

Abstract
The bacterium Helicobacter pylori causes peptic ulcers and gastric cancer in human beings by mechanisms yet not fully understood. H. pylori produces urease which neutralizes the acidic medium permitting its survival in the stomach. We have previously shown that ureases from jackbean, soybean or Bacillus pasteurii induce blood platelet aggregation independently of their enzyme activity by a pathway requiring platelet secretion, activation of calcium channels and lipoxygenase-derived eicosanoids. We investigated whether H. pylori urease displays platelet-activating properties and defined biochemical pathways involved in this phenomenon. For that the effects of purified recombinant H. pylori urease (HPU) added to rabbit platelets were assessed turbidimetrically. ATP secretion and production of lipoxygenase metabolites by activated platelets were measured. Fluorescein-labelled HPU bound to platelets but not to erythrocytes. HPU induced aggregation of rabbit platelets (ED50 0.28 μM) accompanied by ATP secretion. No correlation was found between platelet activation and ureolytic activity of HPU. Platelet aggregation was blocked by esculetin (12-lipoxygenase inhibitor) and enhanced ∼3-fold by indomethacin (cyclooxygenase inhibitor). A metabolite of 12-lipoxygenase was produced by platelets exposed to HPU. Platelet responses to HPU did not involve platelet-activating factor, but required activation of verapamil-inhibitable calcium channels. Our data show that purified H. pylori urease activates blood platelets at submicromolar concentrations. This property seems to be common to ureases regardless of their source (plant or bacteria) or quaternary structure (single, di- or tri-chain proteins). These properties of HPU could play an important role in pathogenesis of gastrointestinal and associated cardiovascular diseases caused by H. pylori.
Keywords: Helicobacter pylori, urease, platelet activation, eicosanoids, lipoxygenase
Introduction
Ureases (EC 3.5.1.5) are highly homologous nickel-dependent enzymes widespread among plants, bacteria and fungi, that hydrolyse urea into ammonia and carbon dioxide [1, 2]. Plant and fungal ureases are homotrimers or hexamers of a ∼90 kD subunit, while bacterial ureases are multimers of two or three subunits complexes [3–4]. The N-terminal halves of plant or fungal urease single chain align with the primary sequence of the small subunits of most bacterial enzymes (e.g.β and γ chains of Bacillus pasteurii urease or the A subunit of Helicobacter pylori urease). The C-terminal portions of plant and fungal chains resemble the large subunits of bacterial ureases (e.g.α chain of B. pasteurii urease or the B subunit of H. pylori enzyme). Considering the similarity in their sequences, all ureases are likely to possess similar tertiary structures and catalytic mechanisms indicating they are variants of the same ancestral protein [2]. H. pylori urease (1E9Z) and jackbean (Canavalia ensiformis) major urease (P07374), share about 50% identity despite differences in their quaternary structures. The 3D crystallographic structures of three bacterial ureases were successfully resolved: Klebsiella aerogenes (1FWJ), B. pasteurii (4UBP) and H. pylori (1E9Z).
Urease activity enables bacteria to use urea as a sole nitrogen source [2, 5]. Some bacterial ureases play an important role in the pathogenesis of human and animal diseases such as those from H. pylori or Proteus mirabilis. H. pylori, a Gram-negative bacterium that colonizes the human stomach mucosa, causes gastric ulcers and gastric adenocarcinoma by mechanisms not completely understood [6, 7]. This bacterium produces factors that damage gastric epithelial cells, such as the vacuolating cytotoxin VacA, the cytotoxin-associated protein CagA, and a urease (up to 10% of bacterial protein) that neutralizes the acidic medium permitting its survival in the stomach. The gastroduodenal illness induced by H. pylori depends on the host inflammatory response elicited by the several virulence factors produced by the microorganism. There are reports showing that H. pylori whole cells or extracts of its water-soluble proteins promote inflammation, activate neutrophils and release cytokines. The biology of H. pylori and its involvement in stomach illness were recently reviewed [2, 8–10].
The physiological role of urease in plants is still largely unknown despite its ubiquity in virtually all plants [3, 4]. Jackbean and soybean ureases display fungicidal [11] and insecticidal activity, suggestive of a role in plant defence [12, 13]. The insecticidal activity is due to a ∼10 kD internal peptide released from plant ureases upon digestion by insect cathepsins [14, 15].
We have previously reported that canatoxin [16], an isoform of jackbean (C. ensiformis) urease [17], presents biological properties that are independent of its enzyme activity, as binding to sialylated glycoconjugates, activation of blood platelets [18–20] and pro-inflammatory effect [21]. Submicromolar concentrations of canatoxin-induced exocytosis in a number of cell system in vitro including platelets, synaptosomes, pancreatic islets, macrophages, neutrophils and mast cells [19, 22]. Canatoxin also induced hypothermia, bradycardia, hypoglycaemia, hyperinsulinemia, hypoxia and paw oedema in rats and mice, preceding convulsions and death of the animals [23]. Canatoxin disrupted Ca2+ transport across membranes [20, 24] and lipoxygenase metabolites were shown to modulate most of its pharmacological effects [18, 19, 21] either in vivo or in vitro. More recently we reported that jackbean, soybean and B. pasteurii ureases are also able to induce aggregation of platelets at nanomolar concentrations independently of enzyme activity [13, 25].
Blood platelets are anucleated disc-shaped cells derived from megakaryocytes. Upon vascular injury or exposition to agonists such as adenosine diphosphate (ADP), collagen or thrombin, non-stimulated platelets become spherical (shape change) and adherent to each other and to surrounding tissues [26, 27]. Stimulated platelets may undergo release reaction, with exocytosis of α-granules and dense granules, whose contents contribute to haemostasis. Primary reversible platelet aggregation induced by direct agonists such as ADP, platelet-activating factor (PAF)-acether or thromboxane A2 does not require the release reaction. When platelets secrete ADP it amplifies the aggregation response [26, 28]. Elevated intracellular levels of Ca2+ are necessary for platelet aggregation and secretion resulting from external Ca2+ influx through voltage-dependent channels, inhibition of Ca2+ ATPases and/or release of intracellular Ca2+ pools by the action of phosphatidylinositol-triphosphate [26–30].
Platelet membrane-bound phospholipase A2 activated by agonist-coupled receptors hydrolyses phospholipids into free arachidonic acid, which serves as substrate for the synthesis of eicosanoids either resulting from the cyclooxygenase pathway, such as thromboxane A2, or the lipoxygenase pathway, such as 12-hydroxyperoxy-eicosatetranoic acid (12-HPETE), which in turn mediate platelet’s response to the agonist [31, 32]. Platelets also synthesize PAF-acether (1-o-alkyl-2-acetyl-sn-glycero-3-phosphocholine) from arachidonic acid which interacts with its own receptors on platelets [33].
In the present work we characterized the platelet aggregating activity of a purified recombinant H. pylori urease (HPU), studied the pathways recruited by the protein to induce platelet activation and compared the data to that previously reported for the plant urease canatoxin and for Bacillus pasteurii urease.
Materials and methods
Materials
The following drugs were obtained from Sigma Chemical Co., St Louis, MO, USA: reagents for electrophoresis, ADP, potato apyrase (A-6535, a kind gift from Dr. Ana Maria O. Batastini, Department of Biochemistry, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil), esculetin, dexamethasone, indomethacin, bovine acid soluble collagen type I, luciferin and luciferase. PAF-acether (platelet-aggregating factor: 1-O-alkyl-2-acetyl-sn-glycero-phosphocholine) and Web 2170 (Bepafant; 5-(2-chloro-phenyl)-3,4-dihydro-10-methyl-3-[(4-morpholinyl) carbonyl]-2H, 7H-cyclopenta (4,5) thieno [3,2-f][1,2,4 triazolo-[4,3-a][1, 4] diazepine]) were a kind gift from Dr. João Baptista Calixto, Department of Pharmacology, Universidade Federal de Santa Catarina, Florianópolis, SC, Brazil. D-methoxy-verapamil (Verapamil hydrochloride) was from Sandoz Laboratory (Saluta Pharma GmbH, Germany). Solutions were prepared as follows: dexamethasone and esculetin were dissolved in absolute ethanol and diluted in saline to give final concentrations of ethanol in the platelet assay of no more than 0.2% v/v; indomethacin was first dissolved in 0.1 M Na2CO3 then diluted with saline and adjusted to pH 6.0; ADP was diluted in Tris buffer pH 8.2; PAF-acether was dissolved in 0.1 w/v% bovine serum albumin solution and used on the same day; Web 2170 and verapamil were dissolved in saline; apyrase was dissolved in phosphate buffered saline.
Helicobacter pylori recombinant urease
HPU was produced by heterologous expression in Escherichia coli SE5000 transformed with plasmid pHP8080 [34], kindly provided by Dr. Harry T Mobley, University of Michigan Medical School. HPU was purified from bacterial extracts as follows: after cultivation, cells were harvested by centrifugation, suspended in 20 mM sodium phosphate, pH 7.5 containing 1 mM ethylenediaminetetraacetic acid (EDTA), 5 mM 2-mercaptoethanol (extracting buffer, EB) and lysed using a Ultrasonic Homogenizer 4710, 10 pulses of 30 sec. in an ice bath. After centrifugation (20 min., 20,000 ×g, 4°C), the supernatant was fractionated by ammonium sulphate precipitation. The precipitate formed between 0.3 and 0.7 saturation was dissolved in EB and dialysed to remove the excess of salt. This material was then submitted to anion exchange chromatography in Q-Sepharose (GE Healthcare, Uppsala, Sweden) at a ratio of 10 mg protein per 1 ml resin equilibrated in EB, pH 7.8. After removing the unbound proteins, the column was eluted stepwise and the urease-enriched fraction was recovered with EB containing 200 mM NaCl, pH 7.8. After dialysis to remove excess of salt and concentration on Centriprep (Millipore, Bedford, MA, USA) cartridges, the material was applied into a size exclusion Superose 6 HR column equilibrated in EB pH 7.8, mounted on a FPLC apparatus, at a flux of 0.3 ml/min. Figure 1 illustrates the elution pattern of purified HPU, with all fractions within the peak showing similar specific activity with a mean value of 264.4 ± 11.4 U/mg of protein. SDS-PAGE of purified HPU showed two major bands of 60 and 30 kD (not shown). The fractions with urease activity were pooled and freeze-dried (in EB buffer). For the experiments, the freeze-dried protein was solubilized to give 0.5 mg protein/ml solution in 20 mM sodium phosphate, pH 7.5, containing 1 mM EDTA and 5 mM 2-mercaptoethanol.
Fig 1.
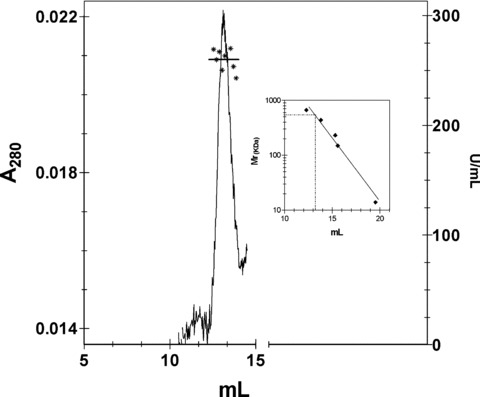
Purification of HPU. The figure shows the chromatographic pattern of HPU obtained in the last purification step consisting of a gel-filtration in a Superose 6 HR column. Fractions within the peak eluted at 13.2 ml all showed similar values of urease activity (asterisks), mean value of 264.4 ± 11.4 U/mg of protein, denoting the homogeneity and high purity of the enzyme. The inset shows the calibration curve of the Superose 6 HR column (Mr standard: tyreoglobulin 669 kD, ferritin 440 kD, catalase 232 kD, alcohol dehydrogenase 150 kD; cytochrome C 12.8 kD). A molecular mass of 540 kD was estimated for HPU.
For fluorescence microscopy experiments, HPU was labelled with fluorescein isothiocyanate (FITC, Sigma Aldrich, St Louis, MO, USA). HPU solutions (1 mg/ml) in EB buffer were incubated with 0.1% FITC for 2 hrs at 4°C. The mixture was exhaustively dialysed against EB buffer and then applied into a Fast-Desalting column (Amersham Biosciences, Uppsala, Sweden) to remove any unbound FITC.
Protein determination
The protein content of samples was determined by their absorbance at 280 nm or by the Coomassie dye binding method.
Urease activity
The ammonia released was measured colorimetrically by the alkaline nitroprussiate method [35]. One unit of urease releases one μmol of ammonia per minute, at 37°C, pH 7.5.
Platelet aggregation
Platelet-rich plasma (PRP) was prepared from rabbit blood collected from the ear central artery in the presence of sodium citrate to a final concentration of 0.313% (v/v). Blood samples were centrifuged at 200 ×g for 20 min. at room temperature, to give a PRP suspension. Platelet aggregation and shape change were monitored turbidimetrically as described [18], using a Lummi-Aggregometer (Chrono-Log Corporation, Havertown, PA, USA). Aggregation resulted in an increase in light transmission across PRP, registered on a chart recorder for 3 min. Platelet aggregation assays were also performed on a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA, USA) as described [36]. Briefly, urease samples in 96-wells flat-bottomed plates were completed to a final volume of 50 μl with saline. Aggregation was triggered by the addition of 100 μl of platelet suspension. The plate was incubated for 2 min. at 37°C before beginning of agitation and readings were performed at 650 nm every 11 sec., during 20 min. When testing potential inhibitors, platelets and the compounds were pre-incubated in the microplate wells for 2 min. at 37°C under stirring, or 10 min. at room temperature without stirring in the case of apyrase, and aggregation was triggered by addition of HPU or control inducer (ADP). Change in turbidity was measured in absorbance units, and results are expressed as area under the aggregation curves.
Fluorescence and scanning electron microscopy
Sample preparation for scanning electron microscopy was done using PRP samples pre-warmed at 37°C and then exposed to saline or HPU under low stirring for 2 min. Platelets were then fixed by adding glutaraldehyde in 0.1 M sodium cacodylate pH 7.2 to a final 2.5% concentration and incubated overnight at 4°C. The samples were washed twice for 30 min. in 0.1 M sodium cacodylate and filtered on 0.4 μm polycarbonate membranes (Millipore). The fixed platelets were sequentially dehydrated in 30%, 50%, 70% and 90% (v/v) acetone, for 5 min. each, and finally twice in 100% acetone for 10 min. Critical-point drying and gold coating treatments were performed at our University’s Center of Electron Microscopy (CEM-UFRGS, Brazil). Specimens were visualized in a JEOL-JSM 6060 scanning electron microscope with automated image digitalization and archiving.
For fluorescence microscopy rabbit PRP was incubated with 300 nM FITC-labelled HPU under vortex stirring for 5 min. at room temperature. PRP without exposition to HPU was submitted to the same stirring and filtering process and used as negative control. Platelet aggregates were recovered by centrifugation; the pellets were smeared on glass slides and observed under a Zeiss-Axiovert-200 (Carl Zeiss, Jena, Germany) fluorescence microscopy.
Platelet secretion
ATP release from PRP suspension was measured as a change in bioluminescence in the presence of the Chromolume® reagent containing a luciferin-luciferase mixture (Chrono-Log Corporation) according to manufacturer instructions.
12-HETE detection
The method described by Coffey and co-authors [37] was applied to investigate the activation of 12-lipoxigenase in HPU-aggregated platelets. Rabbit platelet rich-plasma suspensions (2 ml) were exposed to 300 nM HPU for 2 min. at room temperature under stirring. Platelet aggregates were harvested by centrifugation (900 ×g, 15 min.), washed three times in cold phosphate buffered saline and then lysed by suspending in 200 μl of cold 100% methanol, followed by centrifugation at 10,000 ×g for 10 min. The supernatant was collected and applied into a C-18 column (CLC-ODS(M), Shimadzu, Kyoto, Japan) mounted in a high-performance liquid chromatography system (Shimadzu). The column was previously equilibrated in 50% of solvent A (water-acetonitrile-acetic acid; 75:25:0.1 proportion) and 50% of solvent B (methanol-acetonitrile-acetic acid; 60:40:0.1 proportion). Elution consisted of a 50% to 90% gradient of solvent B in 20 min., at a flow rate of 1 ml/min., monitored at 235 nm. Authentic 12-hydroxide-eicosatetraenoic acid (12-HETE; Cayman Biochemicals, Ann Arbor, MI, USA) was dissolved in 100% methanol and submitted to chromatography under the same conditions.
Statistical analysis
Data were analysed by ANOVA followed by the Tukey–Kramer test using the Instat Graph Pad software and values of P < 0.05 were considered statistically significant.
Results
Purified H. pylori urease activates platelets
Figure 2 illustrates the pattern of aggregation of platelet-rich rabbit platelet suspensions triggered by purified H. pylori urease (HPU) and two physiological agonists, ADP and collagen. HPU induced aggregation of rabbit platelets with an ED50 of ca. 150 μg/ml (0.28 μM), with a time course and collagen-type shape change reaction very similar to those induced by canatoxin or the major jackbean urease (ED50 15.8 μg/ml) [13, 17, 18]. Scanning electron microscopy of platelets exposed to HPU showed a typical morphology of activated platelets with numerous pseudopodia (Fig. 3A). The cells clumped together (Fig. 3C) with no evidence of cell lysis and fluorescein-conjugated H. pylori urease were seen bound to platelets aggregates but not to erythrocytes (Fig. 3D).
Fig 2.
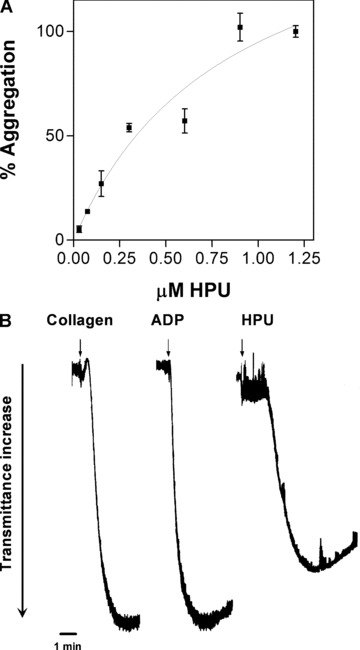
Aggregation of rabbit PRP suspensions induced by purified HPU. (A) Rabbit platetet-rich plasma suspension in microwell plates were exposed to increasing concentrations of HPU or 5 μM ADP (100% aggregation). Aggregation of platelets was monitored every 11 sec. during 20 min. in a SpectraMax plate reader. Results (means ± S.D.) are expressed as percentage of maximal aggregation for four replicates. (B) Comparison of aggregation tracings of platelets stimulated by collagen (30 μg/ml), ADP (5 μM) or HPU (0.3 μM). The arrows indicate addition of the agonists to the platelet suspension.
Fig 3.
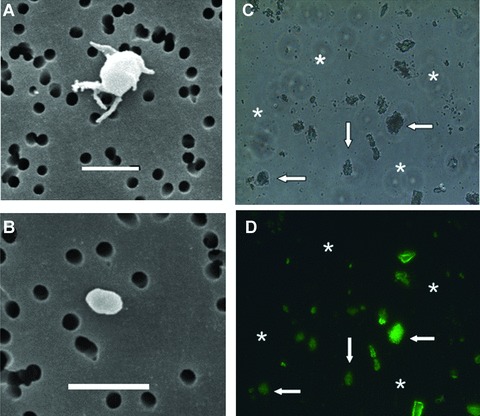
Scanning electronic, light and fluorescence microscopy of platelets treated with HPU. (A) Scanning electronic microscopy of a single rabbit platelet after exposition to 0.3 μM HPU for 2 min. under low stirring (to avoid aggregation). The morphology is typical of an activated platelet showing multiple pseudopodia. (B) Scanning electronic microscopy of a resting platelet. The white bars in (A) and (B) correspond to 2 μm. In (C) and (D) rabbit platelets were aggregated by 0.3 μM fluorescein-labelled HPU and aggregates (arrows) were observed under light (B) and fluorescence microscopy (C) at 400× magnification. Note that the few erythrocytes (stars) present in C do not appear as fluorescein-labelled in (D).
Figure 4(A) shows that platelets stimulated by HPU undergo release reaction and secrete ATP from dense granules. Platelets stimulated by 0.3 μM HPU secreted about 60% of the ATP measured for a collagen-induced release reaction, with a slower kinetics and longer lag phase, peaking after 3 min. As previously described for canatoxin [18] and B. pasteurii urease [25] HPU-induced platelet aggregation is completely dependent on this secreted ADP and therefore inhibited by the scavenger activity of apyrase (Fig. 4B).
Fig 4.

HPU induces release reaction and ADP-induced aggregation of rabbit platelets. (A) Time course of release reaction of platelets activated by HPU (300 nM) or collagen (30 μg/ml) was detected following ATP secretion as light emitted in the presence of a luciferin-luciferase mixture. L.R.U. – light relative units. A typical result out of three replicates is shown. (B) Platelet aggregation induced by HPU (300 nM) or 5 μM ADP (inset) is completely abolished in the presence of 1 U/ml of potato apyrase. Platelets were pre-incubated with apyrase for 10 min. at room temperature without stirring, and then aggregation was triggered by addition of HPU or ADP (time zero) and aggregation was monitored turbidimetrically for 8 min. Typical results are shown.
Involvement of 12-lipoxygenase in HPU-induced platelet activation
To elucidate the pathway(s) recruited by HPU to induce platelet aggregation we investigated the involvement of arachidonic acid metabolites in platelets pre-treated with dexamethasone (a phospholipase A2 inhibitor), or indomethacin (a cyclooxygenase inhibitor) or esculetin (a 12-lipoxygenase inhibitor). Table 1 shows that dexamethasone treatment reduced HPU-induced aggregation indicating a requirement of free arachidonic acid. In indomethacin-treated platelets, HPU-induced aggregation was augmented up to 3-fold, excluding the participation of thromboxane A2, an indirect product of cyclooxygenase activity in the aggregation response. On the other hand HPU-induced aggregation was reduced in esculetin pre-treated platelets, indicating that product(s) of the 12-lipoxygenase, which is inhibited by this compound [38], mediated the platelet’s response to the protein (Table 1 and Fig. 5A). Likewise the observed enhancement of HPU-induced aggregation in indomethacin-treated platelets reflects the increased availability of arachidonic acid as substrate for the 12-lipoxygenase. HPU-activated platelets produced 12-HPETE, which could be measured as 12-hydroxy-eicosatetraenoic acid (12-HETE) in supernatants of lysed platelets (Fig. 5B). Thus, similar to what we described previously for canatoxin [18, 19], and Bacillus pasteurii urease [25], platelet aggregation induced by H. pylori urease is also mediated by lipoxygenase-derived eicosanoids.
Table 1.
Effect of inhibitors of phospholipase A2 and arachidonic acid metabolism on H. pylori urease-induced platelet aggregation
| Platelet pre-treatment | % platelet aggregation mean ± S.D. (N) |
|---|---|
| None | 100.00 ± 10.24 (4) |
| Dexamethasone (50 μM) | 62.64*± 6.06 (4) |
| Esculetin (500 μM) | 55.00**± 6.06 (4) |
| Indomethacin (150 μM) | 160.74***± 12.74 (4) |
| (300 μM) | 313.26***± 3.78 (4) |
Rabbit PRP suspensions in microwell plates were incubated for 2 min. at room temperature in the absence or in the presence of the indicated concentrations of the drugs and aggregation was triggered by addition of HPU (0.3 μM). Aggregation of platelets was monitored ever 11 sec. during 20 min. in a SpectraMax plate reader.
Values of P < 0.05*, P < 0.01** or P < 0.001*** were considered statistically significant.
Fig 5.
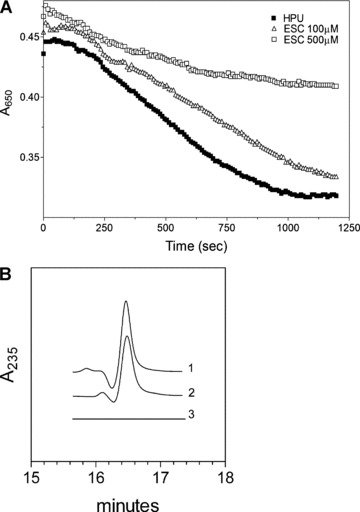
Involvement of lipoxygenase-derived metabolite(s) in HPU-induced platelet aggregation. (A) Rabbit platetet-rich plasma suspensions in microwell plates were incubated for 2 min. at room temperature in the absence or in the presence of the indicated concentrations of esculetin and aggregation was triggered by addition of HPU (0.3 μM). Aggregation of platelets was monitored as decrease in absorbance at 650 nm in a SpectraMax plate reader. A typical result out of four replicates is shown. (B) Fully aggregated platelets induced by 0.3 μM HPU were lysed in 100% methanol and the supernatant was analysed by reverse-phase chromatography according to Coffey et al.[37]. Tracing 1 corresponds to a product isolated from HPU-activated platelets while the elution pattern of authentic standard 12-HETE (∼ 200 ng) is shown in tracing 2, both peaks eluting at approximately at 16.5 min. Tracing 3 is the pattern of elution obtained for supernatant of non-activated platelets.
Similar to what was previously seen for platelets aggregated by canatoxin (jackbean) or B. pasteurii urease, HPU also does not depend on PAF-acether synthesis. Table 2 shows that 2 μM Web2170 inhibited only 16% the aggregation induced by HPU, while it blocked almost 90% of platelets response to a very high non-physiological concentration of PAF-acether. Thus, although statistically significant this inhibition is probably biologically irrelevant, reflecting secondary reactions of HPU-activated platelets. On the other hand, the response of platelets to HPU involved activation of voltage-dependent calcium channels as demonstrated by the inhibition caused by D-methoxy-verapamil (Table 2).
Table 2.
Effect of a calcium channel blocker and of a PAF-acether antagonist on H. pylori urease-induced platelet aggregation
| Platelet treatment | % platelet aggregation mean ± S.D. (N) |
|---|---|
| 0.3 μM HPU | 100.00 ± 7.43 (4) |
| 50 μM Verapamil + HPU | 80.83*± 1.58 (4) |
| 75 μM Verapamil + HPU | 63.35***± 1.19 (4) |
| 2 μM Web2170 + HPU | 83.94*± 6.45 (4) |
| 50 nM PAF-acether | 100.00 ± 3.21 (4) |
| 2 μM Web2170 + PAF acether | 13.1***± 1.62 (4) |
Rabbit platetet-rich plasma suspensions in microwell plates were incubated for 2 min. at room temperature in the absence or in the presence of the indicated concentrations of the calcium channel blocker D-methoxy-verapamil or the PAF-acether antagonist Web2170. Aggregation was triggered by addition of HPU (0.3 μM) or PAF-acether (50 nM) and monitored ever 11 sec. during 20 min. in a SpectraMax plate reader. Results are means ± S.D. of four replicates of each condition.
Values of P < 0.05* or P < 0.001*** were considered statistically significant.
Discussion
We previously reported on the platelet aggregating activity of jackbean [17, 18] and soybean [13] single chain ureases and of the tri-chain urease from Bacillus pasteurii[13]. The jackbean urease isoform canatoxin was shown to induce aggregation of rat, rabbit, guinea pig and human platelets, either in the presence or absence of plasma proteins, but it was inactive upon degranulated platelets [17]. Here we reported the same activity for the two-chain H. pylori urease. Thus, independent of their quaternary structures, the property of activating blood platelets inducing aggregation and release reaction (a model for exocytosis) is a common feature of bacterial and plant ureases (Table 3). The fact that bacterial and plant ureases conserved the property of inducing exocytosis in some cell types may shed new light into the so far poorly understood biological functions of these proteins.
Table 3.
Comparative data on physicochemical and biological properties of the isoform of jackbean urease canatoxin (CNTX), B. pasteurii urease (BPU) and a HPU
| Physicochemical properties | CNTX* | BPU† | HPU‡ |
|---|---|---|---|
| Molecular mass: | |||
| SDS-PAGE | 95 kD | 11–13–61 kD (chains A, B and C, respectively) | 30–62 kD (chains A and B, respectively) |
| Native form | Dimer | Trimer | Hexamer |
| Urease activity (U/ mg protein) | 11.6 | 194.0 | 264.4 |
| Binding to polysialogangliosides | Yes | ND | Yes |
| Biological properties | |||
| Toxicity to: | |||
| Mouse, i.p. | Toxic | Not toxic | ND |
| Treated with pHMB | 100% active | ||
| D. peruvianus, LD50 | 0.01% (w/w) | Not toxic | ND |
| Treated with pHMB | 100% active | ||
| Platelet aggregation, EC50 (rabbit) | 22.2 μg/ml | 400 μg/ml | 150 μg/ml |
| Treated with pHMB | 100% active | ND | ND |
| Platelet secretion | Yes (serotonine) | ND | Yes (ATP) |
| Lipoxygenase inhibitors | Inhibition | Inhibition | Inhibition |
| Cyclooxygenase inhibitors | Potentiation | Potentiation | Potentiation |
| ADP antagonists/scavengers | Inhibition | Inhibition | Inhibition |
| PAF-acether antagonists | No effect | No effect | No effect |
| Ca2+ channel blocker | Inhibition | Inhibition | Inhibition |
Treatment of jackbean ureases [17] with the irreversible inhibitor p-hydroxy-mercury-benzoate abolished their ureolytic activity but did not affect their ability to induce platelet aggregation, clearly demonstrating that these two biological activities are not related. Platelet aggregation inducing property of the B. pasteurii urease did not correlate with the protein’s enzyme activity [17], as shown here also to be the case for HPU (Table 3). Platelet aggregation induced by all three ureases is consequent to the release of ADP from dense granules, stored together with serotonin [18, 19] and ATP (this work) which can be thereafter detected in the medium. As expected, HPU-induced aggregation was completely abolished in the presence of apyrase. PAF-acether is not relevant for urease-induced platelet aggregation [18] (this work). Platelet activation by ureases requires influx of external calcium through voltage-gated Ca2+ channels inhibited by verapamil [18] (this work), and occurs without activation of phospholipase C or production of IP3[20]. The responses of platelets to all three ureases (jackbean, B. pasteurii or H. pylori) were inhibited by different inhibitors of the endogenous phospholipase A2 and of the platelet 12-lipoxygenase and metabolite(s) of this enzyme were produced by HPU-activated platelets. In agreement with an activation induced by lipoxygenase metabolites, indomethacin pre-treated platelets, in which more arachidonic acid is available as substrate for the lipoxygenases, showed significantly enhanced reactivity to ureases [18, 20] (this work). This same increase of platelet reactivity following indomethacin treatment was seen in platelets activated by Chlamydia pneumonia, a human respiratory pathogen linked to cardiovascular disease [39]. Figure 6 summarizes our present knowledge on the signalling pathway recruited by ureases to induce platelet aggregation and release reaction.
Fig 6.
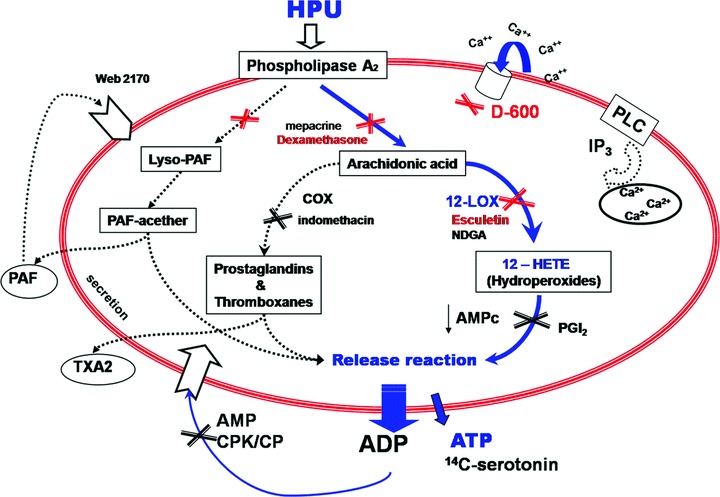
Proposed mechanism of platelet aggregation induced by Helicobacter pylori urease. The biochemical pathways that underlie the platelet-aggregating activity of HPU as well as all ureases studied so far [13, 17–20, 25] are indicated as continuous lines. As depicted in blue, HPU activates platelets through a phospholipase A2 and calcium-dependent pathway that makes arachidonic acid available for the 12-lipoxygenase enzyme and leads to secretion of platelet’s dense granules. The released ADP then triggers aggregation of platelets. Dotted lines indicate other pathways tested for HPU or other ureases that are not relevant to platelet aggregation as induced by ureases. Inhibition sites of pathways are marked by (X). Modified from [25].
In some aspects, urease induced-platelet aggregation resembles collagen-activated platelets. As demonstrated by Coffey and coworkers, collagen interaction with its platelet receptor, glycoprotein VI, results in activation of platelet 12-lipoxygenase with 12-H(P)ETE synthesis [37, 40]. Interestingly, we previously observed that canatoxin-stimulated platelets become refractory to a second exposure to this protein or to collagen, but are still responsive to ADP, PAF-acether or arachidonic acid [18], suggesting that ureases and collagen may be recruiting the same signalling cascade to exert their actions.
It is well known that platelets participate in the inflammatory response by modulating the activity of other inflammatory cells and ischemic lesions due to vascular insufficiency may lead to ulcers within the gastric mucosa [41]. Fluorescent in vivo microscopy studies have shown that H. pylori infection alters blood flow, the endothelial lining of the vessels, leucocyte activity and often induces the formation of circulating or adherent platelet aggregates [42–46], consistent with epidemiological studies that suggest a possible association between H. pylori infection and the incidence of cardiovascular diseases [43]. H. pylori aqueous extracts (which contain HPU) were shown to aggregate platelets [46]. Through a mechanism very different from the one we describe here for purified HPU, whole cell H. pylori also promotes platelet aggregation binding directly to von Willebrand factor and to platelet glycoprotein GPIb [47].
To our knowledge this is the first study demonstrating a direct effect of purified H. pylori urease on platelets. A recent report [48] showed that an active H. pylori urease is pivotal to the gastric epithelial barrier dysfunction that underlies inflammation leading to tissue damage. This mechanism could also lead to liberation of HPU into the vascular bed, where it would directly stimulate platelets.
H. pylori urease is a cytoplasmic enzyme. Upon bacterial lysis urease is released and adsorbed onto the extracellular surface of viable bacteria where it represents about 30% of the total cell urease content [49]. The cell wall bound enzyme facing the external acidic medium is enzymatically inactive and the distribution of urease within the bacterium is dependent on external pH [45, 46]. It is not known whether cell wall bound urease H. pylori displays any biological activity. On the other hand, purified H. pylori urease has been shown to bind mucin and other glycogonjugates [50] and to contribute directly to the gastric inflammatory process by stimulating macrophages and cytokine production [41, 49, 51, 52]. An important aspect to be investigated is whether or not these other biological activities of H. pylori urease depend on its ureolytic activity. Moreover, our finding of a direct platelet-activating activity of H. pylori urease and the modulation of its platelet-activating properties by lipoxygenase-derived eicosanoids could be particularly relevant to the elucidation of mechanisms leading to the gastrointestinal and associated cardiovascular diseases caused by this bacterium and may have to be taken into consideration in the development of more efficient therapeutic approaches.
Acknowledgments
This work was supported by grants from Brazilian agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq; Coordenação de Aperfeiçoamento de Pessoal do Ensino Superior – CAPES; Financiadora de Estudos e Projetos – FINEP and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul – FAPERGS.
References
- 1.Dixon NE, Gazzola C, Blakeley RL, et al. Jack bean urease (EC 3.5.1.5): a metalloenzyme. A simple biological role for nickel. J Amer Chem Soc. 1975;97:4131–3. doi: 10.1021/ja00847a045. [DOI] [PubMed] [Google Scholar]
- 2.Mobley HL, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev. 1995;59:451–80. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Follmer C. Insights into the role and structure of plant ureases. Phytochemistry. 2008;69:18–28. doi: 10.1016/j.phytochem.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 4.Sirko A, Brodzik R. Plant ureases: roles and regulation. Acta Biochim Pol. 2000;47:1189–95. [PubMed] [Google Scholar]
- 5.Carlini CR, Polacco JC. Toxic properties of urease. Crop Sci. 2008;48:1665–72. [Google Scholar]
- 6.Fischbach W, Chan AOO, Wong BCY. Helicobacter pylori and gastric malignancy. Helicobacter. 2005;10:34–9. doi: 10.1111/j.1523-5378.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki H, Marshall BJ, Hibi T. Overview: Helicobacter pylori and Extragastric Disease. Int J Hematol. 2006;84:291–300. doi: 10.1532/IJH97.06180. [DOI] [PubMed] [Google Scholar]
- 8.Clyne M, Dolan B, Reeves EP. Bacterial factors that mediate colonization of the stomach and virulence of Helicobacter pylori. FEMS Microbiol Lett. 2007;268:135–43. doi: 10.1111/j.1574-6968.2007.00648.x. [DOI] [PubMed] [Google Scholar]
- 9.Franco AT, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by helicobacter pylori virulence factors. Cancer Res. 2008;68:379–87. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki H, Hibi T, Marshall BJ. Helicobacter pylori: present status and future prospects in Japan. J Gastroenterol. 2007;42:1–15. doi: 10.1007/s00535-006-1990-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker-Ritt AB, Martinelli AHS, Mitidieri S, et al. Antifungal activity of plant and bacterial ureases. Toxicon. 2007;50:971–83. doi: 10.1016/j.toxicon.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Carlini CR, Grossi-de-Sa MF. Plant toxic proteins with insecticidal properties. A review on their potentialities as bioinsecticides. Toxicon. 2002;40:1515–39. doi: 10.1016/s0041-0101(02)00240-4. [DOI] [PubMed] [Google Scholar]
- 13.Follmer C, Real-Guerra R, Wasserman GE, et al. Jackbean, soybean and Bacillus pasteurii ureases – Biological effects unrelated to ureolytic activity. Eur J Biochem. 2004;271:1357–63. doi: 10.1111/j.1432-1033.2004.04046.x. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira-DaSilva CT, Gombarovits MEC, Masuda H, et al. Proteolytic activation of canatoxin, a plant toxic protein, by insect cathepsin-like enzymes. Arch Insect Biochem Physiol. 2000;44:162–71. doi: 10.1002/1520-6327(200008)44:4<162::AID-ARCH3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Mulinari F, Staniscuaski F, Bertholdo-Vargas LR, et al. Jaburetox-2Ec: an insecticidal peptide derived from an isoform of urease from the plant Canavalia ensiformis. Peptides. 2007;28:2042–50. doi: 10.1016/j.peptides.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Carlini CR, Guimaraes JA. Isolation and characterization of a toxic protein from Canavalia ensiformis (jack bean) seeds, distinct from concanavalin A. Toxicon. 1981;19:667–75. doi: 10.1016/0041-0101(81)90104-5. [DOI] [PubMed] [Google Scholar]
- 17.Follmer C, Barcellos GBS, Zingali RB, et al. Canatoxin, a toxic protein from jack beans (Canavalia ensiformis), is a variant form of urease (EC 3.5.1.5): biological effects of urease independent of its ureolytic activity. Biochem J. 2001;360:217–24. doi: 10.1042/0264-6021:3600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlini CR, Guimaraes JA, Ribeiro JM. Platelet release reaction and aggregation induced by canatoxin, a convulsant protein: evidence for the involvement of the platelet lipoxygenase pathway. Br J Pharmacol. 1985;84:551–60. doi: 10.1111/j.1476-5381.1985.tb12940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barja-Fidalgo C, Guimaraes JA, Carlini CR. Lipoxygenase-mediated secretory effect of canatoxin, the toxic protein from Canavalia ensiformis seeds. Toxicon. 1991;29:453–60. doi: 10.1016/0041-0101(91)90019-n. [DOI] [PubMed] [Google Scholar]
- 20.Ghazaleh FA, Francischetti IM, Gombarovits ME, et al. Stimulation of calcium influx and platelet activation by canatoxin: methoxyverapamil inhibition and downregulation by cGMP. Arch Biochem Biophys. 1997;339:362–7. doi: 10.1006/abbi.1997.9898. [DOI] [PubMed] [Google Scholar]
- 21.Benjamin CF, Carlini CR, Barja-Fidalgo C. Pharmacological characterization of rat paw edema induced by canatoxin, the toxic protein from Canavalia ensiformis (jack bean) seeds. Toxicon. 1992;30:879–85. doi: 10.1016/0041-0101(92)90386-j. [DOI] [PubMed] [Google Scholar]
- 22.Barja-Fidalgo C, Guimaraes JA, Carlini CR. Canatoxin, a plant protein, induces insulin release from isolated pancreatic islets. Endocrinology. 1991;128:675–9. doi: 10.1210/endo-128-2-675. [DOI] [PubMed] [Google Scholar]
- 23.Olivera-Severo D, Wasserman GE, Carlini CR. Ureases display biological effects independent of enzymatic activity. Is there a connection to diseases caused by urease-producing bacteria. Braz J Med Biol Res. 2006;39:851–61. doi: 10.1590/s0100-879x2006000700002. [DOI] [PubMed] [Google Scholar]
- 24.Alves EW, Ferreira AT, Ferreira CT, et al. Effects of canatoxin on the Ca(2+)-ATPase of sarcoplasmic reticulum membranes. Toxicon. 1992;30:1411–8. doi: 10.1016/0041-0101(92)90516-8. [DOI] [PubMed] [Google Scholar]
- 25.Olivera-Severo D, Wassermann GE, Carlini CR. Bacillus pasteurii urease shares with plant ureases the ability to induce aggregation of blood platelets. Arch Biochem Biophys. 2006;452:149–55. doi: 10.1016/j.abb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Andrews RK, Gardiner EE, Shen Y, et al. Platelet interactions in thrombosis. IUBMB Life. 2004;56:13–8. doi: 10.1080/15216540310001649831. [DOI] [PubMed] [Google Scholar]
- 27.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circulation Res. 2007;100:1673–85. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 28.Gachet C, Hechler B, Leon C, et al. Activation of ADP receptors and platelet function. Thromb Haemost. 1997;78:271–5. [PubMed] [Google Scholar]
- 29.Belville JS, Bennett WF, Lynch G. A method for investigating the role of calcium in the shape change, aggregation and serotonin release of rat platelets. J Physiol. 1979;297:289–97. doi: 10.1113/jphysiol.1979.sp013040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blache D, Ciavatti M, Ojeda C. The effect of calcium channel blockers on blood platelet function especially calcium uptake. Biochim Biophys Acta. 1987;923:401–12. doi: 10.1016/0304-4165(87)90048-1. [DOI] [PubMed] [Google Scholar]
- 31.Moncada S, Vane JR. Arachidonic-acid metabolites and the interactions between platelets and blood-vessel walls. N Eng J Med. 1979;300:1142–7. doi: 10.1056/NEJM197905173002006. [DOI] [PubMed] [Google Scholar]
- 32.Hawiger J. Mechanisms involved in platelet-vessel wall interaction. Thromb Haemost. 1995;74:369–72. [PubMed] [Google Scholar]
- 33.Benveniste J, Chignard M, Lecouedic JP, et al. Biosynthesis of Platelet-Activating Factor (Paf-acether).2. Involvement of phospholipase-A2 in the formation of Paf-acether and lyso-Paf-acether from rabbit platelets. Thromb Res. 1982;25:375–85. doi: 10.1016/0049-3848(82)90128-1. [DOI] [PubMed] [Google Scholar]
- 34.McGee DJ, May CA, Garner RM, et al. Isolation of Helicobacter pylori genes that modulate urease activity. J Bacteriol. 1999;181:2477–84. doi: 10.1128/jb.181.8.2477-2484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weatherburn MW. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem. 1967;39:971–4. [Google Scholar]
- 36.Francischetti IMB, Ribeiro JMC, Champagne D, et al. Purification, cloning, expression, and mechanism of action of a novel platelet aggregation inhibitor from the salivary gland of the blood-sucking bug, Rhodnius prolixus. J Biol Chem. 2000;275:12639–50. doi: 10.1074/jbc.275.17.12639. [DOI] [PubMed] [Google Scholar]
- 37.Coffey MJ, Jarvis GE, Gibbins JM, et al. Platelet 12-lipoxygenase activation via glycoprotein VI – involvement of multiple signaling pathways in agonist control of H(P)ETE synthesis. Circulation Res. 2004;94:1598–605. doi: 10.1161/01.RES.0000132281.78948.65. [DOI] [PubMed] [Google Scholar]
- 38.Sekiya K, Okuda H, Arichi S. Selective inhibition of platelet lipoxygenase by esculetin. Biochim Biophys Acta. 1982;713:68–72. [PubMed] [Google Scholar]
- 39.Kalvegren H, Andersson J, Grenegard M, et al. Platelet activation triggered by Chlamydia pneumoniae is antagonized by 12-lipoxygenase inhibitors but not cyclooxygenase inhibitors. Eur J Pharmacol. 2007;566:20–7. doi: 10.1016/j.ejphar.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Coffey MJ, Coles B, Locke M, et al. Interactions of 12-lipoxygenase with phospholipase A(2) isoforms following platelet activation through the glycoprotein VI collagen receptor. FEBS Lett. 2004;576:165–8. doi: 10.1016/j.febslet.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–84. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobin NP, Henehan GT, Murphy RP, et al. Helicobacter pylori-induced inhibition of vascular endothelial cell functions: a role for VacA-dependent nitric oxide reduction. Am J Physiol-Heart C. 2008;295:H1403–13. doi: 10.1152/ajpheart.00240.2008. [DOI] [PubMed] [Google Scholar]
- 43.Manolakis A, Kapsoritakis AN, Potamianos SP. A review of the postulated mechanisms concerning the association of Helicobacter pylori with ischemic heart disease. Helicobacter. 2007;12:287–97. doi: 10.1111/j.1523-5378.2007.00511.x. [DOI] [PubMed] [Google Scholar]
- 44.Corcoran PA, Atherton JC, Kerrigan SW, et al. The effect of different strains of Helicobacter pylori on platelet aggregation. Can J Gastroenterol. 2007;21:367–70. doi: 10.1155/2007/490852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki H, Mori M, Seto K, et al. Polaprezinc attenuates the Helicobacter pylori-induced gastric mucosal leucocyte activation in Mongolian gerbils – a study using intravital videomicroscopy. Aliment Pharmacol Ther. 2001;15:715–25. doi: 10.1046/j.1365-2036.2001.00960.x. [DOI] [PubMed] [Google Scholar]
- 46.Kalia N, Bardhan KD. Of blood and guts: association between Helicobacter pylori and the gastric microcirculation. J Gastroenterol Hepatol. 2003;18:1010–7. doi: 10.1046/j.1440-1746.2003.03062.x. [DOI] [PubMed] [Google Scholar]
- 47.Byrne MF, Kerrigan SW, Corcoran PA, et al. Helicobacter pylori binds von Willebrand factor and interacts with GPlb to induce platelet aggregation. Gastroenterology. 2003;124:1846–54. doi: 10.1016/s0016-5085(03)00397-4. [DOI] [PubMed] [Google Scholar]
- 48.Wroblewski LE, Shen L, Ogden S, et al. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology. 2009;136:236–46. doi: 10.1053/j.gastro.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montecucco C, Papini E, De Bernard M, et al. Molecular and cellular activities of Helicobacter pylori pathogenic factors. FEBS Lett. 1999;452:16–21. doi: 10.1016/s0014-5793(99)00652-3. [DOI] [PubMed] [Google Scholar]
- 50.Icatlo FC, Goshima H, Kimura N, et al. Acid-dependent adherence of Helicobacter pylori urease to diverse polysaccharides. Gastroenterology. 2000;119:358–67. doi: 10.1053/gast.2000.9372. [DOI] [PubMed] [Google Scholar]
- 51.Gobert AP, Mersey BD, Cheng YL, et al. Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol. 2002;168:6002–6. doi: 10.4049/jimmunol.168.12.6002. [DOI] [PubMed] [Google Scholar]
- 52.Harris PR, Mobley HL, Perez-Perez GI, et al. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology. 1996;111:419–25. doi: 10.1053/gast.1996.v111.pm8690207. [DOI] [PubMed] [Google Scholar]