

Abstract
Vascular calcification is prevalent in patients with chronic kidney disease and leads to increased cardiovascular morbidity and mortality. Although several reports have implicated mitochondrial dysfunction in cardiovascular disease and chronic kidney disease, little is known about the potential role of mitochondrial dysfunction in the process of vascular calcification. This study investigated the effect of α-lipoic acid (ALA), a naturally occurring antioxidant that improves mitochondrial function, on vascular calcification in vitro and in vivo. Calcifying vascular smooth muscle cells (VSMCs) treated with inorganic phosphate (Pi) exhibited mitochondrial dysfunction, as demonstrated by decreased mitochondrial membrane potential and ATP production, the disruption of mitochondrial structural integrity and concurrently increased production of reactive oxygen species. These Pi-induced functional and structural mitochondrial defects were accompanied by mitochondria-dependent apoptotic events, including release of cytochrome c from the mitochondria into the cytosol, subsequent activation of caspase-9 and -3, and chromosomal DNA fragmentation. Intriguingly, ALA blocked the Pi-induced VSMC apoptosis and calcification by recovery of mitochondrial function and intracellular redox status. Moreover, ALA inhibited Pi-induced down-regulation of cell survival signals through the binding of growth arrest-specific gene 6 (Gas6) to its cognate receptor Axl and subsequent Akt activation, resulting in increased survival and decreased apoptosis. Finally, ALA significantly ameliorated vitamin D3-induced aortic calcification and mitochondrial damage in mice. Collectively, the findings suggest ALA attenuates vascular calcification by inhibiting VSMC apoptosis through two distinct mechanisms; preservation of mitochondrial function via its antioxidant potential and restoration of the Gas6/Axl/Akt survival pathway.
Keywords: vascular smooth muscle cells, vascular calcification, mitochondria, apoptosis, survival, redox status, chronic kidney disease
Introduction
Vascular calcification (VC), a common consequence of aging, atherosclerosis, diabetes and chronic kidney disease (CKD), is a strong independent predictor of increased cardiovascular morbidity and mortality [1–3]. VC is an active cellular process regulated by an imbalance between pro-mineralizing factors including inflammation, oxidative stress and high phosphate and calcium, and calcification inhibitory factors including fetuin-A, matrix gla protein and pyrophosphate [1–4]. Among the pro-mineralizing factors, hyperphosphatemia is a persistent and prevalent problem in CKD patients and has emerged as a major contributor to VC [5, 6].
Previous in vitro studies have demonstrated that vascular smooth muscle cell (VSMC) calcification by elevated inorganic phosphate (Pi) uptake via a sodium-dependent phosphate cotransporter (Pit-1) is caused by both phenotypic transition from VSMCs to osteoblast-like cells and apoptotic cell death [7–12]. Osteoblastic differentiation of VSMCs is mediated by the up-regulation of several osteogenic genes, including core-binding factor-1 (Cbfa-1, also known as Runx2), osteopontin and osteocalcin [8, 12]. In parallel with phenotypic transition of VSMCs into osteoblast-like cells, VSMC apoptosis plays a crucial role in the development of Pi-induced VSMC calcification [7, 9–11]. VC is initiated by apoptotic bodies and matrix vesicles, which are derived from apoptotic and viable VSMCs, respectively, and may serve as a calcification nidus [3, 9, 13]. Apoptotic bodies and matrix vesicles were known to be implicated in VSMC calcification by nucleating insoluble basic calcium phosphate [9, 13, 14]. Furthermore, recent studies have demonstrated that the Pi-induced VSMC apoptosis and subsequent calcification are dependent on the down-regulation of the Gas6/Axl/Akt survival pathway that inhibits apoptosis and increases survival of VSMCs [10, 11]. For instance, 3-hydroxy-3-methylglutaryl CoA reductase inhibitors (statins) protect VSMCs from Pi-induced calcification by suppressing apoptosis via restoration of Gas6/Axl/Akt survival pathway [11].
Mitochondria, in addition to supplying cellular energy, play a central role in the intrinsic apoptotic pathway. Mitochondria-mediated apoptosis involves the release of cytochrome c from the inner membrane space to the cytosol, which in turn triggers the activation of caspase-9 and -3 cascades [15, 16]. These apoptotic events are closely linked to mitochondrial dysfunction, which exhibits changed mitochondrial membrane potential (ΔΨm), increased oxidant generation as a result of the perturbation of electron transport chain reaction, and decreased intracellular ATP content because of oxidant-insulted low respiratory activity [17–19]. Although the precise mechanisms for mitochondria-mediated apoptosis remain to be elucidated, oxidative stress caused by endogenously and exogenously excessive oxidant insults and/or impaired oxidant defenses is generally believed to be key in both mitochondrial dysfunction and cellular apoptosis [20]. Mitochondria-targeted antioxidants could inhibit the peroxidation of mitochondrial components including cytochrome c and consequently block apoptosis [21]. Among the various antioxidants, α-lipoic acid (1,2-dithiolane-3-pentanoic acid, ALA), a naturally occurring antioxidant with anti-apoptotic property [22–25], is a cofactor for mitochondrial metabolic enzymes, pyruvate dehydrogenase and α-ketoglutarate dehydrogenase [22, 24, 26]. ALA is considered the most potent and ideal antioxidant in that it is soluble in both fat and water and is capable of not only directly scavenging oxidants but also boosting levels of other antioxidants such as glutathione, vitamin C and vitamin E [23, 24]. Moreover, ALA has been demonstrated to improve age-associated decline in mitochondrial function and structure and inhibit intrinsic mitochondrial apoptotic pathway in endothelial cells through its antioxidant function [22, 25, 27]. Owing to the multiple beneficial effects of ALA, this compound has been suggested as a potential therapeutic agent for the prevention and treatment of various pathologies including cardiovascular disease, diabetes, liver damage, atherosclerosis and neurodegenerative diseases [23, 24, 28, 29]. In addition, several studies have reported that oxidants are one of major causative factors of VSMC calcification and antioxidants have beneficial effects on therapy in hypertension and CKD [30–33]. Despite the cumulative data, there is little empirical evidence that mitochondrial dysfunction in conjunction with oxidative stress may be implicated in Pi-induced VSMC apoptosis and calcification.
This study found that Pi-induced VSMC apoptosis and calcification and vitamin D3-induced aortic calcification in mice are connected to mitochondrial dysfunction, and that ALA inhibits Pi-induced VSMC calcification by attenuating mitochondrial-mediated apoptosis because of its antioxidant activity and by restoring Gas6/Axl/Akt survival pathway. The data suggest that one of the pleiotropic effects of ALA is a preventive action against VC by maintaining a steady-state mitochondrial function as a consequence of its high reduction capacity.
Materials and methods
Animals and reagents
Six to seven-week-old C57BL6N male mice were purchased from Hyochang Company (Daegu, Korea). The ALA (R/S-α-lipoic acid) was kindly donated by Viatris GmbH & Co. KG (Frankfurt, Germany). Antibodies to Gas6, Axl and cytochrome c oxidase 2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); p-Akt (Ser 473) and Akt from Cell Signaling Technology (Waltham, MA, USA); cytochrome c from Abcam (Cambridge, UK); and β-actin from Sigma-Aldrich (St. Louis, MO, USA). All other reagents except mentioned were acquired from Sigma-Aldrich.
Cell culture and Pi-induced VSMC calcification
VSMCs were isolated from aortas of 5- to 6-week-old male C57BL6N mice by the explant method as previously described [49], with modifications.
The cells were incubated in Dulbecco’s Modified Eagle’s Medium (DMEM; high glucose, 25 mM; Invitrogen, Carlsbad, CA, USA) supplemented with 50% foetal bovine serum (FBS) and antibiotics at 37°C in a humidified atmosphere with 5% CO2. Cells were then maintained in DMEM containing 10% FBS and used for the experiments at four to five passages. To induce in vitro VSMC calcification, VSMCs were grown to confluence and treated with 2.6 mM of Pi (pH 7.4) for 8 days. Fresh media containing Pi were replaced every 2 days until assayed unless otherwise mentioned.
Calcium deposits
After induction of in vitro calcification, calcium deposition in cells was visualized by von Kossa staining and alizarin red S staining as previously described [50, 51]. For calcium quantification, cells were washed with phosphate buffered saline (PBS) and calcium deposited in cells was extracted with 0.6 N HCl for 24 hrs, after which the calcium content was determined colorimetrically using a QuantiChrome™ Calcium Assay Kit (BioAssay Systems, Hayward, CA, USA). The remaining cells were washed three times with PBS and solubilized in a solution containing 0.1 N NaOH and 0.1% sodium dodecyl sulphate (SDS). The protein concentration was estimated using the Bio-Rad DC protein assay (Bio-Rad, Hercules, CA, USA). The calcium content was normalized to protein concentration. For calcium quantification in aortic tissues, whole aortas were obtained from naive or challenged mice, fixed with 4% formaldehyde in PBS for 2 days, washed with PBS three times and water twice, and then processed using von Kossa staining to visualize the deposited calcium in tissues. The aortas were soaked in 5% silver nitrate (AgNO3) solution, exposed to ultraviolet light for 1 hr, incubated in 5% sodium thiosulphate solution for 2 min, and washed with water. Whole aortas were photographed using a FujiFilm S3pro camera. In addition, aortic segments were weighed and decalcified with 0.6 N HCl for 24 hrs, after which calcium content was determined according to the above-mentioned procedure and was expressed as μg/mg of wet aortic tissues.
Measurement of mitochondrial membrane potential and intracellular ATP content
VSMCs were seeded in a 6-well culture plate at a density of 1 × 105 cells per well, grown to confluence, and were further incubated in the absence or presence of Pi for the indicated times. To assess mitochondrial membrane potential, cells were loaded with 2 μg/ml rhodamine 123 for 30 min, washed with PBS, trypsinized, centrifuged and resuspended in culture medium. At least 10,000 events of the stained cells were analysed using a FACSCalibur flow cytometer (Becton-Dickinson, Sydney, Australia) with an excitation wavelength of 488 nm and an emission wavelength of 530 nm and the fluorescence index was represented as a fold-difference relative to control. To measure ATP contents, cells were washed twice with ice-cold PBS and kept at −70°C until assayed, after which cells were lysed by passage several times through a 31-gauge syringe and subjected to centrifugation at 10,000 ×g for 10 min at 4°C. The resulting supernatant was freed of protein by adding trichloroacetic acid to a final concentration of 2.5%, neutralized to pH 7.75 with 10 N NaOH, and assayed for ATP with an ENLITEN® Total ATP Rapid Biocontamination Detection Kit (Promega, Madison, WI, USA) according to the manufacture’s instruction and a SpectraMAX Gemini EM (Molecular Devices, Sunnyvale, CA, USA) to measure the luminescence. ATP contents were normalized by the protein concentration of the resulting supernatant determined with Bradford reagents.
Electron microscopic analysis of mitochondrial morphological change
The specimens were fixed with 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.5) for 4 hrs at 4°C and washed with 0.1 M sodium phosphate buffer. After fixation with 2% osmium tetroxide in 0.1 M sodium phosphate buffer for 90 min at 4°C, the samples were dehydrated in a series of ethanol solutions and then immersed overnight in a mixture (1:1) of propylene oxide and Epon resin to allow the infiltration of Epon into the samples at room temperature, followed by embedment in Epon resin at 60°C. The Epon-embedded specimens were sectioned at 80 nm with an ultramicrotome (Leica, Heerbrugg, Switzerland), adhered to copper grids, stained with uranyl acetate and lead citrate (Polyscience, Warrington, PA, USA) and then examined using a model H-7000 transmission electron microscope (Hitachi, Tokyo, Japan) at an acceleration voltage of 75 kV.
TUNEL assay
VSMCs were seeded on glass cover slips placed in wells of six-well culture plates and treated with Pi in the presence or absence of ALA (300 μM) for 6 days. After cells were fixed with 1% formaldehyde in PBS for 15 min at room temperature, the fragmented apoptotic DNA was labelled with ApopTag® Fluorescein Direct In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA) according to the manufacturer’s instructions. Cell nuclei were counterstained with 1 μg/ml propidium iodide and visualized using a TCS SP2 AOBS fluorescence microscope (Leica). In vivo TUNEL assays were using ApopTag Peroxidase In Situ Apoptosis Detection kit (Chemicon, Temecula, CA, USA), as previously described [52]. In brief, cross-sections (4 μm thickness) of aortas were deparaffinized, rehydrated and digested by proteinase K method. Sections were then incubated with fresh 3% hydrogen peroxide for 10 min, washed with PBS-Tween 20, incubated with TUNEL reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and digoxigenin-conjugated dUTP for 1 hr at 37°C followed by incubation of anti-digoxigenin conjugate (peroxidase) and DAB kit solution. TUNEL-stained sections were visualized under light microscopy.
Validation for cytochrome c release from mitochondria
For preparation of cytosolic and mitochondrial fraction, cells were washed with PBS, resuspended in a hypotonic solution containing 20 mM HEPES (pH 7.4), 10 mM KCl, 2 mM MgCl2 and 1 mM EDTA, and incubated on ice for 15 min. After cells were homogenized by several passages through a 25-gauge syringe and centrifuged at 10,000 ×g for 10 min, the resultant supernatant and pellet were used as a cytosolic and mitochondrial fraction, respectively. The release of cytochrome c from mitochondria into the cytosol was determined by Western blot using antibodies against cytochrome c, cytochrome c oxidase 2 and β-actin.
Caspase activity assay
After cells were lysed and centrifuged as described in a caspase-9 and -3 colorimetric assay kit (R&D Systems, Minneapolis, MN, USA), the resulting supernatant (100–200 μg) was assayed for caspase-9 or -3 activity in the presence of colorimetric substrate and the absorbance was measured at 405 nm.
Western blot analysis
Whole cell extracts were prepared with a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 2 mM β-glycerophosphate and 1× protease inhibitor cocktail (Roche, Basel, Switzerland) and resolved by 10% SDS-polyacrylamide gel electrophoresis. The separated proteins were electroblotted onto an Immobilon-P membrane (Millipore, Billerica, MA, USA) and probed with primary antibodies and appropriate horseradish peroxidase-conjugated second antibodies. The specific band was detected by enhanced luminol-based chemiluminescent substrate (Lab Frontier, Seoul, Korea).
Determination of oxidant generation and thiol content
To estimate oxidant generation, VSMCs were incubated in the presence of 20 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA) for 10 min and washed with PBS, and the formation of the fluorescent product was monitored by a Leica TCS SP2 AOBS microscope with an excitation wavelength of 488 nm and an emission wavelength of 510–550 nm. To ascertain total thiol contents, the cytosolic fraction was extracted and supplied with 5,5′-dithio-bis(2-nitrobenzoic acid; DTNB) as previously described [53]. Briefly, cytosolic extracts were reacted in a buffer containing 80 mM sodium phosphate (pH 8.0), 250 μM DTNB, 2 mM EDTA and 2% SDS for 30 min and the absorbance was read at 405 nm. Each value was normalized by protein contents.
Murine model of vitamin D3-induced aortic calcification
A vitamin D3-induced aortic calcification mouse model was established. Briefly, 6-week-old C57BL6N male mice were injected intraperitoneally with PBS as control or ALA (40 mg/kg/day) at 1 day intervals for 13 days. At day 3 after the initial injection of ALA, vitamin D3 (cholecalciferol, 5.5 × 105 IU/kg/day) was administrated subcutaneously into mice daily for 3 days. Six days after the final exposure to vitamin D3, mice were anaesthetized with pentobarbital and the whole aortas were collected by surgical dissection to determine calcium content using von Kossa staining and calcium assay.
In situ aortic superoxide anion detection
To estimate oxidant generation in aortic tissues, dihydroethidium (DHE) staining was performed as previously described [31]. DHE is a fluorescent dye that is permeable to cells and specifically reacts with intracellular superoxide anion. Cryosections of aortas (25 μm thickness) were placed in glass slides, incubated with DHE (3 μM), coverslipped and maintained in a light-protected humidified chamber at 37°C for 30 min. To verify if fluorescence signal was superoxide-derived, slides of adjacent sections were incubated with 500 U/ml polyethylene glycol-conjugated superoxide dismutase (Peg-SOD) for 30 min. Images were obtained with an Olympus fluoview FV1000 confocal laser scanning microscope and FV10-ASW ver2.0 software (Olympus, Germany).
Statistical analysis
All results are represented as means ± S.D. from at least three independent experiments. Statistical analyses were performed using Student’s t-test. The level of significance was set at P < 0.05.
Results
Pi-induced calcification causes mitochondrial dysfunction in VSMCs
To induce VSMC calcification, cells were cultured with Pi at a final concentration of 2.6 mM, which is similar to Pi level of hyperphoshatemia in CKD. Pi-induced VSMC calcification was verified by morphological change (nodule formation) and increased calcium deposits in VSMCs, as visualized by von Kossa staining and Alizarin red S staining, and determined by quantification of calcium content (Fig. 1A and B). Next, to examine whether mitochondrial dysfunction is involved in VSMC calcification, changes in mitochondrial membrane potential and cellular ATP levels were examined during Pi-induced VSMC calcification. Intriguingly, mitochondrial membrane potential was gradually decreased during Pi-induced VSMC calcification, accompanied by decline in intracellular ATP levels (Fig. 1C and D). Oxygen consumption was also modestly reduced at the late stage of Pi-induced calcification (Fig. S1). These results suggest that mitochondrial metabolic function is perturbed during Pi-induced VSMC calcification.
Fig 1.
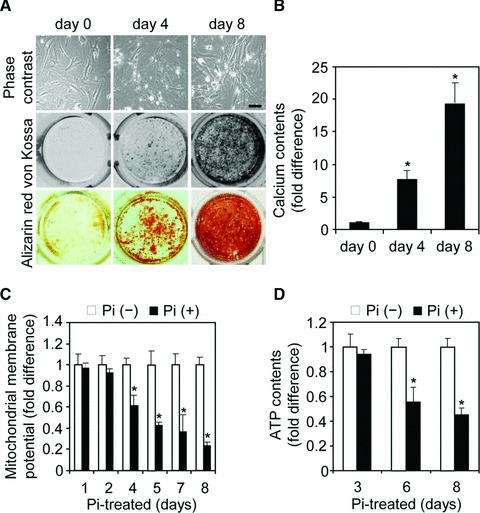
Impairment of mitochondrial function during Pi-induced VSMC calcification. (A, B) Pi-induced VSMC calcification. VSMCs (2 × 104 cells/well) were seeded in a 48-well culture plate and cultured until confluence. After cells were treated with 2.6 mM Pi for the indicated times, calcium deposits were visualized by staining with von Kossa (brown or black) and alizarin red S (red) (A), and calcium content was measured (B). A representative experiment is presented in von Kossa and alizarin red S analysis. Scale bar in (A) indicates 200 μm. (C, D) Declines in mitochondrial membrane potential and intracellular ATP levels during Pi-induced VSMC calcification. VSMCs (1 × 105 cells/well in a six-well culture plate) were grown to confluence and then treated with Pi for the indicated times. Mitochondrial membrane potentials (C) and intracellular ATP levels (D) were measured by flow cytometric analysis of rhodamine 123 fluorescence and by a bioluminescence assay, respectively. Data are expressed as the mean ± S.D. (n= 3). *P < 0.01 versus Pi-untreated control.
ALA decelerates Pi-induced VSMC calcification via reversal of mitochondrial dysfunction
It is well known that ALA plays a fundamental role in enhancing mitochondrial metabolism and ATP production [23, 24, 26, 27]. Appropriately, the effect of ALA on disturbed mitochondrial function caused by Pi in VSMCs was evaluated. The decline in mitochondrial membrane potential and ATP production in Pi-induced VSMC calcification was restored by ALA treatment (Fig. 2A and B). Moreover, electron microscopic analysis revealed that ultrastructural alterations in mitochondria with disrupted cristae by Pi were considerably reversed by ALA (Fig. 2C). Consistent with the beneficial effects of ALA on mitochondrial metabolism and structure, ALA significantly attenuated Pi-induced VSMC calcification (Fig. 2D). Taken together, these results support the suggestion that ALA inhibits Pi-induced VSMC calcification via recovery of mitochondria metabolism and structure.
Fig 2.

ALA attenuates Pi-induced VSMC calcification via reversal of mitochondrial dysfunction and structural damage. (A–C) ALA blocks Pi-induced mitochondrial perturbation. Cells were treated with Pi for 6 days in the presence or absence of ALA (300 μM) and mitochondria membrane potential (A) and cellular ATP levels (B) were assessed. Data are expressed as the mean ± S.D. (n= 3). To verify the changes in mitochondrial structure, specimens were processed for transmission electron microscopy (C). A representative experiment is presented. Scale bar indicates 250 nm. (D) Inhibitory effect of ALA on Pi-induced VSMC calcification. After VSMCs (2 × 104 cells/well in a 48-well culture plate) were treated with Pi in the presence or absence of ALA (300 μM) for 4 and 8 days, calcium content was measured (upper) and calcium deposits were visualized by von Kossa and alizarin red S staining (lower). Data are expressed as the mean ± S.D. (n= 3). *P < 0.01; †P < 0.05.
ALA suppresses Pi-induced mitochondria-mediated apoptosis and restores Gas6/Axl/Akt survival signal down-regulated by Pi
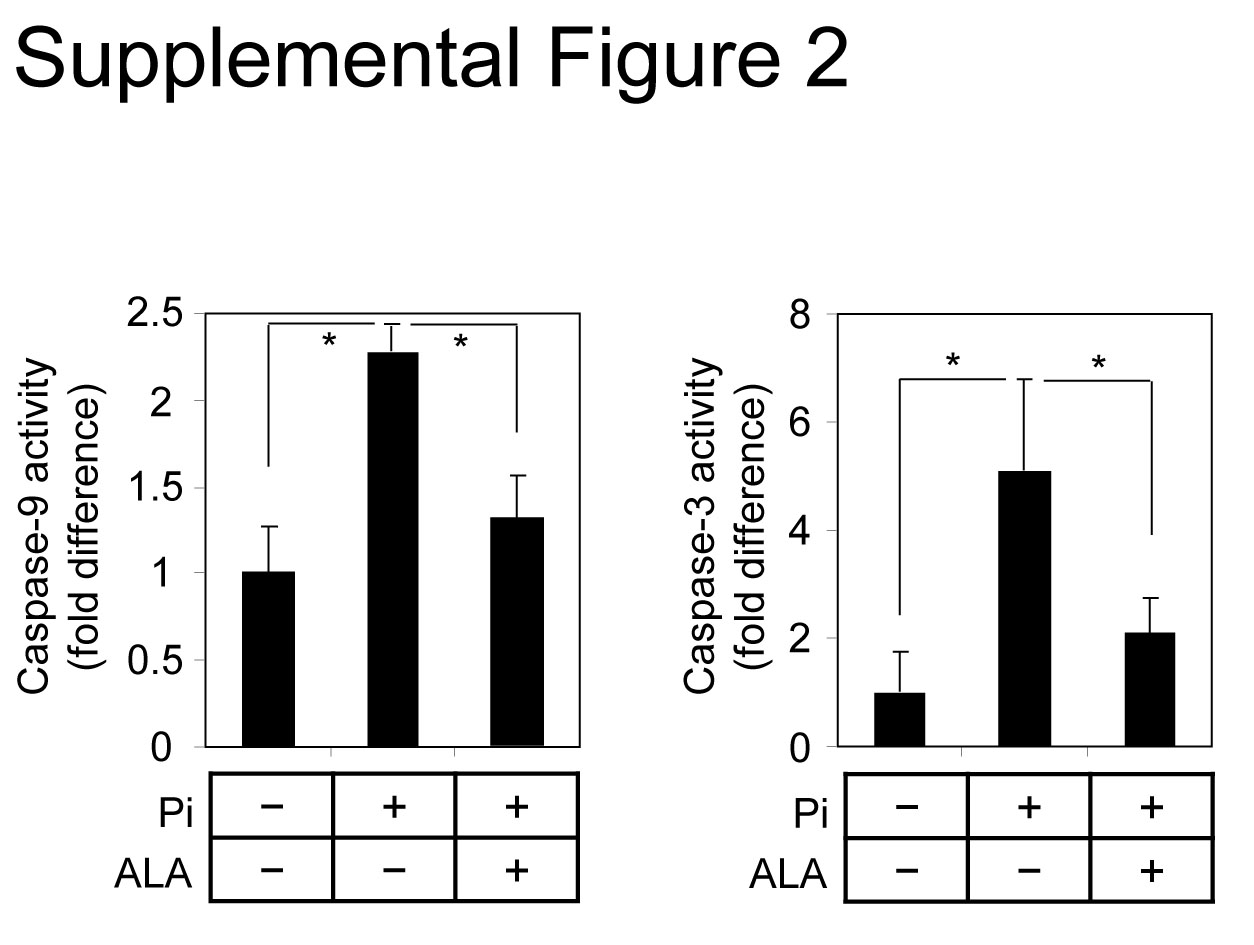
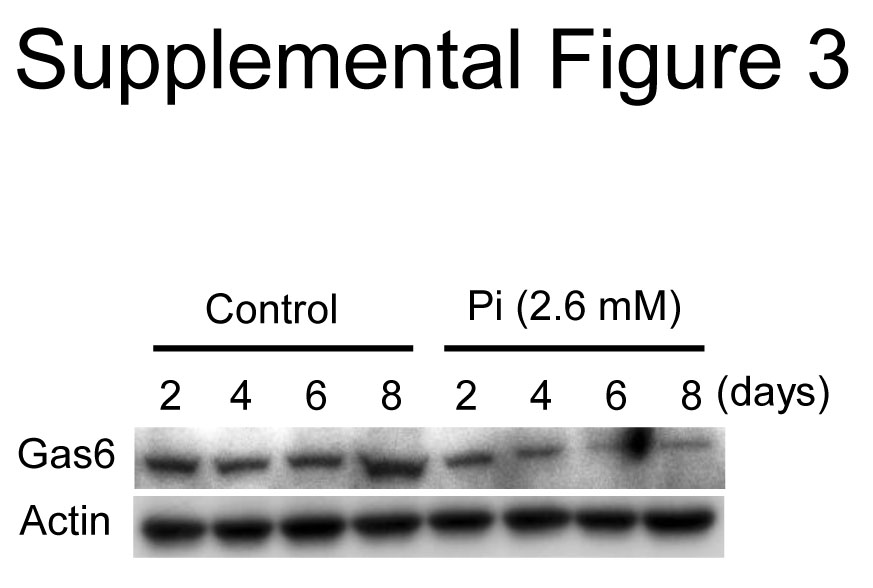
It has been demonstrated that mitochondria dysfunction contributes to cell apoptosis [17–19] and that apoptosis plays a critical role in Pi-induced VSMC calcification [7, 9–11]. Therefore, the effect of ALA on Pi-induced VSMC apoptosis was assessed. As shown in Figure 3A, terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining indicated that apoptotic cell death was obviously increased in Pi-treated VSMCs and that apoptosis was significantly inhibited by ALA. It was also observed that ALA blocked Pi-induced sequential activations of the intrinsic mitochondrial apoptotic pathway (Fig. 3B and C): release of cytochrome c from mitochondria into cytosol, activation of caspase-9, an initiator caspase, and activation of caspase-3, an executioner caspase. Consistent with the inhibitory effect of ALA on Pi-induced caspase activation in mouse aortic VSMCs, ALA inhibited the Pi-induced activation of caspase-9 and -3 in human aortic SMCs (Fig. S2). Pi-induced VSMC apoptosis and calcification have been reportedly shown to be associated with down-regulation of Gas6/Axl/Akt-mediated survival signals [10, 11]. Therefore, the next experiment investigated the effect of ALA on survival signals in VSMCs. Pi markedly suppressed the expression of Gas6 and Axl and led to Akt inactivation, and this down-regulation of Gas6/Axl/Akt signalling pathway was almost completely reversed by ALA (Figs S3 and 3D). Taken together, these findings offer evidence that ALA protects VSMCs from Pi-induced calcification through inhibition of mitochondria-mediated apoptotic pathway by reversal of mitochondrial dysfunction as well as restoration of Gas6/Axl survival pathway.
Fig 3.
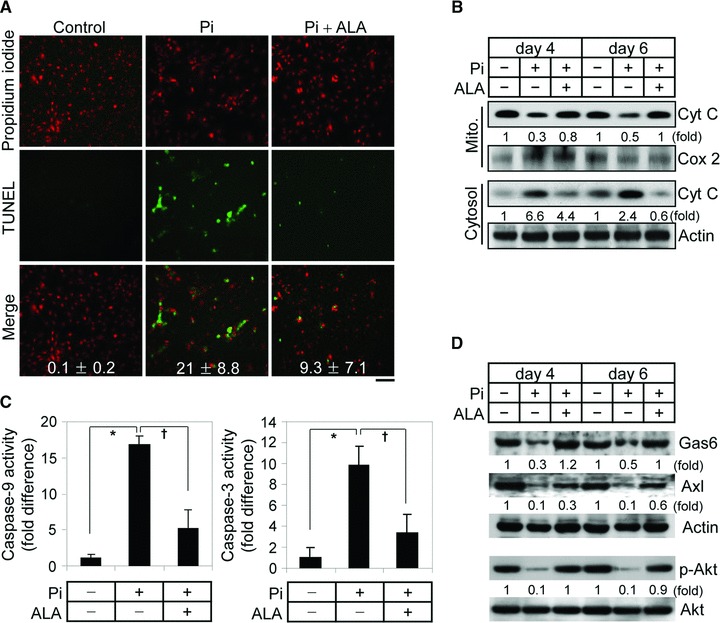
ALA inhibits mitochondria-mediated apoptosis of VSMCs induced by Pi and restores Gas6-Axl survival pathway. (A–C) Inhibition of Pi-induced VSMC apoptosis by ALA. VSMCs were treated with Pi in the presence or absence of ALA (300 μM) for 4 days (A, C) or indicated times (B). Apoptotic cells were detected by TUNEL staining (A). Nuclei were counterstained with propidium iodide and then the two images were merged. TUNEL-positive apoptotic cells were counted as a percentage of the total number of cells. Scale bar indicates 100 μm. To assess the release of cytochrome c from mitochondria into cytosol (B), mitochondrial and cytosolic fractions were subjected to Western blotting with antibodies against cytochrome c (Cyt C), cytochrome c oxidase 2 (Cox 2, a mitochondrial marker protein) and β-actin (a cytosolic marker protein). Cytosolic extracts were analyzed for caspase-9 or caspase-3 activity. The activity was normalized to protein content and expressed as a fold difference relative to control (C). Data are expressed as the mean ± S.D. (n= 3). (D) ALA restored Gas6 and Axl expression and Akt activation, which are down-regulated by Pi. Whole cell lysates were obtained from VSMCs treated with Pi for the indicated times and subjected to Western blotting with specific antibodies to Gas6, Axl, β-actin, phospho-Akt and Akt. *P < 0.01; †P < 0.05.
ALA blocks oxidative stress-induced VSMC calcification by inhibiting apoptosis and sustaining survival
Increasing evidence suggests that oxidants induce the perturbation of electron transport chain reaction of mitochondria and vice-versa, resulting in the accumulation of excessive oxidants and, ultimately, mitochondria-mediated apoptosis through an altered mitochondrial membrane potential and release of apoptotic factors including cytochrome c[15, 16]. Recent studies have potentially implicated exogenous and endogenous oxidants in VSMC calcification [30, 31]. Hence, the effects of mitochondria-originated oxidants on Pi-induced VSMC apoptosis and calcification were assessed. As shown in Figure 4A and B, incubation of VSMCs with Pi resulted in an increased generation of oxidants and the simultaneous oxidation of cellular thiols, showing that intracellular reduction capacity is decreased in Pi-treated VSMCs. In addition, Pi in combination with antimycin A, a mitochondrial complex III inhibitor that is able to produce superoxide anion by the reaction of electrons leaked from mitochondria complex III with oxygen [34], induced a greater increase of oxidant generation and thiol oxidation than Pi alone. In concert with oxidant production under the treatment of Pi or Pi plus antimycin A, VSMC calcification was highly dependent on the extent of oxidation status (Fig. 4C). Consistent with previous findings that ALA directly scavenges oxidants and induces an increase of antioxidants including glutathione [23–25]. The present data show that ALA effectively diminishes Pi-induced oxidant generation and maintains the constant level of cellular thiol groups, and as a result, inhibits Pi and Pi/antimycin A-induced calcification (Fig. 4A–C). ALA totally blocked cytochrome c release by Pi and Pi plus antimycin A, and restored Gas6-Axl, whose expression is down-regulated by Pi and Pi plus antimycin A (Fig. 4D and E). These observations supported the suggestion that ALA possesses antioxidant capacity and the protective effect of ALA on VSMC calcification may be closely connected to the inhibition of mitochondria-mediated apoptosis and restoration of Gas6/Alx survival pathway.
Fig 4.
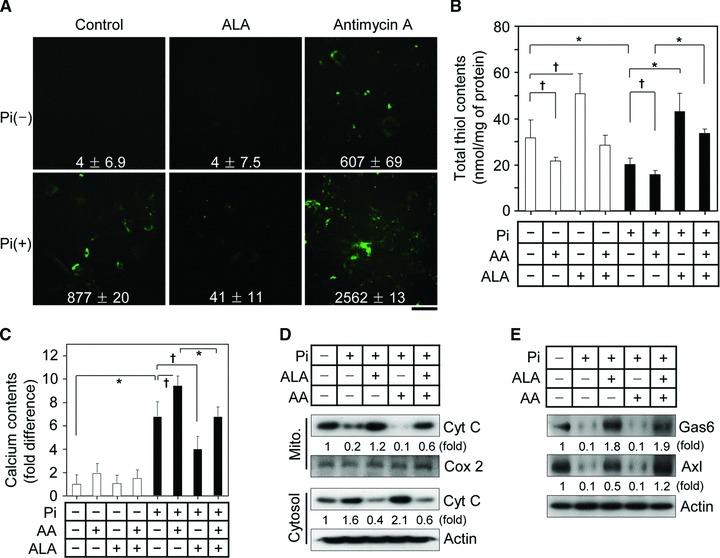
ALA blocks Pi-induced oxidant generation in VSMCs and decelerates Pi-induced VSMC calcification by inhibiting apoptosis and stimulating survival. (A, B) ALA suppresses Pi-induced oxidant production and thiol oxidation in VSMCs. After VSMCs were treated with Pi in the presence or absence of ALA (300 μM) or antimycin A (AA, 20 μM), an oxidant generator by inhibiting mitochondrial complex III, for 4 days, oxidant formation was assessed by confocal microscopy using the fluorescent probe DCFH-DA (A) and intracellular thiol content was measured (B). The fluorescence index was represented as the collected values of fluorescence intensity for the section of photography using Image-Pro Plus version 6.0. Scale bar indicates 100 μm. Calcium content was determined (C). Data in (B) and (C) are expressed as the mean ± S.D. (n= 3). (D, E) After VSMCs were treated with combination between Pi, ALA and AA for 4 days, release of Cyt C from mitochondria to cytosol (D) and cell survival Gas/Axl signal in whole cell extracts (E) were analysed using Western blotting with specific antibodies to Cyt C, Cox-2, Gas6, Axl and β-actin. *P < 0.01; †P < 0.05.
ALA inhibits vitamin D3-induced aortic calcification in mice
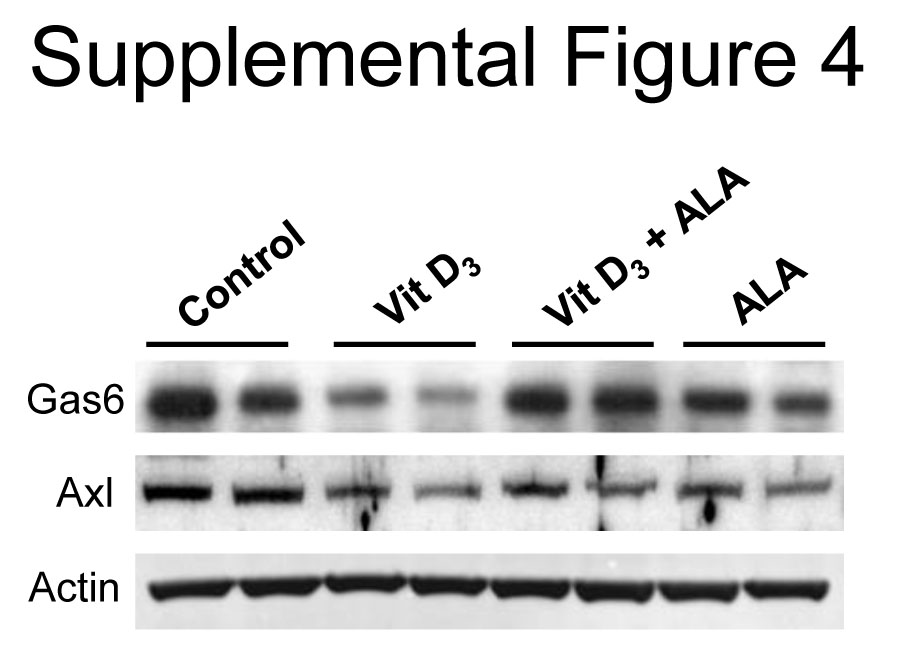
Supra-physiological dosages of vitamin D analogues are associated with ectopic calcification of soft tissues including the aorta [35, 36]. To further confirm the protective effect of ALA against VSMC calcification in vivo, an aortic calcified mouse model was established by subcutaneous administration of vitamin D3 toxic dosages. Vitamin D3-mediated aortic calcification in the mice was visualized by von Kossa staining and quantified by calcium content in a whole aortic tissue. Microscopic examination in the von Kossa-stained cross-sections showed a prominent calcification in the medial layer of the aorta (Fig. 5A and B). This calcification was considerably diminished by the intraperitoneal injection of ALA at a dosage of 40 mg/kg/day. As shown in Figure 5C and D, the superoxide anion production and TUNEL-positive apoptotic cells were strongly increased and the level of Gas6 and Axl expression was decreased in the aorta of vitamin D3-administered mice compared to control. ALA significantly reduced these vitamin-D3-induced superoxide production and apoptotic cell death along with restoration of Gas6 and Axl expression in the aorta (Fig. S4). In line with the beneficial effect of ALA on Pi-induced mitochondrial function in VSMCs, vitamin D3-induced mitochondrial cristae disruption was greatly attenuated by ALA (Fig. 5E).
Fig 5.
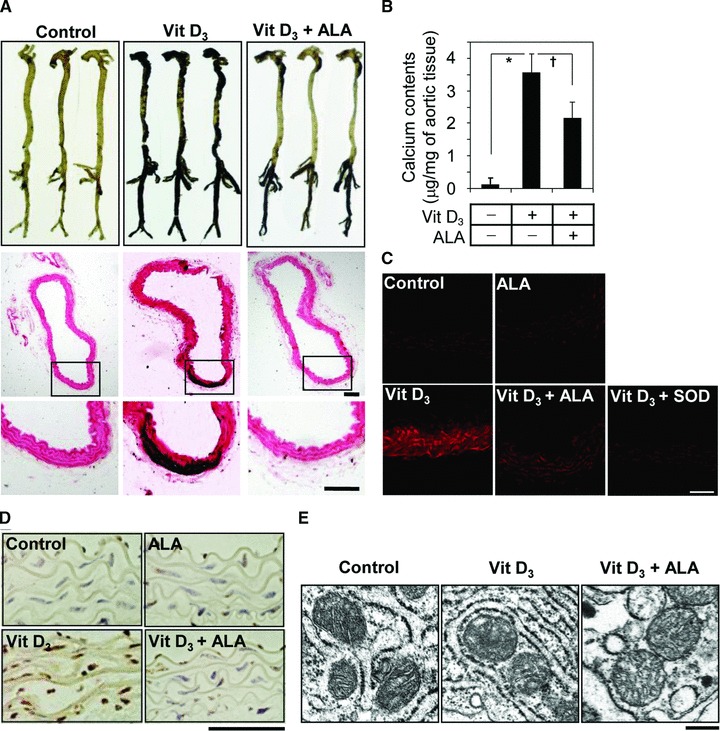
ALA protects vitamin D3-induced aortic calcification in mice. (A) von Kossa staining for mineral deposition in aorta. Whole aortas were dissected from naïve and challenged mice and stained with von Kossa (upper). n= 3. Cross-sectioned specimens of the middle part of thoracic aorta were stained with von Kossa, counterstained with nuclear fast red, and visualized by light microscopy (middle). The boxed area of middle panel is enlarged in lower panel. (B) Calcium content of whole aortas was measured and normalized by semi-wet dried weight of aortic tissues. Data are expressed as the mean ± S.D. (n= 3). (C) To detect superoxide anion in aortic tissues, cryosections of aortas freshly harvested at 6 days after final vitamin D3 injection were stained with DHE and the fluorescence images were captured under confocal microscopy. Peg-SOD was used to verify that the fluorescent signals are specific for superoxide anion. (D) To detect apoptotic cells in aortic tissues, aortas were sectioned, stained with TUNEL, and visualized under light microscopy. (E) Electron microscopic images of mitochondria present on aortic tissues. A representative experiment in (A) and (C–E) is presented. Scale bars in (A) and (C–E) indicate 100, 50, 200 and 500 nm, respectively. *P < 0.01; †P < 0.05.
Discussion
In this study, ALA blocked Pi-induced oxidant generation and thiol oxidation and mitochondrial dysfunction concurrently in VSMCs. Consequently, this affirmative property of ALA prevented Pi-induced VSMC calcification by both the restoration of Gas6/Axl survival signals and suppression of mitochondria-mediated apoptosis (Fig. 6).
Fig 6.
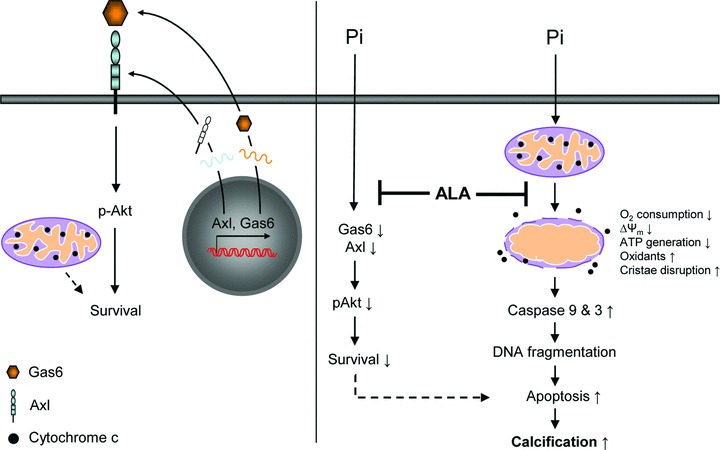
Schematic representation for possible mechanisms underlying the protective role of ALA against vascular calcification. In normal conditions, Gas6/Axl interaction and sequential Akt activation are involved in maintenance of VSMC survival in an autocrine mode. During Pi-induced calcification, VSMCs display a considerable decrease in survival because of down-regulation of Gas6/Axl/Akt signalling and an increase in mitochondria-mediated apoptosis. This apoptosis is thought to cause mitochondria perturbation by insulting accumulated oxidants. ALA may protect against aortic calcification at least in part by rescuing Gas6–Axl survival pathway and by inhibiting mitochondrial-mediated apoptosis in VSMCs through improvement of mitochondria function by its antioxidant potential. The dashed line indicates an unresolved issue to be determined.
Various diseases including aging, atherosclerosis, hypertension, diabetes and CKD that have been linked to a high prevalence of VC are associated with increased oxidative stress [7, 21, 37]. Moreover, recent reports have demonstrated that elevated oxidant signals are mainly observed around the calcifying foci of the aortic valve and exogenous hydrogen peroxide accelerates VSMC calcification in the osteogenic media containing ascorbic acid, β-glycerophosphate and dexamethasone [30, 31]. In general, oxidative stress is generated in cells by diverse sources. One is superoxide anion formation by the reductive reaction of electrons leaked from mitochondria at the level of the respiratory chain with oxygen [34]. Other sources include multiple enzymatic systems including NADPH oxidase, lipooxygenase, cyclooxygenase, xanthine oxidase and uncoupled nitric oxide synthase [38]. Presently, VSMCs treated with Pi, which acts as an inducer for calcification when exposure to excess Pi concentration, represented mitochondrial dysfunction. This mitochondrial perturbation may be blocking the flow of electrons through mitochondrial electron transporters, resulting in oxidant generation and thiol oxidation of cellular constituents. Pi overload induced increased intracellular and mitochondria-derived oxidant production and ALA inhibited the intracellular and mitochondrial oxidant generation induced by Pi. Tempol, a superoxide dismutase-mimetic antioxidant, also showed protective effect against Pi-induced mitochondrial oxidant generation and VSMC calcification (Fig. S5). These results suggest that Pi-induced oxidative stress may trigger mitochondrial oxidant accumulation and VSMC calcification and that there is a causal connection between oxidative stress and the induction of mitochondrial dysfunction [17–19]. To prove this, further study is necessary to reveal the precise causal connection between ALA and mitochondrial function. Presently, increased oxidant generation by antimycin A, which is an inhibitor of complex III that is principle in the leakage of electrons during mitochondrial respiratory chain through complex I–IV, accelerated VSMC calcification. Moreover, treatment of VSMCs with Pi showed an increase of oxidant generation and thiol oxidation. In addition, it has been reported that oxidants could trigger mitochondrial dysfunction [15, 16]. The combined results indicate that an increase of oxidant generation induced by Pi treatment could directly or indirectly contribute to mitochondrial dysfunction and consequently calcification, even if detailed mechanisms underlying the connection between Pi-induced mitochondrial dysfunction and oxidative stress in the process of calcification await further investigation.
Excessive oxidant damages mitochondrial metabolic enzymes and alters mitochondrial membrane permeability, leading to mitochondria dysfunction and cell apoptosis. There is a close link between oxidative stress, mitochondrial dysfunction and apoptosis [17–19]. In the intrinsic mitochondrial-mediated apoptotic pathway, the oxidation of mitochondrial protein thiol groups promotes a decrease in mitochondrial membrane potential, the release of cytochrome c and the activation of caspase-9 and -3, eventually leading to apoptotic cell death [17, 39]. In concert with combined data that antioxidants can inhibit apoptotic serial events [21], antioxidants would be anticipated to have protective effects against Pi-induced VSMC apoptosis, one of the causative factors for calcification. For instance, ALA with reduction potential inhibits apoptosis of various cell types including human bone marrow stromal cells, endothelial cells and hepatocytes [25, 40–42]. The present results indicate that apoptosis during Pi-induced VSMC calcification is associated with intrinsic mitochondrial-mediated apoptotic sequential events, and that ALA blocks both Pi-induced VSMC apoptosis and calcification as a result of its high reduction capacity. In addition, it has been demonstrated that the expression of Gas6 and Axl that is involved in cell survival in a range of cell types [43] is down-regulated during VC, leading to apoptosis and consequent calcification. To maintain cell survival, the binding of Gas6 to its cognate receptor Axl induces phosphatidylinositol 3-OH kinase (PI3K) activation and subsequent Akt activation, showing sequential events of the Gas6/Axl/PI3K/Akt signal. ALA activates PI3K/Akt through direct binding to the tyrosine kinase domain of the insulin receptor [42], but the nature of the ALA-regulated upstream regulators of the PI3K/Akt pathway are unclear. The present results demonstrate that ALA inhibits Pi-induced apoptosis and calcification through the restoration of Gas6 and Axl expression, which is down-regulated by excessive Pi in vitro and vitamin D3 toxicity in vivo and one of the upstream regulators of PI3K/Akt survival pathway. We also found that AMP-activated protein kinase (AMPK) mediates the protective effect of ALA against Pi-induced VSMC calcification through restoration of Gas6 gene expression (Fig. S6). However, further analyses of the possible association of Gas6/Axl/Akt/AMPK survival pathway with mitochondrial dysfunction are necessary.
Both bone-forming osteoblasts and VSMCs are driven from common mesenchymal stem cells [44]. Thus, VSMCs can transdifferentiate into osteoblast-like cells that are able to deposit calcium. Although calcification by osteoblasts in bone is a normal process to maintain bone homeostasis, calcification of VSMCs results in a lethal cardiovascular disease. VC is closely connected to bone decalcification (osteoporosis): (i) the patients with VC who are ageing, diabetic and CKD have a high clinical incidence of osteoporosis [45]; (ii) statins, which are used in the treatment of CKD, have been proven to reduce the coronary calcification as well as to increase bone mineralization [45] and (iii) bisphosphonates, which are used to treat osteoporosis, are effective to prevent VC in CKD patients [3, 46]. However, long-term use of bisphosphonate in stage 4 or 5 CKD raises efficacy and safety concerns [3]. Presently, ALA effectively inhibited Pi-induced VSMC calcification in vitro and vitamin D3-induced aortic calcification in mice. Decreased VC by ALA could alleviate vascular stiffness and as a result, may help reduce a high clinical incidence of cardiovascular-related diseases such as hypertension and cardiac hypertrophy. Moreover, ALA has also been suggested to protect bone loss by inhibition of oxidant and tumour necrosis factor-α-induced apoptosis of bone marrow stromal cells (osteoblast precursors) and suppression of formation of bone-degrading osteoclasts through its antioxidant and anti-inflammatory function [40, 47, 48]. Consequently, the present and previous findings suggest a possibility that ALA might be used for the treatment of VC in CKD patients without bone loss or possibly with enhanced bone formation, although further in vivo studies are required to verify this possibility.
In summary, VC could be determined by cellular redox status and changes in mitochondrial function. ALA, which is known to potentiate reduction capacity and mitochondrial metabolic function, blocks Pi-induced VSMC calcification through inhibition of mitochondria-mediated apoptosis and restoration of the Gas6/Axl/Akt survival pathway. This study provides evidence that targeted candidates, which are able to enhance mitochondrial function and to have antioxidant potential, may offer a prospective therapeutic strategy for the prevention and treatment of VC.
Acknowledgments
This work was supported, in part, by grants from the Korea Science and Engineering Foundation (KOSEF) (Nos. 2010-0019511, 2010-0001742, 2010-0020532 and 2010-0008023 to I.-K.L. and Nos. 2010-0001240 and 2009-0077641 to D.J.) and by the Grant of the Korean Ministry of Education, Science and Technology” (The Regional Core Research Program/Anti-aging and Well-being Research Center). H.K. and J.-Y.K. were supported by the National Research Foundation funded by the Korean Government (NRF-2009-351-C00040) and by Basic Science Research Program through the National Research Foundation of Korea (2010-0023794), respectively.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Oxygen consumption via mitochondrialrespiratory chain is progressively declined during Pi-induced VSMCcalcification. VSMCs (1 x 104 cells/well/200 μl) wereseeded into wells of a 96-well plate with the BD™ OxygenBiosensor System (BD Biosciences, Franklin Lakes, NJ, USA). Aftercells were adapted for 12 h and treated without or with Pi (2.6mM), the value of fluorescence was measured at the indicated timesusing a SpectraMAX Gemini EM (Molecular Devices) with an excitationat 485 nm and an emission at 590 nm. Data are expressed as the mean± S.D. (n = 3). *P < 0.01;†P < 0.05 versus Pi-untreated control.
{kind=link}
ALA inhibits Pi-induced activation of caspase-9and -3 in human aorta-derived SMCs. The aortas from human wereprepared by surgical dissection and applied to the experiments.Cells were treated as in Figure 3C and then assessed for caspase-9and -3 activities. Data are expressed as the mean ± S.D. (n= 3) *P < 0.01.
{kind=link}
The expression of Gas6 is gradually downregulatedduring Pi-induced VSMC calcification. VSMCs were incubated with Pifor the indicated times and whole cell extracts prepared weresubjected to Western blotting with antibodies to Gas6 andβ-actin.
{kind=link}
ALA rescues decreased protein expression of Gas6and Axl in the aortas of vitamin D3-treated mice tonormal level. Four days after final injection of vitaminD3, aortas were harvested from control and vitaminD3-administrated mice and whole lysates were subjectedto Western blot analysis using specific antibodies to Gas6, Axl andβ-actin.
{kind=link}
ALA or tempol inhibits mitochondria-derivedoxidant generation and VSMC calcification by Pi. (A) Todetect mitochondria-derived oxidant, VSMCs treated with Pi in thepresence or absence of ALA or Tempol (100 μM) for 6 days wereincubated in the presence of 50 nM Mitotracker Red CMH2XRos(Invtrogen, Carlsbad, CA) at 37°C for 30 min and washed withPBS, and the formation of the oxidant product was assessed underfluorescence microscopy. The fluorescent intensity per photographysection was measured using Image-Pro Plus version 6.0. (B)Cells as in (A) were stained with von Kossa to visualizecalcium deposits. A representative experiment is presented. Scalebars in (A) and (B) indicate 100 Ìm and 500μm, respectively.
{kind=link}
ALA rescues the Gas6 gene expression and inhibitsVSMC calcification via AMPK. (A) VSMCs (4 x105 cells/well in a 6-well culture plate) were treatedwith Pi in the presence or absence of ALA plus compound C (AMPKinhibitor, 5 μM) and PD98059 (ERK inhibitor, 10 μM) for 6days and stained with von Kossa to detect calcium deposits. Arepresentative experiment is presented. Scale bar indicates 500μm. (B) Total RNA was prepared using TRIzol reagent(Invitrogen, Carlsbad, CA). For semi-quantitative RT-PCR, 2 μgof total RNA was used to synthesize the first strand cDNA using theFirst Strand cDNA synthesis kit (Fermentas, EU). The first strandcDNAs were amplified by PCR according to the following PCRparameters: 94°C for 30 s, 59°C for 30 s, and 72°C for30 s, 25 cycles. Primers used in the PCR were as follows: Gas6forward, 5′-GAA GCA GTT GGT GGT CCT GGC-3′ and reverse,5′-CAT GCA TCC GCG GTA GAA CGC-3′; Axl forward,5′-CAA GAG CGA TGT GTG GTC CTT-3′ and reverse,5′-ATC CAT GTT GAC ATA GAG GAT-3′; -actinforward, 5′-GGC ATC GTC ACC AAC TGG GAC-3′ and reverse,5′-CGA TTT CCC GCT CGG CCG TGG-3′.
{kind=link}
References
- 1.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- 2.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 3.Kapustin A, Shanahan CM. Targeting vascular calcification: softening-up a hard target. Curr Opin Pharmacol. 2009;9:84–9. doi: 10.1016/j.coph.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res. 2004;95:560–7. doi: 10.1161/01.RES.0000141775.67189.98. [DOI] [PubMed] [Google Scholar]
- 5.Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int. 2009;75:890–7. doi: 10.1038/ki.2008.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kestenbaum B, Sampson JN, Rudser KD, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16:520–8. doi: 10.1681/ASN.2004070602. [DOI] [PubMed] [Google Scholar]
- 7.de Cavanagh EM, Inserra F, Ferder M, et al. From mitochondria to disease: role of the renin-angiotensin system. Am J Nephrol. 2007;27:545–53. doi: 10.1159/000107757. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–12. doi: 10.1161/01.RES.0000216409.20863.e7. [DOI] [PubMed] [Google Scholar]
- 9.Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–67. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 10.Son BK, Akishita M, Iijima K, et al. Mechanism of pi-induced vascular calcification. J Atheroscler Thromb. 2008;15:63–8. doi: 10.5551/jat.e545. [DOI] [PubMed] [Google Scholar]
- 11.Son BK, Kozaki K, Iijima K, et al. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur J Pharmacol. 2007;556:1–8. doi: 10.1016/j.ejphar.2006.09.070. [DOI] [PubMed] [Google Scholar]
- 12.Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–54. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 13.Proudfoot D, Skepper JN, Hegyi L, et al. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–62. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds JL, Skepper JN, McNair R, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol. 2005;16:2920–30. doi: 10.1681/ASN.2004100895. [DOI] [PubMed] [Google Scholar]
- 15.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 16.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 17.Bindoli A, Callegaro MT, Barzon E, et al. Influence of the redox state of pyridine nucleotides on mitochondrial sulfhydryl groups and permeability transition. Arch Biochem Biophys. 1997;342:22–8. doi: 10.1006/abbi.1997.9986. [DOI] [PubMed] [Google Scholar]
- 18.Hirsch T, Marzo I, Kroemer G. Role of the mitochondrial permeability transition pore in apoptosis. Biosci Rep. 1997;17:67–76. doi: 10.1023/a:1027339418683. [DOI] [PubMed] [Google Scholar]
- 19.Krishnan KJ, Greaves LC, Reeve AK, et al. Mitochondrial DNA mutations and aging. Ann N Y Acad Sci. 2007;1100:227–40. doi: 10.1196/annals.1395.024. [DOI] [PubMed] [Google Scholar]
- 20.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 21.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–65. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Hagen TM, Liu J, Lykkesfeldt J, et al. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA. 2002;99:1870–5. doi: 10.1073/pnas.261708898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shay KP, Moreau RF, Smith EJ, et al. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta. 2009;1790:1149–60. doi: 10.1016/j.bbagen.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh U, Jialal I. Alpha-lipoic acid supplementation and diabetes. Nutr Rev. 2008;66:646–57. doi: 10.1111/j.1753-4887.2008.00118.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Tharakan B, Holder-Haynes JG, Hunter FA, et al. Alpha lipoic acid attenuates microvascular endothelial cell hyperpermeability by inhibiting the intrinsic apoptotic signaling. Am J Surg. 2008;195:174–8. doi: 10.1016/j.amjsurg.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 26.Liu J. The effects and mechanisms of mitochondrial nutrient alpha-lipoic acid on improving age-associated mitochondrial and cognitive dysfunction: an overview. Neurochem Res. 2008;33:194–203. doi: 10.1007/s11064-007-9403-0. [DOI] [PubMed] [Google Scholar]
- 27.Miquel J. Can antioxidant diet supplementation protect against age-related mitochondrial damage. Ann N Y Acad Sci. 2002;959:508–16. doi: 10.1111/j.1749-6632.2002.tb02120.x. [DOI] [PubMed] [Google Scholar]
- 28.Ghibu S, Richard C, Vergely C, et al. Antioxidant properties of an endogenous thiol: alpha-lipoic acid, useful in the prevention of cardiovascular diseases. J Cardiovasc Pharmacol. 2009;54:391–8. doi: 10.1097/fjc.0b013e3181be7554. [DOI] [PubMed] [Google Scholar]
- 29.Smith AR, Shenvi SV, Widlansky M, et al. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem. 2004;11:1135–46. doi: 10.2174/0929867043365387. [DOI] [PubMed] [Google Scholar]
- 30.Byon CH, Javed A, Dai Q, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–27. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberman M, Bassi E, Martinatti MK, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28:463–70. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 32.Martin CJ, Goeddeke-Merickel CM. Oxidative stress in chronic kidney disease. Nephrol Nurs J. 2005;32:683–5. [PubMed] [Google Scholar]
- 33.Vaziri ND. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr Opin Nephrol Hypertens. 2004;13:93–9. doi: 10.1097/00041552-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 34.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 35.Zittermann A, Koerfer R. Protective and toxic effects of vitamin D on vascular calcification: clinical implications. Mol Aspects Med. 2008;29:423–32. doi: 10.1016/j.mam.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Stubbs JR, Liu S, Tang W, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol. 2007;18:2116–24. doi: 10.1681/ASN.2006121385. [DOI] [PubMed] [Google Scholar]
- 37.Eckert A, Keil U, Marques CA, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem Pharmacol. 2003;66:1627–34. doi: 10.1016/s0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- 38.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(Suppl 2):S170–80. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 39.Kim TS, Jeong DW, Yun BY, et al. Dysfunction of rat liver mitochondria by selenite: induction of mitochondrial permeability transition through thiol-oxidation. Biochem Biophys Res Commun. 2002;294:1130–7. doi: 10.1016/S0006-291X(02)00612-5. [DOI] [PubMed] [Google Scholar]
- 40.Byun CH, Koh JM, Kim DK, et al. Alpha-lipoic acid inhibits TNF-alpha-induced apoptosis in human bone marrow stromal cells. J Bone Miner Res. 2005;20:1125–35. doi: 10.1359/JBMR.050302. [DOI] [PubMed] [Google Scholar]
- 41.Artwohl M, Muth K, Kosulin K, et al. R-(+)-alpha-lipoic acid inhibits endothelial cell apoptosis and proliferation: involvement of Akt and retinoblastoma protein/E2F-1. Am J Physiol Endocrinol Metab. 2007;293:E681–9. doi: 10.1152/ajpendo.00584.2006. [DOI] [PubMed] [Google Scholar]
- 42.Diesel B, Kulhanek-Heinze S, Holtje M, et al. Alpha-lipoic acid as a directly binding activator of the insulin receptor: protection from hepatocyte apoptosis. Biochemistry. 2007;46:2146–55. doi: 10.1021/bi602547m. [DOI] [PubMed] [Google Scholar]
- 43.Binder MD, Kilpatrick TJ. TAM receptor signalling and demyelination. Neurosignals. 2009;17:277–87. doi: 10.1159/000231894. [DOI] [PubMed] [Google Scholar]
- 44.Iyemere VP, Proudfoot D, Weissberg PL, et al. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. 2006;260:192–210. doi: 10.1111/j.1365-2796.2006.01692.x. [DOI] [PubMed] [Google Scholar]
- 45.Persy V, D’Haese P. Vascular calcification and bone disease: the calcification paradox. Trends Mol Med. 2009;15:405–16. doi: 10.1016/j.molmed.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Neven EG, De Broe ME, D’Haese PC. Prevention of vascular calcification with bisphosphonates without affecting bone mineralization: a new challenge. Kidney Int. 2009;75:580–2. doi: 10.1038/ki.2008.663. [DOI] [PubMed] [Google Scholar]
- 47.Kim HJ, Chang EJ, Kim HM, et al. Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumour necrosis factor-alpha. Free Radic Biol Med. 2006;40:1483–93. [Google Scholar]
- 48.Ha H, Lee JH, Kim HN, et al. Alpha-lipoic acid inhibits inflammatory bone resorption by suppressing prostaglandin E2 synthesis. J Immunol. 2006;176:111–7. doi: 10.4049/jimmunol.176.1.111. [DOI] [PubMed] [Google Scholar]
- 49.Passman JN, Dong XR, Wu SP, et al. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci USA. 2008;105:9349–54. doi: 10.1073/pnas.0711382105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anselme K, Broux O, Noel B, et al. In vitro control of human bone marrow stromal cells for bone tissue engineering. Tissue Eng. 2002;8:941–53. doi: 10.1089/107632702320934047. [DOI] [PubMed] [Google Scholar]
- 51.Stanford CM, Jacobson PA, Eanes ED, et al. Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106–01 BSP) J Biol Chem. 1995;270:9420–8. doi: 10.1074/jbc.270.16.9420. [DOI] [PubMed] [Google Scholar]
- 52.Kim HJ, Kim JY, Lee SJ, et al. Alpha-lipoic acid prevents neointimal hyperplasia via induction of p38 mitogen-activated protein kinase/Nur77-mediated apoptosis of vascular smooth muscle cells and accelerates postinjury reendothelialization. Arterioscler Thromb Vasc Biol. 30:2164–72. doi: 10.1161/ATVBAHA.110.212308. [DOI] [PubMed] [Google Scholar]
- 53.Kim H, Yoon SC, Lee TY, et al. Discriminative cytotoxicity assessment based on various cellular damages. Toxicol Lett. 2009;184:13–7. doi: 10.1016/j.toxlet.2008.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oxygen consumption via mitochondrialrespiratory chain is progressively declined during Pi-induced VSMCcalcification. VSMCs (1 x 104 cells/well/200 μl) wereseeded into wells of a 96-well plate with the BD™ OxygenBiosensor System (BD Biosciences, Franklin Lakes, NJ, USA). Aftercells were adapted for 12 h and treated without or with Pi (2.6mM), the value of fluorescence was measured at the indicated timesusing a SpectraMAX Gemini EM (Molecular Devices) with an excitationat 485 nm and an emission at 590 nm. Data are expressed as the mean± S.D. (n = 3). *P < 0.01;†P < 0.05 versus Pi-untreated control.
ALA inhibits Pi-induced activation of caspase-9and -3 in human aorta-derived SMCs. The aortas from human wereprepared by surgical dissection and applied to the experiments.Cells were treated as in Figure 3C and then assessed for caspase-9and -3 activities. Data are expressed as the mean ± S.D. (n= 3) *P < 0.01.
The expression of Gas6 is gradually downregulatedduring Pi-induced VSMC calcification. VSMCs were incubated with Pifor the indicated times and whole cell extracts prepared weresubjected to Western blotting with antibodies to Gas6 andβ-actin.
ALA rescues decreased protein expression of Gas6and Axl in the aortas of vitamin D3-treated mice tonormal level. Four days after final injection of vitaminD3, aortas were harvested from control and vitaminD3-administrated mice and whole lysates were subjectedto Western blot analysis using specific antibodies to Gas6, Axl andβ-actin.
ALA or tempol inhibits mitochondria-derivedoxidant generation and VSMC calcification by Pi. (A) Todetect mitochondria-derived oxidant, VSMCs treated with Pi in thepresence or absence of ALA or Tempol (100 μM) for 6 days wereincubated in the presence of 50 nM Mitotracker Red CMH2XRos(Invtrogen, Carlsbad, CA) at 37°C for 30 min and washed withPBS, and the formation of the oxidant product was assessed underfluorescence microscopy. The fluorescent intensity per photographysection was measured using Image-Pro Plus version 6.0. (B)Cells as in (A) were stained with von Kossa to visualizecalcium deposits. A representative experiment is presented. Scalebars in (A) and (B) indicate 100 Ìm and 500μm, respectively.
ALA rescues the Gas6 gene expression and inhibitsVSMC calcification via AMPK. (A) VSMCs (4 x105 cells/well in a 6-well culture plate) were treatedwith Pi in the presence or absence of ALA plus compound C (AMPKinhibitor, 5 μM) and PD98059 (ERK inhibitor, 10 μM) for 6days and stained with von Kossa to detect calcium deposits. Arepresentative experiment is presented. Scale bar indicates 500μm. (B) Total RNA was prepared using TRIzol reagent(Invitrogen, Carlsbad, CA). For semi-quantitative RT-PCR, 2 μgof total RNA was used to synthesize the first strand cDNA using theFirst Strand cDNA synthesis kit (Fermentas, EU). The first strandcDNAs were amplified by PCR according to the following PCRparameters: 94°C for 30 s, 59°C for 30 s, and 72°C for30 s, 25 cycles. Primers used in the PCR were as follows: Gas6forward, 5′-GAA GCA GTT GGT GGT CCT GGC-3′ and reverse,5′-CAT GCA TCC GCG GTA GAA CGC-3′; Axl forward,5′-CAA GAG CGA TGT GTG GTC CTT-3′ and reverse,5′-ATC CAT GTT GAC ATA GAG GAT-3′; -actinforward, 5′-GGC ATC GTC ACC AAC TGG GAC-3′ and reverse,5′-CGA TTT CCC GCT CGG CCG TGG-3′.