

Abstract
ENDOGLIN/CD105 (ENG) is a transmembrane glycoprotein and an auxiliary unit of the transforming growth factor-β (TGF-β); receptor, expressed predominantly in vascular endothelium. Noteworthy, Eng mRNA expression has been reported also in Kit+ interstitial cells of Cajal (ICC) in the mouse intestine. Gastrointestinal stromal tumours (GIST) are thought to derive from ICC. Here we have investigated Eng expression in the KitK641E mouse GIST model, in human GIST and in the Ba/F3 cell model. In wild type (WT) mouse antrum, Eng immunoreactivity (-ir) was detected in CD34+/CD31+ endothelium and in Kit+ ICC. In KitK641E mice, hyperplasia of Kit+ cells made Eng-ir even more evident. Quantitative PCR confirmed the increased expression of Eng transcript in KitK641E mice. On human GIST TMA, 26/49 cases stained positive for ENG. Strong ENG staining was associated with malignant and high-risk tumours. ENG negative cases were predominantly of the epithelioid type or harboured PDGFRA mutation. In vitro, Eng mRNA was up-regulated in Ba/F3 cell lines stably expressing various oncogenic Kit mutations (K641E, del559, del814). This effect appeared to be independent of Kit activation, as neither the stimulation of WT Kit by its ligand SCF, nor the inhibition of Kit autophosphorylation by imatinib mesylate in oncogenic mutants, altered Eng expression. Elevated Eng expression in Kit oncogenic mutants appeared rather to be indirectly mediated by DNA hypomethylation, because treatment with the demethylating agent 5-Aza/dC increased Eng mRNA expression in KitWT cells. ENG expression in ICC and in GIST deserves further consideration as ENG is emerging as a potential target for cancer therapy.
Keywords: ENDOGLIN, KIT, gastrointestinal stromal tumour, interstitial cells of Cajal
Introduction
ENDOGLIN/CD105 (ENG) is a 180 kD homodimeric transmembrane glycoprotein, expressed predominantly on vascular endothelial and haematopoietic cells. ENG is an auxiliary protein of the transforming growth factor β (TGF-β) receptor complex involved in cellular proliferation, differentiation and migration [1]. Two ENG isoforms, differing in the length of their cytoplasmic domain, have been characterized in human and mouse tissues [2, 3]. The predominant L-ENG isoform contains in its cytosolic domain a consensus PDZ-binding motive, which is lacking in the barely expressed S-ENG isoform. L-ENG has been shown to be involved in epidermal carcinogenesis, whereas no influence of S-ENG could be demonstrated during carcinogenesis in vivo or in vitro[3]. ENG extracellular domain can undergo proteolytic cleavage and shed from the cell membrane into the bloodstream, giving rise to soluble ENG [4]. Elevated levels of soluble ENG have been reported in pre-eclampsia [5, 6] while studies in cancer patients remain conflicting, some studies reporting increased levels [7–9] whereas others reported a decrease [4] or no change [10–12] in circulating soluble ENG.
ENG expression counteracts the potent inhibitory effect of TGF-β on cell proliferation in several cell lines [13, 14]. Mutations in the ENG gene are responsible for the hereditary haemorrhagic telangiectasia type 1 [HHT1, Osler-Weber-Rendu syndrome (OMIM 187300)]. ENG has been largely investigated in hypoxic responses and as a putative diagnostic, prognostic and therapeutic target in tumoural neoangiogenesis. Conversely, ENG expression in non-endothelial cells has so far attracted little attention [1, 15]. Endoglin/CD105 was found to be expressed in non-endothelial cells of various histotypes, including Kit+ interstitial cells of Cajal (ICC) in the mouse small intestine [16]. ICC are specialized mesenchymal cells located within the muscularis propria of the gastrointestinal tract, where they play major roles in coordinating peristalsis through intrinsic pacemaker function and network formation [17]. Gastrointestinal stromal tumours (GIST) are the most common sarcoma of the gastrointestinal tract and are thought to derive from the ICC lineage. Approximately 85% of GIST harbour oncogenic KIT mutations and 7% contain oncogenic platelet-derived growth factor receptor alpha (PDGFRA) mutations. KIT and PDGFRA are members of the receptor tyrosine kinase family type III. Oncogenic mutations lead to their constitutive autophosphorylation, ligand-independent activation of the downstream signal transduction pathways and subsequent deregulation of cell proliferation, survival and migration [18]. KIT K642E, an oncogenic KIT mutation originally identified in sporadic GIST [19], has also been encountered as germ-line mutation in a family with hyperplasia of the ICC layer and GIST formation [20]. Transgenic mice harbouring Kit K641E, the murine homologue of human KIT K642E, have been generated by a knock-in gene targeting strategy, providing an in vivo GIST model with massive hyperplasia of Kit+ cells, especially in antrum and caecum [21, 22].
Here we have identified for the first time ENG immunoreactivity (-ir) in Kit+ ICC by immunofluorescence (IF), in the normal mouse gut, in the KitK641E GIST mouse model, as well as in human GIST. Oncogenic Kit mutations up-regulated Eng mRNA expression in the murine Ba/F3 cell model in vitro. This effect appeared however to be independent of Kit phosphorylation/activation, as neither the stimulation of WT Kit by its ligand SCF, nor the inhibition of Kit autophosphorylation by imatinib mesylate in oncogenic mutants, altered Eng expression.
Materials and methods
The Kit K641E murine GIST model
The knock-in KitK641E murine GIST model has been previously described [21, 22]. Two-week-old (P14) KitK641E/K641E, KitWT/K641E and KitWT/WT wild-type littermates were used. Mice were bred and experiments performed in compliance with protocol #LA1230331/214N approved by the ethics committee for animal well-being of the Faculty of Medicine, Université Libre de Bruxelles, Brussels, Belgium.
Mice were killed by cervical dislocation. The gastrointestinal tract was carefully removed immediately after killing. The antrum, delineated by visual landmarks, was harvested together with pieces of mid small intestine, liver and descending colon. Tissues were subsequently processed as described later.
Ba/F3 cell model
Generation and characterization of Ba/F3 cells stably expressing the full-length mouse Kit cDNA (accession number XM 132123.1) (KitWT) or the oncogenic mutants Kitdel559, KitK641E and Kitdel814 have been reported previously [20, 23]. Untransfected (original, ORI) Ba/F3 and Ba/F3-KitWT cells were cultured at 37°C in RPMI 1640 medium (GIBCO, CA, USA) supplemented with 10% foetal bovine serum (FBS), 2% penicillin/streptomycin, 1% glutamine and 2.5% supernatant of WEHI cell culture (referred to as ‘IL-3 conditioned medium’). The constitutively active Ba/F3-KitK641E, Ba/F3-Kitdel559 and Ba/F3-Kitdel814 mutants were maintained in medium without IL-3 conditioned medium.
Sustained activation of KitWT signalling pathways was achieved by cultivating Ba/F3-KitWT cells in medium without IL3 containing 100 ng/ml recombinant rmSCF (Sigma-Aldrich, St. Louis, MO, USA) for 48 hrs. Inhibition of Kit phosphorylation was achieved by cultivating Ba/F3-KitK641E, Ba/F3-Kitdel559 and Ba/F3-Kitdel814 cells in IL-3 conditioned medium containing 10 μM imatinib mesylate (Novartis, Annandale, NJ, USA) for 48 hrs.
The methyltranferase inhibitor 5′-Aza-2′Deoxycytidine (5-Aza/dC) (Sigma-Aldrich) was dissolved in 0.2% DMSO at 0.1 μM and applied for 72 hrs. DMSO-treated cells were used as control.
Human GIST882 cell line
The human GIST cell line GIST882 [24] was kindly provided by Dr. Jonathan A. Fletcher, Harvard Medical School, Boston, MA, USA. Cells were cultured at 37°C in DMEM (GIBCO, CA, USA) supplemented with 10% FBS, 2% penicillin/streptomycin.
Flow cytometry analysis (FACS)
FACS was used to detect the expression of Kit receptor and presence of phospho-Tyrosine (p-Tyr) in Ba/F3 cells. 2 × 105 cells were fixed and permeabilized using BD cytofix/cytoperm (BD Pharmingen, San Diego, CA, USA) for 20 min. on ice. After washing twice with BD Perm/Wash buffer (BD Pharmingen), the cells were incubated with R-phycoerythrin (R-PE)-conjugated ACK45 antibody directed against c-kit/CD117 and p-Tyr Alexa Fluor 647 conjugated antibodies (Table 2) for 30 min. in the dark on ice. After incubation, the cells were resuspended in 250 μl PBS–BSA 0.1% and analysed by a laser flow cytometer FACSCalibur system (BD Biosciences) and data were analysed with WinMdi 2.8 software (BD Biosciences). Contaminants were removed by forward and side scatter gating. Kit-PE fluorescence was detected through the FL-2 channel, p-Tyr-Alexa641 fluorescence was assessed using the FL-4 channel and data were acquired and analysed using the CellQuest® application on a Power Macintosh G3 computer. For each sample, at least 10,000 individual cells were collected and the mean fluorescence intensity was evaluated.
Table 2.
Table of antibodies
| Antibody | Cat. No. | Supplier | Host | Dilution |
|---|---|---|---|---|
| IF | ||||
| Primary antibodies | ||||
| Endoglin/CD105 | BAF1320 | R&D Systems, Minneapomis, MN | Goat | 1/1000 |
| Kit/CD117 | A4502 | Dako North America, Carpinteria, CA | Rabbit | 1/500 |
| Kit (C-14) | sc-1494 | Santa Cruz Biotechnology, Santa Cruz, CA | Goat | 1/500 |
| CD/31 | 550274 | BD Pharmingen, San Diego, CA | Rat | 1/1000 |
| CD34 | 14-0341 | eBioscience, San Diego, CA | Rat | 1/500 |
| Secondary antibodies | ||||
| Goat Alexa 448 | A11055 | Invitrogen, Eugene, OR | Donkey | 1/200 |
| Rabbit biotinylated | 711065152 | Jackson Immunoresearch, Cambridge, UK | Donkey | 1/200 |
| Rat FITC | 712095150 | Jackson Immunoresearch, Cambridge, UK | Donkey | 1/200 |
| IHC | ||||
| Primary antibodies | ||||
| Endoglin (H-300) | sc-20632 | Santa Cruz Biotechnology, Santa Cruz, CA | Rabbit | 1/100 |
| Secondary antibodies | ||||
| Rabbit biotinylated | 711065152 | Jackson Immunoresearch, Cambridge, UK | Donkey | 1/200 |
| WB | ||||
| Primary antibodies | ||||
| Endoglin/CD105 (mice tissue) | BAF1320 | R&D Systems, Minneapomis, MN | Goat | 1/1000 |
| Endoglin (H-300) (GIST882 cells) | sc-20632 | Santa Cruz Biotechnology, Santa Cruz, CA | Rabbit | 1/500 |
| Gapdh | #2118L | Cell Signaling Technology, Danvers, MA | Rabbit | 1/1000 |
| Secondary antibodies | ||||
| Goat DyLight™ 680 | 35518 | Pierce, Thermo Fisher Scientific, Erembodegem, BE | 1/10000 | |
| Rabbit DyLight™ 800 | 35571 | Pierce, Thermo Fisher Scientific, Erembodegem, BE | 1/10000 | |
| FACS | ||||
| p-Tyr (Alexa Fluor™ 641 conjugated) | #9415 | Cell Signaling Technology Inc., Danvers, MA | 1/50 | |
| Kit/CD117 (R-PE conjugated) | #9415 | BD Pharmingen, San Diego, CA | 1/50 |
Real-time quantitative PCR (qPCR)
qPCR was performed as described [21]. Briefly RNA was extracted from mouse antrum using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA was reverse transcribed with M-MLV Reverse Transcriptase (Invitrogen, Eugene, OR, USA) in a reaction containing random primers (Amersham Bioscience, Piscataway, NJ, USA). Specific primers for long (L-Eng), short (S-Eng) and total (T-Eng) Eng transcripts were used for amplification using SYBR Green chemistry on a 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Transcriptional quantification relative to glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and β-actin housekeeping genes for mouse tissue or Gapdh with hypoxanthine-guanine phosphoribosyl transferase (Hprt) for Ba/F3 cells lysate was performed using Qbase™ software, based on the delta–delta Ct method, calculating relative normalized quantities of mRNA expression (NRQ) [25]. In antrum, the Eng mRNA expression was then normalized by the endothelial marker CD31 mRNA level. Primer sequences are summarized in Table 1.
Table 1.
Table of primers
| T;L;S-ENG F | CAGTCTCCATGCGCCTGAAC |
| T_1Eng R | GGTGATACCCAGTACAGAGGGCA |
| L-ENG R | CTACGCCATGCTGGTG |
| S-ENG R | GGAATGGGGTGGAGGCTT |
| CD/31 F | GGTCATCGCCACCTTAATAGTTG |
| CD/31 R | TCTCTGTGGCTCTCGTTCCC |
| Gapdh F | TGTGTCCGTCGTGGATCTGA |
| Gapdh R | CCTGCTTCACCACCTTCTTGA |
| Hprt F | GCTACTGTAATGATCAGTCAACGGG |
| Hprt R | AAGCTTGCAACCTTAACCATTTTG |
| β-Actin F | AACCGTGAAAAGATGACCCAGAT |
| β-Actin R | GCCTGGATGGCTACGTACATG |
Immunofluorescence staining
Specimens were processed as described [21]. Briefly, slides were brought to RT, rinsed in 10 mM Tris (Merck-Belgolabo, Overijse, Belgium) and 0.15 M sodium chloride, pH 7.4 TBS, containing 0.1% (v/v) Triton-X 100 (TBS-TX), and incubated for 1 hr in 10% normal horse serum (NHS) (Hormonologie Laboratoire, Marloie, Belgium) and TBS-TX. The slides were incubated overnight at RT with the primary antibodies (Table 2), rinsed twice in TBS and incubated in the dark for 1 hr at RT in TBS containing the secondary antibodies (Table 2). Slides were then rinsed twice in TBS and incubated in the dark for 1 hr at RT with streptavidin-coupled NorthernLight™ 557 (NL557) (R&D Systems, Abingdon, Oxon, UK) in TBS.
The optimal working dilution has been determined empirically for each antibody. The protocol used for double IF staining did not modify the distribution or the intensity of each individual labelling observed in corresponding single procedures. Omission of the primary or the secondary antibodies resulted in the absence of the respective fluorescent signal. Nuclei were stained with 5 μM Hoechst in Tris-HCl (Merck-Belgolabo) 0.05 M (pH 7.4), containing 0.5 mg/ml type 1-AS Ribonuclease A for 5 min. in the dark at RT.
Confocal microscopy
After three rinses in TBS, cover slips were mounted with Fluor Save™ Reagent anti-fade mounting medium (Calbiochem, Nottingham, UK) before viewing under a LSM510 NLO multiphoton confocal microscope fitted on an Axiovert M200 inverted microscope equipped with a C-Apochromat 40×/1.2 N.A. water immersion objective (Zeiss, Iena, Germany).
The 488 nm excitation wavelength of the Argon/2 laser, a main dichroic HFT 488 and a band-pass emission filter (BP500–550 nm) were used for selective detection of the green fluorochrome. The 543 nm excitation wavelength of the HeNe1 laser, a main dichroic HFT 488/543/633 and a long-pass emission filter (LP560 nm) were used for selective detection of the red fluorochrome. The nuclear stain Hoechst was excited in multiphotonic mode at 760 nm with a Mai Tai™ tunable broad-band laser (Spectra-Physics, Darmstad, Germany) and detected using a main dichroic HFT KP650 and a band-pass emission filter (BP435–485 nm). Single optical sections, 2 μm thick, were collected for each fluorochrome sequentially. The images generated (pixel size: 0.1 μm) were merged and displayed using the Zeiss LSM510 software. Fluorescence intensity line plots were generated using the same software. Images were exported in .jpg image. All figures show single optical sections across the regions of interest.
Western blotting (WB)
Tissue samples were lysed in sucrose lysis buffer containing 20 mM Tris-HCl (pH 7.9), 250 mM sucrose, 0.01% NaN3, 0.1% Nonidet P-40 and 20 μl/ml proteases inhibitor mixture (Roche, Mannheim, Germany). The antral muscularis propria was cleaned up by sharp dissection and the mucosa was discarded before lysis.
Proteins were solubilized in sample buffer, heated at 95°C for 5 min., separated by SDS-PAGE on 10% polyacrylamide gel and transferred on a 0.2 μm nitrocellulose membrane. Primary antibodies raised in different species and secondary antibodies coupled with different fluorochromes (Table 2), were sequentially combined to specifically label one marker in green (800 DyLight), the other in red (680 DyLight). Gapdh was used as loading control. Liver was used as positive control for Eng expression. The Odyssey™ imaging system (LI-COR Biotechnology, Lincoln, NE, USA) was used to quantify the signals.
Human GIST tissue microarrays (TMA)
A cohort of formalin-fixed, paraffin-embedded human GIST samples was retrieved from the pathology files of the Cleveland Clinic, Cleveland, OH. The collection and analysis of human tissue samples was approved by the Cleveland Clinic Institutional Review Board (IRB 06-977). Consent was not obtained from any participants as it was waived by the IRB because it was a retrospective study involving only collection of tissue cores from paraffin blocks used in the diagnosis of patient tumour samples.
Clinico-pathological characterization of GIST used is summarized in Table 3. Risk assessment was performed using the criteria of Miettinen and Lasota [26], as recommended by the National Cancer Care Network [27]. Mutations in KIT exons 9, 11, 13 and 17 and PDGFRA exons 12 and 18 were examined as described previously [28]. This material was used to construct a TMA with 1 mm cores using a TMarrayer™ (P/N 02110016) semi-automated tissue arrayer (Pathology Devices, Westminster, MD, USA). Each case was present in duplicate in this array.
Table 3.
Clinicopathologic features of cases on the Cleveland Clinic GIST TMA
| Primary tumour site | Total (n) |
|---|---|
| Gastric | 34 |
| Small bowel | 9 |
| Colon | 2 |
| Rectovaginal | 1 |
| Disseminated | 3 |
| Tumour morphology | Total (n) |
| Spindle | 35 |
| Epithelioid | 14 |
| Risk category | Total (n) |
| Malignant | 7 |
| High risk | 10 |
| Moderate risk | 13 |
| Low risk | 8 |
| Very low risk | 6 |
| No risk | 5 |
| Mitotic figures | Total (n) |
| ≤5/5 mm2 | 29 |
| >5/5 mm2 | 13 |
| N.A. | 7 |
| Mutation status | Total (n) |
| KIT mutation | 25 |
| KIT Exon 9 AY 502-503 duplication | 4 |
| KIT Exon 11 duplication | 3 |
| KIT Exon 11 deletion | 8 |
| KIT exon 11 V559D | 6 |
| KIT exon 11 V560D | 2 |
| KIT exon 11 W557G | 1 |
| KIT Exon 13 K642E | 1 |
| PDGF mutation | 9 |
| PDGFRA Exon 12 – V561D | 2 |
| PDGFRA exon 12 deletion | 1 |
| PDGFRA Exon 18 D842V | 4 |
| PDGFRA Exon 18 D842Y | 1 |
| PDGFRA Exon 18 Ins-Del | 1 |
| Wild type | 11 |
| N.A. | 4 |
| KIT-ir | Total (n) |
| Positive | 44 |
| Negative | 4 |
| N.A. | 1 |
N.A.: data not available.
Immunohistochemistry (IHC) on human material
After rehydration through phenol and graded alcohol solutions, TMA slides were heated at 96°C in 1 mM EDTA, 0.05% Tween20 (pH 8.0) antigen retrieval solution for 20 min. to achieve epitope unmasking. Slides were then cooled for 10 min. before being rinsed in TBS. The staining with primary Eng antibody and secondary rabbit biotinylated antibody (Table 2) was performed as mentioned earlier. Then sections were incubated in ABC solution (ABC kit standard PK-4000; Vector Laboratories, Burlingame, CA, USA) for 90 min. at room temperature and peroxidase activity revealed for 5–10 min. with nickel-enhanced DAB (DAB-Ni), resulting in a black precipitate. The DAB-Ni solution was prepared by dissolving 0.2 g of nickel ammonium sulphate (Fluka, Buchs, Switzerland) and 7.5 mg of DAB (Sigma-Aldrich) in 50 ml of 0.05 M Tris/HCl, pH 8. Immediately before use, 10 μl of 30% H2O2 (Merck, Darmstadt, Germany) was added.
GIST tissue present in each core was scored as strong (comparable to intensity of vascular endothelial staining), weak (weaker then staining of vascular endothelia and clearly distinguishable from background), negative (intensity comparable to background) or non-contributive because of poor tissue quality.
Statistical analysis
Student’s t-test for independent samples was performed to compare mean values of qPCR and WB data. Fisher’s exact test was used to assess a possible relationship between ENG expression status and clinicopathological parameters, KIT-ir IHC or mutation status of GIST specimens. Student’s t-test for independent samples was used to compare means of tumour size and mitotic counts between ENG+ and ENG− GIST. Statistical significance was set at a P value of less than 0.05.
In addition, factorial analysis of multiple correspondences (FAMC), followed by an ascending hierarchic classification (AHC) was applied to the subset (n= 34) of GIST cases for which full information was available, using the SPAD version 7.0 statistical software (Coheris, Suresnes, France). These methods provide a graphic representation of the relations between variables, modalities and subjects, close positions indicating similarities [29]. Multiple modalities that could interact with each other were simultaneously analysed and their respective influence was addressed by FAMC, whereas AHC allowed building homogeneous clusters of subjects. By comparing the position of these clusters in the plane with the position of the modalities, subjects belonging to a cluster can be characterized according to these modalities. A test of proportions was performed. Statistical significance was set at a P value of less than 0.05.
Results
Endoglin expression in KitK641E/K641E mouse antrum
Expression of long (L-Eng), short (S-Eng) and total (T-Eng) Eng transcripts in KitWT/WT and KitK641E/K641E antrum was investigated (Fig. 1A). To estimate the proportion of Eng expression in Kit+ cells and not in endothelium, Eng mRNA level was normalized against the endothelial marker CD31 mRNA level (NRQ = 1 and 2.7 for KitWT/WT and KitK641E/K641E, respectively). Relative expression of T-Eng and L-Eng in KitK641E/K641E antrum was significantly increased, compared to KitWT/WT littermates (NRQ = 2.8; P= 0.04 and NRQ = 3.4; P= 0.006, respectively). S-Eng expression in WT antrum was very low compared to L-Eng (0.79 versus 34.49 arbitrary units, respectively) and its relative change in KitK641E/K641E antrum compared to KitWT/WT was not statistically significant (P= 0.22) (Fig. 1B).
Fig 1.
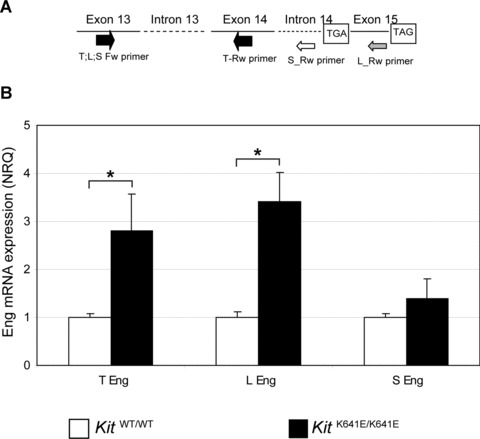
Eng mRNA expression is up-regulated in KitK641E/K641E antrum. (A) Schematic description of primers used to detect expression of long (L-Eng), short (S-Eng) and total (T-Eng) Eng transcripts. (B) In P14 antrum, T-Eng and L-Eng mRNA exhibited significant up-regulation in KitK641E/K641E homozygous compared to KitWT/WT littermates. Relative change of S-Eng mRNA in KitK641E/K641E antrum compared to KitWT/WT was not statistically significant (P= 0.22). Data reported as the mean ± S.E.M. of three independent experiments and presented as normalized relative quantities (NRQ), normalized against the endothelial marker CD31 mRNA level. Student’s t-test, *P≤ 0.05.
A single band at the expected molecular weight of ∼90 kD, representing the predominant L-ENG form was detected by WB on protein extracts of P14 KitK641E/K641E, KitWT/K641E and KitWT/WT mice antrum. No statistically significant difference between genotypes was observed. In the liver, used as control, the L-ENG, S-ENG and soluble Eng isoforms were detected by WB (Fig. S1).
Endoglin is expressed in endothelial cells but also in Kit+ cells in the mouse gut wall
Eng-ir was observed in CD34+/CD31+ endothelium, as expected. Eng-ir was also detected, albeit fainter than in endothelium, in many, but not all, Kit+ ICC (Fig. 2A and C). The same features were observed at different levels (antrum, small intestine, transverse colon) in the WT mouse gastrointestinal tract (Figs S2 and S3).
Fig 2.
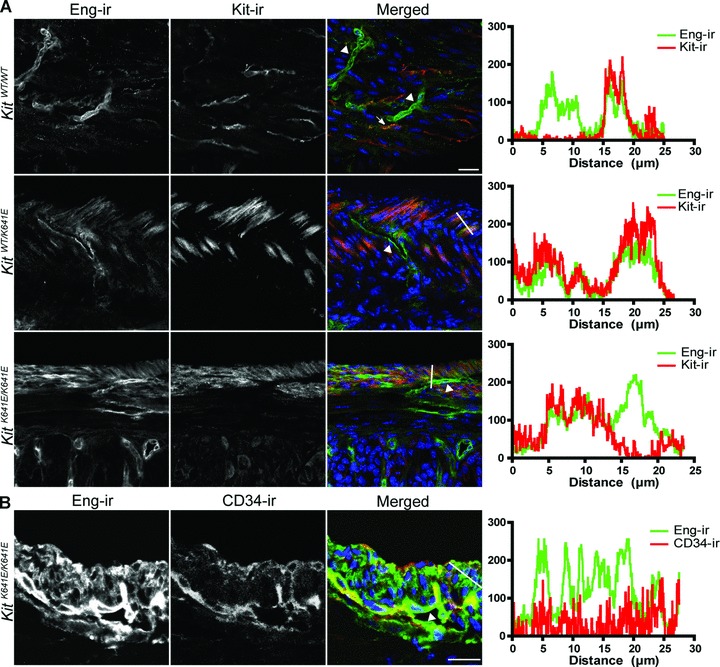
Eng expression in endothelium and in Kit+ ICC in WT and KitK641E antrum. (A) Eng-ir (Alexa 488, green) was observed in Kit-ir ICC (NL559, red) (indicated by arrow) in KitWT/WT antrum. The higher abundance of Kit+ cells in KitK641E heterozygotes and homozygotes made Eng-ir even more evident. Robust Eng-ir was also consistently found in the endothelium of blood vessels (indicated by arrowhead) in all genotypes. (B) Example of Eng-ir (Alexa 488, green) and CD34-ir (NL559, red) in P14 KitK641E/K641E antrum. Blood vessels (indicated by arrowhead) were decorated by both CD34-ir and Eng-ir, although CD34− cells of the hyperplasic longitudinal muscle layer exhibited also Eng-ir. Figures oriented with the serosa facing up and mucosa down. Scale bar: 20 μm. Intensity plots for the green and red fluorochromes were measured along the white line drawn on the respective merged image.
The higher abundance of Kit+ cells in KitK641E heterozygotes and homozygotes made Eng-ir more readily discernible than in the KitWT/WT antrum (Fig. 2A).
Endoglin expression in human GIST
The antibody used for ENG-ir on GIST TMA was validated using the human GIST882 cell line. Two immunoreactive bands of the expected MW for L-ENG and S-ENG were detected by WB. By IF, GIST882 cells stained positively for ENG-ir, whereas omission of the primary antibodies resulted in the absence of signal (Fig. S4).
In GIST TMA, 26 GIST stained positively for ENG-ir (ENG+/++) and 23 were negative (ENG−). Labelling of endothelial cells provided internal positive controls.
Results of correlation between ENG-ir and histopathological GIST characteristics are summarized in Table 4. Median size of the ENG+/++ and ENG− tumours was 5.82 cm (range 0.5–16 cm) and 8.62 (range 0.3–31.5 cm), respectively. Mitotic counts per 5 mm2 were significantly higher in ENG+/++ compared to ENG− GIST (P= 0.013). Strongly positive ENG++ staining was associated with malignant or high-risk cases (P= 0.014) (Fig. 3A). ENG− were associated with PDGFRA mutation status (P= 0.027) and epithelioid histology (P= 0.02) (Fig. 3B).
Table 4.
Association of Eng-ir with clinicopathologic characteristics in 49 GIST cases
| Cohort (n= 49) | ENG+ | ENG− | P value* | |||
|---|---|---|---|---|---|---|
| Mean tumour size | 5.82 (cm) | 8.62 (cm) | 0.07 | |||
| Primary site | ENG++ | ENG+ | Total | ENG− | P value† | |
| n (%) | n (%) | n (%) | n (%) | |||
| Gastric | 8 (16%) | 6 (12%) | 14 (29%) | 20 (41%) | ||
| Small bowel | 2 (4%) | 4 (8%) | 6 (12%) | 3 (6%) | ||
| Colon | 2 (4%) | 0 | 2 (4%) | 0 | ||
| Rectovaginal | 1 (2%) | 0 | 1 (2%) | 0 | ||
| Disseminated | 2 (4%) | 0 | 2 (4%) | 1 (2%) | ||
| Total | 15 (30%) | 10 (20%) | 25 (51%) | 24 (49%) | 0.08 | |
| Mitotic figures | ENG++ | ENG+ | Total | ENG− | P value† | |
| n (%) | n (%) | n (%) | n (%) | |||
| ≤5/5 mm2 | 4 (8%) | 7 (14%) | 11 (22%) | 20 (41%) | ||
| >5/5 mm2 | 7 (14%) | 1 (2%) | 8 (16%) | 4 (8%) | ||
| Total | 11 (22%) | 8 (16%) | 19 (38%) | 24 (49%) | 0.013 | |
| Histological type | ENG++ | ENG+ | Total | ENG− | P value† | |
| n (%) | n (%) | n (%) | n (%) | |||
| Epithelioid | 2 (4%) | 1 (2%) | 3 (6%) | 11 (22%) | ||
| Spindle | 14 (29%) | 9 (18%) | 23 (47%) | 12 (24%) | ||
| Total | 16 (33%) | 10 (20%) | 26 (53%) | 23 (47%) | 0.02 | |
| Mutation Status | ENG++ | ENG+ | Total | ENG− | P value† | |
| n (%) | n (%) | n (%) | n (%) | |||
| KIT mutation | 9 (18%) | 8 (16%) | 17 (35%) | 8 (16%) | ||
| PDGF mutation | 0 | 1 (2%) | 1 (2%) | 8 (16%) | ||
| Wild type | 5 (10%) | 1 (2%) | 6 (12%) | 5 (10%) | ||
| Total | 14 (29%) | 10 (20%) | 24 (49%) | 21 (43%) | 0.027 | |
| Risk classification | ENG++ | ENG+ | Total | ENG− | P value† | |
| n (%) | n (%) | n (%) | n (%) | |||
| Malignant–high risk | 11 (22%) | 1 (2%) | 12 (24%) | 5 (10%) | ||
| Moderate–low risk | 3 (6%) | 6 (12%) | 9 (18%) | 12 (24%) | ||
| Very low–no risk | 2 (4%) | 3 (6%) | 5 (10%) | 6 (12%) | ||
| Total | 16 (33%) | 10 (20%) | 26 (53%) | 23 (47%) | 0.014 | |
Student’s t-test.
Fisher’s exact test.
Fig 3.
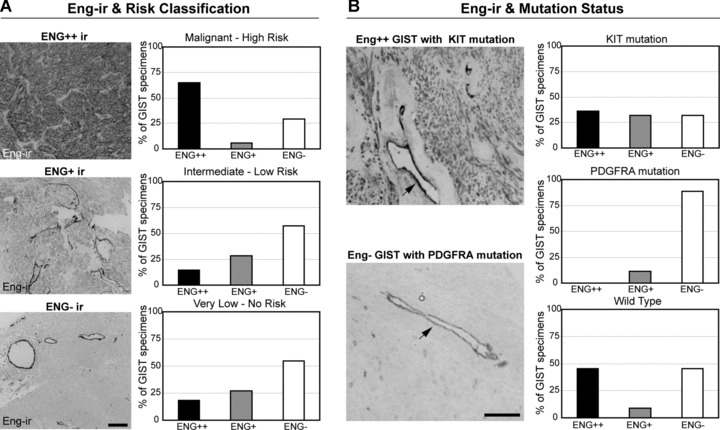
ENG-ir in human GIST tissue arrays. (A) First column: Representative examples of Eng-ir strongly positive, positive and negative (ENG++, ENG+, ENG−, respectively) GIST specimens. Scale bar: 20 μ microns. Second column: Percentage of ENG-ir specimens associated with different risk categories of human GIST. (B) First column: Representative example of ENG++ GIST specimen with KIT mutation (top) and an ENG− specimen with PDGFRA mutation (bottom). ENG-ir in vascular endothelium is indicated by arrow. Scale bar: 20 μm. Second column: Percentage of ENG-ir specimens according to their KIT and PDGFRA mutation status.
Similarly, FAMC and AHC revealed three clusters of GIST specimens. GIST with strong ENG++ staining belong to a cluster with malignant specimens (P < 0.001) and high mitotic counts (>5/5 mm2) (P < 0.001). ENG− GIST belong to a cluster with PDGFRA mutation status (P < 0.001), KIT negative IHC (P= 0.002), epithelioid histological type (P= 0.007), intermediate risk category (P= 0.011), large tumour size (>10 cm) (P= 0.022) and mitotic counts (<5/5 mm2) (P= 0.046) (Fig. S5).
Endoglin mRNA expression in Ba/F3 cell is increased by Kit oncogenic mutants but is not controlled by Kit phosphorylation
Compared to Ba/F3-KitWT cells, T-Eng expression was significantly higher in the constitutively activated mutants Ba/F3-KitK641E (NRQ = 5.5; P= 0.025), Ba/F3-Kitdel559 (NRQ = 8.2; P= 0.019) and Ba/F3-Kitdel814 (NRQ = 5.2; P= 0.001).
Sustained (48 hrs) stimulation of Ba/F3-KitWT by Kit ligand SCF did not modify Eng mRNA expression level (Fig. 4A). Efficiency of SCF treatment was confirmed by the increase of pTyr-ir assessed by flow cytometry. SCF had no effect on pTyr-ir in original Ba/F3 (Fig. S6).
Fig 4.
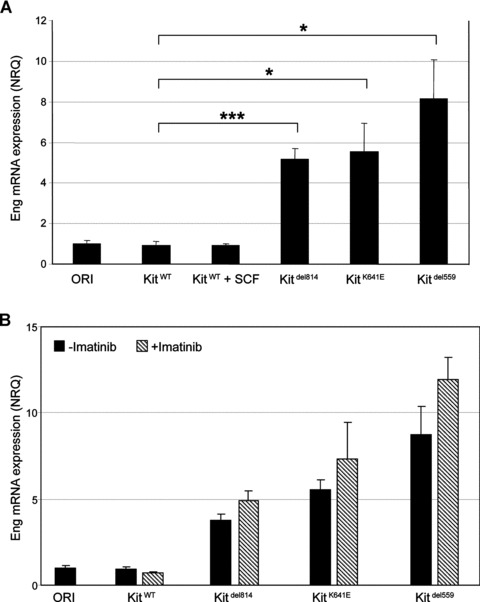
Ba/F3 cell lines, Eng mRNA expression is independent of Kit phosphorylation in vitro. (A) T-Eng expression was significantly higher in constitutive active mutants Ba/F3-Kitdel814, Ba/F3-KitK641E and Ba/F3-Kitdel559, compared to Ba/F3-KitWT. Sustained (48 hrs) stimulation of Ba/F3-KitWT by SCF, did not modify T-Eng mRNA expression level. Data reported as mean ± S.E.M. of three independent experiments and presented as normalized relative quantities (NRQ). Student’s t-test, *P < 0.05, ***P < 0.001. (B) Inhibition of Kit phosphorylation by imatinib mesylate did not significantly alter T-Eng mRNA expression in any of the oncogenic Kit mutants. Data reported as the mean ± S.E.M. of three independent experiments and presented as normalized relative quantities (NRQ).
Similarly, inhibition of Kit phosphorylation by imatinib mesylate for 48 hrs or 7 days did not alter Eng mRNA expression in the oncogenic Kit mutants Ba/F3-KitK641E, Ba/F3-Kitdel559 and in imatinib resistant clone Ba/F3-Kitdel814 (Figs 4A and S7). Efficiency of imatinib treatment was confirmed in Ba/F3-KitK641E in Ba/F3-Kitdel559 and in Ba/F3-Kitdel814 by the drop of pTyr-ir, assessed by flow cytometry (Fig. S8).
Expression of Endoglin mRNA can be activated in Ba/F3WT cells by the DNA methyltransferase inhibiting agent 5-Aza/dC
We wondered whether DNA hypomethylation could be involved in the increase of Eng expression in Ba/F3-KitK641E, BaF3-Kitdel559 and BaF3-Kitdel814 cells. Treatment with the DNA methyl-transferase inhibitor 5-Aza/dC significantly increased Eng mRNA expression in Ba/F3-KitWT (P= 0.003) and in Ba/F3-KitWT cells stimulated with SCF for 48h (P= 0.003). Level of Eng expression in Ba/F3-KitWT after 5-Aza/dC treatment was comparable to Eng expression in Kit oncogenic mutants. Conversely, 5-Aza/dc did not have a statistically significant effect on Eng mRNA expression in Ba/F3-KitK641E, BaF3-Kitdel559 and BaF3-Kitdel814 cells (P= 0.5, 0.09 and 0.2, respectively) (Fig. 5).
Fig 5.
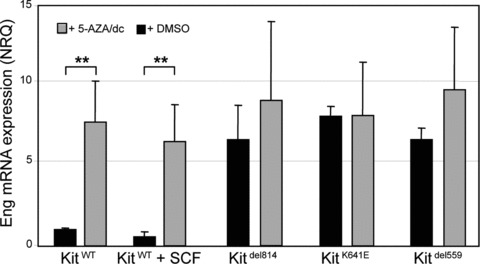
Treatment with the methyltransferase inhibitor 5-Aza/dC increased Eng mRNA expression in Ba/F3WT. Treatment with 5-Aza/dC significantly increased T-Eng mRNA level in Ba/F3-KitWT, reaching a level comparable to T-Eng expression in Kit oncogenic mutants. 5-Aza/dc did not affect T-Eng mRNA expression in Ba/F3-Kitdel814, Ba/F3-KitK641E and Ba/F3-Kitdel559 (P= 0.5, 0.09 and 0.2, respectively). Data reported as the mean ± S.E.M. of three independent experiments and presented as normalized relative quantities (NRQ). Student’s t-test, **P≤ 0.01.
Another possible mechanism for the transcriptional activation of Eng by the oncogenic Kit mutants could be the up-regulation of an upstream transcription factor. Expression of Hypoxia inducible factor 1α (Hif1α), a well-characterized transcription factor upstream of Eng, did not differ in Ba/F3-KitWT cells, compared to the constitutive active mutants BaF3-KitK641E, BaF3-Kitdel559 and BaF3-Kitdel814. Similarly, relative expression of Hif1α in KitK641E/K641E and KitWT/K641E antrum did not differ from expression in KitWT/WT littermates (Fig. S9).
Discussion
We first focused on Eng-ir in the murine gut as Eng mRNA expression had been reported in Kit+ ICC isolated by FACS from mouse small intestine [16]. Here, we have demonstrated, for the first time, by IF the presence of Eng-ir in the Kit+ ICC of the normal mouse gut.
As GIST are thought to derive from the KIT+ ICC lineage [18], we next investigated Eng expression in the KitK641E GIST model. The higher abundance of Kit+ cells in KitK641E heterozygous and homozygous antrum made Eng-ir even more readily discernible than in KitWT/WT ICC. However, WB (this study) and microarrays [21] performed on the entire antrum wall did not demonstrate any significant increase of Eng expression in KitK641E mice. Similarly, up-regulation of ENG has not been reported in microarray studies comparing human GIST and normal tissue. This may illustrate an intrinsic limitation of techniques using homogenates of whole tissues, as the robust ENG expression in endothelium could easily overshadow the weaker signal arising from the KIT+ cells. In line with that interpretation, qPCR data, when normalized against expression of the endothelial marker CD31, revealed a significantly higher expression of T-Eng and L-Eng mRNA in KitK641E/K641E antrum of a magnitude (two- to threefold), similar to the increase of Kit mRNA expression observed in this model [21]. Expression of S-Eng transcript was very low and S-Eng protein was hardly detectable on Western blot. Eng antibodies available do not discriminate L-Eng and S-Eng isoforms, precluding study of their respective distribution by IF. S-Eng may play some regulatory role [3], but its distribution could not be resolved in this study.
Next, we analysed ENG expression in human GIST, using TMA and IHC. ENG-ir was detected in half (26/49) of the cases. Strong ENG expression was significantly associated with malignant or high-risk tumours. Moreover, absence of ENG-ir was observed in 8/9 GIST harbouring PDGFRA mutations, raising the hypothesis of a possible link between ENG expression and oncogenic KIT mutants in GIST. Larger prospective series will be needed to confirm the predictive value of ENG-ir in GIST.
Noteworthy, TGF-β/Smad’s signaling pathway plays a central role in the development and differentiation of mesenchymal cells in the digestive tract [30, 31] and lack of Smad3, an intermediate in TGF-β signaling, leads to the lack of ICC in the colonic wall in Smad3-null mice [32]. Taken together, these data indirectly suggest that the TGF-β receptor complex, including ENG, might be involved in the development of the KIT+ ICC in the normal gut and in the malignant behavior of KIT+ GIST, thus expanding the scope of ENG roles beyond its well-established regulatory role in endothelium.
Finally, we analysed the influence of Kit oncogenic mutants on Eng expression in Ba/F3 cell lines, a well-established model to investigate signalling pathways downstream of the Kit receptor tyrosine kinase in vitro[23]. In this model, three different constitutively activated (oncogenic) Kit mutants markedly increased Eng mRNA expression compared to original (untransfected) cells or cells transfected with KitWT. Oncogenic mutations lead to constitutive autophosphorylation, and subsequent ligand-independent activation of the downstream signal transduction. Noteworthy, Eng expression in vitro was not directly dependent of Kit receptor phosphorylation as neither sustained activation of KitWT by its ligand SCF, nor inhibition of Kit phosphorylation by imatinib mesylate in the Kit oncogenic mutants, altered Eng expression level.
Expression of Hypoxia inducible factor 1α (Hif1α), a well-characterized transcription factor upstream of Eng, did not differ in the various cell lines. Elevated Eng expression in Kit oncogenic mutants appeared to be rather indirectly mediated by DNA hypomethylation, because treatment with the demethylating agent 5-Aza/dC increased Eng mRNA expression in KitWT cells to similar level. Aberrant DNA methylation is a frequent phenomenon in cancer, including GIST [33, 34]. The precise mechanism by which Kit oncogenic mutants could lead to alterations of DNA methylation remains however to be determined.
Although important mechanistic issues remain to be elucidated, this study has brought ENG into the spotlight in the field of KIT-ir ICC and GIST. As therapeutic interventions targeting Eng are already being considered [1, 15], ENG deserves further evaluation as a putative novel marker and/or therapeutic target for GIST harbouring KIT oncogenic mutations.
Acknowledgments
We are indebted to P. Hagué and Huy Nguyen Tran for expert technical assistance and preparation of the figures, respectively. We are grateful to Prof. Viviane De Maertelaer, Department of Biostatistics and Medical Computing, Université Libre de Bruxelles, Brussels, Belgium, for stimulating discussion on data analysis.
Grant support
National Fund for Scientific Research (Belgium), Télévie grant 7.4.558.07.F, jointly to J.M.V. and C.E.
National Fund for Scientific Research (Belgium), Fonds de la Recherche Scientifique Médicale grant 3.4.571.07.F to J.M.V.
National Fund for Scientific Research (Belgium), Grand 7.4.529.08.F and Belgian State Policy PAIP (P6/28) to C.E.
J.M.V. is Research Director of the National Fund for Scientific Research (Belgium).
B.P.R. is funded by a generous grant from The Life Raft Group.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Eng protein expression inKitK641E antrum.
Eng expression in endothelium and inKit+ ICC in WT mouse small intestine.
Eng expression in endothelium and inKit+ ICC in WT mouse colon.
ENG expression in GIST882 cell line.
Graphic representation of factorial analysis of multiple correspondences based on TMA variables and their modalities.
Efficiency of SCF treatment was confirmed by the increase of pTyr-ir assessed by flow cytometry.
Sustained imatinib treatment does not affectEng mRNA expression in Ba/F3 cells.
Efficiency of imatinib treatment was confirmed by the drop of pTyr-ir assessed by flow cytometry.
Expression of Hif1α in Ba/F3 cells and in mouse antrum.
References
- 1.Dallas NA, Samuel S, Xia L, et al. Endoglin (CD105): a marker of tumour vasculature and potential target for therapy. Clin Cancer Res. 2008;14:1931–7. doi: 10.1158/1078-0432.CCR-07-4478. [DOI] [PubMed] [Google Scholar]
- 2.Bellon T, Corbi A, Lastres P, et al. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur J Immunol. 1993;23:2340–5. doi: 10.1002/eji.1830230943. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Gomez E, Eleno N, Lopez-Novoa JM, et al. Characterization of murine S-endoglin isoform and its effects on tumour development. Oncogene. 2005;24:4450–61. doi: 10.1038/sj.onc.1208644. [DOI] [PubMed] [Google Scholar]
- 4.Hawinkels LJ, Kuiper P, Wiercinska E, et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumour angiogenesis. Cancer Res. 2010;70:4141–50. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- 5.Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 6.Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–9. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Gardy R, Seon BK, et al. Both high intratumoral microvessel density determined using CD105 antibody and elevated plasma levels of CD105 in colorectal cancer patients correlate with poor prognosis. Br J Cancer. 2003;88:1424–31. doi: 10.1038/sj.bjc.6600874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C, Guo B, Wilson PB, et al. Plasma levels of soluble CD105 correlate with metastasis in patients with breast cancer. Int J Cancer. 2000;89:122–6. doi: 10.1002/(sici)1097-0215(20000320)89:2<122::aid-ijc4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi N, Kawanishi-Tabata R, Haba A, et al. Association of serum endoglin with metastasis in patients with colorectal, breast, and other solid tumours, and suppressive effect of chemotherapy on the serum endoglin. Clin Cancer Res. 2001;7:524–32. [PubMed] [Google Scholar]
- 10.Mysliwiec P, Pawlak K, Bandurski R, et al. Soluble angiogenesis markers in gastric tumour patients. Folia Histochem Cytobiol. 2009;47:81–6. doi: 10.2478/v10042-009-0004-4. [DOI] [PubMed] [Google Scholar]
- 11.Mysliwiec P, Pawlak K, Kuklinski A, et al. Combined perioperative plasma endoglin and VEGF-a assessment in colorectal cancer patients. Folia Histochem Cytobiol. 2009;47:231–6. doi: 10.2478/v10042-009-0074-3. [DOI] [PubMed] [Google Scholar]
- 12.Furstenberger G, von Moos R, Lucas R, et al. Circulating endothelial cells and angiogenic serum factors during neoadjuvant chemotherapy of primary breast cancer. Br J Cancer. 2006;94:524–31. doi: 10.1038/sj.bjc.6602952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lastres P, Letamendia A, Zhang H, et al. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–21. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Hampson IN, Hampson L, et al. CD105 antagonizes the inhibitory signaling of transforming growth factor beta1 on human vascular endothelial cells. FASEB J. 2000;14:55–64. doi: 10.1096/fasebj.14.1.55. [DOI] [PubMed] [Google Scholar]
- 15.Fonsatti E, Maio M. Highlights on endoglin (CD105): from basic findings towards clinical applications in human cancer. J Transl Med. 2004;2:18. doi: 10.1186/1479-5876-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Ordog T, Chen J, et al. Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol Genomics. 2007;31:492–509. doi: 10.1152/physiolgenomics.00113.2007. [DOI] [PubMed] [Google Scholar]
- 17.Mazzone A, Farrugia G. Evolving concepts in the cellular control of gastrointestinal motility: neurogastroenterology and enteric sciences. Gastroenterol Clin North Am. 2007;36:499–513. doi: 10.1016/j.gtc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Liegl-Atzwanger B, Fletcher JA, Fletcher CD. Gastrointestinal stromal tumours. Virchows Arch. 2010;456:111–27. doi: 10.1007/s00428-010-0891-y. [DOI] [PubMed] [Google Scholar]
- 19.Lux ML, Rubin BP, Biase TL, et al. KIT Extracellular and kinase domain mutations in gastrointestinal stromal tumours. Am J Pathol. 2000;156:791–5. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isozaki K, Terris B, Belghiti J, et al. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumours. Am J Pathol. 2000;157:1581–5. doi: 10.1016/S0002-9440(10)64795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gromova P, Ralea S, Lefort A, et al. Kit K641E oncogene up-regulates Sprouty homolog 4 and trophoblast glycoprotein in interstitial cells of Cajal in a murine model of gastrointestinal stromal tumours. J Cell Mol Med. 2009;13:1536–48. doi: 10.1111/j.1582-4934.2009.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubin BP, Antonescu CR, Scott-Browne JP, et al. A knock-in mouse model of gastrointestinal stromal tumour harboring kit K641E. Cancer Res. 2005;65:6631–9. doi: 10.1158/0008-5472.CAN-05-0891. [DOI] [PubMed] [Google Scholar]
- 23.Vanderwinden JM, Wang D, Paternotte N, et al. Differences in signaling pathways and expression level of the phosphoinositide phosphatase SHIP1 between two oncogenic mutants of the receptor tyrosine kinase KIT. Cell Signal. 2006;18:661–9. doi: 10.1016/j.cellsig.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumour c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–8. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 25.Hellemans J, Mortier G, De Paepe A, et al. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miettinen M, Lasota J. Gastrointestinal stromal tumours: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83. doi: 10.1053/j.semdp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumour (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw. 2007;5(Suppl 2):S1–29. ; quiz S30: S1–29. [PubMed] [Google Scholar]
- 28.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumour. J Clin Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 29.Greenacre M. Correspondence analysis of raw data. Ecology. 2010;91:958–63. doi: 10.1890/09-0239.1. [DOI] [PubMed] [Google Scholar]
- 30.Hahm KB, Im YH, Parks TW, et al. Loss of transforming growth factor beta signaling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–8. doi: 10.1136/gut.49.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–5. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 32.Vetuschi A, Sferra R, Latella G, et al. Smad3-null mice lack interstitial cells of Cajal in the colonic wall. Eur J Clin Invest. 2006;36:41–8. doi: 10.1111/j.1365-2362.2006.01593.x. [DOI] [PubMed] [Google Scholar]
- 33.Saito K, Sakurai S, Sano T, et al. Aberrant methylation status of known methylation-sensitive CpG islands in gastrointestinal stromal tumours without any correlation to the state of c-kit and PDGFRA gene mutations and their malignancy. Cancer Sci. 2008;99:253–9. doi: 10.1111/j.1349-7006.2007.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.House MG, Guo M, Efron DT, et al. Tumour suppressor gene hypermethylation as a predictor of gastric stromal tumour behavior. J Gastrointest Surg. 2003;7:1004–14. doi: 10.1016/j.gassur.2003.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Eng protein expression inKitK641E antrum.
Eng expression in endothelium and inKit+ ICC in WT mouse small intestine.
Eng expression in endothelium and inKit+ ICC in WT mouse colon.
ENG expression in GIST882 cell line.
Graphic representation of factorial analysis of multiple correspondences based on TMA variables and their modalities.
Efficiency of SCF treatment was confirmed by the increase of pTyr-ir assessed by flow cytometry.
Sustained imatinib treatment does not affectEng mRNA expression in Ba/F3 cells.
Efficiency of imatinib treatment was confirmed by the drop of pTyr-ir assessed by flow cytometry.
Expression of Hif1α in Ba/F3 cells and in mouse antrum.