

Abstract
Calcineurin is a serine/threonine phosphatase originally involved in the immune response but is also known for its role as a central mediator in various non-immunological intracellular signals. The nuclear factor of activated T cell (NFAT) proteins are the most widely described substrates of calcineurin, but ongoing work has uncovered other substrates among which are the cytoskeleton organizing proteins (i.e. cofilin, synaptopodin, WAVE-1). Control over cytoskeletal proteins is of outmost interest because the phenotypic properties of cells are dependent on cytoskeleton architecture integrity, while rearrangements of the cytoskeleton are implicated in both physiological and pathological processes. Previous works investigating the role of calcineurin on the cytoskeleton have focused on neurite elongation, myocyte hypertrophic response and recently in kidney cells structure. Nuclear factor of activated T cell activation is expectedly identified in the signalling pathways for calcineurin-induced cytoskeleton organization, however new NFAT-independent pathways have also been uncovered. The aim of this review is to summarize the current knowledge on the effects of calcineurin on cytoskeletal proteins and related intracellular pathways. These newly described properties of calcineurin on cytoskeletal proteins may explain some of the beneficial or deleterious effects observed in kidney cells associated with the use of the calcineurin inhibitors, cyclosporine and tacrolimus.
Keywords: calcineurin, cytoskeleton, kidney, nephrotoxicity
Introduction
Calcineurin (CaN) is a serine/threonine phosphatase widely distributed in mammalian tissues and unique among phosphatases for its ability to sense Ca2+ through its activation by calmodulin. This latter property makes it a powerful mediator of intracellular signals commonly involved in physiological and pathological processes. Calcineurin is composed of two subunits. A catalytic A subunit, which contains the phosphatase domain and mediates interaction with phosphorylated substrates, and a regulatory B subunit which binds Ca2+ and calmodulin and facilitates the conformational change needed for phosphatase activity [1]. First identified in the late 1970s as an inhibitor of the calmodulin-dependent cyclic nucleotide phosphodiesterase [2], CaN was then recognized as the target of the immunosuppressive drugs cyclosporine A (CsA) and tacrolimus (FK506) and was involved in the activation of nuclear factor of activated T cell (NFAT) proteins [3, 4]. Activation of T cell receptor by extracellular stimuli leads to elevation of intracellular Ca2+ and activation of CaN, which dephosphorylates NFAT, allowing its translocation to the nucleus and the activation of gene expression. CaN was originally thought to have a narrow substrate specificity, mainly limited to NFAT [3], but numerous other substrates were thereafter discovered such as the transcription factors Elk-1 and MEF2 [5, 6], the heat-shock protein Hsp25 [7], the growth factor neuromodulin [8], the NMDA receptor [9] and various cytoskeleton organizing proteins (e.g. Tau factor, MAP-2, cofilin, synaptopodin, WAVE1) [10–13]. These later proteins are of particular interest due to their major role as cytoskeletal proteins, determining cell phenotype.
The cytoskeleton is a three-dimensional filamentous protein network providing structural scaffolding to the cell and thereby determining its size, shape and mechanical properties. The cytoskeleton is composed of three distinct but integrated cytoplasmic fibrous polymers: actin microfilaments, microtubules and intermediate filaments. Cytoskeleton is a key regulator of various cellular and molecular events, such as movement of chromosomes during cell division, vesicular trafficking, cellular adhesion and migration and acquisition of cellular polarity. It also acts as a signalling platform and modulates cellular pathways by controlling the activity and/or the subcellular localization of signalling proteins and their targets.
Several studies have provided evidence that CaN influences the cytoskeleton of neurons, myocytes and recently of kidney cells, potentially contributing to some of the non-immunological effects observed with the use of the CaN inhibitors. The aim of this review is to summarize the current knowledge on the effects of CaN on cytoskeletal proteins and the intracellular pathways involved. These newly described properties of CaN on cytoskeletal organization may explain some of the beneficial or deleterious effects observed in kidney cells with the use of the CaN inhibitors.
Calcineurin effects on neuronal cytoskeleton
Calcineurin was first implicated in the regulation of the structural integrity and the activity-dependent modelling of the neuronal cytoskeleton by Goto et al., who highlighted its ability to control the assembly–disassembly cycle of microtubules [10]. Today, CaN is known to directly control microtubules assembly, through dephosphorylation of tubulin and microtubules associated proteins such as Tau and MAP-2 [10, 14–16], and neurofilaments organization in cultured-neuronal cells [8, 17, 18]. These effects were confirmed in mice lacking neuronal CaN Aα subunit, which displayed abnormal phosphorylation of microtubules, diminution of neurofilament content and abnormal cytoskeleton organization [19].
Calcineurin has also been involved in neuron elongation. This physiological process involves cytoskeletal rearrangements in response to extracellular signals that increase intracellular Ca2+[20]. Growth cones at the distal ends of growing neurites determine the rate and the direction of neuron elongation. Their ability to reorganize rapidly in response to a variety of molecular signals allows the axon to advance, retract, turn or branch mainly through reorganization of actin and microtubule cytoskeleton. CsA and FK506 were shown to inhibit the polarized axon elongation of cerebellar macroneurons [14], shorten the length of neurites produced in culture of chicken dorsal root ganglia neurons on laminin [21] and suppress the neurite outgrowth imposed by Ca2+ waves in cultured Xenopus spinal neurons [22]. Transitory and local inactivation of CaN induced a retraction of the growth cone and a deviation of its progression from the inactivated site, suggesting that CaN controls filopodia motility [21]. Expression of CaN is also dramatically increased during neuron development and its localization varies during neuron elongation [14, 23].
Calcineurin effects on myocytes
Investigations into the possible role of CaN in myocyte hypertrophic response started when increase in intracellular Ca2+ provoked by hypertrophic agonist (e.g. Angiotensin II, Phenylephrine or myocyte stretch) was demonstrated to be a major event leading to cardiac hypertrophy [24–28].
Cardiac hypertrophy is characterized by a reorganization of the microtubule network [29–33] and a transcriptional up-regulation of α-tubulin, β1-tubulin and MAP-4 [34–36]. β-Actin is also increased in different models of cardiac hypertrophy [37]. Finally, the intermediate filament desmin is increased in models of cardiac hypertrophy in the guinea pig [38] and is both increased and disorderly rearranged in human end-stage dilated cardiomyopathy [39].
Calcineurin enzymatic activity and protein levels were found to be significantly up-regulated in hearts from numerous cardiac hypertrophy models [40–52] and in human failing or hypertrophied hearts [53–55].
Molketin et al. generated transgenic mice, expressing a constitutively active cardiac form of either CaN A subunit or NFAT3 protein, and showed a profound hypertrophic response in the transgenic mice compared to the control mice. Cardiomyocytes were highly disorganized and hypertrophic with dramatic karyomegaly and myofibre degeneration [56]. A similar hypertrophic response was observed in cultured cardiomyocytes, expressing a constitutively active form of mouse CaN Aα[57].
Cellular, morphological and molecular changes associated with cardiac hypertrophy in activated-CaN transgenic mice were prevented by administration of CsA and FK506 [56]. Both drugs also blocked the ability of cultured cardiomyocytes to undergo hypertrophy in response to Angiotensin II and phenylephrine [56]. Furthermore, Sussman et al. demonstrated that CaN inhibitors prevented the phenotypic manifestations of hypertrophic cardiomyopathy and the disorganization of myofibrils in transgenic mice or pressure-overload hypertrophy rat model [50]. Numerous other studies using a similar pharmacological approach demonstrated that CaN is a key mediator in the hypertrophic response in pleiotropic rodent models [41, 44, 45, 48, 49, 58–69]. The role of CaN was confirmed in transgenic models expressing negative mutants of CaN or inhibitory domains of CaN-interacting proteins [40, 70, 71]. Transgenic mice lacking the CaN Aβ subunit or expressing a negative mutant of CaN displayed a reduced hypertrophic response to aortic banding or agonist stimulation [52, 72].
Intracellular pathway involved in calcineurin-induced effects on cytoskeletal organization (Table 1)
Table 1.
Proteins involved in calcineurin’s regulation of cytoskeleton
| Calcineurin substrate | Cell’s modifications | References, animal models |
|---|---|---|
| Cofilin | Induction of neurite development | [82, 83] |
| Dendritic spine loss | [84] | |
| Platelet activation | [85] | |
| Tracheal muscle cells activation | [86] | |
| Neurite development | ||
| NFAT | Induction on neuronal elongation | [74, 75]→ NFATc3/c4−/− and c2/c3/c4−/− mice; VIVIT-treated zebrafish embryos |
| Inhibition of neuronal outgrowth | [76]→ NFAT3−/− mice | |
| Induction of cardiac hypertrophy | ||
| NFAT3 | [56, 78]→ NFAT3+/+ mice; | |
| NFAT3−/− transfected cardiomyocytes | ||
| NFAT4 | [77]→ NFAT4−/− mice | |
| Glomerulosclerosis, proteinuria | [102]→ NFATc1 conditional induction | |
| Synaptopodin | Podocytes actin cytoskeleton disorganization | [12] |
| WAVE-1 | Neurite development in response to neurotransmitters | [13] |
CaN: calcineurin; NFAT: nuclear factor of activated T cells; WAVE1: WASP-family verprolin homologous protein 1.
CaN/NFAT pathway was first implicated in cytoskeletal control; however, the discovery of new CaN substrates in the brain, known as cytoskeletal organizing proteins, highlights the fact that CaN may act directly on cytoskeletal organization, in parallel to the well-known NFAT-dependent transcriptional effects.
Calcineurin/NFAT pathway
Nuclear factor of activated T cell family is composed of five proteins (NFAT1/c2, NFAT2/c1, NFAT3/c4, NFAT4/c3 and NFAT5/TonEBP). All of them, except NFAT5, contain a Ca2+ sensor/translocation domain and are specifically activated by CaN-mediated dephosphorylation. Serine dephosphorylation within the amino-terminus domain triggers the cytoplasmic-to-nuclear translocation of NFAT and its binding to the promoter of target genes [73].
Calcineurin/NFAT in neuronal elongation
Graef et al. were the first to provide evidence for the role of NFAT in controlling the neurotrophin-dependent outgrowth of embryonic axons [74]. They demonstrated abnormal sensory axon projection and commissural axon growth in double (c3/c4) and triple (c2/c3/c4) NFAT mutant mice, while no defect was observed in single mutants. Abnormal axonal growth is specific to NFAT activation by CaN because similar defects were found in CaN B mutant mice and in embryos from wild-type pregnant mice treated with CsA [74]. More recently, CaN/NFAT pathway was also demonstrated to regulate morphological remodelling of axon terminals of olfactory sensory neurons in zebrafish [75]. Using growth-associate protein-43-EGFP (GAP-43-EGFP) as in vivo visual marker for axon terminal maturation, Yoshida et al. showed that axon terminal remodelling was prevented by CsA and VIVIT, a specific NFAT inhibitor.
All these results support the idea that NFAT activation is required for proper neural development and functions but, conversely, a recent study reported an unexpected role of NFAT3 in decreasing GAP-43 gene expression during latter part of embryonic neurite development [76]. This work is the first to report a direct control of NFAT proteins on axon outgrowth-related genes in brain and provides an unexpected new role for NFAT3 in negative transcriptional regulation of the neuronal outgrowth program.
Calcineurin/NFAT in cardiac hypertrophy
Nuclear factor of activated T cell activity was found to be increased in primary rat cardiomyocytes subjected to angiotensin II or phenylephrine infusion, and completely abolished by CsA or FK506 [56]. However, the identification of the specific isoform involved remains complex because all of the four CaN-regulated NFAT proteins were identified in the heart and present a high degree of homology within the DNA-binding domain [77, 78].
NFAT3 pathway was first implicated as a pivotal transducer of the cardiac hypertrophy response by interacting with the cardiac transcription factor GATA4 and by activating expression of numerous cardiac genes stimulated during cardiac hypertrophy [56]. Involvement of NFAT3 in cardiac hypertrophy was confirmed in transgenic mice expressing a constitutively activated NFAT3 mutant in the heart and displaying pronounced cardiac hypertrophy with extensive fibrosis [56]. Conversely, cardiomyocytes transfected with a negative NFAT3 transcript displayed neither hypertrophic remodelling nor increase in atrial natriuretic peptide expression in response to hypertrophic stimuli [78]. Together, these results defined a signalling pathway coupling hypertrophic signals to pathological changes in cardiac morphology and gene expression though activation of NFAT3.
However, Wilkins et al. did not reproduce the previous results and supported the idea that NFAT3 function might be compensated by another heart-expressed NFAT protein [77]. Expressing a negative mutant of CaN Aα in wild-type and NFAT3 null mice, they observed that loss of NFAT3 did not diminish the magnitude in CaN-induced hypertrophy. Similar reports were made in NFAT3 null mice exposed to angiotensin II or aortic banding, which showed identical cardiac pathology and morphologic hypertrophy than wild-type mice exposed to the same stimuli [77]. On the other hand, NFAT4 null mice showed a significant and long-standing reduction in CaN-induced hypertrophy, or were compromised in their ability to mount an efficient hypertrophic response following aortic banding or angiotensin II infusion [77]. These results support the idea that NFAT4, which is the closest structural homologue to NFAT3, may also act as a downstream effector of CaN in heart.
The hypothesis that NFAT is a critical mediator of CaN signalling is also supported by studies in which cardiac hypertrophic growth was reduced by overexpression of glycogen synthase kinase 3β (GSK3β), known to directly phosphorylate NFAT2 proteins and, thus antagonizing the action of CaN [79, 80].
These results provide multiple lines of evidence for the role of NFAT factors as necessary mediators of CaN-regulated hypertrophic signalling, but further studies are needed to discriminate the proper role of each NFAT isoform.
Calcineurin/actin-associated proteins pathway
ADF and cofilin are major actin-organizing proteins, which regulate actin assembly/disassembly. ADF/cofilin activity is regulated by the state of phosphorylation at a unique site (ser3). Dephosphorylation/activation is achieved by CaN through slingshot phosphatase. Phosphorylation occurs by LIM kinase and inhibits the binding to actin monomers as well as the actin-depolymerizing activity [81]. Meberg et al. demonstrated that ADF/Cofilin are involved in the dynamic changes of actin filaments during neurite extension through Ca2+-dependent dephosphorylation/activation by CaN [82]. Treatment of HT4 cells or primary cortical neurons with Ca2+ ionophore decreased phospho-ADF/Cofilin [p(ADF/cofilin)] level through CaN activation, and conversely, CsA increased p(ADF/Cofilin) level. Regulation of ADF/cofilin phosphorylation was also found to be an important determinant of growth cone motility and neurite outgrowth because decrease of p(ADF/Cofilin) level was associated with increased process extension, whereas agents increasing p(ADF/Cofilin) level inhibited process extension. Moreover, NGF-induced differentiation was accompanied by decrease of p(ADF/Cofilin) level and accumulation of non-phosphorylated proteins co-localized with actin at the tips of lamilipodia. The control of neuronal actin organization by CaN through cofilin dephosphorylation was confirmed by Homma et al., who highlighted the role of CaN-dependent activation of cofilin in the formation of cofilin–actin rods during neurite outgrowth [83]. Pharmacological inhibition of CaN by cypermethrin or expression of an unphosphorylable variant of cofilin both inhibited the formation of cofilin rods and consequently neurite extension. Finally, CaN inhibitors were also shown to inhibit cofilin dephosphorylation and subsequent depolymerization of actin in a pilocarpine model of status epilepticus [84].
As suggested by a recent study, this effect of CaN on cofilin seems not to be restricted to the brain and neurite outgrowth. Indeed, an effect of CaN on cofilin has also been shown in platelets in response to thrombin and in tracheal smooth muscle cells in response to stimulation with acetylcholine [85, 86]. Interestingly, it has been recently demonstrated that cofilin modifies the length of the primary cilium, an organelle located on the apical surface of many cells, such as endothelial or epithelial renal cells, that is involved in mechanosensing of the cell. This effect is observed after cofilin dephosphorylation by the phosphatase PP-1 and is mediated by a reorganization the actin network [87].
WAVE1 protein was recently identified as a new substrate for CaN, able to modify actin organization [13]. WASP-family verprolin homologous protein 1 (WAVE1) is a WASP protein that stimulates the Arp 2/3 complex and nucleates the de novo synthesis and branching of actin filaments [88]. In brain under basal conditions, WAVE1 is inactive and phosphorylated by cyclin-dependent kinase 5 at multiple sites [89]. Ceglia et al. have recently revealed a complex pattern of regulation of WAVE1 in response to increase in intracellular Ca2+ and CaN activation [13]. Calcineurin was found to dephosphorylate different sites of WAVE1 depending on the neurotransmitter-mediated pathway involved. Activated WAVE1 induces actin polymerization, enabling neurite outgrowth and neuronal plasticity in response to the distinct neurotransmitters.
WAVE1 is predominantly present in the brain but other members of the WAVE family are more ubiquitous, playing a major role in the actin organization of numerous cells. For example, WAVE2 has been involved, through actin organization, in fibroblast or myogenic cell migration, vasculogenesis, cell–cell adhesion or formation of the immunological synapse during T cell activation [90–94]. Ceglia’s results showing WAVE1 regulation by CaN of open thus a large field of investigation if a similar modulation by CaN of WAVE2 phosphorylation could be demonstrated.
Calcineurin effects on kidney cells cytoskeleton
The importance of CaN in the kidney is suggested by the frequently observed nephrotoxicity of CsA and FK506. The physiologic role of CaN in kidney cells remained unknown until the recent exhibition of abnormal kidney development in mice invalidated for the CaN Aα subunit. However, the authors do not forward a mechanistic explanation [95].
More recently, Faul et al. have opened a new field of investigation of the role of CaN in kidney by analysing the intracellular pathway involved in the anti-proteinuric effect of CsA in nephrotic syndrome. They identified synaptopodin as a new target of CaN in podocytes and a non-immunological effect of CsA on the podocyte cytoskeleton [12].
Synaptopodin is an actin-associated protein specifically expressed in differentiated podocytes and in a subpopulation of telencephalic neurones, and involved in actin regulation, synaptic plasticity and organization of podocyte foot processes [96, 97]. Synaptopodin is selectively down-regulated by nephrotic plasma or after puromycin aminonucleoside treatment [98, 99]. Synaptopodin-deficient mice display impaired recovery from protamine sulfate-induced foot process effacement and lipopolysaccharide-induced nephrotic syndrome [100]. Gene silencing of synaptopodin in podocytes causes the loss of stress fibres, the formation of aberrant non-polarized filopodia and impairment of cell migration [101].
In their work, Faul et al. highlighted that CaN dephosphorylates synaptopodin, rendering it unable to bind to chaperone molecules of the 14-3-3 proteins family and thus, facilitating its degradation by cathepsin L [12]. CsA, by inhibiting the dephosphorylation of synaptopodin, blocks its degradation by cathepsin and allows a stabilization of the podocyte actin cytoskeleton. Faul et al. also demonstrated that transgenic mice expressing a constitutively active form of CaN developed significant proteinuria. Such an activation of the CaN pathway may be observed in focal and segmental glomerulosclerosis due to mutation of the Ca2+ channel TRPC6 [102]. However, the effect of CaN on the podocyte cytoskeleton is not solely mediated by synaptopodin dephosphorylation. In fact, Wang et al. recently provided in vivo evidence that NFAT activation, either in utero or post-developmentally, leads to proteinuria and glomerulosclerosis [103]. Ultrastructural studies revealed podocyte foot process effacement and deposition of extracellular matrix whereas NFAT activation did not initially affect expression of synaptopodin.
The results of Faul et al. lead us to hypothesize that the deleterious effects of CaN inhibitors on tubular structure observed in kidney transplantation could be due to a disorganization of tubular cytoskeleton and an incorrect adaptation to the increase in tubular flow. To investigate this, we analysed the effect of CsA on the proximal tubule cytoskeleton and demonstrated that CsA induced a strong reorganization of actin filaments [104]. The stiffening of the actin network impacts the phenotype of proximal tubular cells because it is associated with an inhibition of extracellular matrix protease expression with a decrease in tissue-type Plasminogen Activator (tPA) and urokinase and their inhibitor PAI-1 (Fig. 1). These modifications are similar to what is observed when mechanical strains induced by tubular flow on tubular cells are increased such as after subtotal nephrectomy [105]. As regards to the intracellular pathway involved, we demonstrated that CsA-induced actin reorganization was independent of NFAT inhibition because a specific inhibitor of NFAT dephosphorylation did not reproduce the effect of CsA on the cytoskeleton.
Fig 1.
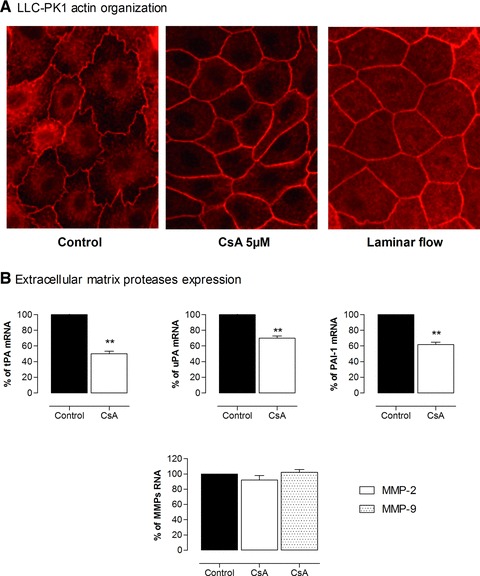
CsA-induced reorganization of actin filaments and decrease in extracellular matrix proteases in proximal tubular cells. (A) LLC-PK1 cells were exposed to CsA 5 μM or laminar flow (1.65 mm/sec.) for 24 hrs then stained with fluorescein-labelled phalloidin and analysed by immunofluorescence microscopy (40×). (B) mRNA of tPA, urokinase, PAI-1 and metalloproteinases 2 and 9 (MMP2, MMP9) were quantified by qPCR. Pictures highlight modification in global cell’s shape, stiffening of the lateral actin network, and a decrease in tPA, urokinase and PAI-1 expression under CsA conditions.
The effect of CaN on renal cells seems thus to be beneficial or detrimental according to the structure triggered. CaN and NFAT activation in podocytes seems to play an important role in the development of podocytes damaged within states of nephrotic syndrome and glomerulosclerosis. In glomerular cells, CsA inhibits CaN acting on actin filament, thus having a beneficial effect by restoring normal podocyte structure. On the contrary, in tubular structure, the stiffening of actin network induced by CsA may alter the response of proximal cells to modifications of tubular flow, thus exerting a detrimental effect on tubular structure.
Conclusion
Calcineurin was initially known for its immunological properties through the activation of NFAT proteins in immune cells. Further studies have underlined its role in the regulation of cytoskeleton organization in other cell types such as neurons, myocytes, and recently podocytes and proximal cells, highlighting some of its non-immunological properties (Fig. 2).
Fig 2.
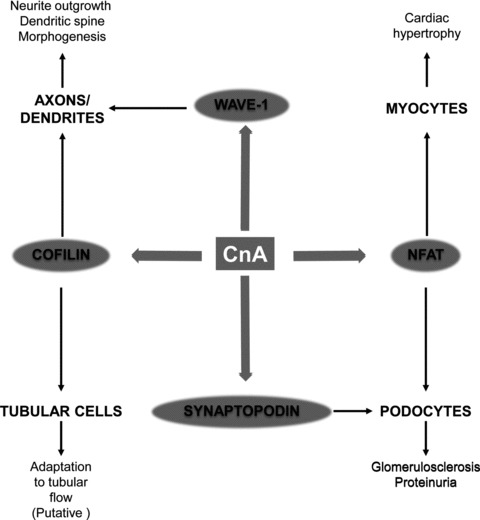
Role of calcineurin-induced organization of cytoskeleton in brain, heart and kidney. CaN: calcineurin; NFAT: nuclear factor of activated T cells; WAVE1: WASP-family verprolin homologous protein 1.
The control of cytoskeleton organization is mediated by both its NFAT-dependent transcriptional effects and by the direct control of filament organization through dephosphorylation of cytoskeletal organizing proteins such as cofilin, WAVE-1 and synaptopodin. The recent identification of these pathways in kidney cells opens new fields of investigation to explain some beneficial or detrimental effects of CaN inhibitors on kidney and the phenotypic changes observed after long-term exposure to these drugs.
Acknowledgments
This study was funded by the Conseil Rgional du Limousin and by INSERM.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 2.Wang JH, Desai R. A brain protein and its effect on the Ca2+ and protein modulator-activated cyclic nucleotide phosphodiesterase. Biochem Biophys Res Commun. 1976;72:926–32. doi: 10.1016/s0006-291x(76)80220-3. [DOI] [PubMed] [Google Scholar]
- 3.Jain J, McCaffrey PG, Miner Z, et al. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–5. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Farmer JD, Jr, Lane WS, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 5.Sugimoto T, Stewart S, Guan KL. The calcium/calmodulin-dependent protein phosphatase calcineurin is the major Elk-1 phosphatase. J Biol Chem. 1997;272:29415–8. doi: 10.1074/jbc.272.47.29415. [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Liu P, Borras A, et al. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. EMBO J. 1997;16:143–53. doi: 10.1093/emboj/16.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaestel M, Benndorf R, Hayess K, et al. Dephosphorylation of the small heat shock protein hsp25 by calcium/calmodulin-dependent (type 2B) protein phosphatase. J Biol Chem. 1992;267:21607–11. [PubMed] [Google Scholar]
- 8.Liu YC, Storm DR. Dephosphorylation of neuromodulin by calcineurin. J Biol Chem. 1989;264:12800–4. [PubMed] [Google Scholar]
- 9.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature. 1994;369:235–9. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 10.Goto S, Yamamoto H, Fukunaga K, et al. Dephosphorylation of microtubule-associated protein 2, tau factor, and tubulin by calcineurin. J Neurochem. 1985;45:276–83. doi: 10.1111/j.1471-4159.1985.tb05504.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280:12683–9. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 12.Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–8. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceglia I, Kim Y, Nairn AC, et al. Signaling pathways controlling the phosphorylation state of WAVE1, a regulator of actin polymerization. J Neurochem. 2010;114:182–90. doi: 10.1111/j.1471-4159.2010.06743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira A, Kincaid R, Kosik KS. Calcineurin is associated with the cytoskeleton of cultured neurons and has a role in the acquisition of polarity. Mol Biol Cell. 1993;4:1225–38. doi: 10.1091/mbc.4.12.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ono T, Yamamoto H, Tashima K, et al. Dephosphorylation of abnormal sites of tau factor by protein phosphatases and its implication for Alzheimer’s disease. Neurochem Int. 1995;26:205–15. doi: 10.1016/0197-0186(94)00135-h. [DOI] [PubMed] [Google Scholar]
- 16.Rahman A, Grundke-Iqbal I, Iqbal K. PP2B isolated from human brain preferentially dephosphorylates Ser-262 and Ser-396 of the Alzheimer disease abnormally hyperphosphorylated tau. J Neural Transm. 2006;113:219–30. doi: 10.1007/s00702-005-0313-5. [DOI] [PubMed] [Google Scholar]
- 17.Fiumelli H, Riederer IM, Martin JL, et al. Phosphorylation of neurofilament subunit NF-M is regulated by activation of NMDA receptors and modulates cytoskeleton stability and neuronal shape. Cell Motil Cytoskeleton. 2008;65:495–504. doi: 10.1002/cm.20278. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T, Takeda M, Niigawa H, et al. Phosphorylated neurofilament accumulation in neuronal perikarya by cyclosporin A injection in rat brain. Methods Find Exp Clin Pharmacol. 1993;15:77–87. [PubMed] [Google Scholar]
- 19.Kayyali US, Zhang W, Yee AG, et al. Cytoskeletal changes in the brains of mice lacking calcineurin A alpha. J Neurochem. 1997;68:1668–78. doi: 10.1046/j.1471-4159.1997.68041668.x. [DOI] [PubMed] [Google Scholar]
- 20.Letourneau PC. The cytoskeleton in nerve growth cone motility and axonal pathfinding. Perspect Dev Neurobiol. 1996;4:111–23. [PubMed] [Google Scholar]
- 21.Chang HY, Takei K, Sydor AM, et al. Asymmetric retraction of growth cone filopodia following focal inactivation of calcineurin. Nature. 1995;376:686–90. doi: 10.1038/376686a0. [DOI] [PubMed] [Google Scholar]
- 22.Lautermilch NJ, Spitzer NC. Regulation of calcineurin by growth cone calcium waves controls neurite extension. J Neurosci. 2000;20:315–25. doi: 10.1523/JNEUROSCI.20-01-00315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polli JW, Billingsley ML, Kincaid RL. Expression of the calmodulin-dependent protein phosphatase, calcineurin, in rat brain: developmental patterns and the role of nigrostriatal innervation. Brain Res Dev Brain Res. 1991;63:105–19. doi: 10.1016/0165-3806(91)90071-p. [DOI] [PubMed] [Google Scholar]
- 24.Karliner JS, Kagiya T, Simpson PC. Effects of pertussis toxin on alpha 1-agonist-mediated phosphatidylinositide turnover and myocardial cell hypertrophy in neonatal rat ventricular myocytes. Experientia. 1990;46:81–4. doi: 10.1007/BF01955423. [DOI] [PubMed] [Google Scholar]
- 25.Leite MF, Page E, Ambler SK. Regulation of ANP secretion by endothelin-1 in cultured atrial myocytes: desensitization and receptor subtype. Am J Physiol. 1994;267:H2193–203. doi: 10.1152/ajpheart.1994.267.6.H2193. [DOI] [PubMed] [Google Scholar]
- 26.Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II-induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ Res. 1993;73:424–38. doi: 10.1161/01.res.73.3.424. [DOI] [PubMed] [Google Scholar]
- 27.Perreault CL, Shannon RP, Shen YT, et al. Excitation-contraction coupling in isolated myocardium from dogs with compensated left ventricular hypertrophy. Am J Physiol. 1994;266:H2436–42. doi: 10.1152/ajpheart.1994.266.6.H2436. [DOI] [PubMed] [Google Scholar]
- 28.Saeki Y, Kurihara S, Hongo K, et al. Tension and intracellular calcium transients of activated ferret ventricular muscle in response to step length changes. Adv Exp Med Biol. 1993;332:639–47. doi: 10.1007/978-1-4615-2872-2_57. ; discussion 47–8. [DOI] [PubMed] [Google Scholar]
- 29.Samuel JL, Bertier B, Bugaisky L, et al. Different distributions of microtubules, desmin filaments and isomyosins during the onset of cardiac hypertrophy in the rat. Eur J Cell Biol. 1984;34:300–6. [PubMed] [Google Scholar]
- 30.Samuel JL, Marotte F, Delcayre C, et al. Microtubule reorganization is related to rate of heart myocyte hypertrophy in rat. Am J Physiol. 1986;251:H1118–25. doi: 10.1152/ajpheart.1986.251.6.H1118. [DOI] [PubMed] [Google Scholar]
- 31.Ishibashi Y, Tsutsui H, Yamamoto S, et al. Role of microtubules in myocyte contractile dysfunction during cardiac hypertrophy in the rat. Am J Physiol. 1996;271:H1978–87. doi: 10.1152/ajpheart.1996.271.5.H1978. [DOI] [PubMed] [Google Scholar]
- 32.Tsutsui H, Ishihara K, Cooper Gt. Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science. 1993;260:682–7. doi: 10.1126/science.8097594. [DOI] [PubMed] [Google Scholar]
- 33.Tsutsui H, Tagawa H, Kent RL, et al. Role of microtubules in contractile dysfunction of hypertrophied cardiocytes. Circulation. 1994;90:533–55. doi: 10.1161/01.cir.90.1.533. [DOI] [PubMed] [Google Scholar]
- 34.Tagawa H, Rozich JD, Tsutsui H, et al. Basis for increased microtubules in pressure-hypertrophied cardiocytes. Circulation. 1996;93:1230–43. doi: 10.1161/01.cir.93.6.1230. [DOI] [PubMed] [Google Scholar]
- 35.Narishige T, Blade KL, Ishibashi Y, et al. Cardiac hypertrophic and developmental regulation of the beta-tubulin multigene family. J Biol Chem. 1999;274:9692–7. doi: 10.1074/jbc.274.14.9692. [DOI] [PubMed] [Google Scholar]
- 36.Sato H, Nagai T, Kuppuswamy D, et al. Microtubule stabilization in pressure overload cardiac hypertrophy. J Cell Biol. 1997;139:963–73. doi: 10.1083/jcb.139.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlyle WC, Toher CA, Vandervelde JR, et al. Changes in beta-actin mRNA expression in remodeling canine myocardium. J Mol Cell Cardiol. 1996;28:53–63. doi: 10.1006/jmcc.1996.0006. [DOI] [PubMed] [Google Scholar]
- 38.Collins JF, Pawloski-Dahm C, Davis MG, et al. The role of the cytoskeleton in left ventricular pressure overload hypertrophy and failure. J Mol Cell Cardiol. 1996;28:1435–43. doi: 10.1006/jmcc.1996.0134. [DOI] [PubMed] [Google Scholar]
- 39.Schaper J, Froede R, Hein S, et al. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–14. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- 40.De Windt LJ, Lim HW, Bueno OF, et al. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3322–7. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eto Y, Yonekura K, Sonoda M, et al. Calcineurin is activated in rat hearts with physiological left ventricular hypertrophy induced by voluntary exercise training. Circulation. 2000;101:2134–7. doi: 10.1161/01.cir.101.18.2134. [DOI] [PubMed] [Google Scholar]
- 42.Kamiya H, Okumura K, Ito M, et al. Calcineurin inhibitor attenuates cardiac hypertrophy due to energy metabolic disorder. Can J Cardiol. 2001;17:1292–8. [PubMed] [Google Scholar]
- 43.Li J, Yatani A, Kim SJ, et al. Neurally-mediated increase in calcineurin activity regulates cardiac contractile function in absence of hypertrophy. Cardiovasc Res. 2003;59:649–57. doi: 10.1016/s0008-6363(03)00471-1. [DOI] [PubMed] [Google Scholar]
- 44.Lim HW, De Windt LJ, Mante J, et al. Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. J Mol Cell Cardiol. 2000;32:697–709. doi: 10.1006/jmcc.2000.1113. [DOI] [PubMed] [Google Scholar]
- 45.Lim HW, De Windt LJ, Steinberg L, et al. Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation. 2000;101:2431–7. doi: 10.1161/01.cir.101.20.2431. [DOI] [PubMed] [Google Scholar]
- 46.Nagata K, Somura F, Obata K, et al. AT1 receptor blockade reduces cardiac calcineurin activity in hypertensive rats. Hypertension. 2002;40:168–74. doi: 10.1161/01.hyp.0000026668.50222.1e. [DOI] [PubMed] [Google Scholar]
- 47.Saito T, Fukuzawa J, Osaki J, et al. Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:1153–60. doi: 10.1016/s0022-2828(03)00234-7. [DOI] [PubMed] [Google Scholar]
- 48.Shimoyama M, Hayashi D, Takimoto E, et al. Calcineurin plays a critical role in pressure overload-induced cardiac hypertrophy. Circulation. 1999;100:2449–54. doi: 10.1161/01.cir.100.24.2449. [DOI] [PubMed] [Google Scholar]
- 49.Shimoyama M, Hayashi D, Zou Y, et al. Calcineurin inhibitor attenuates the development and induces the regression of cardiac hypertrophy in rats with salt-sensitive hypertension. J Cardiol. 2001;37:114–8. [PubMed] [Google Scholar]
- 50.Sussman MA, Lim HW, Gude N, et al. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–3. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 51.Takeda Y, Yoneda T, Demura M, et al. Calcineurin inhibition attenuates mineralocorticoid-induced cardiac hypertrophy. Circulation. 2002;105:677–9. doi: 10.1161/hc0602.104675. [DOI] [PubMed] [Google Scholar]
- 52.Zou Y, Hiroi Y, Uozumi H, et al. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001;104:97–101. doi: 10.1161/01.cir.104.1.97. [DOI] [PubMed] [Google Scholar]
- 53.Haq S, Choukroun G, Lim H, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–7. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 54.Lim HW, Molkentin JD. Calcineurin and human heart failure. Nat Med. 1999;5:246–7. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 55.Ritter O, Hack S, Schuh K, et al. Calcineurin in human heart hypertrophy. Circulation. 2002;105:2265–9. doi: 10.1161/01.cir.0000016044.19527.96. [DOI] [PubMed] [Google Scholar]
- 56.Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Windt LJ, Lim HW, Taigen T, et al. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: an apoptosis-independent model of dilated heart failure. Circ Res. 2000;86:255–63. doi: 10.1161/01.res.86.3.255. [DOI] [PubMed] [Google Scholar]
- 58.Deng L, Huang B, Qin D, et al. Calcineurin inhibition ameliorates structural, contractile, and electrophysiologic consequences of postinfarction remodeling. J Cardiovasc Electrophysiol. 2001;12:1055–61. doi: 10.1046/j.1540-8167.2001.01055.x. [DOI] [PubMed] [Google Scholar]
- 59.Goldspink PH, McKinney RD, Kimball VA, et al. Angiotensin II induced cardiac hypertrophy in vivo is inhibited by cyclosporin A in adult rats. Mol Cell Biochem. 2001;226:83–8. doi: 10.1023/a:1012789819926. [DOI] [PubMed] [Google Scholar]
- 60.Hill JA, Karimi M, Kutschke W, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–9. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 61.Jordan MC, Quednau BD, Roos KP, et al. Cyclosporin A regulates sodium-calcium exchanger (NCX1) gene expression in vitro and cardiac hypertrophy in NCX1 transgenic mice. Ann N Y Acad Sci. 2002;976:259–67. doi: 10.1111/j.1749-6632.2002.tb04748.x. [DOI] [PubMed] [Google Scholar]
- 62.Meguro T, Hong C, Asai K, et al. Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circ Res. 1999;84:735–40. doi: 10.1161/01.res.84.6.735. [DOI] [PubMed] [Google Scholar]
- 63.Mende U, Kagen A, Cohen A, et al. Transient cardiac expression of constitutively active Galphaq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc Natl Acad Sci USA. 1998;95:13893–8. doi: 10.1073/pnas.95.23.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mervaala E, Muller DN, Park JK, et al. Cyclosporin A protects against angiotensin II-induced end-organ damage in double transgenic rats harboring human renin and angiotensinogen genes. Hypertension. 2000;35:360–6. doi: 10.1161/01.hyp.35.1.360. [DOI] [PubMed] [Google Scholar]
- 65.Murat A, Pellieux C, Brunner HR, et al. Calcineurin blockade prevents cardiac mitogen-activated protein kinase activation and hypertrophy in renovascular hypertension. J Biol Chem. 2000;275:40867–73. doi: 10.1074/jbc.M008071200. [DOI] [PubMed] [Google Scholar]
- 66.Oie E, Bjornerheim R, Clausen OP, et al. Cyclosporin A inhibits cardiac hypertrophy and enhances cardiac dysfunction during postinfarction failure in rats. Am J Physiol Heart Circ Physiol. 2000;278:H2115–23. doi: 10.1152/ajpheart.2000.278.6.H2115. [DOI] [PubMed] [Google Scholar]
- 67.Sakata Y, Masuyama T, Yamamoto K, et al. Calcineurin inhibitor attenuates left ventricular hypertrophy, leading to prevention of heart failure in hypertensive rats. Circulation. 2000;102:2269–75. doi: 10.1161/01.cir.102.18.2269. [DOI] [PubMed] [Google Scholar]
- 68.Shimoyama M, Hayashi D, Zou Y, et al. Calcineurin inhibitor attenuates the development and induces the regression of cardiac hypertrophy in rats with salt-sensitive hypertension. Circulation. 2000;102:1996–2004. doi: 10.1161/01.cir.102.16.1996. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z, Nolan B, Kutschke W, et al. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J Biol Chem. 2001;276:17706–11. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- 70.Taigen T, De Windt LJ, Lim HW, et al. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci USA. 2000;97:1196–201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothermel BA, McKinsey TA, Vega RB, et al. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3328–33. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bueno OF, Wilkins BJ, Tymitz KM, et al. Impaired cardiac hypertrophic response in Calcineurin Abeta-deficient mice. Proc Natl Acad Sci USA. 2002;99:4586–91. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Graef IA, Gastier JM, Francke U, et al. Evolutionary relationships among Rel domains indicate functional diversification by recombination. Proc Natl Acad Sci USA. 2001;98:5740–5. doi: 10.1073/pnas.101602398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graef IA, Wang F, Charron F, et al. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–70. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida T, Mishina M. Distinct roles of calcineurin-nuclear factor of activated T-cells and protein kinase A-cAMP response element-binding protein signaling in presynaptic differentiation. J Neurosci. 2005;25:3067–79. doi: 10.1523/JNEUROSCI.3738-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen T, Lindner R, Tedeschi A, et al. NFAT-3 is a transcriptional repressor of the growth-associated protein 43 during neuronal maturation. J Biol Chem. 2009;284:18816–23. doi: 10.1074/jbc.M109.015719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilkins BJ, De Windt LJ, Bueno OF, et al. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–13. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Rooij E, Doevendans PA, de Theije CC, et al. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. 2002;277:48617–26. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 79.Haq S, Choukroun G, Kang ZB, et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–30. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Antos CL, McKinsey TA, Frey N, et al. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2002;99:907–12. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 82.Meberg PJ, Ono S, Minamide LS, et al. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–90. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 83.Homma K, Niino Y, Hotta K, et al. Ca(2+) influx through P2X receptors induces actin cytoskeleton reorganization by the formation of cofilin rods in neurites. Mol Cell Neurosci. 2008;37:261–70. doi: 10.1016/j.mcn.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 84.Kurz JE, Moore BJ, Henderson SC, et al. A cellular mechanism for dendritic spine loss in the pilocarpine model of status epilepticus. Epilepsia. 2008;49:1696–710. doi: 10.1111/j.1528-1167.2008.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pandey D, Goyal P, Dwivedi S, et al. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood. 2009;114:415–24. doi: 10.1182/blood-2008-10-183582. [DOI] [PubMed] [Google Scholar]
- 86.Zhao R, Du L, Huang Y, et al. Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J Biol Chem. 2008;283:36522–31. doi: 10.1074/jbc.M805294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abdul-Majeed S, Moloney BC, Nauli SM. Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell Mol Life Sci. 2011 doi: 10.1007/s00018-011-0744-0. doi: 10.1007/s00018-011-0744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 89.Kim Y, Sung JY, Ceglia I, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–7. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- 90.Suetsugu S, Yamazaki D, Kurisu S, et al. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 91.Yamazaki D, Suetsugu S, Miki H, et al. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–6. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 92.Kawamura K, Takano K, Suetsugu S, et al. N-WASP and WAVE2 acting downstream of phosphatidylinositol 3-kinase are required for myogenic cell migration induced by hepatocyte growth factor. J Biol Chem. 2004;279:54862–71. doi: 10.1074/jbc.M408057200. [DOI] [PubMed] [Google Scholar]
- 93.Yamazaki D, Oikawa T, Takenawa T. Rac-WAVE-mediated actin reorganization is required for organization and maintenance of cell-cell adhesion. J Cell Sci. 2007;120:86–100. doi: 10.1242/jcs.03311. [DOI] [PubMed] [Google Scholar]
- 94.Huang Y, Burkhardt JK. T-cell-receptor-dependent actin regulatory mechanisms. J Cell Sci. 2007;120:723–30. doi: 10.1242/jcs.000786. [DOI] [PubMed] [Google Scholar]
- 95.Gooch JL, Toro JJ, Guler RL, et al. Calcineurin A-alpha but not A-beta is required for normal kidney development and function. Am J Pathol. 2004;165:1755–65. doi: 10.1016/s0002-9440(10)63430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mundel P, Heid HW, Mundel TM, et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kobayashi N. Mechanism of the process formation; podocytes versus neurons. Microsc Res Tech. 2002;57:217–23. doi: 10.1002/jemt.10077. [DOI] [PubMed] [Google Scholar]
- 98.Coward RJ, Foster RR, Patton D, et al. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J Am Soc Nephrol. 2005;16:629–37. doi: 10.1681/ASN.2004030172. [DOI] [PubMed] [Google Scholar]
- 99.Rico M, Mukherjee A, Konieczkowski M, et al. WT1-interacting protein and ZO-1 translocate into podocyte nuclei after puromycin aminonucleoside treatment. Am J Physiol Renal Physiol. 2005;289:F431–41. doi: 10.1152/ajprenal.00389.2004. [DOI] [PubMed] [Google Scholar]
- 100.Asanuma K, Kim K, Oh J, et al. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–98. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–91. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 102.Schlondorff J, Del Camino D, Carrasquillo R, et al. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol. 2009;296:C558–69. doi: 10.1152/ajpcell.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Y, Jarad G, Tripathi P, et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–66. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Merindol V, Mestre M, Essig M. Cyclosporine-induced modifications of proximal tubule actin organization and tPA expression may contribute to the interstitial fibrosis during chronic allograft dysfunction. 43rd Annual Meeting of the American Society of Nephrology, Denver. J Am Soc Nephrol. 2010;21:793A. [Google Scholar]
- 105.Essig M, Terzi F, Burtin M, et al. Mechanical strains induced by tubular flow affect the phenotype of proximal tubular cells. Am J Physiol Renal Physiol. 2001;281:F751–62. doi: 10.1152/ajprenal.2001.281.4.F751. [DOI] [PubMed] [Google Scholar]