

Abstract
Although chemotherapeutic drugs could theoretically target all metastatic sites, current treatments do not provide complementary therapeutics. Therefore, the development of an alternative approach replacing the traditional therapy is urgently needed. To assess the killing efficiency of the functionally identified apoptosis-related protein (APR)-1 in melanoma cells, we established a system for the regulated expression of APR-1. The induction of APR-1 expression caused apoptosis of melanoma cells via the interaction with the juxtamembrane region of p75 neurotrophin receptor (p75NTR), and possible also via the competition with tumour necrosis factor receptor-associated factor-6 (TRAF6) and the catalytic receptor of neurotrophin (Trk) for the same p75NTR interacting site. The accumulation of APR-1 in melanoma cells may block the physical association of p75NRT with TRAF6 and/or Trk, leading to the disruption of both NF-κB and extracellular signal-regulated kinase (ERK) pathways. Also, accumulation of APR-1 protein enhanced the activity of both c-Jun-N-terminal kinase (JNK) and p38 pathways. However, the analysis of APR-1-modulated pathways demonstrated the involvement of apoptosis-regulating kinase 1-JNK/p38 pathway in the induction of Bax expression leading to both mitochondrial dysregulation [as demonstrated by the loss of mitochondrial membrane potential, the release of both cytochrome c and apoptosis-inducing factor into cytoplasm, and cleavage of caspase-9, caspase-3 and poly (ADP-ribose) polymerase (PARP)] and endoplasmic reticulum stress as demonstrated by the increase of intracellular Ca2+ release. Thus, besides the analysis of its pro-apoptotic function, our data provide insight into the molecular mechanism of APR-1-induced apoptosis of melanoma cells.
Keywords: APR-1, apoptosis, p75NTR, ASK1, JNK
Introduction
Presently no effective therapy is available for metastatic melanoma [1]. Although chemotherapeutic drugs could theoretically target all metastatic sites, current treatments do not provide significant therapeutic effects [2]. Treatment failure in metastatic melanoma is not only caused by an impressive array of cell survival pathways, but is also due to resistance to apoptotic stimuli including radio- and chemotherapy [2].
Metastatic cancers including melanoma are frequently associated with increased resistance to apoptosis induced by various therapeutic modalities [3, 4]. Therefore, the success of systemic therapy for the treatment of metastatic melanoma is minimal [5–7]. Cancer chemotherapeutic efficacy is frequently impaired by either intrinsic or acquired tumour resistance to multiple, structurally unrelated therapeutic drugs with different mechanisms of action [8]. Thus, the development of an alternative approach to overcome chemo-resistance of melanoma metastasis, complementary to current therapies, is urgently needed. Apoptosis-related protein-1 (APR-1) also known as melanoma antigen gene H1 (MAGE H1) is identified by functional screening during CD95-mediated pathways to apoptosis [9].
The p75 neurotrophin receptor (p75NTR) is a tumour necrosis factor receptor (TNFR) superfamily member that is expressed at high levels during embryogenesis and functions as a pro-apoptotic receptor during development of neuronal and non-neuronal tissues [10–12]. However, the expression of p75NTR in melanoma is widely documented [13]. The ligand requirements and downstream signalling cascades activated by p75NTR reveal the ability of p75NTR to induce cell death via mechanisms that are distinct from those activated by other pro-apoptotic TNFR super family members. P75NTR-dependent apoptosis does not involve caspase-8 activation or phosphorylation of Bcl-2 homology (BH)-domain-only family members, release of mitochondrial contents or activation of caspase-9 [14–16].
In addition, the interaction of several members of MAGE protein family with the intracellular region of p75NTR has been reported [17–19]. Neurotrophin receptor-interacting MAGE homolog (NRAGE), a member of MAGE family has been shown to function as an adaptor protein that links p75NTR to c-Jun-N-terminal kinase (JNK) activation and subsequently mediates apoptosis [18, 20]. Thus, based on its similarity to NRAGE, we performed serial deletion mapping of APR-1 protein to investigate its interacting affinity for the intracellular region of p75NRT. In this study, we demonstrated the interaction of APR-1 protein with p75NTR via the juxtamembrane domain (JMD). This interaction was found to play an essential non-redundant role in the modulation of APR-1-induced apoptosis by a mechanism including the activation of apoptosis-regulating kinase 1 (ASK1)-JNK/p38, and disruption of both NF-κB and ERK pathways.
Materials and methods
Cell lines
The melanoma cell lines A375 and BLM were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in DMEM medium containing 10% foetal bovine serum, and 100 U/ml penicillin and 100 μg/ml streptomycin.
Reagents and inhibitors
The inhibitor of ASK1 (thioredoxin) was from MERK (Haar, Germany) and the inhibitors of JNK (SP600125) and p38 (SB-203580) were from Biomol (Loerach, Germany).
Establishment of melanoma cell lines for tightly regulated expression of APR-1
The melanoma (A375-TetOn and BLM-TetOn) cells were generated using the Tet-On expression system (Clontech, Inc., Heidelberg, Germany) as described previously [21, 22]. Briefly, the melanoma cell lines (A375 and BLM) were transfected with pTet-On plasmid. Twenty-four hours later, the transfected cells were challenged with G418 (600 μg/ml) for selection. After 6 weeks, single clones were picked up and expanded. The induction level was examined by the transfection of pTRE2hyg-Luciferase vector and subsequent induction of luciferase expression by the addition of Dox (10 ng/ml) for 24 hrs. The induction levels of each clone were determined using luciferase assay as described [23]. Clones with high induction level and low background were chosen for the transfection with pTRE2 hyg-APR-1. Cells were cultured in the presence of both G418 (600 μg/ml) and Hygromycine (300 μg/ml). Single clones were picked up and expanded for further examination using qRT-PCR and luciferase assay. Clones with high induction levels were chosen for functional analysis of APR-1 protein.
Flow cytometric analysis of apoptosis by annexin V/PI staining
The detection of apoptosis was performed as described [24, 25]. Briefly, the cell lines A375-APR-1 and BLM-APR-1 were allowed to grow for 24 hrs under the recommended conditions prior the induction of APR-1 expression by the addition of Dox (10 ng/ml) for the indicated time periods. The cells were stained with 5 μl of annexin V (Vybrant; Invitrogen, Karlsruhe, Germany) and 1 μl propidium iodide (100 μg/ml) for 15 min. at room temperature as recommended by the manufacturer. Cells being annexin fluorescein isothiocyanate (FITC)+ and PI– were considered as apoptotic, and percentage of apoptotic cells was quantified using a FACSCalibur (Becton Dickinson Biosciences, Heidelberg, Germany) with apoptotic cells being annexin V+/PI–.
Measurement of mitochondrial membrane potential (ΔΨm) using JC-1
The loss of ΔΨm was assessed by flow cytometric analysis using JC-1 staining as described [21, 24]. Briefly, A375-APR-1 and BLM-APR-1 cells were allowed to grow for 24 hrs under the recommended conditions before the induction of APR-1 expression by the addition of Dox (10 ng/ml) for the indicated time periods. The cells were stained with JC-1 (10 μM) for 30 min. at room temperature in phosphate-buffered saline (PBS; Biotrend, Cologne, Germany). The intensities of green (520–530 nm) and red fluorescence (550 nm) of 50,000 individual cells were analysed on a FACSCalibur (Becton Dickinson Biosciences).
Staining of intracellular calcium
The intracellular calcium staining was performed as described previously [25]. Briefly, after the induction of APR-1 expression BLM-APR-1 cell by the addition of Dox (10 ng/ml) for 24 hrs the medium was replaced by complete medium without phenol red, and the cells were incubated for further 2 hrs before the addition of the calcium-sensitive dye Fluo3-AM (1.5 μM) and Mitotraker Red (200 nM, Invitrogen). Thirty minutes later, life pictures were taken under standard cell culture conditions on a Leica TCS SP2 AOBS with a 40× oil immersion objective using Leica Confocal software (Leica, Wetzlar, Germany).
Assessment of cell survival
The melanoma (A375-APR-1 and BLM-APR-1) cells in exponential growth phase were allowed to grow in the presence of G418 (600 μg/ml) and Puromycin (10 μg/ml) before the induction of APR-1 protein by the addition of Dox (10 ng/ml). The cell viability was determined using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide salt (MTT) assay as described previously [26].
Determination of cell viability by counting using trypan blue
The viability of either A375-APR-1 or BLM-APR-1 cells before and after the addition of doxycycline was determined by direct counting using trypan blue staining. Briefly, after the induction of APR-1 expression by the addition of Dox (10 ng/ml) for the time periods (0, 12, 24 and 48 hrs), the cells were harvested and stained with 1% trypan blue for 10 min. at room temperature, after the centrifugation for 5 min. at 1000 rpm, the supernatant was removed and the cells were resuspended in PBS. Both stained (dead cells) and unstained (viable cells) were counted using a haemocytometer chamber. The percentage of both dead and viable cells was calculated as follows:
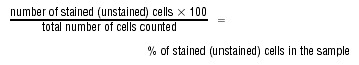 |
Immunoblot
Immunoblot analysis was performed according to standard procedures using the following antibodies and dilutions: anti-APR-1 (Biogenes, Berlin, Germany) 1:500; anti-nerve growth factor receptor (NGFR) p75 (Sc-8317), 1:1000; anti-ASK1 (Sc-7931), 1:1000; anti-p-ASK1 (Sc-101633),1: 1000; anti-JNK (Sc-474), 1: 1000; anti-p-JNK (Sc-6254), 1:1000; anti-p38 (Sc-535),1: 1000; anti-p-p38 (Sc-7973), 1:1000; anti-ERK (Sc-154-G),1: 1000; anti-p-ERK (Sc-7383), 1:1000; anti-IκB kinase (IKK)α (Sc-7182), 1:1000; anti-p-IKKα (Sc-21661), 1:1000; anti-caspase-4 (Sc-1229), 1: 1000; anti-Tom20 (Sc-11415), 1:100; anti-Bap31 (Sc-17764), 1:500; anti-actin (Sc-1615), 1: 2000; anti-caspase 3 (#7190), 1: 1000; anti-caspase 9 (#9501), 1: 1000; anti-PARP [#9542], 1: 1000; anti-Bax (#2772) 1:1000; Bak (#3814), 1:1000; anti-AIF (apoptosis-inducing factor) antibody (#4642), 1:1000 (each Cell Signaling Technology, Inc., Danvers, MA, USA) and anti-cytochrome c (ab1357–100, Abcam, Cambridge, MA, USA), 1:1000; calpain 1 (Sc-7530, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), 1:1000.
Cell lysis and subcellular fractionation
Total and nuclear cell extracts were prepared from both melanoma clones A375-APR-1 and BLM-APR-1 clones as described [21, 24, 27]. The preparations of mitochondrial and endoplasmic reticulum (ER) fractions were performed as described previously [25]. Briefly, APR-1-melanoma transfectants cultured in the presence or in the absence of Dox (10 ng/ml) were scraped off with 5 ml of PBS. Collected cells were precipitated by centrifugation at 600 ×g for 5 min., washed, resuspended and homogenized in PBS by ultrathorax and then centrifuged at 600 ×g for 5 min. The post-nuclear supernatant was layered over a discontinuous gradient of 40 and 60% sucrose. After centrifugation at 100,000 ×g for 3 hrs, aliquots were precipitated with 10% trichloroacetic acid and subjected to SDS-PAGE and immunoblotting using antibodies for mitochondria and ER, respectively.
Electrophoretic mobility shift assay
The details of electrophoretic mobility shift assay (EMSA) have been described [26, 27]. The double-stranded synthetic oligonucleotides carrying a binding site for AP-1, ELK-1, NF-κB, p53 (Santa Cruz Biotechnology, Inc.) were end-labelled with [γ-32P] ATP (Hartmann Analytika, Munich, Germany) in the presence of T4 polynucleotide kinase (Genecraft, Munster, Germany). The competition assay was performed in the same manner, except that unlabeled probes containing AP-1, ELK-1, NF-κB and p53 sequences were incubated with nuclear extracts for 20 min. before adding the labelled probes. Visualization of bands was performed following electrophoresis and exposure to high-performance autoradiography film.
In vitro translation
The cDNA encoding for APR-1 protein (amino acids 1–219) was cloned into PT7 vector (Promega, Mannheim, Germany) and in vitro transcription and translation was performed in a total reaction volume of 50 μl with 2 μg of plasmid DNA in the presence of 01 μCu [35S] methionine (Hartman Analytika) and the buffer specified by the manufacturer of the TNT kit (Promega).
Construction, cloning and production of recombinant proteins
The cDNAs constructs encoding for the full-length (amino acids 1–427) and truncated recombinant proteins (amino acids: 1–341, 1–311, 1–274) of the human p75NTR, and the cDNA encoding for full-length Fas-Associated Death Domain (FADD) protein were cloned into pGEX-5X-1 (Amersham Biosciences, Glattbrugg, Switzerland), and the production of p75NTR and FADD proteins in Escherichia coli L21 was induced for 4 hrs at room temperature by 0.5 mM isopropyl-β-D- thiogalactopyranoside, respectively. The peptides of death domain (amino acids 340–427) of p75NTR, and the intracellular domains of TNFR1, TNFR2, Fas, TrkA, TrkB, TrkC, mouse p75NTR, rat p75NTR and Death Associated protein (DAP) kinase were provided by Peptides&elephants GmbH (Postdam, Germany).
Far Western blot
P75NTR protein was separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membrane (Bio-RAD, Munche, Germany). The blot was placed in TEN 50 buffer (10 mM Tris-Hcl pH 8.0, 1 mM ethylenediaminetetraacetic acid [EDTA], 50 mM NaCl) and stored at 4°C overnight to allow in situ renaturation of the protein. Then the membrane was blocked with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-hybridization buffer (20 mM HEPES, pH 7.7, 75 mM KCl, 0.1 mM EDTA, 2.5 mM MgCl2, 1 mM dithiothreitol, 0.05% Triton X-100) supplemented with 5% non-fat dry milk overnight and incubated with 35S-labelled APR-1 protein in hybridization buffer supplemented with 0.05% non-fat dry milk overnight. The membranes were washed three times for 30 min. each in hybridization buffer. The visualization of bands was performed by the exposure to high-performance autoradiography film.
Application of siRNAs
The knockdown of p75NTR was performed by its specific siRNA (D-009340–03; Dharmacon RNA Technology, Lafayette, CO, USA) as recommended by manufacturer’s protocol.
Results
Tightly regulated expression of APR-1 triggers apoptosis of melanoma cells
To examine the function of APR-1 protein as apoptotic mediator in melanoma cells and to elucidate the underlying mechanisms, whereby APR-1 triggers cell death, we established A375 and BLM cell lines for the inducible expression of APR-1 protein using tetracycline system [21, 27].
Both A375/TetOn-APR-1 and BLM/TetOn-APR-1 cells were, tested for their high induction efficiency and low background in a time course experiments (data not shown). Figure 1A demonstrates tightly regulated expression of APR-1 protein by the addition of Dox (10 ng/ml) to the culture media for regulated time intervals. The induction of APR-1 protein was noted at 12 hrs after the addition of Dox (10 ng/ml) and increased thereafter up to 48 hrs (Fig. 1A). In addition, we performed a time course experiment by the addition of Dox (10 ng/ml) to the culture media of both A375-APR-1 and BLM-APR-1 at different time periods (0, 12, 24 and 48 hrs). Next, we determined the cell viability by either MTT assay (Fig. 1B) or by direct counting using trypan blue staining (Fig. 1C). Although the addition of Dox to culture media does not influence the viability of the parental cells (A375 and BLM), the induction of APR-1 expression, in both A375-APR-1 and BLM-APR-1 was found to reduce markedly the cell viability in a time-dependent manner. Accordingly, the reduction of cell viability was noted at 12 hrs past treatment and increased thereafter to reach a maximum at 48 hrs in both cells (Fig. 1B, C).To check whether APR-1-induced melanoma cell death is mediated by an apoptotic mechanism, we confirmed APR-1-induced cell death by flow cytometry using annexin V/PI staining. Twenty four hours following the induction of APR-1 protein, both melanoma cells were stained with annexin V-FITC/PI and subjected for flow cytometric analysis. The induction of APR-1 protein was found to kill melanoma cells by an apoptotic mechanism as demonstrated by annexin V/PI staining (Fig. 1B). Also, the role of mitochondria in APR-1-induced apoptosis was analysed. Flow cytometric analysis using JC-1 staining (Fig. 1C) demonstrated the loss of ΔΨm by the induction of APR-1 protein in both melanoma cells, suggesting a central role for mitochondrial pathway(s) in the modulation of APR-1-induced apoptosis. Next, we confirmed APR-1-induced apoptosis of melanoma cells at molecular levels. Data obtained from Western blot analysis (Fig. 1D) demonstrated the induction of both mitochondrial AIF and cytochrome c release into the cytoplasm, when analysed in both cytosolic and mitochondrial fractions. The purity of both fractions was assessed using anti-Tom 20 antibody as marker for mitochondrial fraction. Also, the induction of APR-1 expression was found to enhance the cleavage of caspase-9, caspase-3 and PARP (Fig. 1E) providing evidence for the involvement of the mitochondrial apoptotic pathway(s) in the modulation of APR-1-induced apoptosis.
Fig 1.
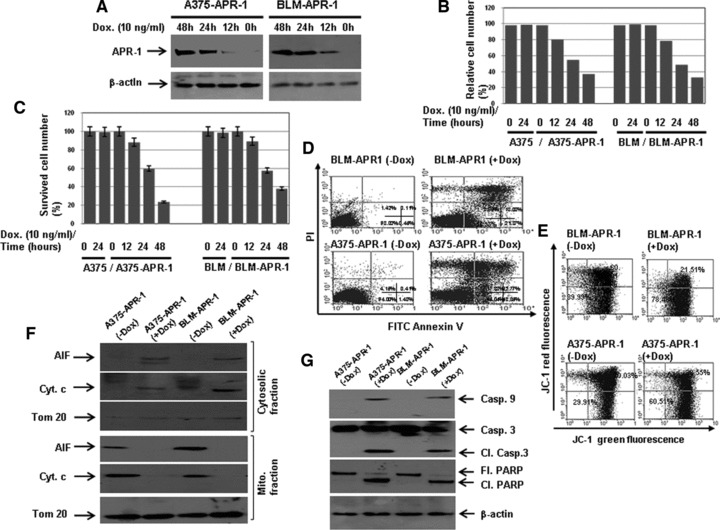
(A) Time-dependent induction of APR-1 protein in both A375-APR-1 and BLM-APR-2 cells. Both A375-APR-1 and BLM-APR-1 cells were allowed to grow in the absence or in the presence of Dox (10 ng/ml) for regulated time intervals up to 48 hrs. The cells were harvested and subjected to Western blot analysis using anti-APR-1 antibody. Time course experiment demonstrates APR-1-induced reduction of cell viability in both A375-APR-1 and BLM-APR-1 cells as assessed by MTT assay (B) or by direct counting using trypan blue staining (C). Both parental cell lines were used as control to assess the toxicity of Dox on melanoma cells. (D) Flow cytometric analysis using annexin V/PI staining demonstrates the induction of apoptosis in melanoma cells by the expression of APR-1 protein. (E) Flow cytometry using JC-1 staining shows the loss of ΔΨm by the induction of APR-1 protein. Cells with intact mitochondria displayed high red and high green fluorescence and appear in the upper right quadrant of the scatterplots. In contrast, cells that had lost their ΔΨm displayed high green and low red fluorescence and appear in the lower right quadrant. (F) Western blot analysis demonstrates AIF and cytochrome c release in both cytosolic and mitochondrial fractions. Tom 20 (mitochondrial marker) was used as specific control for the purity of mitochondrial fractions. (G) Western blot analysis demonstrates cleavage of caspase 9, caspase-3 and PARP by the induction of APR-1 protein in melanoma cells. β-actin was used as internal control for loading and transfer. Data are representative of three independent experiments.
Interaction of APR-1 protein with juxtamembrane domain of p75NTR
Based on the fact that the identification of APR-1 was made during Fas-induced pathway to apoptosis, we suggested that APR-1 protein may achieve its pro-apoptotic function via direct interaction with the intracellular domains of TNFR superfamily members. Next, we screened a set of candidate receptors including TNFR1, TNFR2, Fas, human p75NTR, mouse p75NTR, rate p75NTR, TrkA, TrkB, TrkC, FADD and DAP kinase (Fig. 2A). Data obtained from dot blot analysis of in vitro transcribed and translated [35S] APR-1 protein with the candidate proteins revealed the interaction of APR-1 protein with p75NTR proteins derived from human beings, mice or rats. However, a weak interaction was noted with TNFR1, when compared with the other candidates. Next, we performed co-immune precipitation to confirm the interaction of APR-1 with p75NTR. The detection of APR-1 in immune-precipitated p75NTR protein as well as the detection of p75NTR in immune-precipitated APR-1 derived from APR-1-expressing cells (Fig. 2B) confirmed further the interaction of APR-1 with p75NTR. Moreover, we aimed to localize the APR-1-interaction domain of the human p75NTR protein. We generated a series of deletion covering the cytoplasmic region of p75NTR. Besides the amplification of the cDNA encoding the full-length p75NTR protein (1–427), the different cDNAs encoding for the following peptides (aa 1–341), (aa 1–311), (aa 1–274), JMD (aa 275–341) and death domain (aa 341–427) were also performed (Fig. 2C). Data obtained from Far Western blot using the fusion proteins of the different domains of p75NTR and in vitro transcribed and translated APR-1 protein (Fig. 2D) revealed the interaction of APR-1 with the intracellular domains of the p75NTR. Furthermore, the hybridization of APR-1 to both death domain and JMD identified JMD as APR-1 interaction domain (Fig. 3E), suggesting that APR-1-induced apoptosis may be mediated by direct interaction of APR-1 with the JMD of p75NTR.
Fig 2.
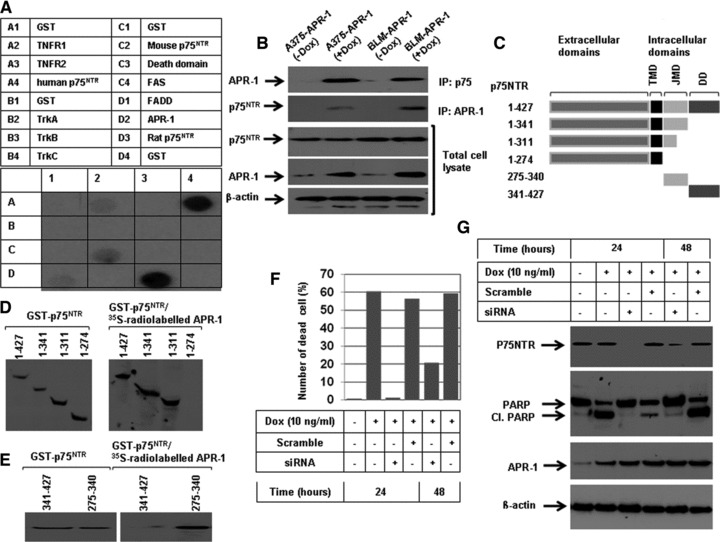
(A) Dot blots analysis of the interaction of APR-1 protein with TNFR1 human p75NTR, mouse p75NTR, FADD and rat p75NTR. A total of 5 μg of glutathione S-transferase (GST)-TNFR1 (100.0 pmol), TNFR2 (111.0 pmol), human p75NTR (106.4 pmol), mouse p75NTR (106 pmol), FADD (92.6 pmol), TrKA (113.6 pmol), TrKB (113.6 pmol), TrKC (113.6 pmol), Fas 142.8 pmol), death domain (142.8 pmol), APR-1 (100 pmol), rat p75NTR (106 pmol) or GST (192.0 pmol) were diluted in PBS and blotted onto nitrocellulose membrane and subsequently incubated overnight with in vitro transcribed and translated [35S] APR-1 protein. (B) Interaction of APR-1 with P75NTR. The total cell lysates prepared from A375-APR-1 and BLM-APR-1 before and after the induction of APR-1 protein were subjected for either electrophoresis (for the detection of APR-1 and P75NTR) or for co-immunoprecipitation (IP) with either anti-P75NTR antibody or with anti-APR-1 antibody. Western blotting of IP: p75NTR for APR-1 revealed the interaction of APR-1 to P75NTR, whereas Western blotting of IP: APR-1 for P75NTR revealed the interaction of P75NTR to APR-1. β-actin was used as internal control for loading and transfer. (C) Schematic diagram of the extracellular and intracellular domains of p75NTR. Transmembrane domain, JMD and death domain. (D) GST-P75NTR recombinant proteins 1–427aa (106.4 pmol), 1–341aa (135.1 pmol), 1–311aa (147 pmol), 1–274 aa (166 pmol), 275–340aa (694.3) and 341–427aa (526.2 pmol), were separated by SDS-PAGE, and blotted on PVDF membrane and probed with in vitro transcribed and translated [35S] APR-1. The interaction of APR-1 with the P75NTR domains was detected by exposing the membrane to X-ray films. The coomassie-stained gel shows the amount and the position of P75NTR recombinant proteins (left panel). (E) GST-JMD and death domain of P75NTR were separated by SDS-PAGE, and blotted on PVDF membrane and probed with in vitro transcribed and translated [35S] APR-1. The interaction of APR-1 with both domains was detected by exposing the membrane to X-ray films. The coomassie-stained gel shows the amount of both JMD and death domains (left panel). (F) Western blot analysis demonstrates the expression of APR-1 by the addition of Dox to the culture medium of BLM-APR- 1, the knockdown of p75NTR by its specific siRNA and the suppression of APR-1-induced cleavage of PARP by the p75NTR siRNA. β-actin was used as internal control for loading and transfer. (G) Analysis of cell viability by counting using trypan blue staining. Rescue of APR-1-induced reduction of cell viability by the knockdown of p75NTR by siRNA for 24 or 48 hrs. Data are mean of three experiments performed separately.
Fig 3.
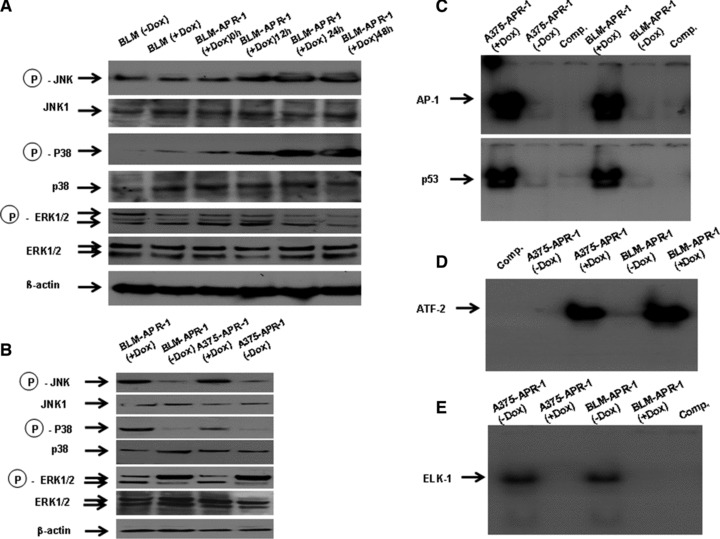
Activation of both JNK and p38, and inhibition of ERK pathways by the induction of APR-1 protein. (A) Western blot analysis of JNK, p38 and ERK pathways demonstrates the enhancement of JNK and p38, and inhibition of ERK phosphorylation by the induction of APR-1 protein, in BLM-APR-1 cells, is time dependent. Western blot (B): the analysis of the total cell lysates of A375-APR-1 and BLM-APR-1 cells using phospho-specific antibodies demonstrated the phosphorylation of both JNK and p38, and the inhibition of phospho-ERK by the induction of APR-1-expression. Whereas, rehybridization of the same blots with antibodies recognizing the total proteins of JNK, p38, and ERK showed no alteration at their expression levels by the induction of APR-1 protein. β-actin was used as internal control for loading and transfer. EMSA demonstrates the enhancement of the DNA-binding activities of the transcription factors AP-1 and p53 (C), and ATF-2 (D), and the inhibition of DNA-binding activity of the transcription factor ELK-1 (E) by the induction of APR-1 protein. Data are representative of three independent experiments performed separately.
Suppression of APR-1-induced apoptosis of melanoma cells by the knockdown of p75NTR
To confirm the functional role of p75NTR in the modulation of APR-1-induced apoptosis of melanoma cells, the BLM-APR-1 cells were pre-treated with either scramble or with p75NRT siRNA before the addition of Dox (10 ng/ml). The cells were harvested at 24 and 48 hrs following the addition of Dox to the culture medium. The harvested cells were either subjected to trypan blue staining or Western blot analysis. Western blot analysis (Fig. 2F) demonstrated the efficiency of siRNA to knockdown the basal expression level of p75NR, although data obtained from cell counting using trypan blue staining (Fig. 2G) demonstrated the rescue of APR-1-induced reduction of cell viability by the knockdown of p75NTR, when compared with control cells. Also, the inhibition of APR-1-induced PARP cleavage in response to the treatment with p75NTR siRNA (Fig. 2F) confirmed the involvement of p75NTR in the modulation of APR-1-induced apoptosis of melanoma cells.
Activation of JNK and p38, and inhibition of ERK pathways by the induction of APR-1 protein in melanoma cells
To show whether APR-1-induced effects on the three mitogen activated protein (MAP) kinases JNK, p38 and ERK are correlated with the expression level of APR-1 protein in melanoma cells. Next, we allowed the BLM-APR1 cells to grow in the presence of Dox (10 ng/ml) for different time intervals (0, 12, 24 and 48 hrs), whereas the parental cells were allowed to grow in the absence or in the presence of Dox (10 ng/ml) for 24 hrs, and used as control for Dox mediated effects. The analysis of total cell lysates by Western blot using specific antibodies for phospho- and total JNK, p38 and ERK proteins (Fig. 3A) demonstrated the induction of both phospho-JNK and p38, and inhibition of ERK proteins at 12 hrs, and the observed effects increased thereafter at 24 and 48 hrs when compared to BLM-APR-1 treated with Dox for 0 hr or with parental cells cultured in the presence or in the absence of Dox. Based on the data of time course experiments, we verified the effect of APR-1-induced effects on MAP kinase following the addition of Dox to the culture media of both A375-APR-1 and BLM-APR-1 for 24 hrs. Although no alteration was noted at the expression levels of JNK, p38 or ERK (Fig. 3A), the induction of APR-1 protein was found to enhance the phosphorylation of both JNK and p38, and to inhibit the phosphorylation of ERK (Fig. 3A). Next, we analysed the DNA-binding activity of the transcription factors AP-1, p53, activating transcription factor (ATF) and ELK-1, possible physiological substrates of JNK, p38 and ERK pathways. The analysis of the DNA-binding activities of the transcription factors using EMSA demonstrated the activation of transcription factors AP-1 and p53 (Fig. 3B), and ATF-2 (Fig. 3C), and the suppression of the transcription factor ELK-1 (Fig. 3D) confirming APR-1-induced effects on both JNK/p38 and ERK pathways.
Inhibition of NF-κB pathway in APR-1-expressing cells
To investigate the effect of APR-1 accumulation on NF-κB pathway, we performed a time course experiments to show whether APR-1-induced effects on NF-κB pathways are correlated with the induced level of APR-1 protein. Both A375-APR-1 and BLM-APR-1 cells were allowed to grow in the presence of Dox (10 ng/ml) at different time intervals (0, 12, 24 and 48 hrs), although the parental cells A375 and BLM cells were allowed to grow in the presence or in the absence of Dox (10 ng/ml) for 24 hrs and used as control for cytotoxic effects of Dox. The total cell lysates were subjected to Western blot analysis using specific antibodies for total and phospho-IκBα protein. Although no marked effect was noted on the viability of the parental cells cultured in presence or in the absence of Dox, the induction of APR-1 expression for 12 hrs was found to suppress the basal phospho IκBα. However, the noted APR-1-induced inhibition of IκBα increased thereafter following the induction of APR-1 for 24 and 48 hrs by the addition of Dox to the culture medium of A375-APR-1 (Fig. 4A) and BLM-APR-1 (Fig. 4B) cells. In addition, the inhibition of IκBα phosphorylation by APR-1 in both A375-APR-1 (Fig. 4A) and BLM-APR-1(Fig. 4B) cells was correlated with the increase of the stability of IκBα protein, an evidence for the inhibition of NF-κB pathway. Also, EMSA assay (Fig. 4C and D) demonstrated that APR-1-induced inhibition of NF-κB activity is time dependent.
Fig 4.
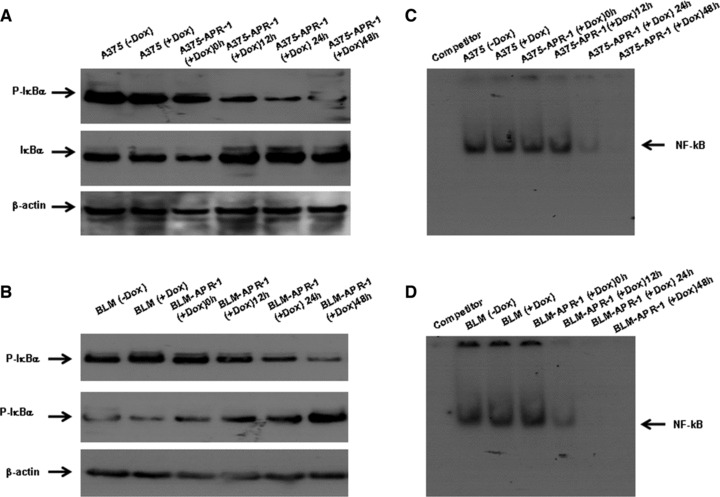
Inhibition of NF-κB pathway by the induction of APR-1 expression in melanoma cells. (A, B) Western blot analysis demonstrates time-dependent inhibition of p-IκBα that is correlated with the increase of IκBα protein stability by the induction of APR-1 in both A375-APR-1and BLM-APR-1 cells. (C, D) EMSA demonstrates time-dependent inhibition of the transcription factor NF-κB by the expression of APR-1 in both A375-APR-1 and BLM-APR-1cells, when compared to control parental cell cultured in the absence or in the presence of Dox. Data are representative of three independent experiments.
The induction of APR-1 protein in melanoma cells influences both pro- and anti-apoptotic factors
Next, we investigated the effect of APR-1 protein on both pro-and anti-apoptotic proteins, by the analysis of the expression levels of pro-apoptotic proteins (Bax and Bak), and the anti-apoptotic proteins (Mcl-1 and Bcl-xL), as well as the inhibitor of apoptosis (XIAP) and survivin proteins. Figure 5A demonstrates the induction of Bax, but not Bak expression, and the suppression of Mcl-1, XIAP and survivin in APR-1-expressing cells, suggesting the involvement of both pro-and anti-apoptotic proteins in the modulation of APR-1-induced apoptosis. In order to show the role of Bax protein in the context of APR-1-induced apoptosis, we analysed the subcellular localization of the pro-apoptotic protein Bax in both ER and mitochondrial fractions. Data obtained from Western blot analysis (Fig. 5B) revealed the accumulation of Bax protein in both ER and mitochondrial fractions prepared from APR-1-expressing cells. This clearly indicates the localization of Bax protein at both ER and mitochondria, suggesting a functional role for Bax in the modulation of APR-1-induced apoptosis. In addition, we investigated whether the localization of APR-1-induced Bax protein is associated with intracellular Ca2+ release. Therefore, BLM-APR-1 cells were induced for the expression of APR-1 protein by addition of Dox (10 ng/ml) to the culture medium for 24 hrs, and then incubated with the Ca2+−sensitive dye Fluo3 (Fig. 5C). As expected, the induction of APR-1 protein resulted in the increase of intracellular Ca2+ release into the cytoplasm when compared to BLM-APR-1 control cells.
Fig 5.
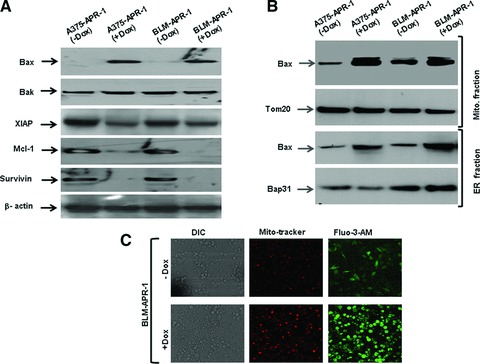
(A) Western blot analysis demonstrates induction of Bax and inhibition of XIAP, Mcl-1 and surviving by the induction of APR-1 protein without influencing the basal expression level of Bak. β-actin was used as internal control for loading and transfer. (B) Western blot analysis of Bax protein in both mitochondrial and ER fractions. The purity of both mitochondrial and ER fractions was assessed by the detection of Tom20 (mitochondrial marker) and Bap31 (ER marker) in the corresponding fractions. (C) Intracellular Ca2+ levels in BLM-APR-1 cells before and after the addition of Dox (10 ng/ml) to the culture medium for 24 hrs as assessed by staining with Ca2+-sensitive dye Fluo3-AM and Mitotracker red. Differential interference contrast images (DIC) are shown. Data are representative of three independent experiments.
APR-1-induced apoptosis of melanoma cells is mediated by Bax via ASK1-JNK/p38 pathway
To investigate the molecular mechanism of APR-1-induced Bax expression and to address the role of Bax in the context of APR-1-induced apoptosis of melanoma cells, both A375-APR-1 and BLM-APR-1 cells, were pre-treated with the inhibitors of ASK1 (thioredoxin) and JNK (SP600125) before induction of APR-1 expression for 24 hrs, and the total cell lysates and nuclear extracts were prepared. Data obtained from EMSA (Fig. 6) demonstrated the inhibition of APR-1-induced AP-1 (Fig. 6A and B), p53 (Fig. 6C and D) and ATF-2 (Fig. 6E and F) by the corresponding inhibitors of ASK1, JNK and p38 pathways. Next, we examined the effect of both ASK1 and JNK inhibitors, on APR-1-induced Bax expression. Interestingly, the inhibition of either ASK1 or JNK by their specific inhibitors was found to block APR-1-induced Bax as demonstrated by Western blot analysis (Fig. 7A and B), suggesting that APR-1-induced Bax expression is mediated by ASK1-JNK-p53/AP-1 pathway in both melanoma cells. In addition, we analysed the effect of ASK1 and JNK inhibitors on the extent of APR-1-induced apoptosis in both melanoma cells. Next, we evaluated the effect of both Dox and inhibitors (thioredoxin and Sp600125) on the extent of the viability of the parental cells; both A375 and BLM cells were treated with the recommended concentration of both Dox and inhibitors for the indicated time (24 hrs) before the measurement of their viability using MTT assay. Data obtained from cell viability assay (Fig. 7C and D) demonstrated no marked effect on the viability of the parental cells in response to the treatment with either Dox or the inhibitors. In contrast, the abrogation of APR-1-induced cell death by the inhibitors of ASK1 (thioredoxin) or JNK (SP600125) as shown (Fig. 7C and D), provides evidence for the involvement of the ASK1-JNK-AP-1/p53-Bax pathway in the modulation of APR-1-induced apoptosis of melanoma cells.
Fig 6.
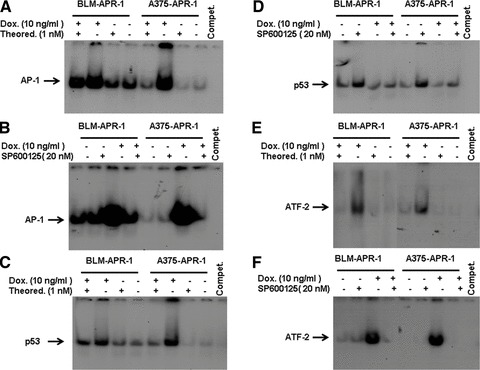
Inhibition of ASK1, JNK and p38 by their specific inhibitors blocks APR-1-induced activation of the transcription factors AP-1, ATF-2 and p53. EMSA demonstrates the inhibition of APR-1-induced DNA-binding activities of AP-1 (A, B), p53 (C, D) and ATF-2 (E, F) by the corresponding inhibitors of ASK1, JNK and p38. Data are representative of three independent experiments.
Fig 7.
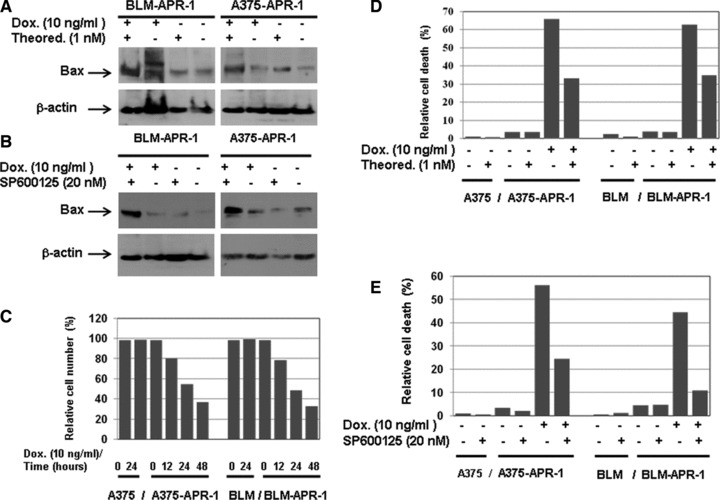
Suppression of APR-1-induced Bax expression by the inhibition of ASK1 and JNK. Western blot analysis demonstrates the inhibition of APR-1-induced Bax expression by the inhibitors of ASK1 (A) or JNK (B). β-actin was used as internal control for loading and transfer. MTT assay demonstrates the inhibition of APR-1 induced cell death by the inhibitors of ASK1 (C) or JNK (D). Data are the mean values of three independent experiments.
Discussion
APR-1 (also known as Restin or MAGE H1) is a recently identified protein that belongs to the MAGE proteins family [9]. The overexpression of APR-1 of this protein in melanoma cells was found to overcome melanoma resistance to current therapies via the interaction with the JMD of p75NTR.
Although the activation and expression of MAGEs were reported in various human cancers with highest frequency in melanoma, the physiological function of MAGEs remains largely unknown [28]. However, the involvement of MAGEs in the regulation of cell cycle progression [29] and apoptosis [18] has been suggested. Thus, the determination of the mechanisms, which are responsible for the regulation of MAGEs functions, may help for the development of a therapeutic approach for the treatment of melanoma metastasis.
Based on our data, the interaction of APR-1 with p75NTR seems to be responsible for the disruption of both NF-κB and ERK pathways, as well as the activation of both JNK and p38 pathways. APR-1 may function as an adaptor protein that links p75NTR to JNK and p38 activation, and subsequently mediates apoptosis. This phenomenon relies, at least in part, on the competition of TNFR-associated factor-6 (TRAF6), and APR-1 for the same p75NTR interacting domain. Thus, the overexpression of APR-1 may block the physical association of p75NTR with TRAF6, and thereby cause disruption of the TRAF6/NF-κB pathway that subsequently, in turn, results in the enhancement of APR-1-induced apoptosis. However, the mechanism of APR-1- mediated inhibition of ERK pathway still remains to be determined in detail.
Two parallel signalling pathways are coupled to p75NTR: one pathway leads to apoptosis [30] and the other pathway leads to survival [31]. The activation of p75NTR results in the activation of JNK and, in turn, to apoptotic cell death. However, this event depends on the cell type and its differentiation state [29]. The outcome of p75NTR signalling is modified by the orchestration of several downstream adaptor proteins [10, 32]. Some adaptor proteins are associated with cell death such as neurotrophin receptor interaction factor and p75NTR-associated cell death executor, whereas others appear to promote cellular survival (Fas-associated phosphatase-1, receptor-interacting protein-2 and TNFR-associated factor 6) [32, 33].
In the present study, we found that APR-1 triggers apoptosis of melanoma cells via a mechanism mediated by mitochondrial-dependent pathway(s). Using pharmalogical inhibitors, we could demonstrate that APR-1-induced apoptosis of melanoma cells is mediated by the activation of the ASK1-JNK/p38 pathway leading to the overexpression of Bax that subsequently localizes to both mitochondria and ER resulting in mitochondrial dysregulation and ER stress. Mitochondrial dysregulation was characterized by the release of both AIF and cytochrome c into the cytoplasm, and cleavage of casapase-9, caspase-3 and PARP, whereas ER stress was characterized by the increase of intracellular Ca2+ release into the cytoplasm. Also, the inhibition of the survival pathways NF-κB and ERK by the expression of APR-1 in melanoma cells suggests an important role for these pathways in the modulation of APR-1-induced apoptosis.
A variety of signalling pathways have been detected for p75NTR. These include the activation of JNK that has been observed in cells undergoing cell death [34]. The activity of JNK can be up-regulated by neurotrophins under apoptotic conditions through p75NTR. Another signalling response is the activation of the nuclear transcription factor NF-κB. However, increasing evidence has demonstrated that many TNFR family members interact with TRAFs to modulate JNK and NF-κB activity as well as apoptosis [35, 36]. Thus, based on our data and in agreement with the above-mentioned reports, the involvement of both JNK and NF-κB pathways in the modulation of APR-1-induced apoptosis is considered.
In addition to the investigation of the molecular mechanisms, which are implicated in the regulation of APR-1-induced apoptosis, we demonstrated that APR-1-induced apoptosis is mediated via the interaction of APR-1 protein with the intracellular region of p75NTR. The intracellular domains of p75NTR are 154 amino acids in length and contain a highly conserved juxtamembrane region and a death domain sequence in the C terminus [37]. Data obtained from Far Western blot revealed the interaction of APR-1 protein with the highly conserved region of p75NTR cytoplasmic region, JMD. This JMD is completely conserved between human, rat and chicken p75NTR genes [38].
The identification of juxtamembrane region of p75NTR as an interacting domain for APR-1 was initially unexpected, because p75NTR contains a C-terminal death domain that has been shown to be relevant for apoptosis [39]. Death domains are responsible for apoptosis induced by the canonical death receptors such as TNFR-1 and Fas [40, 41]. Therefore, it may be more appropriate to think of the death domain of p75NTR as an APR-1 interaction domain.
Thus, the overexpression of APR-1 may initiate a strong apoptotic signalling cascade via the interaction with the JMD of p75NTR that leads to activation of both JNK and p38, and disruption of pro-survival signalling NF-κB and ERK pathways, together with the induction of pro-apoptotic protein Bax and degradation of anti-apoptotic proteins Mcl-1, XIAP and survivin.
In the present study, APR-1 was found to interact with P75NTR to induce the activation of JNK and p38, and disruption of both NF-κB and ERK pathways, and subsequently leads to the release of apoptosis. However, it is not known whether these two mechanisms operate independently, perhaps in different cell types, or whether APR-1 can induce multiple signals to ensure cell death. Thus, based on our finding we suggested a model for APR-1-induced apoptosis of melanoma cells (Fig. 8).
Fig 8.
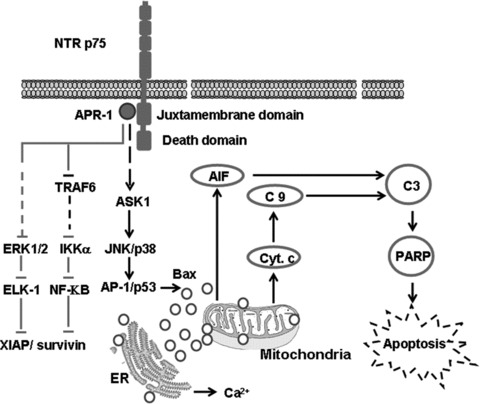
Proposed model for APR-1-induced apoptosis of melanoma cells. The interaction of APR-1 with the JMD of P75NTR leading to the activation of ASK1-JNK/p38 and the disruption of ERK and NF-κB pathways. The activation of ASK1-JNK/p38 pathway enhances the expression of Bax protein that, in turn, triggers mitochondrial dysregulation leading to the release of cytochrome C and AIFs into the cytoplasm, and the cleavage of caspase-9, caspase-3 and PARP, whereas the contribution of both ERK and NF-κB pathways to APR-1-induced apoptosis may be mediated by the inhibition of survival proteins including XIAP and survivin.
In conclusion, we confirmed the pro-apoptotic function of APR-1 in melanoma-derived cells lines. APR-1 triggers apoptosis of melanoma cells through the interaction with the JMD of p75NTR via a molecular mechanism including the activation of both JNK and p38, and disruption of both NF-κB and ERK pathways.
Acknowledgments
This study was supported by a grant from German Research Foundation (HA 5081/3–1) to M.H. We gratefully acknowledge W. Hoth-Hannig (Research Laboratory of the Clinic of Operative Dentistry, Periodontology and Preventive Dentistry, Saarland University, Homburg/Saar, Germany) for flow cytometry analysis and MTT assay; G. Koenig (Institut National de la Santé et de la Recherché Médicale, U 977, University of Strasbourg) and A. Chesspot (Dental Faculty, University of Strasbourg) for cell culture experiments.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Li Y, McClay EF. Systemic chemotherapy for the treatment of metastatic melanoma. Semin Oncol. 2002;29:413–26. doi: 10.1053/sonc.2002.35237. [DOI] [PubMed] [Google Scholar]
- 2.Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–51. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 3.Glinsky GV, Glinsky VV, Ivanova AB, et al. Apoptosis and metastasis: increased apoptosis resistance of metastatic cancer cells is associated with the profound deficiency of apoptosis execution mechanisms. Cancer Lett. 1997;115:185–93. doi: 10.1016/s0304-3835(97)04738-1. [DOI] [PubMed] [Google Scholar]
- 4.Röckmann H, Schadendorf D. Role of apaf-1 in melanoma drug resistance and apoptosis. J Invest Dermatol. 2005;125:386–7. doi: 10.1111/j.0022-202X.2005.23821.x. [DOI] [PubMed] [Google Scholar]
- 5.Serrone L, Zeuli M, Sega FM, et al. Dacarbazine-based chemotherapy for metstatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19:21–34. [PubMed] [Google Scholar]
- 6.Bhatia S. Treatment of metastatic melanoma: an overview. Oncology. 2009;6:1–5. [PMC free article] [PubMed] [Google Scholar]
- 7.Atkins MB, Gollob JA, Sosman JA, et al. A phase II pilot trial of concurrent biochemotherapy with cisplatin, vinblastine, temozolomide, interleukin 2, and IFN-alpha 2B in patients with metastatic melanoma. Clin Cancer Res. 2002;8:3075–81. [PubMed] [Google Scholar]
- 8.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;22:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 9.Hassan M, Mirmohammadsadegh A, Selimovic D, et al. Identification of functional genes during Fas-mediated apoptosis using a randomly fragmented cDNA library. Cell Mol Life Sci. 2005;62:2015–26. doi: 10.1007/s00018-005-5172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. 2002;67:203–33. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 11.Nykjaer A, Willnow TE, Peterson CM. P75NTR-live or let to die. Current Opin Neurobiol. 2005;15:49–57. doi: 10.1016/j.conb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Teng KK, Hempstead BL. Neurotrophins and their receptors: signaling trios in complex biological systems. Cell Mol Life Sci. 2004;61:35–48. doi: 10.1007/s00018-003-3099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trúzzi F, Marconi A, Lotti R, et al. Neurotrophins and their receptors stimulate melanoma cell proliferation and Migration. J Invest Dermat. 2008;128:2031–40. doi: 10.1038/jid.2008.21. [DOI] [PubMed] [Google Scholar]
- 14.Beattie MS, Harrington AW, Lee R, et al. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–86. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrington AW, Leiner B, Blechschmitt C, et al. Second proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Pros Natl Acad Sci USA. 2004;101:6226–30. doi: 10.1073/pnas.0305755101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Bauer JH, Li Y, Shao Z, et al. Characterization of a p75 (NTR) apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–20. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- 17.Kuwako K, Taniura H, Yoshikawa K. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J Biol Chem. 2004;279:1703–12. doi: 10.1074/jbc.M308454200. [DOI] [PubMed] [Google Scholar]
- 18.Salehi AH, Roux PP, Kubu CJ, et al. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–88. doi: 10.1016/s0896-6273(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 19.Tcherpakov M, Bronfman FC, Conticello SG, et al. The p75 neurotrophin receptor interacts with multiple MAGE proteins. J Biol Chem. 2002;277:49101–4. doi: 10.1074/jbc.C200533200. [DOI] [PubMed] [Google Scholar]
- 20.Bhakar AL, Howell JL, Paul CE, et al. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of bad. J Neurosci. 2003;23:11373–81. doi: 10.1523/JNEUROSCI.23-36-11373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassan M, Selimovic D, Ghozlan H, et al. Hepatitis C virus (HCV) core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology. 2009;49:1469–82. doi: 10.1002/hep.22849. [DOI] [PubMed] [Google Scholar]
- 22.Erhardt A, Hassan M, Heintges T, et al. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292:272–84. doi: 10.1006/viro.2001.1227. [DOI] [PubMed] [Google Scholar]
- 23.Hassan M, Selimovic D, Ghozlan H, et al. Induction of high-molecular-weight (HMW) tumour necrosis factor (TNF) alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3) in liver cells is AP-1 and NF-kappaB-dependent activation. Cell Signal. 2007;19:301–11. doi: 10.1016/j.cellsig.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Cetindere T, Nambiar S, Santourlidis S, et al. Induction of indolamine 2, 3-deoxygenase by death receptor activation contributes to apoptosis of melanoma cells via mitochondrial damage-dependent ROS accumulation. Cell Signal. 2010;22:197–211. doi: 10.1016/j.cellsig.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Hassan M, Alaoui A, Feyen O, et al. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene. 2008;27:4557–68. doi: 10.1038/onc.2008.90. [DOI] [PubMed] [Google Scholar]
- 26.Hassan M, Ghozlan H, Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology. 2005;333:324–36. doi: 10.1016/j.virol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Hassan M, Ghozlan H, Abdel-Kader O. Activation of RB/E2F signaling pathway is required for the modulation of hepatitis C virus core protein-induced cell growth in liver and non-liver cells. Cell Signal. 2004;16:1375–85. doi: 10.1016/j.cellsig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Sang M, Wang L, Ding C, et al. Melanoma-associated antigen genes-An update. Cancer Lett. 2011;302:85–90. doi: 10.1016/j.canlet.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 29.Saburi S, Nadano D, Akama TO, et al. The trophinin gene encodes a novel group of MAGE proteinsmagphinins, and regulates cell proliferation during gametogensis in the mouse. J Biol Chem. 2001;276:49378–89. doi: 10.1074/jbc.M108584200. [DOI] [PubMed] [Google Scholar]
- 30.Yaar M, Zhai S, Pilch PF, et al. binding of beta-amyliod to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J Clin Invest. 1997;100:2333–40. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein RS, Avivi C, Geffen R. In vivo NGF treatment increases proliferation on the primary sympathetic ganglia of chick embryos. Dev Biol. 1997;181:116–20. doi: 10.1006/dbio.1996.8456. [DOI] [PubMed] [Google Scholar]
- 32.Arevalo JC, Wu SH. Neurotrophin signaling: many exciting surprises. Cell Mol Life Sci. 2006;63:1523–37. doi: 10.1007/s00018-006-6010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hempstead BL. The many faces of p75NTR. Curr Opin Neurobiol. 2002;12:260–7. doi: 10.1016/s0959-4388(02)00321-5. [DOI] [PubMed] [Google Scholar]
- 34.Bemis LT, Geske FJ, Strange R. Use of the yeast two-hybrid system for identifying the cascade of protein interactions resulting in apoptotic cell death. Methods Cell Biol. 1995;46:139–51. doi: 10.1016/s0091-679x(08)61928-7. [DOI] [PubMed] [Google Scholar]
- 35.Rothe M, Pan MG, Henzel WJ, et al. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–52. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 36.Song HY, Régnier CH, Kirschning CJ, et al. Tumour necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci USA. 1997;94:9792–6. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liepinsh E, Ilag LL, Otting G, et al. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J. 1997;16:4999–5005. doi: 10.1093/emboj/16.16.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Large TH, Weskamp G, Helder JC, et al. Structure and developmental expression of the nerve growth factor receptor in the chicken central nervous system. Neuron. 1989;2:1123–34. doi: 10.1016/0896-6273(89)90179-7. [DOI] [PubMed] [Google Scholar]
- 39.Llambi F, Causeret F, Bloch-Gallego E, et al. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 2001;20:2715–22. doi: 10.1093/emboj/20.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tartaglia LA, Ayres TM, Wong GH, et al. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845–53. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 41.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]