

Abstract
Previous studies have shown that the transcription factor signal transducer and activator of transcription 1 (STAT1) activation is increased in primary cardiac myocytes exposed to simulated ischaemia/reperfusion injury. This promotes apoptotic cell death by enhancing the expression of pro-apoptotic proteins. Autophagy has been demonstrated to play a cardioprotective role in the heart following myocardial infarction (MI). We therefore investigated the role of STAT1 in the intact heart subjected to MI and examined the contribution of autophagy in modulating the protective effect of STAT1 after MI injury. STAT1-deficient hearts had significantly smaller infarcts than wild-type hearts and this correlated with increased levels of autophagy shown by light chain 3 (LC3)-I/LC3-II conversion, and up-regulation of Atg12 and Beclin 1. Moreover, pre-treatment with the autophagy inhibitor 3-methyladenine reversed the cardioprotection observed in the STAT1-deficient hearts. These results reveal a new function of STAT1 in the control of autophagy and indicate a cross-talk between the cardioprotective versus the damaging effects of STAT1 in the intact heart exposed to MI injury.
Keywords: STAT1, autophagy, cardioprotection, ischaemia
Introduction
Macroautophagy (herein autophagy) refers to an intracellular catabolic pathway that involves the delivery of cytoplasmic cargo to the lysosome. Autophagy is characterized by the appearance of double membrane cytoplasmic vesicles (autophagosomes) that fuse and are then destroyed by the lysosomal system [1, 2]. Autophagy occurs at low basal levels in virtually all cells to perform homeostatic functions such as protein and organelle turnover [1, 2]. It is rapidly increased when cells need to generate intracellular nutrients and energy such as those encountered during starvation, growth factor withdrawal or high bioenergetic demands [1, 2]. Neonates deficient in autophagy-related gene (Atg)5 die within 12 hrs after birth compared to wild-type (wt) counterparts, suggesting autophagy is essential between the time of birth and suckling [3]. The molecular cascade that regulates and executes autophagy has been extensively studied in yeast and has resulted in the identification of a number of Atgs [1, 2]. One of the key pathways that promotes autophagy requires two ubiquitin-like systems, one leading to the conjugation of Atg12 to Atg5 and the second converting the microtubule-associated protein 1 light chain 3 (LC3 or Atg8) soluble form (LC3-I) to the autophagic vesicle-associated form (LC3-II) [1, 2].
The role of autophagy in maintaining macromolecular synthesis and ATP production is likely to be a critical mechanism underlying its evolutionarily conserved prosurvival function. There is evidence that autophagy may be an alternative and adaptive response to maintain cell survival under different stress conditions [1–3]. For instance, autophagy may provide the cell with amino acids under unfavourable conditions. An apparent paradox is that autophagy is also considered a form of non-apoptotic programmed cell death called ‘type II’ or ‘autophagic’ cell death [4]. There are reports that autophagy may antagonize apoptosis and inhibition of autophagy may increase the sensitivity of the cells to apoptosis [4]. Similarly, inhibition of the apoptotic pathway (via caspase inhibitors) may enhance autophagic-induced death [4]. Thus, apoptosis and autophagy may not be mutually exclusive and may be interconnected in some settings depending on the context and severity of the stress response.
Ischaemia/reperfusion (I/R) injury or myocardial infarction (MI) in the heart results in the death of irreplaceable cardiac myocytes [5]. Although apoptosis is recognized as one of the mechanisms of cell death during I/R injury in the heart [6, 7], autophagy may also constitute an alternative pathophysiological response to ischaemia [8, 9]. The heart may uniquely rely on stress-induced up-regulation of autophagy to ensure the availability of energy substrates and to promote cellular remodelling when exposed to ischaemia and this adaptive response may indeed protect the heart [10]. However, the mechanism for such a process in the stress-induced myocardium is not known.
Signal transducers and activators of transcription (STAT) factors are a family of transcription factors that mediate intracellular signalling [11]. The transduction pathway is generally initiated at cytokine cell surface receptors, and results in STAT phosphorylation, dimerization and translocation to the nucleus [11]. STATs have important roles in homeostasis, development and cell fate decisions [11]. Previously, we reported that STAT1 plays an important role in enhancing apoptotic cell death in cardiac myocytes following I/R injury [12, 13].
Here we have investigated the role of STAT1 in an MI model and assessed whether STAT1 has any link to the autophagic pathway using wt versus STAT1-deficient hearts. Interestingly, not only did we observe that STAT1−/− null hearts had significantly smaller infarcts, but also that this was also associated with increased levels of autophagic markers. Moreover, inhibiting autophagy pharmacologically reverted the smaller infarct size in the STAT1-deficient hearts. These results reveal a new function of STAT1 in the reduction of autophagy and add more complexity to the role of STAT1 in modulating the stress response to I/R.
Materials and methods
Mouse isolated heart perfusion protocol
STAT1 Knockout (KO) [14] was purchased from Taconic Farms, Inc. (Bomholtvej, Denmark) Wt hearts and STAT1 KO from 10-week-old male mice were isolated and perfused as described previously [15]. Following 20 min. of stabilization, hearts were exposed to one of three protocols, control (Con), ischemia (I) or Ischemic preconditioning (IPC) plus I. Hearts were reperfused for 45 min. following ischemia after which infarct size was assessed by using triphenyl tetrazolium chloride staining as described previously [15]. Infarct size was assessed using computerized planimetry (planimerty, NIH Image 1.63 software package; National Institutes of Health, USA) by a researcher blinded to the groups.
Simulated ischemia in isolated cardiomyocytes
Isolated cardiomyocytes were isolated as previously described [7, 12] and exposed to simulated ischemia in a modified Esumi buffer [7, 12] containing: 118 mM NaCl, 3.58 mM KCl, 0.46 mM MgCl2, 0.9 mM CaCl2, 4 mM HEPES and 20 mM 2-deoxy-d-glucose 20, pH 6.2 for a period of up to 26 hrs, at 37°C, in a humidified environment containing 1% O2, 5% CO2 and balance N2. Normoxic myocytes were maintained as previously described [7, 12] media, pH 7.4, in a humidified environment containing 20% O2, 5%CO2 and balance N2.
Western blotting
Western blotting was performed as described [12] using specific antibodies against beclin 1 (Abcam, Cambridge, UK), LC3 (Abcam), Atg12 (Abcam) and anti-STAT1, (Santa Cruz Biotechnology Inc., Delaware, CA, USA).
Plasmid and cell culture
Mouse embryonic fibroblasts (MEF) containing wt STAT1 or STAT1 null were cultured in Dulbecco’s modified Eagle medium containing 10% foetal bovine serum. Transfections were performed with GeneJuice (Merck Biosciences, Gibbstown, NJ, USA) and cells were harvested 24 hrs after transfection. The polyQ80 luciferase plasmid was a kind gift from Conrad Weihl, Washington University School of Medicine. CMV-renilla was used as internal control for all luciferase experiments. Dual-luciferase assays were performed according to the manufacturer’s instructions (Promega, Madison, WI, USA) and analysed using a luminometer. Data were expressed as mean values ± S.E.M. and analysed with a Student’s t-test with a 95% confidence level using Graphpad Prism (GraphPad Software, San Diego, CA, USA).
Electron microscopy
Tissue samples were fixed and embedded for electron microscopy analysis, as previously described, with minimal modifications [6].
Statistical analysis
Results are expressed as mean S.E.M. (analyzed by the GraphPad Software). Differences were considered statistically significant at values of P < 0.05.
Results
STAT1 deficiency reduces infarct size hearts and show increased autophagy after I/R in the intact heart following I/R
We have shown previously that activated STAT1 can promote apoptotic cell death in isolated cardiac myocytes and in the isolated perfused heart exposed to I/R injury [12]. To confirm the cytotoxic effects of STAT1 during I/R, we first measured infarct sizes in the hearts of wt and STAT1-deficient mice exposed to I/R ex vivo. In confirmation of the in vitro data, hearts from STAT1-deficient animals had significantly smaller infarcts than wt hearts subjected to ex vivo I/R (Fig. 1A). To assess whether the reduced infarct size in the STAT1-deficient hearts was simply due to reduced apoptosis or involved other cell death/survival mechanisms, we pre-treated hearts from wt and STAT1-deficient mice with the autophagy inhibitor, 3-MA, prior to performing I/R in vivo. 3-MA increased infarct size in hearts from wt mice, and completely reversed the reduced infarct size in hearts from STAT1-deficient animals (Fig. 1B), suggesting that autophagy has a protective role in the ischaemic heart, and is a major mechanism resulting in the reduced infarct size in STAT1 knockout mice. To confirm this protective effect, we pre-treated hearts with the mTOR inhibitor, rapamycin, which induces autophagy. Rapamycin reduced infarct size in the wt animals, but did not have a significant effect in hearts from the STAT1-deficient mice (Fig. 1C).
Fig 1.
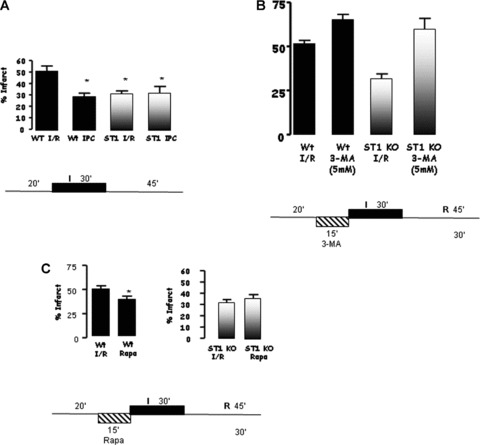
(A) Mice lacking STAT1 (STAT1−/−) showed significantly smaller infarct areas than wt littermates when subjected to ex vivo I/R. (B) Inhibiting autophagy using 3-methyladenine (3-MA, 5 nM) in STAT1−/− hearts abrogated the protective effects of STAT1. 3-MA treatment also enhanced the infarct size in wt animals. (C) Pre-treatment with the mTOR inhibitor, rapamycin (Rapa, 5 nM), followed by ex vivo I/R reduced infarct size in wt animals suggesting activation of autophagy elicits cardioprotective effects.
STAT1−/− hearts have increased levels of autophagic markers following I/R
To further explore the involvement of STAT1 in modulating autophagy, we first assessed the number of double-membrane autophagosomes by transmission electron microscopy, a gold standard assay for measuring autophagic activity. As shown in Figure 2A, we found more double membrane autophagosomes in STAT1−/− than in wt hearts after I/R. To validate this further we also assessed the levels of LC3 and the ratio of LC3-II to LC3-I. Immunoblot analysis showed an increase of LC3-I and -II during the I/R phases in wt and STAT1-deficient hearts. However, the LC3-II/LC3-I ratio was significantly higher in the STAT1−/− hearts (Fig. 2B). We also assessed other autophagic markers including Beclin 1 and Atg12 (a regulatory protein that is important for the initiation of autophagosomes). Beclin 1 expression was increased in wt and STAT1−/− hearts during I/R, which was significantly greater in the hearts from STAT1 knockout mice (Fig. 2C). However, whereas Atg12 levels were unchanged or even decreased in wt STAT1+/+ hearts exposed to I/R, ischaemia produced a robust increase in Atg12 expression in STAT1−/− hearts (Fig. 2D). These results further indicate that STAT1 may have suppressive activities on autophagy.
Fig 2.
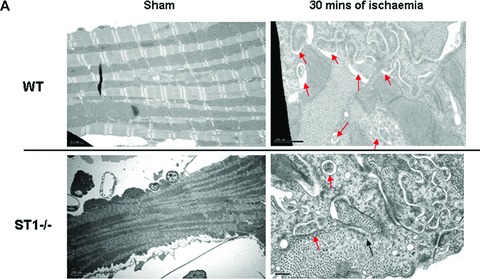
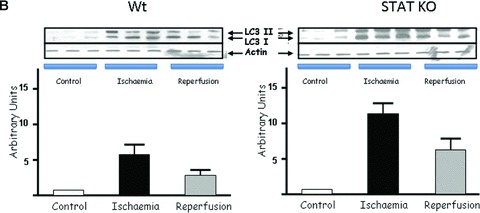
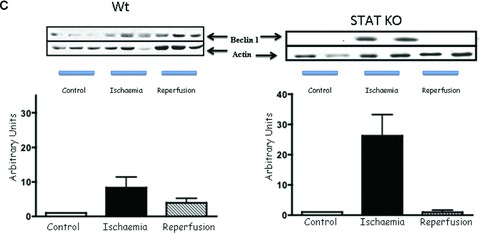
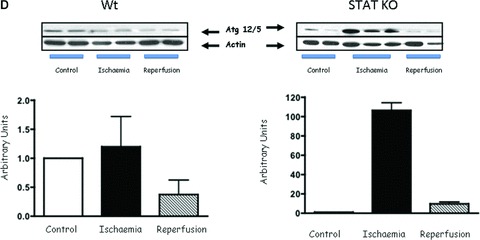
(A) Electron scanning microscopy of wt (STAT+/+) and STAT1−/− hearts subjected to ex vivo I/R. As indicated by the arrow heads STAT1−/− hearts subjected to I/R contained more number of double-membrane autophagosomes than the wt STAT1+/+ hearts. (B) wt (STAT1+/+) and STAT1−/− hearts were subjected to ex vivo I/R. LC3-I and LC3-II Western blotting of LC3-II. Levels appeared raised during ischaemia in the STAT1−/− animals. (C) Western blot analysis of Beclin 1 levels is enhanced during ischaemia in STAT1−/− compared to wts. Densitometry demonstrated significant increase of Beclin 1 protein levels in STAT1−/− hearts (lower panel). (D) Western blots analysis of Atg12 levels in wt (STAT+/+) and STAT1−/− hearts subjected to ex vivo I/R. Densitometry demonstrated significant increase of Atg12 protein levels in STAT1−/− hearts (lower panel).
To further explore whether the potential suppressive effect of STAT1 on autophagy, we assessed whether STAT1 may have inhibitory effects on the expression of autophagic genes. PCR analysis demonstrated that the levels of Beclin 1, were similar in wt and STAT1−/− hearts exposed both to I alone and to I/R (Fig. 3A). This lack of change in the mRNA levels was also validated by qPCR (Fig. 3B). Similar results were also observed with Atg12 and Atg5 mRNA levels (data not shown). These results therefore suggest that the inhibitory effects of STAT1 on autophagy are not trasncriptionally mediated.
Fig 3.
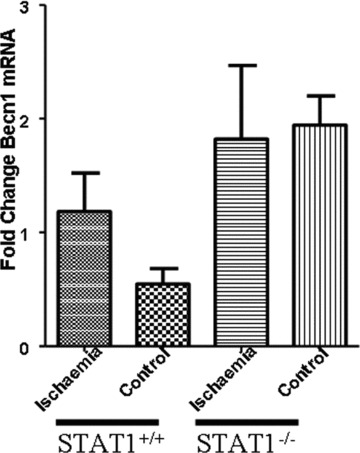
Quantitative PCR (qPCR) was assessed as to whether Beclin1 was being regulated at the mRNA level. ΔΔCT was used to calculate fold change and one-way ANOVA was used to calculate statistical significance. No significance in mRNA levels was detected suggesting Beclin1 stability is regulated at the protein level.
Inhibition of STAT1 enhances autophagy and autophagic flux in cardiac myocytes
Previously we have shown that inhibiting STAT1 activity with the polyphenol epigallocatechin-3-gallate (ECGC) protects both primary cardiac myocytes and the intact heart from I/R injury [16]. We next tested whether EGCG not only inhibits STAT1 but also enhances autophagy in primary cardiac myocytes. As shown in Figure 4A, primary cardiac myocytes exposed to simulated I/R induced LC3-II levels after 90 min. Moreover, interferon (IFN)-γ-treated primary cardiac myocytes activated STAT1 and reduced LC3-II levels. In contrast, ECGC known to inhibit STAT1 phosphorylation [16], significantly enhanced the levels of LC3-II levels (Fig. 4B). Thus, these data demonstrate that autophagy can be modulating by regulating STAT1 activity in cardiac myocytes. To further evaluate the role of STAT1 in modulating autophagic activity/flux, we used STAT1-deficient MEFs and took advantage of a recently described quantitative method which relies on the preferential degradation of a long-lived polyglutamine protein aggregate, polyQ80-luciferase, compared with a non-aggregating polyQ19-luciferase construct. By using such a reporter system, the induction of autophagy by starvation results in a selective reduction in polyQ80-luciferase activity [17]. We found that, after 6 hrs of nutrient deprivation, no significant difference in autophagic turnover of the polyQ80 aggregates was seen either in STAT1-deficient MEFs (ST1−/−) or wt STAT1 expressing MEFs (ST1+/+). However, with 18 hrs of nutrient deprivation, ST1−/− MEFs showed a 59% decrease in polyQ80 luciferase activity compared to ST1+/+ (Fig. 4D). Therefore, these studies strongly support a suppressive role for STAT1 on autophagic activity and confirms our ex vivo MI experiments in the heart.
Fig 4.
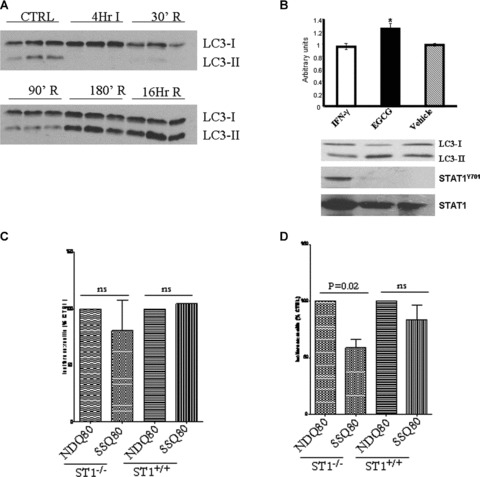
(A) Simulated I/R of isolated primary cardiac myocytes showed LC3-II protein levels being induced from 90 min. reperfusion. Western blot analysis at various time-points and immunoblotting with the indicated antibodies (B) IFN-γ-STAT1 activation reduced and epigallocatechin-3-gallate-reduce STAT1 activation enhanced conversion of LC3-I to II following simulated I/R of isolated primary cardiac myocytes. Primary cardiac myocytes were treated with either IFN-γ (50 ng/ml) or epigallocatechin-3-gallate (EGCG 50 ng/ml) and exposed to simulated I/R and cells harvested after 180 min. and processed for Western blot with the indicated antibodies (lower panel). Upper panel shows densitometric analysis of LC3-I over LC3-II ratio. (C) Enhanced autophagic flux in STAT1-deficient (ST1−/−) cells following serum starvation (SS) using the ployQ80 luciferase reporter assay. Wt (ST1+/+ or STAT1-deficient (ST1−/−) MEF cells were transfected with polyQ80luc and subjected to serum starvation (SSQ80) or control conditions (NDQ80). At 6 hrs of nutrient deprivation, no significant difference in autophagic turnover of the polyQ80 aggregates was seen. (D) After 18 hrs of nutrient deprivation the ST1−/− MEFs showed nearly 59% decrease in polyQ80 luciferase activity, whereas the ST1+/+ MEFs showed no statistically significant decrease in polyQ80 luciferase activity The data above show pooled data from three experiments with the NDQ80 set at 100% and the data analysed using a paired Student’s t-test to compare NDQ80 versus SSQ80.
Discussion
Autophagy is a physiological process which has been shown to be essential for neonatal survival and is involved in the normal recycling of damaged or old intracellular organelles. Autophagy is a highly conserved process found in yeast, plants and mammals and is activated under conditions of cellular stress, such as nutrient deprivation. Therefore, increased autophagy might be expected to be a feature of I/R in the heart, where nutrient and oxygen deprivation threaten cellular survival, and its provision of a source of intracellular nutrients may provide an, at least temporary, reprieve for a threatened myocardium. If intracellular sources of intracellular energy are no longer adequate, however, autophagic processes become a particular form of cell death called autophagic or type II cell death [4].
Our earlier work had established that STAT1 plays a pro-apoptotic role in cardiac myocytes exposed to I/R injury [12]. We have now shown that STAT1 has an additional role in reducing autophagy under stress-induced conditions such as I/R. Thus, STAT1−/− null hearts not only have smaller infarcts but also have enhanced levels of autophagy. Moreover, inhibiting autophagy abrogated the cardioprotection seen in the STAT1−/− hearts following I/R. This shows that STAT1 modulates autophagy and also that autophagy, at least during the time kinetics used in these experiments, is cardioprotective. In vitro studies using cells that express or lack STAT1 confirmed that autophagic activity is inhibited by STAT1. The regulatory function of STAT1 on autophagic proteins is not dependent on its known transcriptional activity, because expression levels of a number of autophagy genes are not altered in STAT1−/− hearts compared to wt hearts under both normal conditions and after I/R.
Reactive oxygen species (ROS) are generated following I/R [18], and these free radicals promote the onset of cell death, resulting in characteristic cellular changes including lipid peroxidation, protein denaturation and DNA double-strand breaks [18]. The generation of ROS during I/R has been shown both to activate STAT1 [19] and to induce apoptosis, and ROS have been implicated in I/R-induced cardiac myocyte apoptosis [7]. Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl) is a low molecular weight agent capable of scavenging superoxide anions, which offers an advantage over many other ROS scavengers as it is able to permeate biological membranes in vitro. We previously demonstrated that tempol was able to significantly reduce infarct size after in vivo IR injury [20]. In addition, we showed that IFN-γ, a potent activator of STAT1, not only enhanced infarct size but also could overcome the protective effects of tempol [20]. The results suggest a potential mechanism of action for the cardioprotective effects of tempol by partly inhibiting the ROS-STAT1 activated pathway and thus potentially enhancing cardiac autophagy [20].
IFN-γ has also been reported to play an important role in promoting autophagy and removal of intracellular pathogens and therefore links the inflammatory and autophagic pathways in the innate immune system via inducing IFN-γ inducible proteins [21, 22]. The inflammatory response is also enhanced following IR and this causes cardiac injury [23], because inhibiting the inflammatory response reduces myocardial injury following IR. Whether IFN-γvia JAK-STAT1 activation is required for promoting autophagy in immune cells is unclear because IFN-γ can also mediate effects independent of STAT1 [24].
Flavonoids exhibit a variety of beneficial effects in cardiovascular diseases and this has been attributed mainly to their antioxidant action. Recently, we also demonstrated additional cardioprotective mechanisms of some flavonoids through their inhibition of STAT1 activation [16, 25]. Furthermore, biochemical and computer modelling analysis indicated a direct interaction between STAT1 and flavonoids which correlated with their anti-STAT1 activity [26] Thus, those flavonoids (myricetin and delphinidin) that had higher affinity binding to STAT1 were associated with more pronounced protective effects against both cardiac injury and haemodynamic impairment. Resveratrol is another flavanoid that is attracting increased attention due to its diverse health benefits, especially in the case of cardiovascular disease, and has also been found to induce autophagy which may be mediated by its reported inhibition of STAT1 activity [27]. Together, these data provide further evidence that STAT1 could be a relevant target for protection against I/R injury in the heart and further suggest that agents that inhibit STAT1 are also agents that may promote autophagy.
The p53 transcription factor is a key regulator of the cellular response to acute stress and also a tumour suppressor [28]. Recent studies have shown that p53 can modulate autophagy in a dual fashion depending on the sub-cellular localization. Thus, nuclear and transcriptional actions of p53 are known to transactivate pro-apoptotic and autophagic genes [28, 29]. In contrast, cytoplasmic p53 also acts at the mitochondrial level to induce apoptosis but it inhibits autophagy [30]. Moreover, inhibiting p53 by RNA interference also induces autophagy [30]. In the present study the molecular mechanism on how STAT1 inhibits autophagy is still unclear. Our original findings that STAT1 is able to interact directly with p53 and modulates its functional activity [31] may suggest that STAT1 may act together with p53 to modulate autophagy. Similarly, STAT1 may also interact with specific autophagic proteins that may also determine autophagic activity. Further studies are required to elucidate how STAT1 is able to modulate autophagy.
Acknowledgments
We thank the British Heart Foundation for funding the study (N.S.: PG/05/118/19834; J.M.: PG/08/091/26002). We also thank Conrad Weihl (Washington University School of Medicine) for providing the polyQ80 reporter plasmid.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 4.Baehrecke EH. Autophagy:dual roles in life and death. Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 5.Olivevetti G, Quani F, Sala R, et al. Acute myocardial infarction in humans is associated with the activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1994;28:2005–16. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- 6.Scarabelli TM, Stephanou A, Rayment NB, et al. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischaemia/reperfusion injury. Circulation. 2001;104:253–6. doi: 10.1161/01.cir.104.3.253. [DOI] [PubMed] [Google Scholar]
- 7.Scarabelli TM, Stephanou A, Pasini E, et al. Different signalling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischaemia/reperfusion injury. Circ Res. 2002;90:745–8. doi: 10.1161/01.res.0000015224.07870.9a. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 10.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 11.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 12.Stephanou A, Brar B, Knight RA, et al. Ischaemia-induced STAT1 expression and activation plays a critical role in cardiac myocyte apoptosis. J Biol Chem. 2000;275:10002–8. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- 13.Stephanou A. Role of STAT-1 and STAT-3 in ischaemia/reperfusion injury. J Cell Mol Med. 2004;8:519–25. doi: 10.1111/j.1582-4934.2004.tb00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meraz MA, White JM, Sheehan KC, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–42. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 15.Smith RM, Suleman N, Lacerda L, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res. 2004;63:611–6. doi: 10.1016/j.cardiores.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Townsend PA, Scarabelli TM, Pasini E, et al. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J. 2004;18:1621–3. doi: 10.1096/fj.04-1716fje. [DOI] [PubMed] [Google Scholar]
- 17.Ju JS, Miller SE, Jackson E, et al. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy. 2010;5:511–9. doi: 10.4161/auto.5.4.7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semenza GL. Cellular and molecular dissection of reperfusion injury: ROS within and without. Circ Res. 2000;86:117–8. doi: 10.1161/01.res.86.2.117. [DOI] [PubMed] [Google Scholar]
- 19.Simon AR, Rai U, Fanburg BL, et al. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 20.McCormick J, Barry SP, Sivarajah A, et al. Free radical scavenging inhibits STAT phosphorylation following in vivo ischemia/reperfusion injury. FASEB J. 2006;20:2115–7. doi: 10.1096/fj.06-6188fje. [DOI] [PubMed] [Google Scholar]
- 21.Al-Zeer MA, Al-Younes HM, Braun PR, et al. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One. 2009;4(e4588):1–13. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang YP, Tsai CC, Huang WC, et al. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. J Biol Chem. 2010;285:28715–22. doi: 10.1074/jbc.M110.133355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 24.Ramana CV, Gil MP, Schreiber RD, et al. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 25.Scarabelli TM, Mariotto S, Abdel-Azeim S, et al. Targeting STAT1 by myricetin and delphinidin provides efficient protection of the heart from ischemia/reperfusion-induced injury. FEBS Lett. 2009;583:531–41. doi: 10.1016/j.febslet.2008.12.037. [DOI] [PubMed] [Google Scholar]
- 26.Darra E, Shoji K, Mariotto S, et al. Protective effect of epigallocatechin-3-gallate on ischemia/reperfusion-induced injuries in the heart: STAT1 silencing flavonoid. Genes Nutr. 2007;2:307–10. doi: 10.1007/s12263-007-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das M, Das DK. Resveratrol and cardiovascular health. Mol Aspects Med. 2010;31:503–12. doi: 10.1016/j.mam.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–16. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 29.Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 30.Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsend PA, Scarabelli TM, Davidson SM, et al. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004;279:5811–20. doi: 10.1074/jbc.M302637200. [DOI] [PubMed] [Google Scholar]