

Abstract
Elucidation of gene regulatory complexity holds much promise towards aiding therapeutic interventions in medical research. It has become progressively more evident that the characterization of highly conserved regulatory modules within promoters may assist in the elucidation of distinct cis-motif and trans-element regulatory interactions, shared in response to stimulus-evoked pathological changes. With special emphasis on the promoter, accurate analyses of cis-motif architecture combined with integrative in silico modelling might serve as a more refined approach for prediction and study of regulatory targets and major regulators governing transcriptional control. In this review, we have highlighted key examples and recent advances implementing in silico promoter models that could serve as essential contributions for future research in molecular medicine.
Keywords: In silico, promoter model, conserved cis-module, target prediction
Introduction
Transcriptional regulation is the first and vital step in the unified flow of biological information and is governed by (i) the context of cis-regulatory regions (cis-motifs residing within promoters, distant enhancers and silencers) and (ii) functional interactions between the products of specific regulatory genes (transcription factors-TFs) and cis-motifs [1]. Gaining insight into the orchestrated assembly and synergistic interplay of transcriptional regulatory mechanisms have been a challenging and burgeoning effort and much progress has been made since the preliminary deciphering of the human genome [2, 3]. Advances in high-throughput microarray and chromatin immunoprecipitation (ChIP) technologies have gained much momentum [4], but these systems are unable on their own to reveal new insights into the combinatorial and conserved nature of transcription. Whole-genome sequence data integrated with high-throughput technologies and complemented by systematic computational (in silico) strategies have set the stage for functional genomics (Fig. 1). Genome-wide functional analysis has allowed researchers to gain and predict a holistic view of the regulatory networks controlling gene expression and, although at a slower pace than anticipated, holds much promise in advancing post-genomic biomedical research [5–9]. Endeavours to decipher the principles of transcriptional regulation involve comprehensive interactions from different data systems on different levels that are managed, processed and modelled by integrative in silico tools combining database-assistance and motif detection algorithms. Numerous systematic integration and modelling strategies have been developed to elucidate the factors that contribute to the complexity of gene regulation within a network of genomic circuits. However, most of these approaches comprise of a relative general integration design combining high-throughput gene expression analysis, promoter data and bioinformatics. The scope of high-throughput technologies and transcriptional regulatory network analysis is too large to be covered here, and has been reviewed elsewhere [4, 10–12]. With special emphasis on cis-motif logic and promoter architecture, it is apparent that the complexity in identifying and predicting the presence, abundance, orientation and particular order of true (or over-represented) cis-motifs, poses a major challenge for understanding the functional relevance within a specific environment (e.g. tissue-specificity), condition (e.g. health- or disease-state) and/or in response to a specific compound (e.g. drug) or stimulus (e.g. stress) (Fig. 2). In this article, we focus on an integrative in silico modelling approach with special emphasis on promoter models in the context of regulatory target prediction in medical research. We have not attempted to summarize all the related literature, instead a limited number of the most relevant references have been used and we have highlighted a few concepts and results that more recent key studies have generated. On the basis of these observations and information gained, we present simplified integrative promoter modelling-strategies.
1.
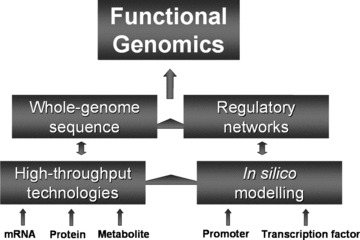
Simplified ‘building-block’ representation of integrated platforms constituting functional genomics.
2.
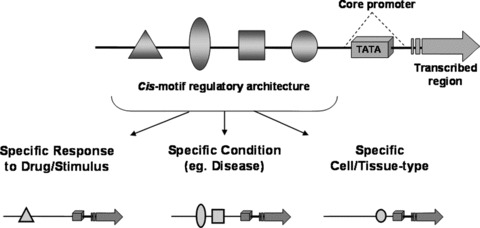
Cis-motif logic. Accurate dissection of promoter architecture upstream of the TATA-box containing core-promoter, allows modelling of cis-motif context particular to a specific stimulus, micro-environment or condition.
Combined in silico strategies contribute to accurate deciphering
Promoters hold the key to understanding and functional interpretation of the regulatory factors in cis and trans that control the site and level of gene activity [13–18]. It has become progressively more evident that accurate analysis and in silico modelling of promoter architecture and regulatory networks could assist in the study and prediction of disease-state regulatory processes, novel therapeutic targets and consequently facilitate pharmaceutical drug design [9, 12, 19–25]. Numerous other elements i.e. co-regulators, chromatin modulators and the presence of bi-directional and alternative promoters, contribute to the complexity (and diversity) of transcriptional regulation [18] and poses a significant challenge for accurate promoter detection and analysis. Therefore, the reliability and accuracy of promoter modelling strategies relies heavily on computational methods to detect over-representation of ‘true’cis-motifs in highly conserved modules that in turn could be used to study or accurately predict possible TFs modulating a group of genes expressed during a defined condition [11, 12, 21]. The in silico promoter model can be defined as the representation of a specific framework of DNA sequences (motifs detected by computational tools), within cis-context, that could provide essential information on a mechanism regulating transcriptional activity within a unique biological process, pathway or environment [21]. Comprehensive advances in the development of in silico strategies have shed light on the limitations in our understanding of complex regulatory processes by providing means to visualize gene regulation as a holistic event rather than a linear series of events. There are currently numerous motif and/or module detection tools, TF-binding-site databases and modelling platforms available.
These computational tools are evaluated on a regular basis and comprehensive overviews and assessments are provided elsewhere [26–31]. Here we highlight specific examples of three computational components; (i) motif detection algorithm, (ii) conserved non-coding sequence identification and (iii) TF-binding site database assistance—that are needed to integrate high-throughput molecular data in a systematic modelling strategy. We illustrate this strategy combining in silico modelling and experimental extraction/validation in a simplified representation (Fig. 3). High-throughput gene expression analyses (i.e. microarrays) of a particular disease-state tissue reveal a cluster of co-expressed genes of which the promoter sequences contain conserved cis-motifs. Probabilistic alignment-based methods such as MEME (Multiple Expectation Maximization for Motif Elicitation [32]) and Gibbs-sampling [33] are some of the most powerful and widely used algorithms (Fig. 3A) to detect motifs within a so-called ‘noisy’ background. These methods perform maximum likelihood estimates to identify statistical over-represented motifs in the highest scoring sequence alignments. In parallel, multi-species conservation analysis combined with predicted cis-motif clustering can be performed using the regulatory visualization tool for alignment (rVISTA) [34] computational platform (Fig. 3B). This is a hypothesis-driven strategy, which states that cis-regulatory motifs within evolutionary conserved sequences are more likely to be functional compared to cis-motifs in non-conserved regions [35]. Currently the two major databases comprising comprehensive sets of TF-binding profiles are TRANSFAC [36] and JASPAR [37]. TF-binding site database assistance allows for large-scale cis-motif comparisons to consensus sequences or energy binding scores of experimentally validated TF-binding sites (Fig. 3C). Binding scores are calculated from positional weight matrices (PWMs) that are derived from log-scale converted positional frequency matrices (PFMs) and these scores are directly related to DNA-protein binding energy interactions [27]. The extent to which the identified regulatory modules/motifs contribute to a specific interaction can be evaluated by modified array technologies (reviewed by Hoheisel [4]) such as ChiP assays [29]. Putative identification and comparison of newly discovered TF-targets is restricted to the experimentally verified database entries, therefore it is imperative that TF-binding site databases are continuously updated. Nevertheless, the combined employment of computational tools integrated with biological data (extrapolated from high-throughput variations of validation techniques) is powerful and allows for a refined elucidation, comparison and prediction of cis-regulatory context [29].
3.
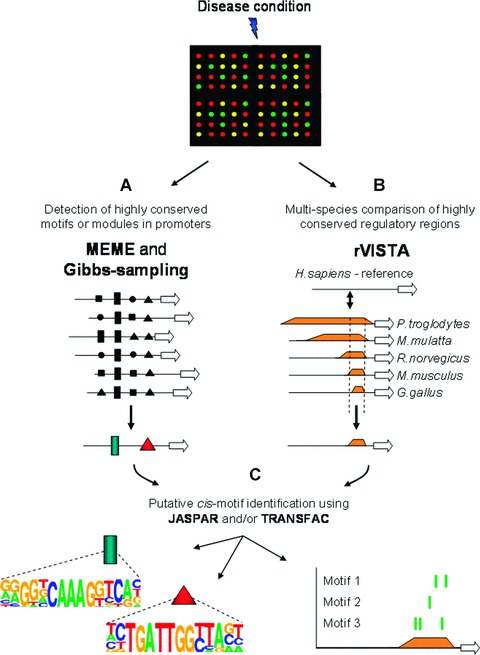
Accurate deciphering of regulatory context. Extrapolation of promoter data from genes expressed during a particular disease (transcriptomic profile represented by microarray) using (A) probabilistic motif detection algorithms i.e. MEME and Gibbs-sampling or (B) comparative phylogenetic promoter analysis across different species. (C) Combining before-mentioned strategies with TF-database assistance (using TRANSFAC and/or JASPAR) for putative motif identification and representation.
Systematic module analysis leads to target discovery
A well-established view on gene regulation is the fact that the promoter regions of co-expressed genes usually contain conserved areas that are comprised of single and/or a compact arrangement (a.k.a. module) of specific cis-motifs that are likely to be regulated by similar TFs. Therefore, if the deciphering of regulatory context is defined and well characterized, it is possible to predict novel genes or functional interactions of regulatory networks within a specific biological environment (e.g. tissue-specificity), process (e.g. biological pathway) or condition (e.g. disease). This traditional view allows for systematic modelling of promoter architecture and could expedite the discovery of biomarker or pharmaceutical cis- and/or trans-acting targets [7, 23, 24, 38]. Studies in model organisms S. cerevisiae, E. coli and D. melanogaster demonstrated how the analysis of promoter sequence information could serve as a platform for integration and successful prediction of transcriptional synergistic and regulatory events [39–42]. Recently integrative strategies combining phylogenetic footprinting, content-driven bioinformatics and gene expression profiles (and/or knowledge of gene function) have been applied in higher eukaryotes to predict transcriptional targets in cholesterol biosynthesis [43], regulatory single nucleotide polymorphisms (SNPs) influencing antioxidant response elements [44] and tissue specificity [17, 45–47]. Several studies have successfully accentuated the strategy of systematic modelling in clinical applications to predict transcriptional targets by integration of in silico analysis and high-throughput technologies. These investigations identified the conserved organization of regulatory modules or targets that were implicated in distinct conditions and/or processes such as reovirus infection of human embryonic kidney cells [48], androgen receptor binding [49], antibacterial response [50], enterocyte differentiation [51], human erythropoiesis [52], alcohol-related apoptosis and cell proliferation [53] and methylation in prostate cancer [54]. Within this context, we specifically highlight a study by Freebern et al. [23] that implemented an integrated ‘profiling of transcriptional targets’ (PTT)-strategy by extrapolating information from (i) multiple high-throughput data sets, (ii) computational cis-regulatory evaluation, (iii) mapping of signalling pathways and (iv) functional promoter validation during mitogen- and drug-induced activation of T cells. Conserved cis-regulatory module data combined with computational interpretation from different data sets, and functional promoter analyses of the candidate genes involved in immune cell function, led to the discovery of co-modulation by insulin-like growth factor 1 (IGF-1) [23]. Subsequently, focussed screening assays on T cells, co-stimulated with different IGF-1 concentrations, over short-time intervals and in the presence of different mitogen combinations, confirmed the regulation of immune cell function genes in response to IGF-1 induction on both a transcriptional and proteomic level. Although complex signalling of IGF-1 and multiple dataset analysis is not discussed here, results from this study underscore the importance of using evolutionary conserved promoter data, integrated with PTT, as a robust route to screen potential drug targets [23]. Clinically, although information of the underlying regulatory mechanisms in several pathological and cellular processes remains obscure, it is evident that highly conserved promoter regions could serve as ‘building-blocks’ for implementation of a systems approach, combining several elements (multi-dimensional datasets) on different levels, to assist in identifying the conserved nature of transcriptional targeting during a particular disease or pathological process.
Applications of in silico promoters based on conserved regulatory context
Accurate interpretation of promoter architecture is dependent on underlying regulatory commonalities that exist for genes that are similar on the basis of expression, regulation and/or function. Moreover, (i) combining expression data with functional annotation and (ii) the use of cis-regulatory module, rather than individual motif-information, can lead to a more defined predictive model design [16, 55]. An early model-based study by Gailus-Durner et al. [19] illustrated how in silico promoter models can be generated from literature data-mining in the absence of sequence similarity. A promoter model was constructed from a previously identified specific Sp1-cis-motif arrangement in the proximal promoter region of the muscle-specific cardiac/slow twitch sarcoplasmic reticulum Ca2+-ATPase (SERCA2) rabbit gene. This model was compared to the sequences in the rodent section of the European Molecular Biology Laboratory (EMBL) nucleotide sequence database [56]. Out of 28 possible matches, 14 were associated with muscle expression and 6 of the 14 showed high muscle-expression specificity [19]. Overall results of this study showed relative accurate prediction of co-regulated muscle-specific genes based on a single experimentally verified model that was used as reference for comparison and prediction [19]. A similar study, combining database assistance, generated in silico cell type-specific submodels based on the functional context of experimentally verified (stimulated or unstimulated) cis-regulatory information of the human RANTES/CCL5 promoter as reference [20]. The RANTES/CCL5 gene is a chemokine involved in the pathology of inflammatory disease and transcriptional regulation is governed by a module comprised of six functionally characterized cis-motifs within <300 nucleotides of the core promoter [20]. Elucidation and subsequent comparison of regulatory context (or framework) allowed for the characterization of 53 additional target genes that either shared co-regulation with RANTES/CCL5 or were associated with inflammation [20]. This work was highlighted in a review by Werner et al. [21] emphasizing the use of in silico promoter modelling that could serve as a valuable tool to identify and predict co-regulated target genes sharing conserved organizational features within their promoters [21]. These investigations provided an exciting and relatively new strategy for elucidation of transcriptional regulatory complexity, furthermore demonstrating that in silico promoter analyses could facilitate systematic modelling and understanding of the synergistic interplay between expression arrays, regulatory networks and gene function [16, 21, 38]. Two studies performed with similar in silico promoter comparative strategies revealed how the conserved organization of promoter motifs that are linked to a disease [22] or a tissue-specific micro-environment [57] can be successfully used to detect novel tissue-specific or disease-associated genes, based on reconstructive promoter modelling. The strategy used by Döhr et al. [22] combined promoter frameworks of (i) orthologous genes and (ii) co-regulated genes associated with a similar biological function, disease or pathway. Models generated from these cross-referenced frameworks were used to identify signalling pathways co-ordinating the interaction of co-regulated genes associated with maturity onset diabetes of the young (MODY- [22]). Cohen et al.[57] expanded the comparative promoter strategy by showing that accurate analysis of the promoter framework (module organization) derived from genes distinctively expressed in the functional unit that contributes to the complex phenotype of the podocyte/slit-diaphragm, can provide direct links to identify a novel regulatory network by interaction of co-regulation and a related event (podocyte-directed expression). Based on the hypothesis that podocyte-specific genes may share conserved promoter features, Cohen et al.[57] used a comparative computational promoter strategy to search for shared TF-binding sites in the promoters of 47 podocyte-associated genes in human beings, mouse and rat species. The initial analysis revealed that two genes, nephrin (NPHS1) and zonula occludens (ZO-1), shared a similar promoter context or so-called ‘framework’, phylogenetically conserved across all three species. Experimental gene expression analysis of NPHS1 and ZO-1 confirmed significant co-regulatory activity in micro-dissected human glomeruli taken from renal biopsies representing various disease conditions that have an effect on the glomerular filtration barrier i.e. benign nephrosclerosis, membranous glomerulosclerosis and diabetic nephropathy [57]. A subsequent second round promoter model screening revealed the presence of the shared NPHS1/ZO-1 promoter framework in 79 of 50,145 human promoter sequences screened [57]. However, only one novel candidate gene, cadherin-5 (CDH5), was identified to share the NPHS1/ZO-1 promoter model in all three species (human beings, mouse and rat) and, more interestingly, has not previously been associated with podocyte-specific gene expression [57]. Experimental gene expression analysis performed with biopsy glomeruli samples from 76 patients representing human glomerular disease confirmed predicted co-regulation of all three genes (NPHS1, ZO-1 and CDH5) [57], thus validating the accuracy of the promoter model. Findings from the investigations described above expanded hypothesis-driven research by combining phylogenetic conserved regulatory context with shared biological function. Predicting co-regulated genes of functional significance derived from the conserved organization of promoter modules is not restricted to phylogenetic analyses. Additionally, integrative promoter modelling strategies can be used to predict novel transcriptional targets or genes based solely on the functional interaction and/or association within a specific biological environment, process or condition without inter-species comparison. In a most recent investigation, Moss et al. [58] elegantly demonstrated the use of in silico promoter models by successfully predicting novel colon cancer-associated genes based on the compact arrangement of highly conserved cis-motifs associated with cell proliferation. This study utilized a range of different bioinformatic tools on all levels ranging from extrapolation of transcriptomic data and characterization of promoter architecture to the prediction of novel co-regulated genes sharing a common promoter module associated with cell proliferation in colon cancer [58]. This example showed how the conserved nature of cis-motif promoter architecture can be implemented to identify additional molecular targets in a systematic top-down approach (Fig. 4A). By further accentuation of this strategy, we suggest that a bottom-up approach based on cis-motif logic and promoter sequence availability can be used to predict so-called master-regulators that operate as activators, repressors, modifiers and/or co-activators by modulating the regulation of several genes in a biological pathway, disease or cellular environment (Fig. 4B). In the midst of studies described here, research efforts that report on the use of in silico promoter models are relatively limited. Although different modelling strategies exist, it is evident that refined analysis of promoter organization serves as the major objective within all methods. In silico and comparative analysis (i.e. shared promoter framework within specific tissue, condition and/or species) of cis-regulatory architecture could (i) provide further insight into defining the relationship of genes that are co-expressed and/or co-regulated, (ii) assist in the identification of functionally related promoter elements in the absence of gene sequence similarity, (iii) predict novel disease-associated genes sharing a unique regulatory mechanism or biological pathway and (iv) subsequently allow for identification and representation of transcriptional targets for the development of therapeutic agents [19–23, 51, 57, 58]. Contrary to the advantages offered by promoter modelling, several drawbacks such as (i) limited experimental validation of promoter function and protein-DNA interactions, (ii) the lack of high-throughput biological data, (iii) variation in the accuracy of computational tools and (iv) overall complexity of regulatory mechanisms (in addition to transcriptional control) pose significant challenges for future modelling strategies. Nevertheless, examples highlighted underscore the importance of (i) integration, (ii) the conserved nature of a regulatory framework and (iii) the use of in silico promoter models as valuable tools to study the complex mechanisms governing transcriptional regulation in the context of disease and potential target discovery.
4.
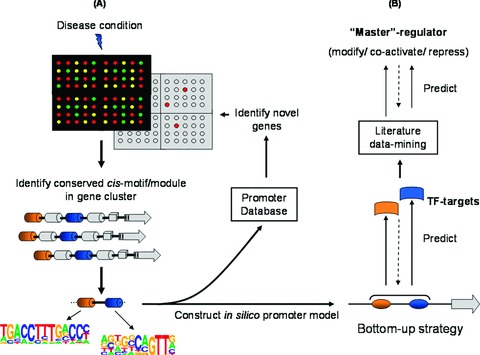
In silico modelling strategy and implementation. (A) Combination of highly conserved promoter cis-motifs identified from genes expressed during a particular disease (transcriptomic profile represented by microarray) and used to construct promoter model. Specific promoter model can be implemented to identify novel genes (represented by red dots on underlying grey microarray) that can otherwise not be identified by conventional gene expression profile. (B) In addition, promoter model can be used to predict putative (i) regulatory pathways and (ii) TF-targets. This in silico bottom-up strategy is based on the concept of using a conserved regulatory promoter area (represented as a model) to predict (combining literature database mining) a central TF-target and/or so-called ‘master regulator’ (regulating a specific cascade of genes during a particular biological process). This information could be valuable for the development of a therapeutic agent affecting a central molecular regulator.
Conclusion
The principles of several regulatory mechanisms have been well characterized individually; however, gaining a holistic insight into the complexity of orchestrated regulatory events remains a challenge. While gaps in our appreciation of transcriptional regulation still remain, advances in bioinformatics and high-throughput technologies such as ChIP-on-chip have greatly extended our reach into the discovery of novel promoters as well as enhancer elements, allowing a more accurate modelling of the regulatory code in cis-context. Consequently such models can be used to identify and/or predict transcriptional activation and signalling in fundamental research endeavours and biopharmaceutical applications. The studies described in this review have laid the groundwork for future investigations integrating the concept of promoter modelling as a tool in molecular medicine.
Acknowledgments
The authors would like to thank the Medical Research Council (MRC) of South Africa for financial support.
References
- 1.Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–51. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang HM, Yu J, Wang J, Huang GY, Gu J, Hood L, Rowen L, Madan A, Qin SZ, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan HQ, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JGR, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang WH, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz JR, Slater G, Smit AFA, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XQH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang JH, Miklos GLG, Nelson C, Broder S, Clark AG, Nadeau C, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng ZM, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge WM, Gong FC, Gu ZP, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke ZX, Ketchum KA, Lai ZW, Lei YD, Li ZY, Li JY, Liang Y, Lin XY, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue BX, Sun JT, Wang ZY, Wang AH, Wang X, Wang J, Wei MH, Wides R, Xiao CL, Yan CH, Yao A, Ye J, Zhan M, Zhang WQ, Zhang HY, Zhao Q, Zheng LS, Zhong F, Zhong WY, Zhu SPC, Zhao SY, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An HJ, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi HY, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays AD, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu XJ, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen MY, Wu D, Wu M, Xia A, Zandieh A, Zhu XH. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 4.Hoheisel JD. Microarray technology: beyond transcript profiling and genotype analysis. Nat Rev Genet. 2006;7:200–10. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 5.Joos L, Eryüksel E, Brutsche MH. Functional genomics and gene microar-rays—the use in research and clinical medicine. Swiss Med Wkly. 2003;133:31–8. doi: 10.4414/smw.2003.10007. [DOI] [PubMed] [Google Scholar]
- 6.Kramer R, Cohen D. Functional genomics to new drug targets. Nat Rev Drug Discov. 2004;3:965–72. doi: 10.1038/nrd1552. [DOI] [PubMed] [Google Scholar]
- 7.Fischer HP. Towards quantitative biology: integration of biological information to elucidate disease pathways and to guide drug discovery. Biotechnol Annu Rev. 2005;11:1–68. doi: 10.1016/S1387-2656(05)11001-1. [DOI] [PubMed] [Google Scholar]
- 8.Collins CD, Purohit S, Podolsky RH, Zhao HS, Schatz D, Eckenrode SE, Yang P, Hopkins D, Muir A, Hoffman M, McIndoe RA, Rewers M, She JX. The application of genomic and proteomic technologies in predictive, preventive and personalized medicine. Vascul Pharmacol. 2006;45:258–67. doi: 10.1016/j.vph.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Werner T, Nelson PJ. Joining high-throughput technology with in silico modelling advances genome-wide screening towards targeted discovery. Brief Funct Genomic Proteomic. 2006;5:32–6. doi: 10.1093/bfgp/ell010. [DOI] [PubMed] [Google Scholar]
- 10.Blais A, Dynlacht BD. Constructing transcriptional regulatory networks. Genes Dev. 2005;19:1499–511. doi: 10.1101/gad.1325605. [DOI] [PubMed] [Google Scholar]
- 11.Wingender E, Crass T, Hogan JD, Kel AE, Kel-Margoulis OV, Potapov AP. Integrative content-driven concepts for bioinformatics “beyond the cell”. J Biosci. 2006;32:169–80. doi: 10.1007/s12038-007-0015-2. [DOI] [PubMed] [Google Scholar]
- 12.Goutsias J, Lee NH. Computational and experimental approaches for modeling gene regulatory networks. Curr Pharm Des. 2007;13:1415–36. doi: 10.2174/138161207780765945. [DOI] [PubMed] [Google Scholar]
- 13.Smale ST. Core promoters: active contributors to combinatorial gene regulation. Gene Dev. 2001;15:2503–8. doi: 10.1101/gad.937701. [DOI] [PubMed] [Google Scholar]
- 14.Butler JEF, Kadonaga JT. The RNA poly-merase II core promoter: a key component in the regulation of gene expression. Gene Dev. 2002;16:2583–92. doi: 10.1101/gad.1026202. [DOI] [PubMed] [Google Scholar]
- 15.Hochheimer A, Tjian R. Diversified transcription initiation complexes expand promoter selectivity and tissue-specific gene expression. Gene Dev. 2003;17:1309–20. doi: 10.1101/gad.1099903. [DOI] [PubMed] [Google Scholar]
- 16.Werner T. Promoters can contribute to the elucidation of protein function. Trends Biotechnol. 2003;21:9–13. doi: 10.1016/s0167-7799(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 17.Smith AD, Sumazin P, Zhang MQ. Tissue-specific regulatory elements in mammalian promoters. Mol Syst Biol. 2007;3:73. doi: 10.1038/msb4100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heintzman ND, Ren B. The gateway to transcription: identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol Life Sci. 2007;64:386–400. doi: 10.1007/s00018-006-6295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gailus-Durner V, Scherf M, Werner T. Experimental data of a single promoter can be used for in silico detection of genes with related regulation in the absence of sequence similarity. Mamm Genome. 2001;12:67–72. doi: 10.1007/s003350010219. [DOI] [PubMed] [Google Scholar]
- 20.Fessele S, Maier H, Zischek C, Nelson PJ, Werner T. Regulatory context is a crucial part of gene function. Trends Genet. 2002;18:60–3. doi: 10.1016/s0168-9525(02)02591-x. [DOI] [PubMed] [Google Scholar]
- 21.Werner T, Fessele S, Maier H, Nelson PJ. Computer modeling of promoter organization as a tool to study transcriptional coregulation. FASEB J. 2003;17:1228–37. doi: 10.1096/fj.02-0955rev. [DOI] [PubMed] [Google Scholar]
- 22.Döhr S, Klingenhoff A, Maier H, Hrabé de Angelis M, Werner T, Schneider R. Linking disease-associated genes to regulatory networks via promoter organization. Nucleic Acids Res. 2005;33:864–72. doi: 10.1093/nar/gki230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freebern WJ, Haggerty CM, Montano I, McNutt MC, Collins I, Graham A, Chandramouli GV, Stewart DH, Biebuyck HA, Taub DD, Gardner K. Pharmacologic profiling of transcriptional targets deciphers promoter logic. Pharmacogenomics J. 2005;5:305–23. doi: 10.1038/sj.tpj.6500325. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh D, Papavassiliou AG. Transcription factor therapeutics: long-shot or lodestone. Curr Med Chem. 2005;12:691–701. doi: 10.2174/0929867053202197. [DOI] [PubMed] [Google Scholar]
- 25.Bussemaker HJ, Foat BC, Ward LD. Predictive modeling of genomewide mRNA expression: from modules to molecules. Annu Rev Biophys Biomol Struct. 2007;36:329–47. doi: 10.1146/annurev.biophys.36.040306.132725. [DOI] [PubMed] [Google Scholar]
- 26.Pavesi G, Mauri G, Pesole G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform. 2004;5:217–36. doi: 10.1093/bib/5.3.217. [DOI] [PubMed] [Google Scholar]
- 27.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–87. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 28.Tompa M, Li N, Bailey TL, Church GM, de Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Régnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–44. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 29.Elnitski L, Jin VX, Farnham PJ, Jones SJM. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006;16:1455–64. doi: 10.1101/gr.4140006. [DOI] [PubMed] [Google Scholar]
- 30.Li N, Tompa M. Analysis of computational approaches for motif discovery. Algorithms Mol Biol. 2006;1:8. doi: 10.1186/1748-7188-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolchanov NA, Merkulova TI, Ignatieva EV, Ananko EA, Oshchepkov DY, Levitsky VG, Vasiliev GV, Klimova NV, Merkulov VM, Hodgman TC. Combined experimental and computational approaches to study the regulatory elements in eukaryotic genes. Brief Bioinform. 2007;8:266–74. doi: 10.1093/bib/bbm027. [DOI] [PubMed] [Google Scholar]
- 32.Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using Expectation Maximization. Mach. Learn. 1995;21:51–80. [Google Scholar]
- 33.Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AN, Wootton J. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–14. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- 34.Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 2002;12:832–9. doi: 10.1101/gr.225502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16:369–72. doi: 10.1016/s0168-9525(00)02081-3. [DOI] [PubMed] [Google Scholar]
- 36.Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Prüß M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–9. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–4. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werner T. Cluster analysis and promoter modelling as bioinformatics tools for the identification of target genes from expression array data. Pharmacogenomics. 2001;2:25–36. doi: 10.1517/14622416.2.1.25. [DOI] [PubMed] [Google Scholar]
- 39.Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–9. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- 40.Beer MA, Tavazoie S. Predicting Gene Expression from Sequence. Cell. 2004;117:185–98. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- 41.Guido NJ, Wang X, Adalsteinsson D, McMillen D, Hasty J, Cantor CR, Elston TC, Collins JJ. A bottom-up approach to gene regulation. Nature. 2006;439:856–60. doi: 10.1038/nature04473. [DOI] [PubMed] [Google Scholar]
- 42.Pierstorff N, Bergman CM, Wiehe T. Identifying cis-regulatory modules by combining comparative and compositional analysis of DNA. Bioinformatics. 2006;22:2858–64. doi: 10.1093/bioinformatics/btl499. [DOI] [PubMed] [Google Scholar]
- 43.Chang LW, Nagarajan R, Magee JA, Milbrandt J, Stormo GD. A systematic model to predict transcriptional regulatory mechanisms based on overrepresentation of transcription factor binding profiles. Genome Res. 2006;16:405–13. doi: 10.1101/gr.4303406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Tomso DJ, Chorley BN, Cho H-Y, Cheung VG, Kleeberger SR, Bell DA. Identification of polymorphic antioxidant response elements in the human genome. Hum Mol Genet. 2007;16:1188–1200. doi: 10.1093/hmg/ddm066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith AD, Sumazin P, Xuan Z, Zhang MQ. DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc Nat Acad Sci USA. 2006;103:6275–80. doi: 10.1073/pnas.0508169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez MJ, Smith AD, Li B, Zhang MQ, Harrod KS. Computational prediction of novel components of lung transcriptional networks. Bioinformatics. 2007;23:21–9. doi: 10.1093/bioinformatics/btl531. [DOI] [PubMed] [Google Scholar]
- 47.van Deursen D, Botma GJ, Jansen H, Verhoeven AJM. Comparative genomics and experimental promoter analysis reveal functional liver-specific elements in mammalian hepatic lipase genes. BMC Genomics. 2007;8:99. doi: 10.1186/1471-2164-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lapadat R, DeBiasi RL, Johnson GL, Tyler KL, Shah I. Genes induced by Reovirus infection have a distinct modular cis-regulatory architecture. Curr Genomics. 2005;6:501–13. doi: 10.2174/138920205775067675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masuda K, Werner T, Maheshwari S, Frisch M, Oh S, Petrovics G, May K, Srikantan V, Srivastava S, Dobi A. Androgen receptor binding sites identified by a GREF_GATA model. J Mol Biol. 2005;353:763–71. doi: 10.1016/j.jmb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Shelest E, Wingender E. Construction of predictive promoter models on the example of antibacterial response of human epithelial cells. Theor Biol Med Model. 2005;2:2. doi: 10.1186/1742-4682-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stegmann A, Hansen M, Wang Y, Larsen JB, Lund LR, Ritie L, Nicholson JK, Quistorff B, Simon-Assmann P, Troelsen JT, Olsen J. Metabolome, transcriptome, and bioinformatic cis-element analyses point to HNF-4 as a central regulator of gene expression during enterocyte differentiation. Physiol Genomics. 2006;27:141–55. doi: 10.1152/physiolgenomics.00314.2005. [DOI] [PubMed] [Google Scholar]
- 52.Keller MA, Addya S, Vadigepalli R, Banini B, Delgrosso K, Huang H, Surrey S. Transcriptional regulatory network analysis of developing human erythroid progenitors reveals patterns of coregulation and potential transcriptional regulators. Physiol Genomics. 2006;28:114–28. doi: 10.1152/physiolgenomics.00055.2006. [DOI] [PubMed] [Google Scholar]
- 53.Uddin RK, Singh SM. cis-Regulatory sequences of the genes involved in apoptosis, cell growth, and proliferation may provide a target for some of the effects of acute ethanol exposure. Brain Res. 2006;1088:31–44. doi: 10.1016/j.brainres.2006.02.125. [DOI] [PubMed] [Google Scholar]
- 54.Perry AS, Loftus B, Moroose R, Lynch TH, Hollywood D, Watson RWG, Woodson K, Lawler M. In silico mining identifies IGFBP3 as a novel target of methylation in prostate cancer. Br J Cancer. 2007;96:1587–94. doi: 10.1038/sj.bjc.6603767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allocco DJ, Kohane IS, Butte AJ. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics. 2007;5:18. doi: 10.1186/1471-2105-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kulikova T, Akhtar R, Aldebert P, Althorpe N, Andersson M, Baldwin A, Bates K, Bhattacharyya S, Bower L, Browne P, Castro M, Cochrane G, Duggan K, Eberhardt R, Faruque N, Hoad G, Kanz C, Lee C, Leinonen R, Lin Q, Lombard V, Lopez R, Lorenc D, McWilliam H, Mukherjee G, Nardone F, Pastor MPG, Plaister S, Sobhany S, Stoehr P, Vaughan R, Wu D, Zhu W, Apweiler R. EMBL Nucleotide Sequence Database in 2006. Nucleic Acids Res. 2007;35:D16–D20. doi: 10.1093/nar/gkl913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, Schmid H, Merkle M, Saleem MA, Koller K-P, Werner T, Gröne H-J, Nelson PJ, Kretzler M. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc Nat Acad Sci USA. 2006;103:5682–7. doi: 10.1073/pnas.0511257103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moss AC, Doran PP, MacMathuna P. In silico promoter analysis can predict genes of functional relevance in cell proliferation: validation in a colon cancer model. Translational Oncogenomics. 2007;2:1–16. [PMC free article] [PubMed] [Google Scholar]