

Abstract
The heart is the main target organ of the parasite Trypanosoma cruzi, the causal agent of Chagas' disease, a significant public health issue and still a major cause of morbidity and mortality in Latin America. During the acute disease, tissue damage in the heart is related to the intense myocardium parasitism. To control parasite multiplication, cells of the monocytic lineage are highly mobilized. In response to inflammatory and immune stimulation, an intense migration and extravasation of monocytes occurs from the bloodstream into heart. Monocyte differentiation leads to the formation of tissue phagocytosing macrophages, which are strongly activated and direct host defence. Newly elicited monocyte-derived macrophages both undergo profound physiological changes and display morphological heterogeneity that greatly differs from originally non-inflammatory macrophages, and underlie their functional activities as potent inflammatory cells. Thus, activated macrophages play a critical role in the outcome of parasite infection. This review covers functional and ultrastructural aspects of heart inflammatory macrophages triggered by the acute Chagas' disease, including recent discoveries on morphologically distinct, inflammation-related organelles, termed lipid bodies, which are actively formed in vivo within macrophages in response to T. cruzi infection. These findings are defining a broader role for lipid bodies as key markers of macrophage activation during innate immune responses to infectious diseases and attractive targets for novel anti-inflammatory therapies. Modulation of macrophage activation may be central in providing therapeutic benefits for Chagas' disease control.
Keywords: Chagas' disease, inflammation, monocytes/macrophages, innate immunity, lipid bodies, phagocytosis, heart, toll-like receptors, cell activation, electron microscopy
Introduction
Chagas' disease remains a public health problem in Latin America, where approximately 18–20 million people are chronically infected, with ∼ 100 millions still at a risk of acquiring the infection [1]. Unfortunately, there is no vaccine available to prevent this disease. The causative agent of Chagas' disease is the unicelular protozoan parasite Trypanosoma cruzi. Once the infection has established in vertebrate hosts, intracellular parasite replication occurs as amastigotes, followed by the release of trypomastigote forms that can be carried by the bloodstream to infect different organs, especially the heart (reviewed in [2, 3]).
The most common and serious pathology seen in Chagas' disease is a cardiomyopathy characterized by inflammatory infiltrates, necrosis, and fibrosis. Chagasic cardiomyopathy leads to sudden death, complex arrhythmias, ventricular aneurysms, heart failure, and thromboemolism. Despite the importance of the cardiac clinical form, patients infected with T. cruzi may have an asymptomatic form of the disease, named indeterminate. The factors that determine the distinct clinical outcomes, leading to a mild or to a severe form, are not completely understood (reviewed in [4, 5]). In fact, the most intriguing challenge to understanding the pathophisiology of Chagas' heart disease still lies in the complex host-parasite interrelationship [6].
During the acute phase of the infection, the heart is dramatically parasitized. In order to control the parasite multiplication, cells of the monocytic lineage are highly mobilized. There is an intense migration and extravasation of monocytes from the bloodstream into heart. Monocyte differentiation leads to the formation of phagocytosing macrophages, strongly activated and involved in inhibiting parasite replication in the myocardium [7, 8]. The ability of monocyte-derived macrophages to process and present antigens, produce cytokines, and provide costimulatory signals demonstrates their pivotal role in initiating immune responses. The importance of these cells to the host defence has been pointed out by us and other groups during the in vivo acute T. cruzi infection in both human beings and experimental models [2, 7–9].
Through use of a rat model of Chagas' disease, we have been investigating migration, accumulation and activation of monocytic lineage cells within the heart following T. cruzi infectionin a series of in vivo studies, with special interest in characterizing the ultrastructure and function of heart inflammatory macrophages [7, 8, 10–13]. Compared to macrophages from non-infected animals, inflammatory macrophages undergo profound physiological changes and exhibit a distinct and remarkable morphology, which underlie their functional activities as activated cells [7]. This review covers functional and ultrastructural aspects of the heart macrophages triggered by the acute Chagas' disease, including recent discoveries on morphologically distinct, inflammation-related organelles, termed lipid bodies, which are actively formed in vivo within macrophages in response to T. cruzi infection. These findings are defining a broader role for lipid bodies as key markers of macrophage activation during infectious diseases and attractive targets for novel anti-inflammatory therapies [13–15].
Acute T. cruzi infection induces accentuated mobilization/migration of monocytic lineage cells into heart
Peripheral blood monocytes (PBM) recruitment is a rapid and remarkable phenomenon during the acute infection with T. cruzi . At this time, we have demonstrated, in vivo, a dramatic increase in the numbers of monocytes in the blood [8, 12]. PBM increase ∼1.0-fold at day 6 post-infection, reaching maximum values at day 12 (∼10.0-fold higher compared to control values and ∼5.0-fold higher compared to data from day 6) and decreasing by day 20 of infection (compared to day 12, but significantly higher that the values found at day 6 of infection) (Fig. 1A).
1.
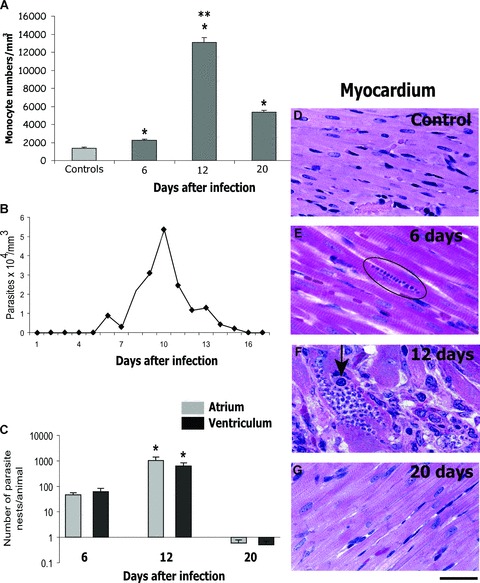
Number of monocytes in the peripheral blood (A), parasitemia (B), heart parasitism (C) and light micrographs of myocardium in uninfected (D) and infected (E, F, G) rats, at different time-points of the acute infection with Trypanosoma cruzi. (A) Monocyte numbers increased (P < 0.05) compared to control values (*). At day 12, these numbers increased (P< 0.05) in comparison to numbers found at day 6 (**) while at day 20, they decreased (P < 0.05) compared to day 12 (**). (C) Parasite numbers were significantly higher (P < 0.05) at day 12 compared to day 6. At day 20, the parasitism decreased dramatically. Parasite nests showed higher size at day 12 (F) compared to day 6 (E, circle). A macrophage-like cell (arrow) is seen within a parasite nest in F. Data are expressed as mean ± S.E.M. and are representative of three independent experiments. Scale Bar, 15 μm. Reprinted from ref. [8] with permission.
Analyses using transmission electron microscopy (TEM) revealed morphological signs of activation in PBM during different time-points of the acute infection (6, 12 and 20 days) [8]. These cells are voluminous with a striking increase of surface ruflings and cytoplasmic organelles, especially the Golgi complex, a finding associated with the increased ability of cell secretion [8]. Moreover, PBM show a significant increase in size at day 12 of infection [8]. Interestingly, the lymphocyte, neutrophil and eosinophil numbers and the total number of peripheral blood leukocytes are not significantly different from the control values on days analysed [12].
The rapid mobilization of monocytes from bone marrow precursor cells is consistent with a sharp increase in the plasma concentration of the granulocyte-macrophage colony-stimulating factor (GM-CSF) in mice acutely infected with T. cruzi[16]. Experiments using whole-body irradiation of rats have also provided evidence for an important mobilization of monocytic lineage cells induced by the T. cruzi infection. In rats immunosuppressed by a single, high dose of gamma irradiation, there is an initial fall in the number of PBM, but the recovery of these cells is faster if the animals are irradiated and infected [10, 11].
In vitro, PBM are able to destroy the parasite after ingestion [17] and exhibit activated morphology when cultured with T. cruzi[18]. In addition, intracellular replication of T. cruzi was inhibited within PBM stimulated with GM-CSF or interferon-gamma (INF-γ) [19]. in vivo, circulating monocytes from patients with differential clinical forms of Chagas' disease display distinct phenotypic and functional profiles. Analysis of the expression of immunoregulatory cytokines showed that while monocytes from indeterminate-disease patients are committed to IL-10 expression, a higher percentage of monocytes from cardiac-disease patients express the proinflammmatory cytokine tumour necrosis factor-alpha (TNF-α) after exposure to live parasites, suggesting a role for these cells in the outcome of the pathology [9]. PBM are also able to secrete hydrogen peroxide when challenged with the parasite in vivo[12] and seem to undergo consistent remodelling of arachidonic acid pools as they mature or enter into the tissue from the blood so to play a role in inflammation as macrophages [20]. However, the in vivo production of nitric oxide (NO) by PBM seems to be inhibited during the acute Chagas' disease [8].
Although the physiological relevance of blood monocyte heterogeneity is not completely understood, it is now becoming apparent that these cells consist of several subpopulations of cells which differ both morphologically and functionally and that can be influenced by infection (reviewed in [21, 22]). Differential expression of antigenic markers on monocytes such as CD14, CD16, CD32, CD86, CX3C-chemokine receptor 1 (CX3CR1) and CCR2 seems to have important functional consequences for these cells [21–24]. For example, a considerable increase in the number of CD14+ CD16+ monocytes has been described in the human blood in response to infections [22]. This monocyte subset, while still in circulation, has acquired features in common with mature tissue macrophages [25]. In mice, the CX3CR1 + CCR2+ subpopulation is the population that is increased during infections. This subset of monocytes also express Ly6C (Gr1) (granulocyte antigen 1) and is known as ‘inflammatory’ subset because of their rapid recruitment to areas of damaged or inflamed tissue while the CX3CR1hi CCR2− Ly6C− subset is referred to as ‘resident’ monocytes and seems to replenish tissue-resident macrophages [21–23].
The receptors and signalling pathways that initiate and promote the inflammatory response have become increasingly well characterized. Chemokines and their cognate receptors direct the extravasation of leukocytes and monocytes from the bloodstream into tissues and orchestrate the positional migration of these cells within the tissues. The CC chemokine receptor 5 (CCR5), which binds CCL3, CCL4 and CCL5, is prominently expressed within the heart during the acute Chagas' disease [26]. CCR5 is involved in directing the migration of mononuclear cells to the heart early in acute infection with T. cruzi, and activate macrophages, contributing to the control of parasite replication [26]. The accentuated mobilization of monocytes in the peripheral blood occurs in concert to a prominent influx of macrophages into the heart, target organ of the disease [7, 8, 27].
Heart inflammatory macrophages show a distinct morphology and are actively involved in parasite phagocytosis
Macrophages are widely distributed immune system cells with essential functions in homeostasis and defence [28]. Activated macrophages exert critical activities in immunity to parasites, playing a pivotal role in the mechanism for halting the acute T. cruzi infection. Activation of macrophages by parasite antigens results in pro-inflammatory cytokine production, and consequent control of parasitemia and mortality (reviewed in [2, 29]).
The accumulation of newly recruited macrophages in the heart is one of the main findings of the acute T. cruzi infection and it is correlated with the intense myocardium parasitism, which occurs during the early infection in both experimental models and human beings (reviewed in [4, 5]). Heart inflammatory macrophages have been studied by us in Holtzman rats infected with the virulent Y strain of T. cruzi . These animals have been proved to be a reliable model for T. cruzi infection studies, mimicking myocarditis, heart parasitism and nerve lesions characteristic of the human acute Chagas' disease [7, 11, 12, 30, 31]. Parasitemia in Y strain-infected Holtzman rats peaks on day 10 of infection (Fig. 1B) and these animals rarely die during the acute phase. In human beings, parasites may be found in the blood during the acute disease, which lasts approximately 2–3 months. Often, the acute phase is not perceived by the patient; 95% of the cases are asymptomatic and less than 5% of patients with symptoms (fever, swollen lymph glands and, occasionally, an inflammatory reaction at the biting site or a swollen eye) die during the disease's acute stage [32, 33].
At day 12 of infection in rats, is observed the most intense inflammatory process and parasitism in the heart compared to other points during the acute phase (Fig. 1C-G). Extensive histopathological studies using semi-serial sections of both atria and ventricles showed a total of 112.0 ± 28.6 parasites/animal within cardiomyocytes at day 6 compared to 1890.0 ± 618.7 at day 12 (mean ± S.E.M., n= 4–6 rats/group) (Figs 1C and 2A). In addition, the size of the parasite nests is clearly increased at day 12 [8] (compare Fig. 1E and 1F). The myocarditis is predominantly mononuclear (Fig. 2A) and large, macrophage-like cells are always observed by light microscopy in the myocardium of the infected animals, sometimes in close apposition to amastigote nests (Fig. 1F, arrow) [7, 8, 11, 12]. Accordingly, TEM has confirmed the presence of a large number of typical macrophages infiltrated in the heart (Fig. 2B and C).
2.
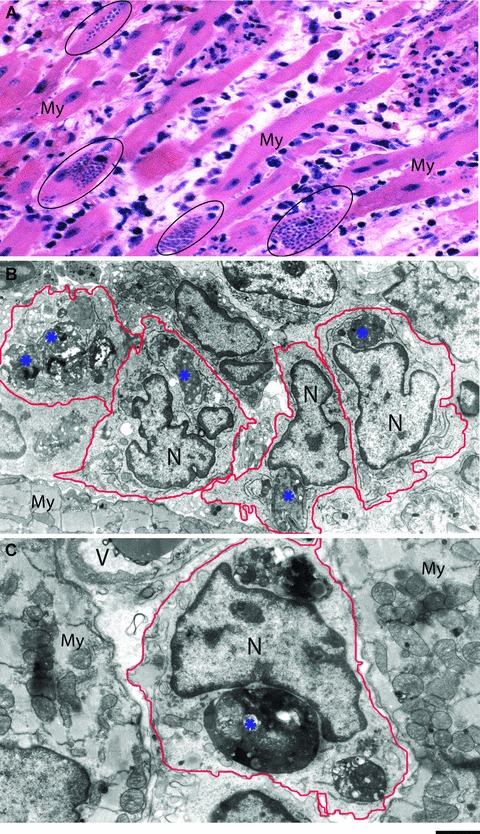
Myocarditis elicited by the acute phase of experimental Chagas' disease. (A) Histopathological analyses show myocarditis predominantly mononuclear and diffuse, dissociated myocardial fibres (My) and edematous intersticial tissue. Parasite nests are encircled. (B, C) Transmission electron microscopy reveals a large number of inflammatory macrophages infiltrated in the myocardium (My). Degenerating parasite amastigote forms (*) are seen within these cells. Samples were processed at day 12 of infection with Trypanosoma cruzi as before [12]. N, nucleus; V, blood vessel. Scale bar, 10 μm (A), 2 μm (B), 1 μm (C).
The tissue damage that occurs in the heart during the acute phase has been related to the intense parasitism, which induces cell alteration and destruction. At day 12, we observed that parasitized cardiomyocytes are frequently dissociated and show degenerative signs mainly in the atria, in addition to the presence of edematous interstitial tissue [8, 12] (Fig. 2A). By TEM, cardiomyocytes in far advanced stages of disintegration with cytoskeleton changes are seen adjacent to normal cells [10]. Other experimental models such as dogs [27] or Calomys callosus[34] show the same cardiomyocyte alterations by both light and/or electron microscopy.
We have been studying the ultrastructure of heart inflammatory macrophages in more detail during acute T. cruzi infection [7, 10–13, 20]. These cells have a distinct and notable morphology (Figs 2C, 3A, B and 4B), compared to non-inflammatory macrophages from noninfected rats (Fig. 4A). In these animals, a few macrophages are seen close to blood vessels or among cardiomyocytes. These macrophages usually show smaller size, few surface extensions, lesser amount of cytoplasmic organelles, and more heterochromatic nuclei in comparison with most macrophages from infected animals (compare Fig. 4A with 4B). In contrast, inflammatory macrophages, individually (Fig. 3A) or in clusters (Fig. 2B), are frequently observed in the vicinity of the cardiomyocytes, parasitized or not, and blood vessels. These macrophages exhibit a significant increase in their organelle apparatus, mainly Golgi complex, rough endoplasmic reticulum, polysomes, lysosomes and vesicles (Figs 3B and 4B). Moreover, phagolysosomes with varying sizes and electron densities containing amorphous or granular materials, cell debris and parasites are often found in the macrophage cytoplasm (Figs 3A and 4B). The nucleus of inflammatory macrophages is irregular in outline, more euchromatic and occasionally multinucleated (Fig. 4B) [7]. We have measured the diameter of heart macrophages in non-infected and infected animals and inflammatory macrophages show a significant increase in size compared to macrophages from non-infected animals [12]. Morphological signs of activation in inflammatory macrophages within the heart also include a striking increase in surface projections and pseudopodia (Figs 3B and 4B) [7, 12]. Morphological characteristics of activation as above described are also present in activated macrophages during the experimental infection with Trypanosoma congolense[35], and have been recognized as an accurate indication of high phagocytic and microbicidal activities of macrophages [7, 35–37].
3.
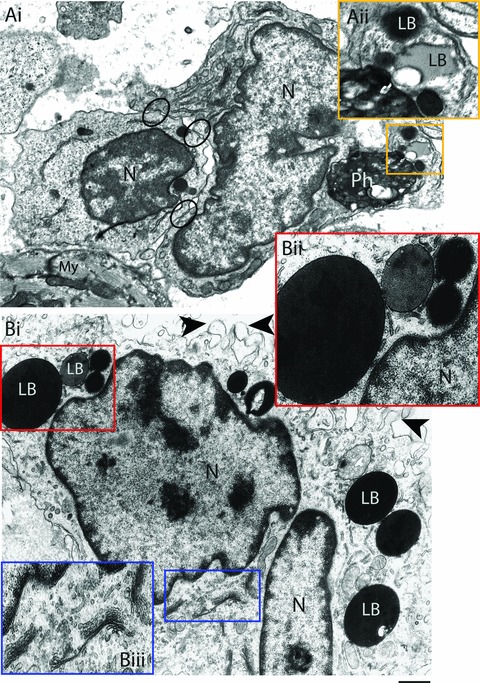
Activated heart macrophages triggered by the acute Trypanosoma cruzi infection. A clear interaction between an activated macrophage and a lymphocyte is observed in (Ai). Note areas of cell-to-cell attachments (circles). A phagolysosome is seen within the macrophage cytoplasm in close apposition to lipid bodies (LB) (box). (Aii) Higher magnification of the boxed area shows LBs with different electron densities and sizes. (Bi) Other features of macrophage activation include increase of both surface projections (arrowheads) and LB numbers and prominent Golgi complex profiles. Boxed areas show details of LBs and Golgi in higher magnification in Bii and Biii, respectively. Heart samples were processed for transmission electron microscopy at day 12 of infection. N, nucleus; My, myocardium. Scale bar, 600 nm (Ai); 400 nm (Aii); 750 nm (Bi) and 500 nm (Bii and Biii).
4.
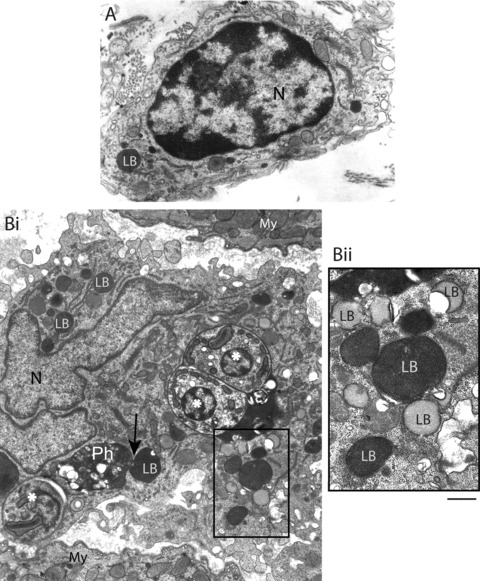
Ultrastructural features of a control and an inflammatory macrophage in the heart following infection with Trypanosoma cruzi (12 days). (A) A non-activated macrophage shows smaller size, few surface extensions, lesser amount of cytoplasmic organelles and more heterochromatic nucleus in comparison to a Chagas' disease-activated macrophage (Bi). This cell exhibits cytoplasm voluminous, rich in organelles, including phagolysosomes (Ph) and lipid bodies (LB), and parasite amastigote forms (*). Note a close association between a LB and a phagolysosome (arrow). The nucleus (N) is irregular in outline and very euchromatic. Bii corresponds to the boxed area in Bi and shows LBs imaged as heterogeneous organelles. Electron lucent LBs show a peripheral electron dense rim. My, myocardium. Scale bar, 1 μm (A, B); 500 nm (Bii). Panel A was reprinted from ref. [7] and panel B from [12] with permission.
In addition to large macrophages showing a variety of morphological features of activation, undifferentiated cells and macrophages in intermediary stages of activation, i.e. with some morphological signs of activation, are observed in the heart during the acute phase of Chagas' disease. Such ultrastructural diversity was absent in hearts of control uninfected animals and probably represents different stages of differentiation, maturation and activation of the monocyte/macrophage system [11]. Indeed, exposure of macrophages to microbial products and cell-derived cytokines such as INF-γ and IL-4, in vitro and in vivo, has been shown to induce different activation states in these cells, adding to further physiological heterogeneity [21, 38–41]. Therefore, the nature of the stimulus, or the combination of stimuli, can exert a profound effect in terms of macrophage morphology and functional capabilities [7, 40, 42].
TEM has also revealed a remarkable ability of heart inflammatory macrophages to establish cell-to-cell interactions, mostly macrophage-monocyte, macrophage-macrophage or macrophage-lymphocyte interactions in response to the acute phase of Chagas' disease (Fig. 3A) [7, 13]. These interactions are characterized by multiple areas of close apposition of the plasma membrane of the 2 cells or cell-to-cell attachments (Fig. 3A), and may have relevance for macrophage and/or T-cell activation [7].
The biology of T. cruzi-macrophage interaction has been investigated (reviewed in [43]). Macrophages from different origins [38] including heart macrophages [44] are able to express the surface receptor sialodhesin (Sn). This receptor recognizes sialic acid, present in high amounts at the parasite surface and seems to have an important role in the adherence process during T. cruzi phagocytosis [45]. In fact, phagocytosis is the main process for parasite internalization by macrophages. In vivo, heart inflammatory macrophages are actively involved in T. cruzi phagocytosis and killing and degenerating amastigotes are frequently observed within these macrophages by TEM [7, 12] (Fig. 2B and C). Experiments based on macrophage depletion by silica, a selective cytotoxic agent for macrophages, emphasize the importance of newly formed monocyte-derived macrophages in the resistance to the T. cruzi infection [7]. Depletion of macrophages by silica leads to a marked increase in the number of amastigote nests in the heart [7, 10]. Histoquantitative studies have demonstrated that parasite nests correspond to 16.5 ± 12.5% of the myocardium in silica-treated/infected animals compared to 1.9 ア 12.5% in rats only infected [7]. On the contrary, IFN−/− treated rats show reduced heart parasitism [46]. Moreover, when mice deficient of macrophage migration inhibitory factor (MIF−/−), a pleiotropic cytokine that favours the production of several pro-inflammatory cytokines such as IL-12, TNF-α and INF-γ, are infected with T. cruzi, it is also detected a higher parasitism in the heart compared to MIF+/+ mice [47]. Accordingly, mice deficient of INF-γ or IL-12 [48] or CCR5 [26] show increased heart parasitism compared to control animals. The findings that CCR5−/− mice have increased parasitism may result from the fact that CCR5-deficient macrophages have reported defects in activation [49, 50]. Therefore, as pointed out before, macrophage activation plays a critical role in the initiation of the inflammatory response and parasite killing. In fact, it is long recognized that inflammatory but not non-activated macrophages are able to kill the T. cruzi[36].
Classically activated macrophages (termed M1) develop in response to two signals: INF-γ, produced by natural killer cells in the early infection and by what has been termed ‘pathogen-associated molecular patterns’ (PAMPS), provided by a microbe and implicated in macrophage stimulation through pattern recognition receptors (PRRs). Several classes of PRRs, including Toll-like receptors (TLRs) and cytoplasmic receptors, recognize distinct microbial components and directly activate immune cells (reviewed in [51, 52]). The TLRs are pattern recognition receptors essential for the host to initiate an immune response. This receptor family comprises an extremely important arm of the immune system, whose function is to provide detection of microbial infection and linkage of innate to adaptative immunity (reviewed in [53]). TLR2 and TLR9 on the macrophage surface have been implicated in the recognition of T. cruzi antigens – GPI (glycosylphosphatidylinositol) anchors, a dominant glycolipid distributed at the surface membrane of T. cruzi, and parasite DNA, respectively (Fig. 5) [54–56].
5.
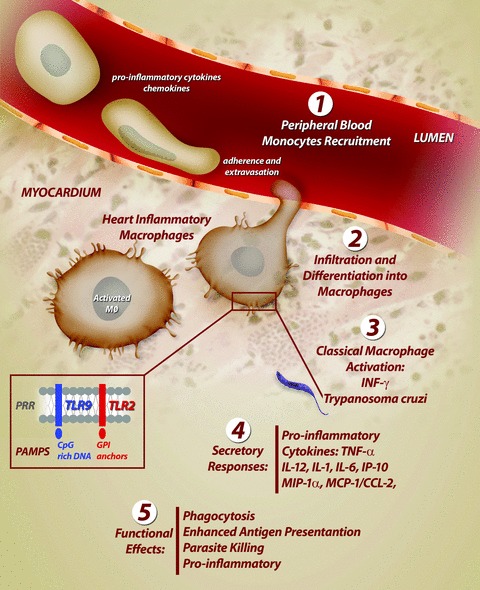
Activation of heart inflammatory macrophages during the acute infection with Trypanosoma cruzi. In response to acute infection, a cascade of events recruit cells derived from monocytic lineage from the peripheral blood into heart. This cascade culminates in a strong activation of macrophages. Classical activation of macrophages involves the key cytokine interferon gamma (INF-γ) and T. cruzi components (GPI anchors and CpG-rich DNA). These parasite products are recognized at the macrophage surface by Toll-like receptors (TLRs), a class of pattern recognition receptors (PRRs), which initiate an immune response and directly activate macrophages. Activated macrophages kill the parasite through various effector mechanisms such as TNF-α production. PAMPS, pathogen-associated molecular patterns; GPI, glycosylphosphatidylinositol; TNF-α, tumour necrosis factor-alpha; MIP-1α, macrophage inflammatory protein1α; MCP-1/CCL-2, monocyte chemoattractant protein-1; IP-10, inducible protein 10.
Classical activation induces profound metabolic changes in macrophages such as increased activity of inducible nitric oxide synthase (iNOS or NOS2) and respiratory burst, and secretory responses such as production of pro-inflammatory cytokines and chemokines (TNF-α, IL-12, IL-1, IL-6, macrophage inflammatory protein1α -MIP-1α, MCP-1) which lead to phagocytosis, killing of intracellular pathogens, antigen presentation and costimulation (Fig. 5) (reviewed in [38]).
A second population of activated macrophages, termed alternatively activated macrophages (M2 or nonclassical), arise in response to the Th2 cytokines IL-4 and/or IL-13 and are functionally and biochemically distinct from classically activated macrophages. Alternatively activated macrophages, through arginase-1 activity, shift the metabolism of L-arginine, a substrate for NOS2, to polyamine synthesis, impairing NOS2-mediated reactive nitrogen intermediates (NO, nitrite, nitrate) production. These cells can exert a growth-promoting effect on parasites that might result in uncontrolled infection. The available data suggest that subtypes of M2 are induced during protozoan infections and differentially affect the course of disease. For example, macrophages subtypes can be generated in the presence of immune complexes or apoptotic cells and/or induced by IL-10 or TGF-β, produced by T cells. Macrophages are also able to produce IL-10, TGF-p and prostaglandin E2 (PGE2) in response to phagocytosis of apoptotic host cells (reviewed in [39, 41]).
Classically and alternatively activated macrophages can be identified by a variety of biological markers. Classically activated macrophages present, for example, increased expression of major histocompatibility complex (MHC) and CD86 and decreased expression of manose receptor. Alternatively activated macrophages express CD163 and higher amounts of CD23 and mannose and scavenger receptors [41]. Interestingly, using an experimental model of murine trypanosomiasis, it was also demonstrated that the genes FIZZ1 and Ym1 are expressed differentially in alternatively activated macrophages versus classically activated macrophages. The expression of FIZZ1 and Ym1 is induced strongly in alternatively activated macrophages as compared with classically macrophages. FIZZ1 and Ym1 may thus constitute useful markers for the identification of alternatively activated macrophages [57] .
Despite extensive experimental investigations, the mechanisms used by activated macrophages to kill T. cruzi are not fully understood. NO secreted by INF-γ-activated macrophages, for instance, has been claimed as a relevant molecule for host defence against the parasite during the experimental murine infection [58–61]. In contrast, it has also been demonstrated that neither NO production [62] nor iNOS [63] are essential for the in vivo control of T. cruzi in mouse models. Of note, in our rat model of T. cruzi infection, there is no significant in vivo production of NO by both splenic and peritoneal macrophages during the acute disease, in spite of the control of the parasitemia and heart parasitism (Fabrino, unpublished data). In investigating the respiratory burst responses of macrophages from different origins to the in vivo infection in rats, we also observed that the infection increases the hydrogen peroxide (H2O2) production by splenic but not by peritoneal macrophages [12], indicating that the in vivo production of antimicrobial molecules seems related to specific types of macrophages and/or the parasite ability to activate these cells [12].
Lipid bodies as structural markers of inflammatory macrophages
A distinguishing ultrastructural feature of Chagas' disease-triggered macrophages is the presence of increased numbers of distinct cytoplasmic organelles termed lipid bodies (also known as lipid droplets) [20]. Lipid bodies are lipid-rich organelles found in small numbers in most eukaryotic cells as roughly spherical organelles (Fig. 4A), comprised of an outer monolayer of phospholipids which have a unique fatty acid composition, a core containing neutral lipids such as triglycerides and cholesterol, and variable protein composition. In the past, lipid bodies were largely associated with lipid storage, but it has become apparent that lipid bodies are dynamic and functionally active organelles [13, 64–67].
It is well documented that upon cell activation during different conditions, a significant increase of the LB numbers occurs in the cytoplasm, especially in activated leukocytes (reviewed in [15, 68, 69]). These organelles have been recognized as cytoplasmic domains for synthesis of inflammatory mediators (eicosanoids) in leukocytes from natural and experimental inflammatory processes such hypereosinophilic syndrome, acute respiratory distress syndrome and Crohn's disease (reviewed in [15, 70]). Several eicosanoid-forming enzymes pertinent to arachidonate mobilization and oxidative metabolism including lipoxygenases and cyclooxygenases are associated intracellularly with lipid bodies [71–73].
Lipid body formation was identified by us in both peritoneal and heart macrophages during different times of acute T. cruzi infection in vivo (Figs 3A,B and 4B) [20]. While control peritoneal macrophages present ∼2.19 ± 0.4 (mean ± S.E.M.) lipid bodies/macrophage, peritoneal macrophages from infected animals show ∼ 18.09 ± 1.4 at day 12 of infection. Accordingly, inflammatory heart macrophages, evaluated by quantitative electron microscopy, exhibit a mean of 8.3 lipid bodies/cell section (range of 1–25) at the same time of infection whereas control non-inflammatory macrophages show a mean of 2.6 lipid bodies/cell section (range of 0–3) (compare Fig. 4A and 4B) [20].
Since the first report of lipid bodies within Chagas' disease-elicited macrophages in vivo by our group in 2003 [20], we have been investigating lipid body structure and interactions by TEM within inflammatory macrophages during acute pathogen infections [13–15]. Lipid bodies are not surrounded by a typical bilayer membrane, but by a monolayer of phospholipids. The lack of a peripheral true membrane facilitates the identification of these organelles by TEM compared to other intracellular membranous organelles [64]. We have demonstrated that in vivo lipid body formation within inflammatory macrophages is strongly associated with the host response not only during the acute infection with T. cruzi[20] but also with other pathogens such as Mycobacterium bovis bacillus Calmette-Guérin – BCG [14]. For this reason, our group has recently highlighted these distinct organelles as structural markers of the innate immune responses in phagocytic cells [13].
In contrast to inflammatory macrophages, peripheral blood monocytes from T. cruzi-infected animals show low number of lipid bodies [20]. This may be associated with the fact that these cells are not directly involved or have a minor participation in the modulation of inflammation in target tissues where the eicosainoids function as paracrine mediators. On the other hand, the maturation of peripheral blood monocytes to tissue macrophages followed by activation of these cells is likely involved in lipid body formation and eicosanoid release [20].
One intriguing aspect of lipid bodies is their osmiophilia, which is dependent on the cell type and can change during elicited inflammatory responses (reviewed in [15]). In inflammatory macrophages, the lipid body density can consistently change during pathogen infections, as revealed by ultrastructural studies [13, 14, 20]. Based on osmiophilia, lipid bodies were recently identified and quantitated as three main types (light, electron and strongly electron-dense) within inflammatory macrophages from different origins, especially from the heart [13]. T. cruzi infection induces a significant increase in the numbers of light dense lipid bodies compared to non-infected controls which show lipid bodies preferentially as electron dense structures (Fig. 4A and B). Of note, lipid bodies change consistently their osmiophilia in macrophages stimulated in vivo with higher parasite load in irradiated-infected rats (Fig. 6B). These animals were exposed to a single, high dose of gamma irradiation 1 day before infection, which depletes the humoural and cellular immune responses except for the phagocytic activity of macrophages. Inflammatory macrophages from irradiated-infected animals show an increase of the numbers of both light dense and strongly electron dense lipid bodies compared to infection alone [13]. During the in vivo infection with Mycobacterium bovis Bacillus Calmette-Guérin (BCG), macrophage lipid bodies also change their osmiophilia. After 1 hr of infection, they appear as electron-dense organelles, morphologically similar to lipid bodies from control cells, but become predominantly light-dense organelles with a peripheral, prominent electron-dense rim after 24 hrs of infection [14]. Lipid body morphological changes may reflect differences in lipid composition, stages of formation of new lipid bodies, mobilization and/or neutral lipids/phospholipids ratio within lipid bodies. In addition, these morphological changes highlight lipid bodies as dynamic organelles, able to consistently change their structure in concert with cell activation [13]. In fact, lipid bodies in activated macrophages can be imaged as heterogeneous organelles, with lucent areas, granular and/or membranous internal structures (Fig. 4B) [13]. Of interest, by proteomic and ultrastructural studies we have recently defined lipid bodies as organelles with internal endoplasmic reticulum (ER)-like membranes and ER luminal proteins, suggesting a model by which enveloped ER-membranes and domains form lipid bodies [67].
6.
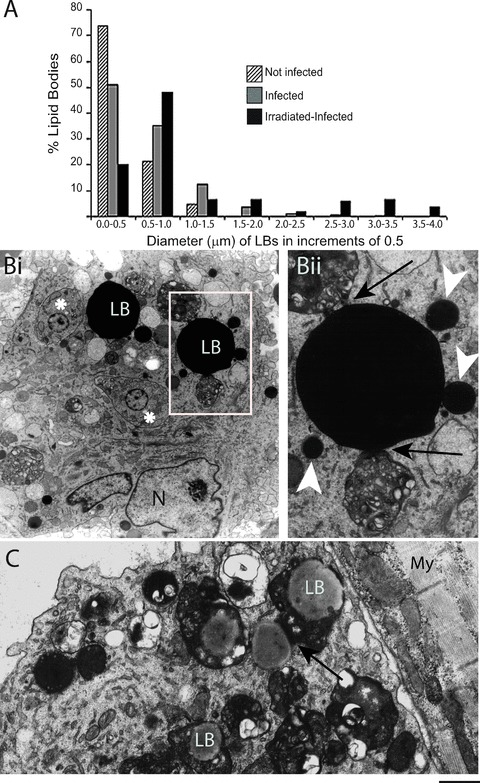
Lipid bodies (LB) within heart inflammatory macrophages increase in size and interact with phagolysosomes in response to acute Trypanosoma cruzi infection and parasite load. (A) LB diameter variation in different groups. A significant increase of LB occurred in infected alone compared to uninfected and in irradiated-infected compared to infected alone groups (P < 0.05). Irradiation induced-increased parasite load triggered formation of giant lipid bodies. (Bi) Strongly electron dense LBs from an irradiated-infected animal are observed in cytoplasmic areas in conjunction with many phagolysosomes. A close association between an LB and a phagolysosome is indicated by arrows in (Bii). (Bii) corresponds to the boxed area in (Bi). Arrowheads point to small LBs. Amastigotes (*) are also observed in the cytoplasm. (C) LBs are seen within or in contact (arrow) to phagolysosomes containing degenerating material. N, nucleus; My, myocardium. Before infection, rats were irradiated or not and heart samples processed at day 12 of infection [13]. Scale bar, 1.25 μm (Bi); 700 nm (Bii); 600 nm (C). Reprinted from ref. [13] with permission.
Another morphological feature of lipid bodies is their considerable size variation (Fig. 6A and B). For example, in scoring the diameters of lipid bodies within macrophages from rats experimentally infected with T. cruzi, 74% of lipid bodies had size <0.5 μm in non-infected whereas 54% of lipid bodies from infected animals were >0.5 μm, reaching up to 3 μm. Increase of the parasite burden induced by gamma irradiation triggered significant formation of gigantic lipid bodies within inflammatory macrophages, with diameters around 4 μm (Fig. 6A) [13]. These findings reveal that not only the number, but also the osmiophilia and size of lipid bodies represent structural indicatives of the participation of these organelles in innate immune responses [13, 74].
There has been increasing evidence that formation of lipid bodies in the course of infections involves well-regulated mechanisms and may have implications for microbial pathogenesis. However the detailed molecular mechanisms controlling inducible lipid body biogenic process within leukocytes remain to be defined (reviewed in [15]). In both migrating neutrophils and macrophages, in vivo stimulation with pathogen-related molecules (lipopolysaccharide and lipoarabinomannan), bacterial sepsis or BCG causes lipid body biogenesis through direct activation of CD14, TLR2 and TLR4 [14, 75, 76]. Indirect activation by endogenous mediators, like platelet activating factor (PAF), also contributes to lipid body biogenesis during infected-driven inflammation [75]. Both Mycobacterium bovis BCG [14] and T. cruzi (D'Avila, unpublished data) induce lipid body formation in a TLR-2-dependent pathway, but TLR4 seem not to be involved in this mechanism [14]. Moreover, it was recently demonstrated that MCP-1 acts as a key endogenous mediator for rapid biogenesis of Leukotriene B4-synthesizing lipid bodies within macrophages in infection-driven inflammatory disorders (sepsis or endotoxemia). The MCP-1-elicited lipid body formation was dependent on activation of a CCR2-elicited ERK and P13K signalling [77].
One important aspect of newly formed lipid bodies in inflammatory macrophages from animals infected with T. cruzi or BCG is their ability to interact with phagolysosomes and to relocate in the cytoplasm suggesting an association between these structures (Figs 3A, 4B and 6B) [14, 20]. Quantitative TEM revealed that 47% of lipid bodies were associated with phagolysosomes within Chagas' disease-elicited inflammatory macrophages, in addition to be noted within phagolysosomes (Fig. 6C) [13]. Similar association between lipid bodies and latex bead-containing phagosomes has been confirmed by high-resolution Raman microscopy in neutrophilic granulocytes [78]. The clear association lipid body-phagosome points to a yet ill-understood role of lipid bodies in pathogen control. It has been demonstrated that arachidonic acid has important functions in the induction of apoptosis of mycobacterium-infected macrophages, and in actin recruitment to the phagosome, enabling phagosome maturation and increased mycobacterial killing [79, 80]. The lipid body-phagosome association as well as the internalization of lipid bodies by phagosomes during the acute T. cruzi infection may, therefore, represent different stages of phagosome maturation leading to killing of the parasite [74].
Of interest, we [67] and other groups [81–84] have been localizing several GTPases of the Rab family and Kinases, including Rab 5, Rab 7 and PI3 kinase at lipid bodies. These molecules are involved in membrane trafficking and intracellular signalling, playing a key role in the control of sequential interactions of early and late endosomes; and have also been implicated in the maturation process of phagosomes containing intracellular pathogens [85, 86].
On the other hand, the interactions between lipid bodies and phagosomes maybe related to parasite survival. Intracellular pathogens exploit host cell nutrients for their growth and this close apposition of host lipid bodies to phagosomes may represent zones of privileged exchange of lipid body lipids favouring pathogen growth (reviewed in [15]). Moreover, macrophage lipid bodies have been directly implicated in the accumulation of iron complexes (mycobactin), which would enable iron delivery to mycobacteria through the association lipid body-phagosome [87]. This pathway could represent a new target for the control of mycobacterial infection [87]. Interestingly, lipid bodies function as platform sites for hepatitis C virus proteins (reviewed in [15]) and are involved in the production of infectious virus particles, which also open new avenues for antiviral intervention [88].
Formation of lipid bodies within inflammatory macrophages was positively correlated with augmented generation of prostaglandin E2 (PGE2), indicating a role for these organelles in the heightened eicosanoid production observed during the acute Chagas' disease [20]. Moreover, by intracellular immunofluores-cent detection of immobilized newly formed eicosanoid we have demonstrated that lipid bodies were the predominant sites for PGE2 synthesis in macrophages infected with BCG [14]. The enhanced capacity of macrophages to generate PGE2 in the course of pathogen infection due to increased lipid body formation and compartmentalization of signalling to eicosanoid production within lipid bodies may contribute to the mechanisms that intracellular pathogens have evolved to survive in host cells. Indeed, high concentration of PGE2 is a potent inhibitor of Th1 type response and of NO production favouring intracellular parasite growth [89–91]. Alternatively, lipid bodies could function as a draining compartment to rapidly uptake and re-acetylate free arachidonic acid with potentially detrimental outcome for the host cell.
In addition to the function of lipid bodies in prostaglandin synthesis, lipid bodies may have other roles in host response to pathogen infection. Cytokines, chemokines and growth factors seem to be localized in lipid bodies formed in activated leukocytes (reviewed in [15]). For instance, evidence for localization of the pro-inflammatory cytokine TNF-α in mast cells, eosinophils and macrophages lipid bodies was provided by immunogold electron microscopy in colonic Crohn's disease [92], and by immunofluo-rescence microscopy after in vivo LPS stimulation of peripheral blood neutrophils and monocytes from septic patients [76]. However, to date, the association of cytokines with lipid bodies is still limited and whether and how lipid body-stored cytokines are released, and whether they have signalling functions within lipid bodies are questions that remain to be addressed.
Macrophages and the resolution of acute inflammation and heart repair
Histopathological analyses of both atria and ventricles show an apparent inflammation resolution in concert to myocardium repair at the end of the acute phase of T. cruzi infection (Fig. 1G) [8]. Are macrophages involved in these phenomena? In fact, macrophages are recognized for taking part in the resolution of acute inflammation and in wound healing in a range of diseases and injuries [93–97]. The macrophage plasticity is particularly relevant to the resolution of inflammation. These cells can respond to a variety of stimuli and undergo distinct physiological changes. If the environment in which the macrophage resides changes, so too will the functional properties of these cells change [51]. Therefore, components present in the local microenvironment can prime macrophages to adopt unique phenotype to promote resolution of inflammation, i.e. macrophages that had previously been the major contributor to the inflammatory process can later participate in its resolution through the production of cytokines, growth factors and angiogenic factores [51].
Although the participation of macrophages in the inflammation resolution and subsequent tissue repair at the end of the acute Chagas' disease remain to be defined, the large number of activated macrophages present in the myocardium may likely play a role in these processes. At day 20 of T. cruzi infection in rat models, both parasitism and inflammation (Fig. 1C and G) are dramatically decreased in the heart in parallel to a drop of monocyte numbers in the blood (Fig. 1A) [8]. Moreover, TEM analyses revealed that at day 20 of infection, most cardiomyocytes do not show morphological alterations as seen in the early stage of the disease (Fig. 1G), indicating that myocardium repair is a rapid and consistent phenomenon by the end of the acute Chagasic infection [8]. Indeed, it is becoming apparent that the heart responds to T. cruzi infection with DNA repair and cell multiplication in the inflamed sites and with hypertrophy of the unaffected myocardium [98]. Of note, skeletal muscle regeneration is also documented following infection with T. cruzi[99].
In a different perspective though, the excessive activation of innate immunity, leading to an uncontrolled inflammatory cytokine production by macrophages, can result in a variety of pathological processes and autoimmune diseases (reviewed in [51, 55]). In fact, the immune response against T. cruzi has been hypothesized to contribute to the acute cardiac damage [27], but the occurrence of autoimmunity during Chagas' disease in both acute and chronic phases is still a matter of debate (reviewed in [5, 100, 101]). In our hands, in vivo depletion of macrophages or immunosuppression by gamma irradiation, during the acute infection exacerbated cardiomyocyte damage compared to the lesions seen in the infected controls [10, 11]. Our results coupled to findings from other groups do not support the autoimmune aetiology for acute Chagas' disease and indicate that the host immune system protects, rather than induces, tissue injury in the acute phase of infection [10, 99, 100].
The restoration of the inflamed myocardium to its prior physiological functioning, however, does not eliminate completely the parasite nor prevent the development of a progressive chronic symptomatic phase, characterized by irreversible pathological changes in the heart, the chronic Chagas' heart disease, observed in 30–40% of infected individuals (reviewed in [5]). Chagasic cardiomyopathy is a major life-threatening complication of T. cruzi infection in human beings [5]. This cardiomyopathy has been associated with the host immune response. On the other hand, the parasite is becoming increasingly detected in chronically infected hosts and may also be the cause of pathology either directly or through parasite-specific mediated inflammatory responses (reviewed in [5, 101]). The parasite persistence hypothesis supports research into immune enhancing therapies, including vaccine development with the primary goal of reducing the parasite load [102].
Concluding remarks
Chagas' disease remains a prominent problem with a major impact on public health in Latin America. The outcome of this protozoan infection, which highly affects the heart, is crucially dependent on activation events of the monocyte/macrophage system. Newly recruited monocyte-derived macrophages are actively involved in parasite phagocytosis in the heart and exhibit varying degrees of morphological heterogeneity and mixed activation states [7, 8]. Recent findings show that the in vivo host response to T. cruzi infection involves the formation of morphologically distinct inflammatory organelles termed lipid bodies within activated macrophages [13, 15] as observed in a variety of immunopathological conditions [15]. Lipid bodies are sites of eicosanoid production, key markers of macrophage activation and may be involved in pathogen control or favour intracellular parasite growth [15]. Better characterization of the roles of lipid bodies in the pathogenic mechanisms of infections may not only contribute to the understanding of pathogen-host interactions but may also identify new targets for intervention.
For the development of appropriate therapeutic interventions, it is also required a better understanding of the functional capabilities of monocyte subsets –‘inflammatory’ and ‘resident’ monocytes – and their contribution to the macrophage population in heart inflammation. It is now apparent that resident and inflammatory monocytes are defined by distinct sets of adhesion molecules and chemokine receptors, which suggests different modes of tissue trafficking and may represent a target for treatment [21–24].
Future studies are therefore important to understand the complexity of the mechanisms involved in the macrophage recruitment and activation and host resistance, as well as in pathogenesis during Chagas' disease for the successful development of new strategies to control disease severity [55]. Modulation of macrophage activation, including lipid body formation by induction of stronger and more persistent immune responses that protect the host against parasite replication and/or prevent excessive and deleterious activation may be instrumental in providing therapeutic benefits for Chagas' disease control.
Acknowledgments
Supported by Conselho Nacional de Desenvolvimento Cientìfico e Teenológico (CNPq, Brazil) and Fundaçào de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG). I acknowledge Bruno Menon for the fine illustration (Fig. 5) used in this paper. I am grateful to present and past members of the Laboratory of Cellular Biology (Federal University of Juiz de Fora, Brazil) for important contributions.
References
- 1.Dias JC, Silveira AC, Schofield CJ. The impact of Chagas' disease control in Latin America: a review. Mem Inst Oswaldo Cruz. 2002;97:603–12. doi: 10.1590/s0074-02762002000500002. [DOI] [PubMed] [Google Scholar]
- 2.Ropert C, Ferreira LR, Campos MA, Procopio DO, Travassos LR, Ferguson MA, Reis LF, Teixeira MM, Almeida IC, Gazzinelli RT. Macrophage signaling by glycosylphosphatidylinositol-anchored mucin-like glycoproteins derived from Trypanosoma cruzi trypomastigotes. Microbes Infect. 2002;4:1015–25. doi: 10.1016/s1286-4579(02)01609-x. [DOI] [PubMed] [Google Scholar]
- 3.Teixeira AR, Nascimento RJ, Sturm NR. Evolution and pathology in Chagas' disease–a review. Mem Inst Oswaldo Cruz. 2006;101:463–91. doi: 10.1590/s0074-02762006000500001. [DOI] [PubMed] [Google Scholar]
- 4.Brener Z, Gazzinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol. 1997;114:103–10. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]
- 5.Marin-Neto JA, Cunha-Neto E, Maciel BC, Simoes MV. Pathogenesis of chronic Chagas' heart disease. Circulation. 2007;115:1109–23. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- 6.Kierszenbaum F. Where do we stand on the autoimmunity hypothesis of Chagas' disease? Trends Parasitol. 2005;21:513–6. doi: 10.1016/j.pt.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Melo RCN, Machado CRS. Trypanosoma cruzi: peripheral blood monocytes and heart macrophages in the resistance to acute experimental infection in rats. Exp Parasitol. 2001;97:15–23. doi: 10.1006/expr.2000.4576. [DOI] [PubMed] [Google Scholar]
- 8.Fabrino DL, Leon LL, Parreira GG, Genestra M, Almeida PE, Melo RCN. Peripheral blood monocytes show morphological pattern of activation and decreased nitric oxide production during acute Chagas' disease in rats. Nitric Oxide. 2004;11:166–74. doi: 10.1016/j.niox.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Souza PE, Rocha MO, Rocha-Vieira E, Menezes CA, Chaves AC, Gollob KJ, Dutra WO. Monocytes from patients with indeterminate and cardiac forms of Chagas' disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun. 2004;72:5283–91. doi: 10.1128/IAI.72.9.5283-5291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melo RCN. Depletion of immune effector cells induces myocardial damage in the acute experimental Trypanosoma cruzi infection: ultrastructural study in rats. Tissue Cell. 1999;31:281–90. doi: 10.1054/tice.1999.0040. [DOI] [PubMed] [Google Scholar]
- 11.Melo RCN, Machado CR. Depletion of radiosensitive leukocytes exacerbates the heart sympathetic denervation and parasitism in experimental Chagas' disease in rats. J Neuroimmunol. 1998;84:151–7. doi: 10.1016/s0165-5728(97)00248-8. [DOI] [PubMed] [Google Scholar]
- 12.Melo RCN, Fabrino DL, D'Avila H, Teixeira HC, Ferreira AP. Production of hydrogen peroxide by peripheral blood monocytes and specific macrophages during experimental infection with Trypanosoma cruzi in vivo. Cell Biol Int. 2003;27:853–61. doi: 10.1016/s1065-6995(03)00173-2. [DOI] [PubMed] [Google Scholar]
- 13.Melo RCN, Fabrino DL, Dias FF, Parreira GG. Lipid bodies: structural markers of inflammatory macrophages in innate immunity. Inflamm Res. 2006;55:342–8. doi: 10.1007/s00011-006-5205-0. [DOI] [PubMed] [Google Scholar]
- 14.D'Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol. 2006;176:3087–97. doi: 10.4049/jimmunol.176.5.3087. [DOI] [PubMed] [Google Scholar]
- 15.Bozza PT, Melo RCN, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther. 2007;113:30–49. doi: 10.1016/j.pharmthera.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Olivares Fontt E, Heirman C, Thielemans K, Vray B. Granulocyte-macrophage colony-stimulating factor: involvement in control of Trypanosoma cruzi infection in mice. Infect Immun. 1996;64:3429–34. doi: 10.1128/iai.64.8.3429-3434.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villalta F, Kierszenbaum F. Role of inflammatory cells in Chagas' disease. II. Interactions of mouse macrophages and human monocytes with intracellular forms of Trypanosoma cruzi: uptake and mechanism of destruction. J Immunol. 1984;133:3338–43. [PubMed] [Google Scholar]
- 18.Van Voorhis W. Coculture of human peripheral blood mononuclear cells with Trypanosoma cruzi leads to proliferation of lymphocytes and cytokine production. J Immunol. 1992;148:239–48. [PubMed] [Google Scholar]
- 19.Reed SG, Nathan CF, Pihl DL, Rodricks P, Shanebeck K, Conlon PJ, Grabstein KH. Recombinant granulocyte/macrophage colony-stimulating factor activates macrophages to inhibit Trypanosoma cruzi and release hydrogen peroxide. Comparison with interferon gamma. J Exp Med. 1987;166:1734–46. doi: 10.1084/jem.166.6.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melo RCN, D'Avila H, Fabrino DL, Almeida PE, Bozza PT. Macrophage lipid body induction by Chagas' disease in vivo: putative intracellular domains for eicosanoid formation during infection. Tissue Cell. 2003;35:59–67. doi: 10.1016/s0040-8166(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 21.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 22.Strauss-Ayall D, Conrad SM, Mosser DM. Monocyte subpopulations and their differentiation patterns during infection. J Leukoc Biol. 2007;82:244–52. doi: 10.1189/jlb.0307191. [DOI] [PubMed] [Google Scholar]
- 23.Gelssmann F, Jung S, Llttman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 24.Auffray C, Fogg D, Garfa M, Elaln G, Join-Lambert O, Kayal S, Sarnackl S, Cumano A, Lauvau G, Gelssmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 25.Zlegler-Heltbrock HW, Flngerle G, Strobel M, Schraut W, Stelter F, Schutt C, Passllck B, Pforte A. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993;23:2053–8. doi: 10.1002/eji.1830230902. [DOI] [PubMed] [Google Scholar]
- 26.Hardlson JL, Wrlghtsman RA, Carpenter PM, Kuzlel WA, Lane TE, Manning JE. The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi. Infect Immun. 2006;74:135–43. doi: 10.1128/IAI.74.1.135-143.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrade ZA, Andrade SG, Correa R, Sadlgursky M, Ferrans VJ. Myocardial changes in acute Trypanosoma cruzi infection. Ultrastructural evidence of immune damage and the role of microangiopathy. Am J Pathol. 1994;144:1403–11. [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon S. The macrophage: past, present and future. Eur J Immunol. 2007;37:S9–17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 29.Savlno W, Villa-Verde DM, Mendes-da-Cruz DA, Silva-Monteiro E, Perez AR, Aoki Mdel P, Bottasso O, Guinazu N, Silva-Barbosa SD, Gea S. Cytokines and cell adhesion receptors in the regulation of immunity to Trypanosoma cruzi. Cytokine Growth Factor Rev. 2007;18:107–24. doi: 10.1016/j.cytogfr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Camargos ER, Rocha LL, Rachid MA, Almeida AP, Ferreira AJ, Teixeira AL, Jr, Chiari E, Barton M, Teixeira MM, Machado CRS. Protective role of ETA endothelin receptors during the acute phase of Trypanosoma cruzi infection in rats. Microbes Infect. 2004;6:650–6. doi: 10.1016/j.micinf.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Machado CRS, de Oliveira DA, Magalhaes MJ, Carvalho EM, Ramalho-Pinto FJ. Trypanosoma cruzi infection in rats induced early lesion of the heart noradrenergic nerve terminals by a complement-independent mechanism. J Neural Transm Gen Sect. 1994;97:149–59. doi: 10.1007/BF01277950. [DOI] [PubMed] [Google Scholar]
- 32.Coura JR. Chagas' disease: what is known and what is needed–a background article. Mem Inst Oswaldo Cruz. 2007;102:113–22. doi: 10.1590/s0074-02762007000900018. [DOI] [PubMed] [Google Scholar]
- 33.Teixeira AR, Nitz N, Guimaro MC, Gomes C, Santos-Buch CA. Chagas' disease. Postgrad Med J. 2006;82:788–98. doi: 10.1136/pgmj.2006.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magalhaes-Santos IF, Andrade SG. Participation of cytokines in the necrotic-inflammatory lesions in the heart and skeletal muscles of Calomys callosus infected with Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 2005;100:555–61. doi: 10.1590/s0074-02762005000500017. [DOI] [PubMed] [Google Scholar]
- 35.Anosa VO, Logan-Henfrey LL, Wells CW. The haematology of Trypanosoma con-golense infection in cattle. II- Macrophage structure and function in the bone marrow of Boran cattle. Comp. Haematology Int. 1997;7:23–9. [Google Scholar]
- 36.Nogueira N, Cohn ZA. Trypanosoma cruzi: in vitro induction of macrophage microbicidal activity. J Exp Med. 1978;148:288–300. doi: 10.1084/jem.148.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takemura T, Rom WN, Ferrans VJ, Crystal RG. Morphologic characterization of alveolar macrophages from subjects with occupational exposure to inorganic particles. Am Rev Respir Dis. 1989;140:1674–85. doi: 10.1164/ajrccm/140.6.1674. [DOI] [PubMed] [Google Scholar]
- 38.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 39.Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol. 2007;19:454–9. doi: 10.1016/j.coi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 42.Cook AD, Braine EL, Hamilton JA. The phenotype of inflammatory macrophages is stimulus dependent: implications for the nature of the inflammatory response. J Immunol. 2003;171:4816–23. doi: 10.4049/jimmunol.171.9.4816. [DOI] [PubMed] [Google Scholar]
- 43.De Souza W. Basic cell biology of Trypanosoma cruzi. Curr Pharm Des. 2002;8:269–85. doi: 10.2174/1381612023396276. [DOI] [PubMed] [Google Scholar]
- 44.Crocker PR, Hartnell A, Munday J, Nath D. The potential role of sialoadhesin as a macrophage recognition molecule in health and disease. Glycoconj J. 1997;14:601–9. doi: 10.1023/a:1018588526788. [DOI] [PubMed] [Google Scholar]
- 45.Monteiro VG, Lobato CS, Silva AR, Medina DV, de Oliveira MA, Seabra SH, de Souza W, DaMatta RA. Increased association of Trypanosoma cruzi with sialoadhesin positive mice macrophages. Parasitol Res. 2005;97:380–5. doi: 10.1007/s00436-005-1460-1. [DOI] [PubMed] [Google Scholar]
- 46.Revelli S, Didoli G, Roggero E, Moreno H, Bernabo J, Wietzerbin J, Bottasso O. Macrophage activity, IL-6 levels, antibody response and heart histology in rats undergoing an attenuated Trypanosoma cruzi acute infection upon treatment with recombinant interferon gamma. Cytokines Cell Mol Ther. 1998;4:153–9. [PubMed] [Google Scholar]
- 47.Reyes JL, Terrazas LI, Espinoza B, Cruz-Robles D, Soto V, Rivera-Montoya I, Gomez-Garcia L, Snider H, Satoskar AR, Rodriguez-Sosa M. Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect Immun. 2006;74:3170–9. doi: 10.1128/IAI.01648-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and inter-feron-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosomacruzi infection. Am J Pathol. 2001;159:1723–33. doi: 10.1016/s0002-9440(10)63019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato N, Kuziel WA, Melby PC, Reddick RL, Kostecki V, Zhao W, Maeda N, Ahuja SK, Ahuja SS. Defects in the generation of IFN-gamma are overcome to control infection with Leishmania donovani in CC chemokine receptor (CCR) 5-, macrophage inflammatory protein-1 alpha-, or CCR2-deficient mice. J Immunol. 1999;163:5519–25. [PubMed] [Google Scholar]
- 50.Zhou Y, Kurihara T, Ryseck RP, Yang Y, Ryan C, Loy J, Warr G, Bravo R. Impaired macrophage function and enhanced T cell-dependent immune response in mice lacking CCR5, the mouse homologue of the major HIV-1 coreceptor. J Immunol. 1998;160:4018–25. [PubMed] [Google Scholar]
- 51.Zhang X, Mosser D. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–78. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 53.Sabroe I, Parker L, Dower S, Whyte M. The role of TLR activation in inflammation. J Pathol. 2008;214:126–35. doi: 10.1002/path.2264. [DOI] [PubMed] [Google Scholar]
- 54.Tarleton RL. Immune system recognition of Trypanosoma cruzi. Curr Opin Immunol. 2007;19:430–4. doi: 10.1016/j.coi.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Gazzinelli RT, Ropert C, Campos MA. Role of the Toll/interleukin-1 receptor signaling pathway in host resistance and pathogenesis during infection with protozoan parasites. Immunol Rev. 2004;201:9–25. doi: 10.1111/j.0105-2896.2004.00174.x. [DOI] [PubMed] [Google Scholar]
- 56.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–9. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 57.Raes G, Noel W, Beschin A, Brys L, de Baetselier P, Hassanzadeh GH. FIZZ1 and Ym as tools to discriminate between differentially activated macrophages. Dev Immunol. 2002;9:151–9. doi: 10.1080/1044667031000137629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect Immun. 1994;62:5177–82. doi: 10.1128/iai.62.11.5177-5182.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodrigues MM, Ribeirao M, Boscardin SB. CD4 Th1 but not Th2 clones efficiently activate macrophages to eliminate Trypanosoma cruzi through a nitric oxide dependent mechanism. Immunol Lett. 2000;73:43–50. doi: 10.1016/s0165-2478(00)00205-4. [DOI] [PubMed] [Google Scholar]
- 60.Petray P, Castanos-Velez E, Grinstein S, Orn A, Rottenberg ME. Role of nitric oxide in resistance and histopathology during experimental infection with Trypanosoma cruzi. Immunol Lett. 1995;47:121–6. doi: 10.1016/0165-2478(95)00083-h. [DOI] [PubMed] [Google Scholar]
- 61.Silva JS, Machado FS, Martins GA. The role of nitric oxide in the pathogenesis of Chagas' disease. Front Biosci. 2003;8:s314–25. doi: 10.2741/1012. [DOI] [PubMed] [Google Scholar]
- 62.Marinho CR, Nunez-Apaza LN, Martins-Santos R, Bastos KR, Bombeiro AL, Bucci DZ, Sardinha LR, Lima MR, Alvarez JM. IFN-gamma, but not nitric oxide or specific IgG, is essential for the in vivo control of low-virulence Sylvio X10/4 Trypanosoma cruzi parasites. Scand J Immunol. 2007;66:297–308. doi: 10.1111/j.1365-3083.2007.01958.x. [DOI] [PubMed] [Google Scholar]
- 63.Cummings KL, Tarleton RL. Inducible nitric oxide synthase is not essential for control of Trypanosoma cruzi infection in mice. Infect Immun. 2004;72:4081–9. doi: 10.1128/IAI.72.7.4081-4089.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dvorak AM, Dvorak HF, Peters SP, Shulman ES, MacGlashan DW, Jr, Pyne K, Harvey VS, Galli SJ, Lichtenstein LM. Lipid bodies: cytoplasmic organelles important to arachidonate metabolism in macrophages and mast cells. J Immunol. 1983;131:2965–76. [PubMed] [Google Scholar]
- 65.Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res. 2001;40:325–438. doi: 10.1016/s0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- 66.Weller PF, Ryeom SW, Dvorak AM. Lipid bodies: structurally distinct, non-membranous intracellular sites of eicosanoid formation. In: Bailey JM, editor. Prostaglandins, leukotrienes, lipoxins, and PAF. New York: Plenum Press; 1991. [Google Scholar]
- 67.Wan HC, Melo RCN, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB J. 2007;21:167–78. doi: 10.1096/fj.06-6711com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weller PF, Ackerman SJ, Nicholson-Weller A, Dvorak AM. Cytoplasmic lipid bodies of human neutrophilic leukocytes. Am J Pathol. 1989;135:947–59. [PMC free article] [PubMed] [Google Scholar]
- 69.Dvorak AM, Weller PF. Ultrastructural analysis of human eosinophils. In: Marone G, editor. Human eosinophils: biological and chemical aspects. Basel: Karger: 2000. pp. 1–28. [DOI] [PubMed] [Google Scholar]
- 70.Weller PF, Dvorak AM. Lipid bodies: intracellular sites for eicosanoid formation. J Allergy Clin Immunol. 1994;94:1151–6. doi: 10.1016/0091-6749(94)90325-5. [DOI] [PubMed] [Google Scholar]
- 71.Dvorak AM, Morgan E, Schleimer RP, Ryeom SW, Lichtenstein LM, Weller PF. Ultrastructural immunogold localization of prostaglandin endoperoxide synthase (cyclooxygenase) to non-membrane-bound cytoplasmic lipid bodies in human lung mast cells, alveolar macrophages, type II pneumocytes, and neutrophils. J Histochem Cytochem. 1992;40:759–69. doi: 10.1177/40.6.1316915. [DOI] [PubMed] [Google Scholar]
- 72.Dvorak AM, Morgan ES, Tzizik DM, Weller PF. Prostaglandin endoperoxide synthase (cyclooxygenase): ultrastructural localization to nonmembrane-bound cyto-plasmic lipid bodies in human eosinophils and 3T3 fibroblasts. Int Arch Allergy Immunol. 1994;105:245–50. doi: 10.1159/000236764. [DOI] [PubMed] [Google Scholar]
- 73.Bozza PT, Yu W, Penrose JF, Morgan ES, Dvorak AM, Weller PF. Eosinophil lipid bodies: specific, inducible intracellular sites for enhanced eicosanoid formation. J Exp Med. 1997;186:909–20. doi: 10.1084/jem.186.6.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Melo RCN, Sabban A, Weller PF. Leukocyte lipid bodies: inflammation-related organelles are rapidly detected by wet scanning electron microscopy. J Lipid Res. 2006;47:2589–94. doi: 10.1194/jlr.D600028-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Silva AR, de Assis EF, Caiado LF, Marathe GK, Bozza MT, McIntyre TM, Zimmerman GA, Prescott SM, Bozza PT, Castro-Faria-Neto HC. Monocyte chemoattractant protein-1 and 5-lipoxygenase products recruit leukocytes in response to platelet-activating factor-like lipids in oxidized low-density lipoprotein. J Immunol. 2002;168:4112–20. doi: 10.4049/jimmunol.168.8.4112. [DOI] [PubMed] [Google Scholar]
- 76.Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, Bozza PT. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular Loci involved in eicosanoid metabolism. J Immunol. 2002;169:6498–506. doi: 10.4049/jimmunol.169.11.6498. [DOI] [PubMed] [Google Scholar]
- 77.Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, Silva AR, Bozza MT, Castro-Faria-Neto HC, Bandeira-Melo C, Bozza PT. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol. 2007;179:8500–8. doi: 10.4049/jimmunol.179.12.8500. [DOI] [PubMed] [Google Scholar]
- 78.van Manen HJ, Kraan YM, Roos D, Otto C. Single-cell Raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. Proc Natl Acad Sci USA. 2005;102:10159–64. doi: 10.1073/pnas.0502746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anes E, Kuhnel MP, Bos E, Moniz-Pereira J, Habermann A, Griffiths G. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat Cell Biol. 2003;5:793–802. doi: 10.1038/ncb1036. [DOI] [PubMed] [Google Scholar]
- 80.Duan L, Gan H, Arm J, Remold HG. Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apopto-sis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J Immunol. 2001;166:7469–76. doi: 10.4049/jimmunol.166.12.7469. [DOI] [PubMed] [Google Scholar]
- 81.Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279:3787–92. doi: 10.1074/jbc.M311945200. [DOI] [PubMed] [Google Scholar]
- 82.Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–42. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 83.Umlauf E, Csaszar E, Moertelmaier M, Schuetz GJ, Parton RG, Prohaska R. Association of stomatin with lipid bodies. J Biol Chem. 2004;279:23699–709. doi: 10.1074/jbc.M310546200. [DOI] [PubMed] [Google Scholar]
- 84.Yu W, Cassara J, Weller PF. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymor-phonuclear leukocytes and other myeloid-derived cells. Blood. 2000;95:1078–85. [PubMed] [Google Scholar]
- 85.Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 1997;272:13326–31. doi: 10.1074/jbc.272.20.13326. [DOI] [PubMed] [Google Scholar]
- 86.Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 2001;154:631–44. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol. 2005;1:149–53. doi: 10.1038/nchembio717. [DOI] [PubMed] [Google Scholar]
- 88.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–97. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 89.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146:108–13. [PubMed] [Google Scholar]
- 90.Renz H, Gong JH, Schmidt A, Nain M, Gemsa D. Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2 and cyclic nucleotides. J Immunol. 1988;141:2388–93. [PubMed] [Google Scholar]
- 91.Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, DosReis GA, Lopes MF. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403:199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- 92.Beil WJ, Weller PF, Peppercorn MA, Galli SJ, Dvorak AM. Ultrastructural immunogold localization of subcellular sites of TNF-alpha in colonic Crohn's disease. J Leukoc Biol. 1995;58:284–98. doi: 10.1002/jlb.58.3.284. [DOI] [PubMed] [Google Scholar]
- 93.Vinuesa E, Hotter G, Jung M, Herrero-Fresneda I, Torras J, Sola A. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J Pathol. 2008;214:104–13. doi: 10.1002/path.2259. [DOI] [PubMed] [Google Scholar]
- 94.Baldwin T, Sakthianandeswaren A, Curtis JM, Kumar B, Smyth GK, Foote SJ, Handman E. Wound healing response is a major contributor to the severity of cutaneous leishmaniasis in the ear model of infection. Parasite Immunol. 2007;29:501–13. doi: 10.1111/j.1365-3024.2007.00969.x. [DOI] [PubMed] [Google Scholar]
- 95.Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Chuai M, Imai Y, Takahashi H, Tanaka J. Accumulation of macrophage-like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2008;28:149–63. doi: 10.1038/sj.jcbfm.9600519. [DOI] [PubMed] [Google Scholar]
- 96.Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol. 2006;168:529–41. doi: 10.2353/ajpath.2006.050255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amano H, Morimoto K, Senba M, Wang H, Ishida Y, Kumatori A, Yoshimine H, Oishi K, Mukaida N, Nagatake T. Essential contribution of monocyte chemoattractant protein-1/C-C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J Immunol. 2004;172:398–409. doi: 10.4049/jimmunol.172.1.398. [DOI] [PubMed] [Google Scholar]
- 98.Arnaiz MR, Fichera LE, Postan M. Cardiac myocyte hypertrophy and proliferating cell nuclear antigen expression in Wistar rats infected with Trypanosoma cruzi. J Parasitol. 2002;88:919–25. doi: 10.1645/0022-3395(2002)088[0919:CMHAPC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 99.Maldonado IR, Ferreira ML, Camargos ER, Chiari E, Machado CR. Skeletal muscle regeneration and Trypanosoma cruzi-induced myositis in rats. Histol Histopathol. 2004;19:85–93. doi: 10.14670/HH-19.85. [DOI] [PubMed] [Google Scholar]
- 100.Tarleton RL. Chagas' disease: a role for autoimmunity? Trends Parasitol. 2003;19:447–51. doi: 10.1016/j.pt.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 101.Girones N, Cuervo H, Fresno M. Trypanosoma cruzi-induced molecular mimicry and Chagas' disease. Curr Top Microbiol Immunol. 2005;296:89–123. doi: 10.1007/3-540-30791-5_6. [DOI] [PubMed] [Google Scholar]
- 102.Tarleton RL. Parasite in the aetiology of Chagas' disease. Int J Parasitol. 2001;31:550–4. doi: 10.1016/s0020-7519(01)00158-8. [DOI] [PubMed] [Google Scholar]