

Abstract
The crystal structure of the P450cin substrate-bound nitric oxide complex and the substrate-free form have been determined revealing a substrate-free structure that adopts an open conformation relative to the substrate-bound structure. The region of the I helix that forms part of the O2 binding pocket shifts from α helix in the substrate-free form to a π helix in the substrate-bound form. Unique to P450cin is an active site residue, Asn242, in the I helix that H-bonds with the substrate. In most other P450s this residue is a Thr and plays an important role in O2 activation by participating in an H-bonding network required for O2 activation. The π/α I helix transition results in the carbonyl O atom of Gly238 moving in to form an H-bond with the water/hydroxide ligand in the substrate-free form. The corresponding residue, Gly248, in the substrate-free P450cam structure experiences a similar motion. Most significantly, the oxy-P450cam complex Gly248 adopts a position midway between the substrate-free and -bound states. A comparison between these P450cam and the new P450cin structures provides insights into differences in how the two P450s activate O2. The structure of P450cin complexed with nitric oxide, a close mimic of the O2 complex, shows that Gly238 is likely to form tighter interactions with ligands than the corresponding Gly248 in P450cam. Having a close interaction between an H-bond acceptor, the Gly238 carbonyl O atom, and the distal oxygen atom of O2 will promote protonation and hence further reduction of the oxy-complex to the hydroperoxy intermediate resulting in heterolytic cleavage of the peroxide O-O bond and formation of the active ferryl intermediate required for substrate hydroxylation.
Cytochrome P450s catalyze the monooxygenation of a vast array of compounds described by the following reaction:
O2 is cleaved with one oxygen atom ending up as water while the other is incorporated into the substrate. Reducing equivalents originate from either NADPH or NADH via redox partners and are sequentially transferred to the heme iron. O2 binds the ferrous iron to form the ferrous-oxy-complex, Fe(II)-O-O or its equivalent, the ferric superoxide complex, Fe(III)-O-O-. Subsequent proton transfer and oxygen cleavage generate the compound I species, Fe(IV)=O and a porphyrin cation radical. The peroxy intermediates have been characterized in P450cam by cryo-reduction/annealing and subsequent monitoring by electron paramagnetic resonance and electron nuclear double resonance spectroscopies.1 Previously, the existence of compound I could only be inferred by analogy to peroxidases and catalases.2 However, Rittle and Green recently characterized compound I in CYP119.3
While a reasonable picture of P450 oxygen activation has emerged,4 how the oxy-complex is activated by specific protonation of the leaving O atom and what role conformational dynamics plays remain open questions. Recent oxy-complex crystal structures of P450cam and P450eryF have been helpful in understanding how the oxygen complex is stabilized as well as the probable source of protons necessary for O2 cleavage.5-7 In P450cam O2 binding is accompanied by enlargement of the active site and the appearance of two new waters with one in position for proton delivery to the distal O2 oxygen atom.7 It recently has been proposed6 that the highly conserved Thr252 stabilizes the hydroperoxy intermediate, Fe(III)-OOH, where it most likely acts as a hydrogen bond acceptor. P450eryF is rare among P450s since an Ala replaces the catalytically important Thr and P450eryF does not show P450cam-like structural changes upon O2 binding. P450eryF is thought to utilize substrate-assisted catalysis where the substrate itself provides the OH group normally provided by the conserved active site Thr.8
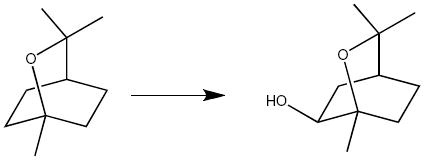
P450cin from the soil bacterium Citrobacter brakki catalyzes the regio- and stereo-selective oxidation of cineole to (R)6-α-hydroxycineole as outlined below.9
P450cin is yet another outlier since it contains neither a Thr nor an Ala, but instead an Asn242 at this position. An Asn242Ala mutant enzyme retains good activity and remains 50% coupled to substrate hydroxylation10 meaning that half of the NADPH derived electrons are responsible for productive product formation while the remaining half futilely reduce O2 to H2O2. In addition P450cin does not use substrate-assisted catalysis.11 While Asn242 is not essential for enzyme activity, it is critical for stereo and regio-selective substrate hydroxylation.10 This is not unexpected considering Asn242 donates an H-bond to the substrate ethereal oxygen and is likely important for proper substrate orientation. Therefore, we do not expect P450cin to adopt the same way structurally to oxygen binding as P450s that either retain the conserved Thr or utilize their substrates to functionally replace the Thr. However, like all other P450s, proton shuttling to the dioxygen must either be provided by water(s) not seen in the ferric substrate-bound structure12 or by a functional group other than Asn242. Comparison of the P450cam-oxy and recent P450cam substrate-free structures13 underscores the relationship between oxygen binding, active site solvent structure, and conformational changes required for proton delivery. In all P450s structures observed thus far, I helix residues contact both substrate and O2. In P450cam there are substantial differences in the I helix between the substrate-free and -bound states.13 These changes are also seen upon O2 binding in P450cam but to a lesser extent. The I helix movement resulting from O2 binding is about midway between that observed in the substrate-free and -bound conformations. This shows that comparison of substrate-free and -bound P450 structures can provide important clues as to the available flexibility that is required not only for substrate binding but also for O2 activation. In order to shed light on the possible conformational changes available to a P450cin-oxy-complex, we will examine and compare structural changes in the substrate-free P450cin crystal structure as well as the ferrous-NO structure, a partial mimic of the oxy-complex.
EXPERIMENTAL PROCEDURES
P450cam purification, crystallization, and data collection
P450cam was purified using an E. coli expression system and crystallized as previously described.14 To prepare the ferrous-NO complex, crystals were transferred into an anaerobic chamber and harvested into mother liquor containing 32% PEG4000 that had been previously purged of oxygen by repeated vacuum and argon flushing. The crystals were then transferred into cryo-protectant solution containing mother liquor and 15% glycerol. The Fe(III) heme center was reduced to Fe(II) by stepwise addition of 1 mM, 5 mM and 10 mM sodium dithionite. Crystals turned from brown to bright red within 5 minutes of this treatment indicating reduction of the heme iron. NO-bound enzyme was prepared by transferring the crystal to 0.25 mL of degassed cryo-protectant solution containing 5 mM dithionite in a septum-sealed 5 mL serum vial. The vial was then brought out of the glove box and purged with NO gas (15-psi gauge pressure) through the septum with venting for 20 minutes followed by non-venting for 5 minutes. After three cycles of this treatment 100 μl of the cryo-protectant solution containing the crystal was transferred by pipette to a nine-well depression plate cooled on ice. The crystal was then flash frozen as quickly as possible in liquid nitrogen prior to data collection.
Diffraction data for the NO complex were collected using an in-house Rigaku Micromax 007HF rotating anode x-ray generator equipped with a Saturn 944+ CCD detector. Crystals were kept frozen in a stream of -160 °C liquid nitrogen (Crystal Logic, Los Angeles, CA) during data collection. The detector to crystal distance was 100 mm and the 2θ angle was -17°. Each data frame was 1° oscillation with 60 second exposures for a total of 180 frames per data scan in ω angle. Two data scans were collected at χ angles of 15° and 30°. All data frames were integrated, merged and scaled using d*TREK.15 Initial 2Fo-Fc and Fo-Fc maps were calculated using rigid body refinement in Phenix.refine from the Phenix package.16 The initial model was taken from a previously published P450cam-CO structure that exhibited identical unit cell dimensions (PDB ID: 1T87). Prior to further calculations the CO coordinates were removed. The 2Fo-Fc map and atomic coordinates were iteratively improved with model adjustment in the graphics program Coot17 followed by restrained coordinate and individual isotropic B-factor refinement with Phenix.refine. Initial rounds of refinement were carried out without the NO ligand resulting in strong Fo-Fc density above the heme iron in molecule B with a more modest effect in molecule A. NO was subsequently added into molecule B but left out of molecule A as it appeared to be present with low occupancy.
P450cin purification and crystallization
Details of P450cin purification are provided in the Supplementary material.
For crystallization of substrate-free P450cin, stock protein was thawed and diluted from 44.5 mg/mL to 34 mg/mL in cineole free buffer consisting of 50 mM KPi pH 7.4 and 50 mM KCl. One μl of protein was mixed with 1 μl of well solution (100 mM Hepes pH 7, 14% PEG 4000 and 80 mM MgCl2) in sitting drop vapor diffusion Cryschem® crystal trays (Hampton). Following a one-week incubation at room temperature, rod shaped crystals were harvested and soaked in cryo-protectant solutions consisting of mother liquor and increasing concentrations of ethylene glycol (5%, 10%, 15%, and 20%). The crystals were flash cooled and stored in liquid nitrogen prior to data collection. It should be noted that 750 μM cineole was carried along throughout purification yet substrate-free P450cin crystallized. However, cineole was present at sub-stoichiometric amounts since the concentration of enzyme in the crystallization drops ranged from 0.8-1.0 mM so substrate-free enzyme should dominate. Even so the presence of cineole was essential for crystallization so cineole must have been serving as an additive that assisted with crystallization.
For substrate-bound P450cin crystal growth, stock protein was thawed and diluted from 42 mg/mL to 25 mg/mL in 50 mM Tris pH 7.5, 20 mM NaCl and 1 mM Cineole. Sitting drop vapor diffusion crystal trials were set up in Cryschem® crystal trays with 1:1 protein to well solution ratio (6-9 % PEG 3350, 100 mM BisTris pH 6.2, 100 mM lithium sulfate, 5 mM cineole). After a four-day incubation period at room temperature well defined thick plates were harvested and transferred into cryo-protectant solutions consisting of mother liquor with 12% PEG 3350 and increasing concentrations of glycerol (5%, 10%, 15%, and 20%). As above the crystals were flash cooled and stored in liquid nitrogen.
P450cin data collection and refinement
Data from the substrate-free and substrate-bound crystals were collected at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA, USA) on beam line 9-2 using a Mar325 CCD detector. Data were integrated with Denzo and scaled with Scalepack from the HKL2000 suite.16 Phasing was accomplished by molecular replacement using Phaser18 in the CCP4 suite19 using a substrate bound P450cin structure (PDB code: 1T2B) as a search model. Molecular replacement was necessary for phasing in both cases due to the space group (P1) difference for the substrate-free structure and large changes in unit cell dimensions for the substrate-bound structure. In both cases refinement was carried out with non-crystallographic symmetry (NCS) restraints in the early stages of refinement, individual coordinate refinement as well as hydrogens added in the riding positions using Phenix.refine from the Phenix package.16 The large data to parameter ratio enabled anisotropic B-factor refinement for all atoms in the substrate-free structure while waters were refined isotropically. Large changes in the F and G helices and B-C loop compared to the substrate bound structure resulted in discontinuous electron density for parts of these regions. Multiple rounds of refinement and model building in the graphics program, Coot,20 along with an averaged “kicked” 2mFo-DFc map calculated in Phenix.refine brought more clarity to these regions. Despite these improvements, a portion of the B-C loop remained disordered in all four chains (Table. 1). In the final rounds of refinement the substrate-free model was polished by automated optimized weighting between x-ray target stereochemistry and x-ray target ADP weights.
Table 1.
Scaling and refinement statistics for P450cin substrate-bound and substrate-free crystal structures as well as P450cin-NO and P450cam-NO ligand complexes.
| Data set | P450cin substrate-free | P450cin substrate-bound | NO-P450cin | NO-P450cam |
|---|---|---|---|---|
|
| ||||
| Radiation source | SSRL 9-1 | SSRL 9-2 | Saturn 944 | Saturn 944 |
|
| ||||
| Space group | P1 | P21 | P21 | P21 |
|
| ||||
| Unit cell dimensions (a,b,c) (Å), (α, β, γ) (°) | 60.38, 83.91, 88.19 | 57.55, 69.26 | 64.18, 68.44, 104.05 | 67.55, 62.27, 95.49 |
| 96.81, 96.39, 89.94 | 122.61 | |||
| β = 97.14 | β = 95.79 | β = 90.06 | ||
|
| ||||
| Resolution Range (Å) (highest shell) | 50-1.37 (1.39-1.37) | 50-1.55 (1.58-1.55) | 50-2.32 (2.40-2.32) | 33-2.20 (2.28-2.20) |
|
| ||||
| Wavelength (Å) | 0.98 | 1.00 | 1.54 | 1.54 |
|
| ||||
| Total observations | 1,334,749 | 445,389 | 187,358 | 120,887 |
|
| ||||
| Unique reflections (highest shell) | 343,124 (16,728) | 136,152 (6,791) | 37,894 (3,636) | 39,581 (8,897) |
|
| ||||
| Completeness (%) (highest shell) | 95.7 (93.1) | 99.2 (99.7) | 97.1 (93.9) | 97.6 (92.7) |
|
| ||||
| Rsym (highest shell) | .081 (0.571) | .074 (0.583) | .071 (0.213) | .075 (0.356) |
|
| ||||
| <I/σ> (highest shell) | 32.4 (2.3) | 20.0 (2.1) | 19.8 (5.7) | 9.6 (2.2) |
|
| ||||
| Redundancy (highest shell) | 3.9 (3.2) | 3.3(3.3) | 5.0 (4.0) | 3.0(2.4) |
|
| ||||
| B factor, Wilson plot (Å2) | 15.3 | 17.1 | 30.1 | 32.6 |
|
| ||||
| Reflections used in refinement | 343,124 | 136,152 | 37,862 | 39,561 |
|
| ||||
| Resolution range (Å) used in refinement | 41.7-1.37 | 35.0-1.55 | 36.0-2.32 | 33.0-2.20 |
|
| ||||
| No. of protein atoms fit | 12,573 | 6,358 | 6,358 | 6,403 |
|
| ||||
| No. of heteroatoms fit | 197 | 121 | 112 | 114 |
|
| ||||
| No. of waters fit | 1,864 | 1,061 | 347 | 409 |
|
| ||||
| Unmodeled residues | Ch. A; 1, 82-85, 404 | Ch. A; 1 | Ch. A; 1 | Ch. A; 1-7 |
| Ch. B; 1, 82-85, 404 | Ch. B; 1 | Ch. B; 1 | Ch. B; 1-10 | |
| Ch. C; 1, 81-85, 404 | ||||
| Ch. D; 1, 82-85, 177-179, 404 | ||||
|
| ||||
| Rwork % | 13.6 | 17.2 | 19.5 | 20.7 |
|
| ||||
| Rfree % | 16.7 | 22.0 | 25.0 | 26.3 |
|
| ||||
| RMSD Bond length (Å) | .012 | .006 | .011 | .007 |
|
| ||||
| RMSD Bond angle (°) | 1.416 | 1.222 | 1.172 | 1.227 |
|
| ||||
| Observation/parameter | 2.51 | 1.98 | 1.41 | 1.43 |
P450cin-NO complex
Substrate-bound crystals were transferred into an anaerobic chamber and harvested into mother liquor containing 12 % PEG3350 that had been previously purged of oxygen. Five mM cineole was added from a degassed 1 M stock as it has a tendency to evaporate from the mother liquor during degassing. The crystals were then transferred into cryo-protectant solutions containing mother liquor and increasing concentrations of PEG400 up to 30% in nine incremental steps over the course of 3 hours. The Fe(III) heme center was reduced to Fe(II) by addition of 1 mM and 5 mM sodium dithionite. Due to crystal cracking dithionite was not added to the soaking solutions until the PEG400 concentration reached 15%. The NO-bound P450cin crystal was prepared exactly analogous to that of P450cam described above.
Diffraction data for the P450cin-NO complex was collected in house as described for P450cam with the following exceptions. The 2θ angle was -15° and exposure time was 30 seconds per frame while the χ angles were 0° and 30°. The data were integrated, merged and scaled using HKL2000.21 Molecular replacement was accomplished using Phaser18 as described above. TLS (translation/ libration/screw) refinement using NCS restraints and isotropic B-factors was carried out using Phenix.refine.16 NO was not included in the first round of refinement resulting in a large Fo-Fc density above the heme iron. All subsequent rounds of refinement included NO.
Stopped flow spectroscopy of the P450cin-oxy-complex
One mM P450cin was reduced in an anaerobic glove box by titrating with a 100 μM solution of dithionite and diluted with buffer in an anaerobic cuvette to generate a Soret peak shift from 392nm to 411nm indicative of P450 heme iron reduction. Absorbance changes associated with oxygen binding to the P450 ferrous iron center were recorded as a shift from 411nm (reduced) to 418nm (oxygen bound) in a SX.18MV stopped flow apparatus (Applied Photophysics) by rapid mixing of the reduced P450cin with an oxygen saturated buffer. One syringe was filled with 20 μM reduced P450cin in deoxygenated buffer (50 mM KPi, pH 7.4, 50 mM KCl, 2 mM cineole, 10 U/mL catalase, 20 U/mL glucose oxidase and 1 mM glucose). The second syringe contained oxygen saturated buffers without P450cin or oxygen scrubbers. The photodiode array rapid spectral scans were taken for 2 and 10 seconds and plotted in Igor Pro (WaveMetrics, Inc.). A 4 °C water bath pumped cold water into the mixing chamber throughout the experiment.
Results and Discussion
Spectroscopic visualization of a transient P450cin ferrous iron oxy-complex
In order to visualize any potential structural changes required for oxygen activation in P450cin, the P450cin-oxy complex structure is highly desirable. Our ability to capture the oxy-complex in a crystal depends on its inherent stability. Therefore, it was important to determine the stability of the P450cin-oxy-complex in solution before attempting to trap it in a crystal. Unfortunately, diode array stopped flow experiments revealed that the instability of the P450cin oxy-complex was likely to present a serious challenge in trapping this intermediate. As seen in Fig. 1, there is a clear Soret peak shift from 410nm to 418nm immediately after mixing Fe(II) P450cin with oxygen. This shift indicates formation of the ferrous-oxy-complex similar to other P450s. However, the ferrous-oxy-complex rapidly auto-oxidizes back to Fe(III) within 10 seconds as evidenced by the Soret shift to 392nm. This instability coupled with many failed attempts at trapping the oxy-complex in crystals led us to pursue complexes with other diatomic ligands such as NO and cyanide.
Figure 1.
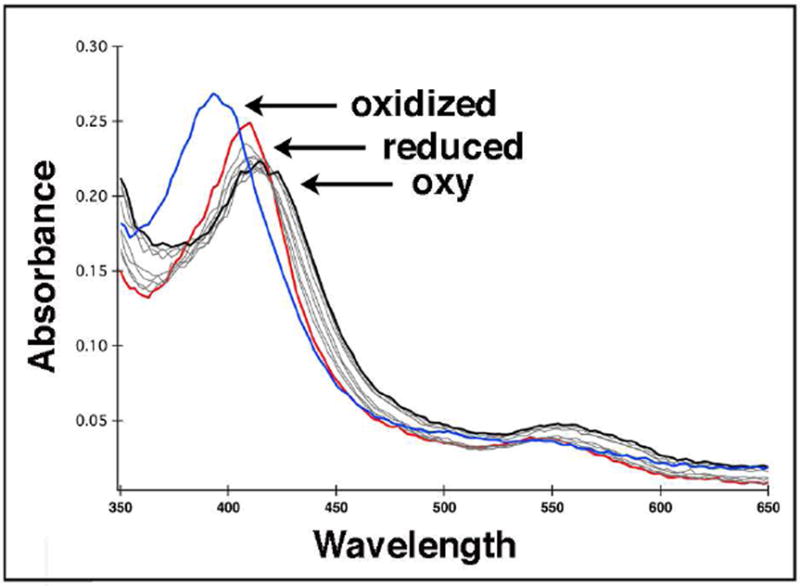
Stopped flow absorption spectra showing oxy-P450cin complex formation. One syringe contained deoxygenated 20 μM P450cin in 50 mM KPi, pH 7.4, 50 mM KCl with oxygen scrubbers. The other syringe contained only oxygen saturated buffer. Upon mixing, the Soret peak of reduced P450cin (red) immediately shifts from 411 nm to 418 nm (black). Autooxidation to Fe(III) occurs rapidly with a Soret peak shift to 392 nm (blue). The oxidized spectrum was recorded 10 seconds after mixing so the lifetime of the oxy-complex is extremely short.
P450cin-NO complex
Molecular oxygen contains 12 valence molecular orbital electrons giving the following molecular orbital configuration with a net bond order of 2:
When oxygen binds the ferrous heme iron the expected σ bond is donated from the lone pair of the ligand to the empty iron dz2 orbital. However, the strength and geometry around the Fe-X-O bond is in large part determined by overlap between the antibonding π* orbitals of the diatomic ligand and the iron dπ orbitals (Fig. S1). This phenomenon is known as ‘π back-bonding’, with electrons being donated from the metal to the ligand π* orbitals. The bending angle between the iron and ligand depends on how many electrons in the complex are filled in the ligand π* orbitals22. While the bending angle between ferrous iron and oxygen has been shown to be approximately 120 degrees with two electrons filling the two π* orbitals, in the CO complex the π* orbitals are empty and thus Fe-CO is in a linear arrangement. Nitric oxide binding represents an intermediate between these two with only one electron in its π* orbitals. Therefore, NO is the best alternative geometric mimic for the oxy-complex.
The NO P450cin complex crystallized in space group P21 with two molecules in the asymmetric unit. Scaling and refinement statistics for this complex as well as other structures presented here are described in Table 1. Both molecules exhibit well defined 2Fo-Fc density just above the heme iron as illustrated in Fig. 2A. The Fe-N-O bond angle and Fe-N bond length refine to 138° and 1.9Å, respectively. These values closely agree with experimental model heme derivative NO compounds22 as well as NO bound NOS structures.23 In P450cin NO is bound in a bent orientation leaning towards the I helix. An overlay of the ferric structure and ferrous-NO complex shows that there is very little change in active site structure with the exception of the substrate. In order to avoid a steric clash the substrate, 1,6-cineole, rotates away from NO using the ethereal oxygen as a pivot point. This movement avoids a potential clash with NO while maintaining a hydrogen bond with the catalytically important Asn242 (Fig. 2A).
Figure 2.
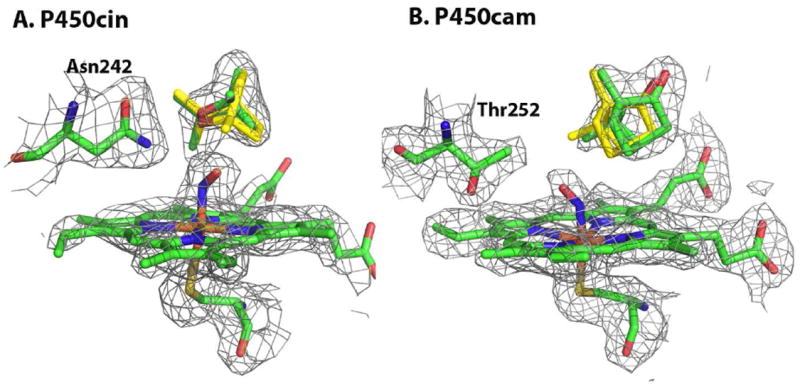
The P450cin-NO and P450cam-NO complexes are illustrated in green with the 2DFo-mFc map colored grey and contoured to 1σ. The substrate, cineole, from the ferric structures is overlaid and colored yellow.
Asn242 protrudes into the active site and is limited in its range of motion by hydrogen bonding with the ethereal substrate oxygen. These restraints cause the NO to bend in the opposite direction from the large and conformationally restrained Asn242 side chain. This is the opposite of what is seen in P450cam-oxy structure where O2 bends toward Thr252. Of course O2 binding in P450cam is also accompanied by significant changes in the I helix and a rearrangement of solvent structure.6, 7 We therefore solved the P450cam-NO structure as a control to validate our justification for extrapolating conclusions drawn for the P450cin-NO structure to a P450cin oxy-complex. Only one of the two molecules in the asymmetric unit exhibits good electron density for NO. Even here the electron density is not as sharp and as well defined as in P450cin which precludes a precise determination of iron-NO bond parameters. Even so NO clearly bends toward Thr252 (Fig. 2B). As is the case with the P450cin-NO structure, there is no I helix rearrangement observed in the P450cam-NO structure and the only significant change is a rotation of the substrate, camphor, away from the NO ligand (Fig. 2B). However, in P450cam the NO ligand bends toward Thr252 while it bends away from Asn242 in the P450cin-NO structure. There is little doubt that this results from the larger Asn242 side chain compared to Thr252 and Asn242 being restrained from much motion owing to the H-bonding interaction with the substrate. Thus the NO must rotate about 90° away from Asn242 relative to the position of NO in P450cam. Despite not exhibiting the oxy-P450cam I helix rearrangement, the NO ligand in P450cam-NO structure bends in the same direction as the dioxygen in the O2 complex. This shows that the P450cam-NO complex can provide an excellent geometric mimic of the oxy-complex even if it falls short of eliciting the O2-induced I helical changes. Therefore, it is not unreasonable to speculate that in the P450cin oxy-complex O2 also bends in the same direction as in the P450cin-NO complex. This would place the distal O2 oxygen atom in a quite distinct position from that found in P450cam suggesting differences in the O2 activation process.
Federov et al24 have shown that the P450cam-cyanide complex is a good mimic of the O2 complex resulting in the same changes in the I helix and solvent as does O2. A cyanide complex, therefore, has the potential to act as an excellent mimic of a P450cin oxy-complex. We therefore attempted to prepare the P450cin-cyanide complex. Unfortunately, adding excess cyanide resulted in an increase in pH that was not tolerated by our crystals. Lower concentrations (up to 30 mM) failed to show cyanide binding to the heme iron.
In the absence of an oxy-complex we cannot directly visualize P450cin structural changes induced by O2 binding. However, a handful of substrate-free P450 crystal structures have revealed large range of motions available to P450s13, 25-27 that are relevant to oxygen binding. All of these structures exhibit an ‘open’ conformation in which a large retraction of the F and G helices and B-C loop opens the active site to solvent. In P450cam the I helix moves closer to the heme iron enabling the Gly248 carbonyl oxygen to H-bond with the axial water ligand.13 In the P450cam-oxy-complex the Gly248 carbonyl is midway between its position in the substrate-bound and -free structures about 3.1 Å from the distal O atom in dioxygen.5, 7 If, as we expect, P450cin experiences a similar motion then it is important to determine the structure of the substrate-free open conformation of P450cin.
Substrate-free P450cin
The first hint of an open P450 conformation was the P450BM3 heme domain, substrate-free and –bound crystal structures.28 It was postulated that these structures represent snapshots of the equilibrium between open (substrate-free) and closed (substrate-bound) structures. Since then, this view has been confirmed in P450cam by crystallizing a range of substrate analogues with varying linkers that effectively map the transition between open and closed states.13, 29 Recent kinetics with P450eryK illustrate that a mixture of open and closed conformations are likely present in solution.26 We will see that the P450cin open structure displays a greater degree of movement than was initially seen for the open structure of P450BM3 heme domain crystal structure and more recently for P450cam.13, 28
The difference between the open and closed P450cin structure is illustrated in Figs. 3 and 4 and is dominated by changes in the F and G helices and B-C loops. In Fig. 4 the RMSD between Cα carbons for the open and closed structures have been plotted using Lsqkab as implemented in CCP4 for both P450cin and P450cam (PDB ID: 2CPP). For both enzymes the greatest deviations between substrate-bound and substrate-free structures occur in the B-C loop, F and G helices and the I helix. As is the case with P450cam, retraction of the B-C loop and F and G helices result in an opening up of the enzyme to solvent. However, P450cin shows a greater deviation in the hinge region of the H helix and H-I loop. Amino acids 82-85 (analogous to the B’ helix in other P450s) become completely disordered as the B-C loop pulls out of the active site in the substrate-free structure. Tyr81 is buried deep in the active site in the substrate bound structure where it participates in a water bridged H-bonding network that includes Asp241, Asn242, and the substrate (Fig. 5). This hydrogen bonding network may help to shield the active site from solvent. Although some of the atoms in the Tyr81 side chain are disordered, there is strong 2mFo-DFc density for the Cα atom with a B-factor of 31.4 Å2. In the substrate free structure the Tyr81 Cα moves 7.4 Å toward the surface. This widening of the active site opens up a well-defined solvent channel in the substrate-free structure (Fig. 3).
Figure 3.
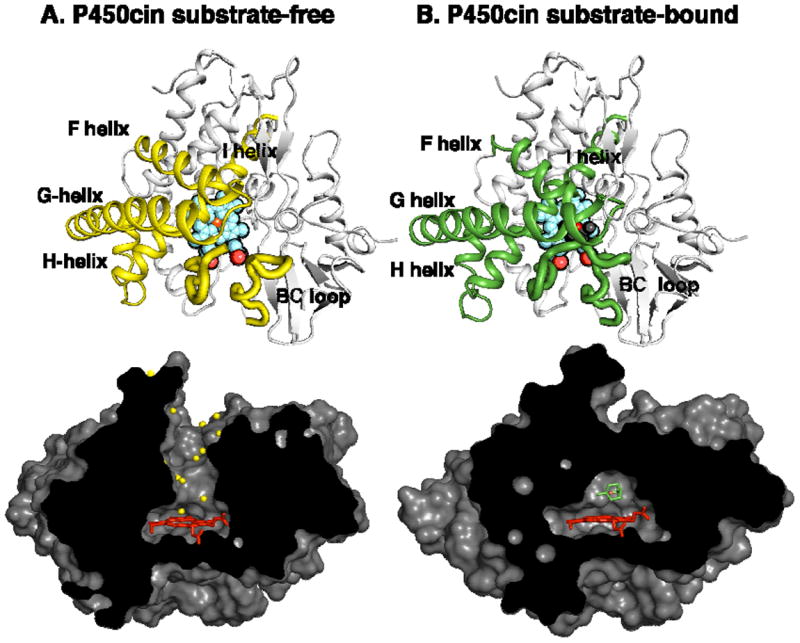
(A) The P450cin substrate-free and (B) -bound structures. The cartoon representations are viewed directly above the heme plane with regions involved in movement colored yellow for substrate-free and green for substrate-bound. The surface representations are viewed along the heme edge. In the substrate-free structure, the substrate access channel opens up and is lined with solvent (yellow spheres).
Figure 4.
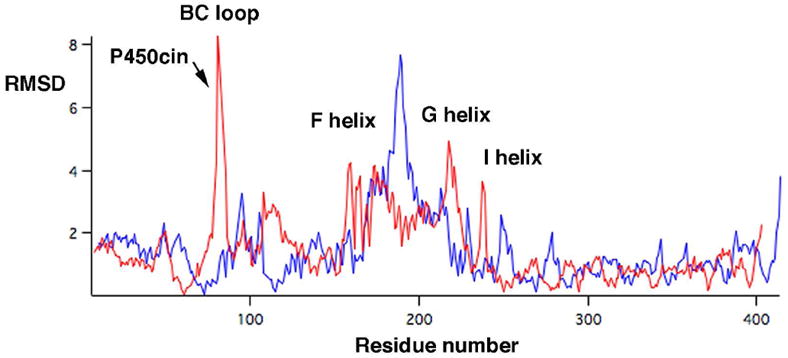
The Cα RMSD’s between substrate-bound and -free structures were calculated with Lsqkab and plotted vs. residue number for both P450cin (red) and P450cam (blue).
Figure 5.
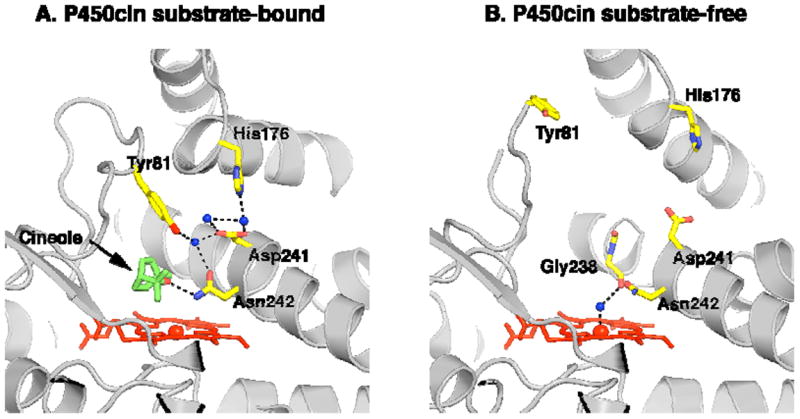
(A) The substrate bound ferric P450cin structure is depicted with active site waters colored as blue spheres and key residues illustrated in yellow. Black dashes represent hydrogen bonding and coordination interactions. (B) The open substrate-free structure with all solvent removed except for the water coordinated to the heme iron.
For the purposes of our current discussion it is instructive to take a closer look at the changes occurring in the I helix. In both P450cin and P450cam structures the I helix displays similar changes in going from substrate-free to the substrate-bound state with a modest RMSD of about 0.7Å. However, both structures also exhibit much larger differences near the active site. For P450cin the Cα carbon movements for Gly238 and Gly239 are 3.6 and 3.3Å, respectively, compared to 2.6Å and 2.1Å for Gly248 and Gly249 in P450cam. In substrate-bound P450cin Gly238 and Gly239 participate in fairly long π-helical H-bonds with distances of 3.1Å for Gly238 to Thr243 and 3.3Å for Gly239 to Ala244. This unique hydrogen bonding pattern in P450cin is enabled by a flipping out of the Ile240 carbonyl so that it does not participate in normal α-helical hydrogen bonding thus allowing Gly238 and Gly239 to form a π-helix (Fig. 6A). Conversely, in the substrate-free structure Ile240 and Gly239 participate in normal α-helical hydrogen bonds while the carbonyl O atom of Gly238 reorients to H-bond with the water coordinated to the heme iron (Fig. 6B). In effect, the transition to an α-helix in the substrate-free structure causes a screw-like rotation bringing Gly238 closer to the heme. An overlay of the P450cin NO-complex with the I helix from the substrate-free structure illustrates the kind of conformational change that is likely to occur in the oxy-complex (Fig. 7). In the open structure the Gly238 carbonyl hydrogen bonds with the water/hydroxide ligand but could conceivably interact with the distal atom of the bound dioxygen much the same way as Gly248 in P450cam. The analogy between the P450cam oxy-complex and the P450cin oxy-complex is further illustrated in Fig. 8 where we can see similar movements for Gly238 (P450cin-open) and Gly248 (P450cam-oxy). In oxy-P450cam the Gly248 carbonyl is close enough to H-bond to both an active site water and a hydroperoxy-complex (Fig. 8A). In Fig. 8B, a similar movement for Gly238 in P450cin substrate-free structure can be seen overlaid on the P450cin-NO complex.
Figure 6.
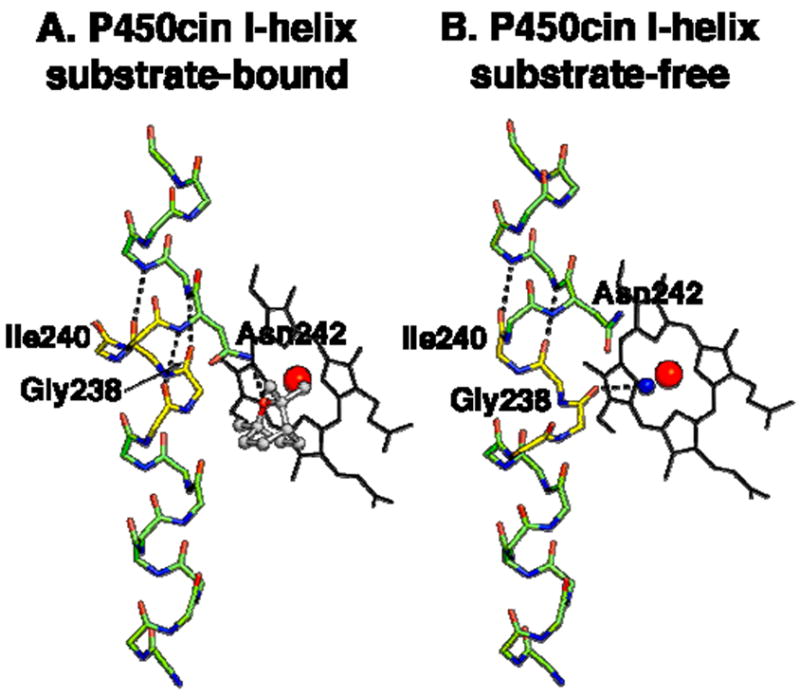
Substrate-bound (A) and –free (B) I helix main-chain atoms are shown as green and yellow sticks. Residues of interest are colored yellow and hydrogen bonds are depicted by black dashes. The heme is depicted as black sticks with the iron colored red and the water nearest the iron colored blue.
Figure 7.
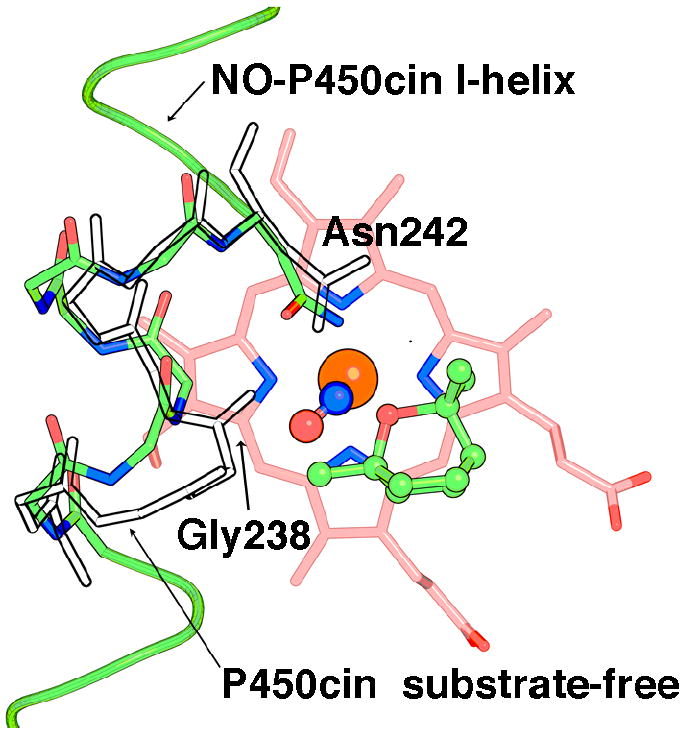
The I helix of NO-P450cin (green) has been superimposed with a portion of the substrate-free I helix (transparent). The heme is colored salmon while NO is depicted as red and blue spheres just above the iron (orange).
Figure 8.
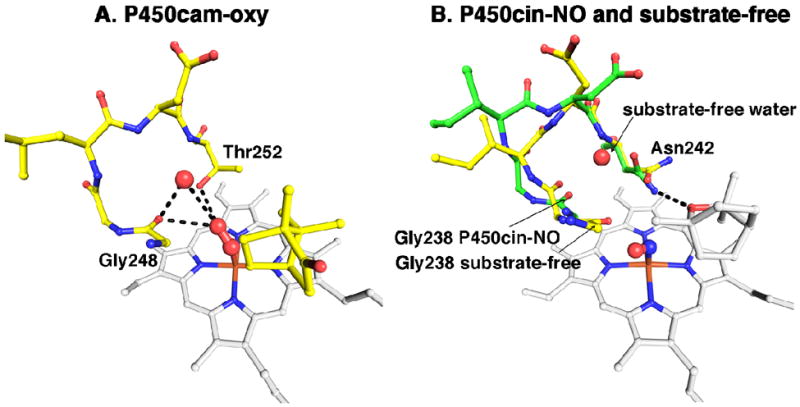
(A) The P450cam oxy complex active site is colored yellow and the heme colored white. The oxygens and water are represented by red spheres. Hydrogen bonds are drawn as black dashes. (B) NO-P450cin (green) is overlaid with substrate-free P450cin (yellow). Both the heme and substrate are colored white. The NO complex is represented as red and blue spheres just above the heme iron. The water coordinating the heme iron in the substrate-free structure is not shown.
Π-helices are rare in proteins since they are not as stable as α-helices and often have functional significance. There are other examples where the ability to transition between a π- and α-helix is an effective way to accommodate substrate binding. In soluble methane monooxygenase, this characteristic allows a Thr that would be involved in a steric clash upon substrate binding to rotate 50° away from the potential clash.30 Likewise, in cytochrome c oxidase, a two-residue π-helix allows a carbonyl group to rotate perpendicular to the helix axis and participate in hydrogen bonding between chains.31 Thus, the ability to transition between an α-helix and even a short π-helix can bestow dynamic flexibility near an active site. It is tempting to speculate that the role of the π-helix in P450cin is for proper positioning of Asn242 for interaction with its substrate. However, this is unlikely since the Cα position of Asn242 does not substantially deviate between the substrate-free and -bound conformations. It is more likely that the α/π helical switch is required for setting up proton delivery to dioxygen.
Although π-helices are less stable, the α-helical backbone hydrogen bonds are weak, all with distances of greater than 3Å near the active site, which should substantially lower the energy barrier between the α/π transition. A P450cin I helix with idealized α-helical geometry near the active site would greatly restrict the ability to undergo conformational changes near the active site. P450cam does not have π-helices but solves this problem in another way. In the ferric substrate-bound state the α-helix is completely disrupted near the active site with the Gly248 carbonyl forming a hydrogen bond with the Thr252 side chain. Upon oxygen binding Thr252 and Gly248 move apart providing room for the catalytic waters involved in proton transfer.
Relevance to O2 Activation
A peculiar feature of P450cam is that cyanide, not CO or NO, is able to induce the same changes in the I helix and local solvent structure as O2. This has been attributed to both the distal O atom of dioxygen and N atom of CN- carrying a negative charge24 so the electrostatic properties of O2 and CN- more closely match each other than they do with NO or CO. The opening of the I helix when O2 or CN- binds enables a water to move into position to H-bond with the ligand thus establishing the H-bonding network required for proper protonation of the distal O atom in dioxygen. As noted, this change is half way between the substrate-bound and -free conformations. The H-bond donor-acceptor relationship between this water and neighboring protein groups indicates that the water would serve as an H-bond donor to the N atom of CN- and the distal O atom of O2. Since O2 is thought to bind as superoxide, Fe(III)-OO-, this water would be in a position to help “solvate” the negative charge on the O2 and CN-. Thus the energetic incentive for the I helix conformational change is to establish H-bonds with the O2 or CN- ligand. Because the oxy-P450cam I helix moves toward the substrate-free conformation we can anticipate that a similar change occurs in P450cin. This would open up the I helix allowing water to enter the active site and H-bond with O2. The unique feature of P450cin compared to P450cam is the switching between a π-helix near the O2 binding site in the substrate-bound structure and an α-helix in the substrate-free structure. This difference is due in part to the Thr252/Asn242 difference in P450cam and P450cin, respectively. It thus appears that the I helix region of P450s is quite adaptable and solves the problem of O2 activation in subtly different ways depending on the requirements of substrate binding. The most important common feature in P450cam and P450cin is that the Gly248/Gly238 peptide carbonyl O atom moves into a more substrate-free-like position in order to accept an H-bond from the ligand although the range of motion available to P450cin is larger than that to P450cam. This should help to promote protonation of the distal O atom of O2 thereby stabilizing the hydroperoxy intermediate. This stabilization would further promote the second protonation step followed by heterolytic cleavage of the O-O bond to give the active ferryl intermediate.
Finally, the interaction between Gly238 and O2 may be especially important in P450cin. As noted earlier the orientation of the NO ligand and, by analogy the O2 ligand, is quite different in P450cam and P450cin owing to the larger Asn242 side chain in P450cin compared to Thr252 in P450cam. In P450cin the O atom of NO is oriented closer to the Gly238 peptide carbonyl than to Gly248 in P450cam suggesting that Gly238 may form a closer and thus stronger H-bonding interaction with the hydroxperoxy intermediate in P450cin. The observation that the range of motion available to Gly238 in P450cin is larger than that to Gly248 in P450cam also suggests a possibly tighter interaction between Gly238 and the iron-linked dioxygen. The greater flexibility of the I helix in P450cin thus may be a requirement to enable optimal interactions between Gly238 and the hydroperoxy intermediate since P450cin has no nearby side chain (i.e. Thr252 as in P450cam) to serve this function.
Supplementary Material
Acknowledgments
We would like to thank Dr. James J. De Voss for the pCW-P450cin construct and Dr. David B. Goodin for his generous advice on making publication quality figures. This paper involves research carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
This work was supported by NIH grant GM33688
Abbreviations
- CYP
cytochrome P450
- CO
carbon monoxide
- CN-
cyanide
- DEAE
diethylaminoethyl cellulose
- DTT
dithiothreitol
- HEPES
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOS
nitric oxide synthase
- PEG
poly(ethylene glycol)
- PMSF
phenylmethanesufonyl fluoride
- RMSD
root-mean-square deviation
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Tris
tris(hydroxymethyl)aminomethane
References
- 1.Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J Amer Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 2.Dolphin D, Forman A, Borg DC, Fajer J, Felton RH. Compounds I of catalase and horse radish peroxidase: pi-cation radicals. Proc Natl Acad Sci U S A. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 4.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 5.Nagano S, Cupp-Vickery JR, Poulos TL. Crystal structures of the ferrous dioxygen complex of wild-type cytochrome P450eryF and its mutants, A245S and A245T - Investigation of the proton transfer system in P450eryF. J Biol Chem. 2005;280:22102–22107. doi: 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]
- 6.Nagano S, Poulos TL. Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 7.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 8.Cupp-Vickery JR, Han O, Hutchinson CR, Poulos TL. Substrate-assisted catalysis in cytochrome P450eryF. Nat Struct Biol. 1996;3:632–637. doi: 10.1038/nsb0796-632. [DOI] [PubMed] [Google Scholar]
- 9.Hawkes DB, Adams GW, Burlingame AL, Ortiz de Montellano PR, De Voss JJ. Cytochrome P450cin (CYP176A), Isolation, expression, and characterization. J Biol Chem. 2002;277:27725–27732. doi: 10.1074/jbc.M203382200. [DOI] [PubMed] [Google Scholar]
- 10.Meharenna YT, Slessor KE, Cavaignac SM, Poulos TL, De Voss JJ. The critical role of substrate-protein hydrogen bonding in the control of regioselective hydroxylation in P450cin. J Biol Chem. 2008;283:10804–10812. doi: 10.1074/jbc.M709722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slessor KE, Farlow AJ, Cavaignac SM, Stok JE, De Voss JJ. Oxygen activation by P450(cin): Protein and substrate mutagenesis. Arch Biochem Biophys. 2011;507:154–162. doi: 10.1016/j.abb.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Meharenna YT, Li H, David BH, Hawkes DB, Pearson AG, De Voss J, Poulos TL. Crystal Structure of P450cin in a complex with its substrate, 1,8-cineole, a close structural homolog to D-camphor, the substrate for P450cam. Biochemistry. 2004;43:9487–9494. doi: 10.1021/bi049293p. [DOI] [PubMed] [Google Scholar]
- 13.Lee YT, Wilson RF, Rupniewski I, Goodin DB. P450cam visits an open conformation in the absence of substrate. Biochemistry. 2010;49:3412–3419. doi: 10.1021/bi100183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshioka S, Takahashi S, Ishimori K, Morishima I. Roles of the axial push effect in cytochrome P450cam studied with the site-directed mutagenesis at the heme proximal site. J Inorg Biochem. 2000;81:141–151. doi: 10.1016/s0162-0134(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 15.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Lehnert N, Scheidt WR. Preface for the inorganic chemistry forum: the coordination chemistry of nitric oxide and its significance for metabolism, signaling, and toxicity in biology. Inorg Chem. 2010;49:6223–6225. doi: 10.1021/ic101080g. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Igarashi J, Jamal J, Yang W, Poulos TL. Structural studies of constitutive nitric oxide synthases with diatomic ligands bound. J Biol Inorg Chem. 2006;11:753–768. doi: 10.1007/s00775-006-0123-8. [DOI] [PubMed] [Google Scholar]
- 24.Fedorov R, Ghosh DK, Schlichting I. Crystal structures of cyanide complexes of P450cam and the oxygenase domain of inducible nitric oxide synthase-structural models of the short-lived oxygen complexes. Arch Biochem Biophys. 2003;409:25–31. doi: 10.1016/s0003-9861(02)00555-6. [DOI] [PubMed] [Google Scholar]
- 25.Lee YT, Glazer EC, Wilson RF, Stout CD, Goodin DB. Three clusters of conformational states in p450cam reveal a multistep pathway for closing of the substrate access channel. Biochemistry. 2011;50:693–703. doi: 10.1021/bi101726d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savino C, Montemiglio LC, Sciara G, Miele AE, Kendrew SG, Jemth P, Gianni S, Vallone B. Investigating the structural plasticity of a cytochrome P450: three-dimensional structures of P450 EryK and binding to its physiological substrate. J Biol Chem. 2009;284:29170–29179. doi: 10.1074/jbc.M109.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilderman PR, Shah MB, Liu T, Li S, Hsu S, Roberts AG, Goodlett DR, Zhang Q, Woods VL, Jr, Stout CD, Halpert JR. Plasticity of cytochrome P450 2B4 as investigated by hydrogen-deuterium exchange mass spectrometry and X-ray crystallography. J Biol Chem. 2010;285:38602–38611. doi: 10.1074/jbc.M110.180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Poulos TL. The structure of the cytochrome p450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat Struct Biol. 1997;4:140–146. doi: 10.1038/nsb0297-140. [DOI] [PubMed] [Google Scholar]
- 29.Dunn AR, Dmochowski IJ, Bilwes AM, Gray HB, Crane BR. Probing the open state of cytochrome P450cam with ruthenium-linker substrates. Proc Natl Acad Sci U S A. 2001;98:12420–12425. doi: 10.1073/pnas.221297998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sazinky MH, Lippard SJ. Product bound structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): protein motion in the α-subunit. J Am Chem Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]
- 31.Riek RP, Graham RM. The elusive pi-helix. J Struct Biol. 2010;173:153–160. doi: 10.1016/j.jsb.2010.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.