was tested in ELISA towards K562 (Fig. S3). Stainings were performed on K562 and lymphocytes mixed together in a ratio of 1:3 (Fig. 5). Negative controls were performed without antibodies or with streptavidin conjugated with fluorochrome alone (data not shown). The three tested scFv antibodies all showed preferential staining of the K562 cells compared with the heterogeneous population of lymphocytes.
Fig 5.
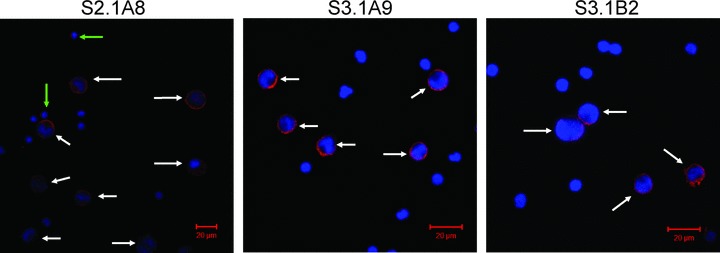
Immunostaining of mixture of K562 and lymphocytes. Cells were mixed in a ratio of 1:3, of K562 and lymphocytes, respectively. Stainings were performed with scFv’s directly conjugated with Cy5 at a concentration of 4 μg/ml (determined from Fig. S3). For all three antibodies it can be seen that they preferentially stain the K562 cells (marked by white arrows). Antibody S2.1A8 also has a staining of some of the lymphocytes, presumably a subpopulation (green arrows). Imaging and scale bar performed with AxioVision software.
Discussion
We wanted to use phage display for selection of antibodies against a single cell in a heterogeneous cell population, in particular for the selection of antibodies towards very rare cells, such as non-blood CTCs and foetal cells from the maternal bloodstream. For foetal cells in the maternal blood stream, FISH can be used as an unrelated way of identification in situations where the mother is expecting a male foetus, making it possible to distinguish the foetal cells by the presence of the Y-chromosome [15]. Identification of CTCs by FISH could be performed by demonstration of cancer cell aneuploidies [16] as altered karyotyping is observed in various cancers [17]. FISH methods are widely used methods that can be made more accurate by reversing the colours of the chromosome specific probes [18] or by using multiple probes from the same chromosome [19].
To develop the method we have taken advantage of a model cell system which allows us easy access to multiple cells similar to the single target cell. This allowed us to perform tradition ELISA using the selected antibodies by coating approximately 10,000 cells per well, which would not have been possible in the case of very rare cells. To resemble a very rare cell in the blood, we chose the myelogenous leukaemia cell line K562 [12], as our target cell and lymphocytes as the mixture in which to place the target cell. Because the K562 originates from a female donor, lymphocytes were used from a male donor. FISH were performed to distinguish the K562 cells from lymphocytes, using X and Y chromosome specific probes. Here we chose to reverse the colours of the X- and Y- chromosomes, thereby imitating an established identification procedure for very rare cells in the peripheral blood.
The success of single cell selection depend to a large extend on the ability to reduce background of non-specially bound phage particles and to deplete phage antibodies binding to common biomarkers. Here we take advantage of UV irradiation and the protease cleavable helper phage KM13 [9] to achieve a reduction of the selection background. The KM13 helper phage introduces a trypsin cleavage site between domain 2 and 3 of coat protein 3 (the protein responsible for host infection). By cleaving of the helper phage introduced protein 3 the background of non-displaying phage is removed and only displaying phage are capable of infection of the host bacteria.
After the logistic problems of the selection method had been solved, the average outcome of the single cell selections was approximately three phage particles per selection. From these later selection 16% of the selected antibodies had more than three times higher ELISA signal against K562 than of lymphocytes; 24% with more than 2.5 times higher signal.
The amount of K562 recognizing antibodies from the optimized single cell selections correspond well with the amount obtained from other selections performed with the KM13 helper phage; namely 10–25% depending on the library type and quality [20, 21]. The phage outcome of one to eight antibodies after the complete development of the selection method is a realistic quantity because the background of non-displaying phage is eliminated due to KM13 helper phage. Without phage rescue with a helper phage in order to lower the background of non-displaying phage, single cell selection would most likely not be feasible.
In addition most antibodies will bind antigens common for multiple cell types, because only few biomarkers will be unique for any given cell type. The excess of 106 times lymphocytes compared to K562 on the slide will most likely give a higher percentage of specific antibodies, compared to a selection performed on K562 alone. The ratio of 1:106 means than most antibodies recognizing non-specific proteins will bind the lymphocytes, known as depletion.
The three scFv antibodies, S2.1A8, S3.1A9 and S3.1B2, all stained the K562 cells in different ways and therefore recognized biomarkers must be localized differently; the two antibodies S3.1A9 and S3.1B2 localizes mainly at the membrane and S2.1A8 more in the cytoplasm. Sequencing of the clones was carried out and confirmed their uniqueness. Western blots were performed with the selected antibodies and revealed that S.31A9 recognize a band in K562 cell extract of approximately 100 kD, but none in lymphocyte extract (Fig. S4). Clone S.2.1A8, also recognized K562-derived proteins in Western blots, staining a large molecular size smear of bands (data not shown). In general selections performed on whole cells yield binders of membrane proteins. It is, however, possible to select antibodies on whole cells towards intracellular proteins also on whole cells [20]. In the present set-up the cells have been subjected to a harsh treatment in relation to the FISH, and are no longer intact, most likely increasing the likelihood of raising antibodies against intracellular epitopes. Clone S.2.1A8 could be recognizing a post-translational modification, which is more prevalent in K562 cells compared to lymphocytes. Furthermore S2.1A8 revealed an additional weak staining in a fraction of the lymphocytes. Antibody S3.1A9 exhibited the highest specificity towards the K562 cells in the ELISA dilution series (Fig. 4). In the cell staining it also produced the most specific staining of the K562 cells. Therefore antibody S3.1A9 is the antibody with the highest specificity towards K562 from the single cell selections. This confirms that the method is capable of selecting antibodies against biomarkers specific for very rare cells in a heterogeneous cell population.
Conclusion
A model system has been developed, in which we have selected specific antibodies from single cells in a heterogeneous cell population. This was achieved by a novel selection method where a minute disc shields the phage bound to cell of interest, while UV irradiation inactivates the phage particles binding to cells outside of the area covered by the disc. Essential for the success of the method is the ability to perform selection in a single round; this is achieved by using a protease sensitive helper phage, which is capable of reducing the background of bound non-display phage. Further we established that it is essential that the cell of interest is prevented from drying out during the procedure.
After optimization, up to eight phage could be achieved from each of the microselections, with an average of three phage per selection. Of selected antibodies around 25% exhibits an increased binding to K562 compared to lymphocytes.
The difference between input titre (1013 c.f.u.) and the average output titre of 3 is much larger than during traditional selections, where the input typically would be 1012 c.f.u. (100 copies of each antibody when the diversity of library is ∼108 and the display level is between 1% and 10%) and the output (KM13 helper phage rescued phage) commonly lies between 103–105 depending on the selection method and stringency of washing. The strength of the selection method is this low output of phage only from the cell of interest.
From 37 selections performed with the novel method; three highly K562 specific antibodies were obtained and further analysed. The three unique clones were tested for specificity both in ELISA, Western blot, and immunostaining. Clone S3.1A9 showed the highest specificity, both in ELISA (45 times higher signal towards K562 than lymphocytes) and Western blot (one unique band of ∼100 kD, Fig. S4). All three antibodies stained K562 cells in immunostaining, showing different localization and indicating different specificities.
One important notion is that selections were performed on cells which had been previously subjected to double FISH analysis. This clearly points to the fact that by careful preparation of the cells, it is possible to keep some of the proteins intact. Furthermore the subsequent staining of K562 and lymphocytes were performed on cells which had only been fixed, thus indicating that it is possible to isolate antibodies recognized epitopes which are preserved during different treatments.
To our knowledge this is the first time successful single cell selections have been reported. Our technique offers a unique method for obtaining specific antibodies against a broad range of rare cells present in a heterogeneous population. The finding of specific biomarkers for such rare cells has many important implications and opens new doors within fields such as regenerative medicine, cancer research and non-invasive prenatal diagnostics.
Especially in cancer and stem cell research specific biomarkers are often lacking; identification of such cells relies on the use of a mixture of antibodies which together can identify the target cell. The method described here offers a possibility of raising antibodies towards the cell type of interest as long as it can be identified under a microscope by morphology and/or staining. In this manner new biological pathways can be identified, revealing more of the biological function of the cells, through identification of novel biomarker.
Following isolation of such antibodies, they can be applied for identification and enrichments. Recently it has been shown that microfluidic devices now are capable of extracting extremely rare cells from whole blood without any processing prior to passing the blood through the microfluidic chip [22]. The combination of single cell selection of antibodies together with microfluidic devices offer a unique mean of obtaining rare cells from blood, for the purpose of diagnosis.
Acknowledgments
This work has been supported by FCMB Aps, The Danish Agency for Science, Technology and Innovation and the FP6 funded Proteomage project (LSH-518230).
Supporting Information
Fig. S1 Flowchart of the microselection. Initially biopanning is performed, by incubation of the phage library displaying the antibody fragments with the slide containing the target cell. Phage diversity illustrated with different colours of the phage. Cells on the slide are shown in green. Excess phage are washed away and the slide is placed in a microscope where the target cell (XY) is relocated; here illustrated by a red circle (target cell identified by FISH; because the target cell of this selection is male, confirmed by the presence of a Y-chromosome specific signal, green dot). A natural case of such target cell would be a foetal cell in the maternal blood of a woman carrying a male foetus. Shadow stick is placed on top of the target cell by micromanipulation equipment, and the slide is irradiated by UV-C, inactivating all phage not shielded by the shadow stick (detailed illustration in Fig. S2). Target cell (illustrated as red) is protected by the shadow stick. Bound phage are eluted and the viable phage are propagated monoclonally for antibody production. Subsequently the antibodies selected by the microselection are tested for specificity by methods such as ELISA and immunostaining.
Fig. S2 Cartoon of the UV irradiation process in the microselection, visualized as observed through the microscope. The cells are illustrated after FISH, with X-chromosomes as red dots, and Y-chromosome as a green dot. The nucleus is visualized by DAPI staining (blue). First the target cell with the XY signal (in the red circle) with the bound phage is identified. The shadow stick is then placed above the target cell, and the slide irradiated by UV-C (purple transparent colour). The shadow stick is subsequently removed, leaving only the shielded phage viable. Illustration with relative proportions.
Fig. S3 ELISA results from with scFv antibodies againstK562 cells. The three antibodies S2.1A8, S3.1A9 and S3.1B2 weretested in ELISA as soluble scFv antibodies from the pKBJ3 vector todetermine concentration for immunostainings. The bars showdecreasing concentration of antibodies. The value 0 is without scFvantibody; only secondary HRP conjugated anti-His. Signal towardslymphocytes with 20 μg/ml shown as negative control.Signals shown relatively to 100 μg/ml antibody towardsK562.
Fig. S4 Western blot with three of the selected clones.The three antibodies S2.1A8, S3.1A9 and S3.1B2 together with acontrol antibody E known not to bind K562 or lymphocytes wereapplied in Western blotting. (A) is ponsceau staining of theblot (B) Western blot using anti-M13 HRP as secondaryantibody. The lanes are marked M for marker, K for K562 cellextract and L for lymphocytes cell extract. A non-specific band isobserved in the lanes of the marker M. Although S2.1A8 recognized asmear of bands on the K562 cell line and S3.1A9 recognized a highMW band in the K562 cell line, the antibody 3.1B2 did not show anyrecognition in Western blot.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Ross AA, Cooper BW, Lazarus HM, et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood. 1993;82:2605–10. [PubMed] [Google Scholar]
- 2.Bischoff FZ, Marquez-Do DA, Martinez DI, et al. Intact fetal cell isolation from maternal blood: improved isolation using a simple whole blood progenitor cell enrichment approach (RosetteSep) Clin Genet. 2003;63:483–9. doi: 10.1034/j.1399-0004.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 3.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 4.Hoogenboom HR. Designing and optimizing library selection strategies for generating high-affinity antibodies. Trends Biotechnol. 1997;15:62–70. doi: 10.1016/S0167-7799(97)84205-9. [DOI] [PubMed] [Google Scholar]
- 5.Lu H, Jin D, Kapila YL. Application of laser capture microdissection to phage display peptide library screening. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:692–7. doi: 10.1016/j.tripleo.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Ruan W, Sassoon A, An F, et al. Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection. Mol Cell Proteomics. 2006;5:2364–73. doi: 10.1074/mcp.M600246-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.De Kruif J, Terstappen L, Boel E, et al. Rapid selection of cell subpopulation-specific human monoclonal antibodies from a synthetic phage antibody library. Proc Natl Acad Sci USA. 1995;92:3938–42. doi: 10.1073/pnas.92.9.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen B, Philip J, Kolvraa S, et al. Fetal cells in maternal blood: a comparison of methods for cell isolation and identification. Fetal Diagn Ther. 2005;20:106–12. doi: 10.1159/000082432. [DOI] [PubMed] [Google Scholar]
- 9.Kristensen P, Winter G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des. 1998;3:321–8. doi: 10.1016/S1359-0278(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 10.Jensen KB, Larsen M, Pedersen JS, et al. Functional improvement of antibody fragments using a novel phage coat protein III fusion system. Biochem Biophys Res Commun. 2002;298:566–73. doi: 10.1016/s0006-291x(02)02484-1. [DOI] [PubMed] [Google Scholar]
- 11.Marks JD, Hoogenboom HR, Bonnert TP, et al. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 12.Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45:321–34. [PubMed] [Google Scholar]
- 13.Shibata D, Hawes D, Li ZH, et al. Specific genetic analysis of microscopic tissue after selective ultraviolet radiation fractionation and the polymerase chain reaction. Am J Pathol. 1992;141:539–43. [PMC free article] [PubMed] [Google Scholar]
- 14.Kurosaki Y, Abe H, Morioka H, et al. Pyrimidine dimer formation and oxidative damage in M13 bacteriophage inactivation by ultraviolet C irradiation. Photochem Photobiol. 2003;78:349–54. doi: 10.1562/0031-8655(2003)078<0349:pdfaod>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 15.Walknowska J, Conte FA, Grumbach MM. Practical and theoretical implications of fetal-maternal lymphocyte transfer. Lancet. 1969;1:1119–22. doi: 10.1016/s0140-6736(69)91642-0. [DOI] [PubMed] [Google Scholar]
- 16.Fehm T, Sagalowsky A, Clifford E, et al. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res. 2002;8:2073–84. [PubMed] [Google Scholar]
- 17.Duesberg P, Li R, Fabarius A, et al. Aneuploidy and cancer: from correlation to causation. Contrib Microbiol. 2006;13:16–44. doi: 10.1159/000092963. [DOI] [PubMed] [Google Scholar]
- 18.Krabchi K, Gros-Louis F, Yan J, et al. Quantification of all fetal nucleated cells in maternal blood between the 18th and 22nd weeks of pregnancy using molecular cytogenetic techniques. Clin Genet. 2001;60:145–50. doi: 10.1034/j.1399-0004.2001.600209.x. [DOI] [PubMed] [Google Scholar]
- 19.Kilpatrick MW, Tafas T, Evans MI, et al. Automated detection of rare fetal cells in maternal blood: eliminating the false-positive XY signals in XX pregnancies. Am J Obstet Gynecol. 2004;190:1571–8. doi: 10.1016/j.ajog.2004.03.055. [DOI] [PubMed] [Google Scholar]
- 20.Jensen KB, Jensen ON, Ravn P, et al. Identification of keratinocyte-specific markers using phage display and mass spectrometry. Mol Cell Proteomics. 2003;2:61–9. doi: 10.1074/mcp.M200049-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Schofield DJ, Pope AR, Clementel V, et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007;8:R254. doi: 10.1186/gb-2007-8-11-r254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagrath S, Sequist LV, Maheswaran S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–9. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Flowchart of the microselection. Initially biopanning is performed, by incubation of the phage library displaying the antibody fragments with the slide containing the target cell. Phage diversity illustrated with different colours of the phage. Cells on the slide are shown in green. Excess phage are washed away and the slide is placed in a microscope where the target cell (XY) is relocated; here illustrated by a red circle (target cell identified by FISH; because the target cell of this selection is male, confirmed by the presence of a Y-chromosome specific signal, green dot). A natural case of such target cell would be a foetal cell in the maternal blood of a woman carrying a male foetus. Shadow stick is placed on top of the target cell by micromanipulation equipment, and the slide is irradiated by UV-C, inactivating all phage not shielded by the shadow stick (detailed illustration in Fig. S2). Target cell (illustrated as red) is protected by the shadow stick. Bound phage are eluted and the viable phage are propagated monoclonally for antibody production. Subsequently the antibodies selected by the microselection are tested for specificity by methods such as ELISA and immunostaining.
Fig. S2 Cartoon of the UV irradiation process in the microselection, visualized as observed through the microscope. The cells are illustrated after FISH, with X-chromosomes as red dots, and Y-chromosome as a green dot. The nucleus is visualized by DAPI staining (blue). First the target cell with the XY signal (in the red circle) with the bound phage is identified. The shadow stick is then placed above the target cell, and the slide irradiated by UV-C (purple transparent colour). The shadow stick is subsequently removed, leaving only the shielded phage viable. Illustration with relative proportions.
Fig. S3 ELISA results from with scFv antibodies againstK562 cells. The three antibodies S2.1A8, S3.1A9 and S3.1B2 weretested in ELISA as soluble scFv antibodies from the pKBJ3 vector todetermine concentration for immunostainings. The bars showdecreasing concentration of antibodies. The value 0 is without scFvantibody; only secondary HRP conjugated anti-His. Signal towardslymphocytes with 20 μg/ml shown as negative control.Signals shown relatively to 100 μg/ml antibody towardsK562.
Fig. S4 Western blot with three of the selected clones.The three antibodies S2.1A8, S3.1A9 and S3.1B2 together with acontrol antibody E known not to bind K562 or lymphocytes wereapplied in Western blotting. (A) is ponsceau staining of theblot (B) Western blot using anti-M13 HRP as secondaryantibody. The lanes are marked M for marker, K for K562 cellextract and L for lymphocytes cell extract. A non-specific band isobserved in the lanes of the marker M. Although S2.1A8 recognized asmear of bands on the K562 cell line and S3.1A9 recognized a highMW band in the K562 cell line, the antibody 3.1B2 did not show anyrecognition in Western blot.