

Abstract
The ‘histone code’ is a well-established hypothesis describing the idea that specific patterns of post-translational modifications to histones act like a molecular ‘code’ recognized and used by non-histone proteins to regulate specific chromatin functions. One modification, which has received significant attention, is that of histone acetylation. The enzymes that regulate this modification are described as lysine acetyltransferases or KATs, and histone deacetylases or HDACs. Due to their conserved catalytic domain HDACs have been actively targeted as a therapeutic target. The pro-inflammatory environment is increasingly being recognized as a critical element for both degenerative diseases and cancer. The present review will discuss the current knowledge surrounding the clinical potential and current development of histone deacetylases for the treatment of diseases for which a pro-inflammatory environment plays important roles, and the molecular mechanisms by which such inhibitors may play important functions in modulating the pro-inflammatory environment.
Keywords: chromatin, histone deacetylase, cancer, disease, targeting
Introduction
The ‘histone code’ is a well-established hypothesis describing the idea that specific patterns of post-translational modifications to histones act like a molecular ‘code’ recognized and used by non-histone proteins to regulate specific chromatin functions [1–3]. One modification that has received significant attention is that of histone acetylation. The enzymes that regulate this modification are described as lysine acetyltransferases or KATs [4, 5], and histone deacetylases or HDACs (Table 1) [6]. Although originally the activities of these enzymes were thought to only occur on histones, it is now well established that these enzymes can also alter non-histone proteins [7, 8]. Due to their conserved catalytic domain HDACs have been actively targeted as a therapeutic target [9, 10].
1.
Current classes of HDACs
In the following sections we will describe the evidence emerging from in vitro (cell culture) and in vivo (animal models) studies which indicate the usefulness of agents targeting HDACs in the treatment of disease using models of cancer, diabetes and neu-rodegenerative disease, and finish by describing the current emerging data from human clinical trials supporting the use of these drugs in the treatment of these diseases.
HATs/HDACs and the pro-inflammatory environment
Inflammation is a critical component that is increasingly being associated with cancer [11, 12], diabetes [13] and neurodegener-ative disease [14].
Histone modifying enzymes such as histone deacetylases have been identified as critical regulators of pro-inflammatory cascades. One of the best-established mechanisms identified concerns the roles of these enzymes in the regulation of nuclear factor κB (NF-κB) activation, as summarized in Fig. 1. The NF-κB-Rel family consists of five subunits, but NF-κB typically consists of a heterodimeric protein comprising a p50 and a p65 (RelA) sub-unit. Early studies identified the lysine acetyltransferases KAT3B and KAT3A as key coactivators in regulating NF-κB driven gene expression [15–17]. These interactions were found to involve the RelA/p65 subunit. Another lysine acetyltransferase KAT13A was found to also potentiate NF-κB transactivation through interactions with the other subunit p50 [18]. Following the identification of interactions between NF-κB and lysine acetyltransferases it was subsequently shown that the RelA/p65 subunit could associate with HDAC1 and HDAC2 to repress expression of NF-κB regulated genes as well as to control the induced level of expression of these genes [19].
1.
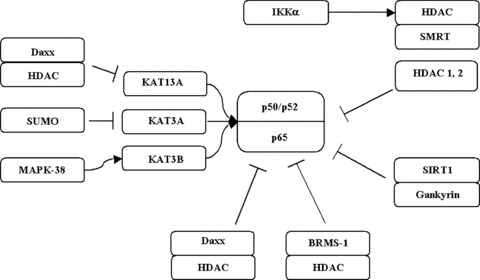
Interplay of KATs and HDACs in the regulation of NF-κB.
It has since been shown that the histone deacetylase Sirtuin 1 (SIRT1) regulates NF-κB transactivation by physically interacting with the RelA/p65 subunit of NF-κB and inhibiting transcription by deacetylating a critical lysine at position 310 [20]. Both in vitro and in vivo exposure to cigarette smoke causes dose- and time-dependent decrease in SIRT1 protein and deacetylase activity resulting in increased NF-κB dependent pro-inflammatory mediator release [21].
One of the critical regulators of NF-κB activation is IκB kinase-α (IKK-α), where NF-κB transcription requires IKK-α to phospho-rylate silencing mediator for retinoic acid and thyroid hormone receptor, which stimulates the exchange of corepressor for coac-tivator complexes. In the initial stage of NF-κB activation, following this phosphorylation event HDAC3 is displaced, and this allows KAT3B to acetylate RelA/p65 [22, 23].
Daxx is another protein that has been shown to regulate NF-κB activation by binding to a region that includes the major sites of acetylation mediated by KAT3B/KAT3A [24]. However, it must be noted that Daxx has also been shown to directly associate with HDAC2 [25], and so may represent a mechanism by which KATs and HDACs compete for critical lysines on NF-κB subunits. In this regard, small ubiquitin-like modifier modification of KAT3A negatively modulates its transcriptional activity by recruiting a Daxx complex that contains HDAC2 [26].
Pro-inflammatory genes associated with NF-κB are inter-leukins (IL)-6 and IL-8 [27–29]. NF-κB has also been shown to utilize the lysine acetyltransferase activity of KAT3A/KAT3B to stimulate the transcription of these genes [30]. Recently, breast cancer metastasis suppressor 1 (BRMS1) has been shown to decrease the transactivation potential of RelA/p65 by promoting binding of HDAC1 to RelA/p65, where it deacetylates lysine K310 on RelA/p65, suppressing transcriptional activity [31].
For a full comprehensive review of the role of HATs and HDACs in the regulation of NF-κB the reader is directed to the recent review by Carine Van Lint and colleagues [32].
HATs/HDACs and endoplasmic reticulum (ER) stress
The ability of a cell to sense, respond to and circumvent stress is essential for maintaining homeostasis. There are many ways in which stress, either endogenous or exogenous, can be manifested in a cell; these include pathogenic infection, chemical insult, genetic mutation, nutrient deprivation and even normal differentiation. The process of mutant protein folding is particularly sensitive to such insults. As such for the cellular compartments in which mutant proteins are processed and folded, there are adaptive programs that enable both their detection and correction for more efficient processing [33].
The ER is a large cellular organelle comprising a network of interconnected, closed membrane-bound vesicles. It is the site of synthesis, folding and modification of secretory and cell-surface proteins and serves many essential functions, including the production of the components of cellular membranes, proteins, lipids and sterols [34]. Only correctly folded proteins are transported out of the ER while incompletely folded proteins are retained in the organelle to complete the folding process or to be targeted for destruction [35]. Due to the important roles of this organelle, its proper functioning is essential to cellular homeostasis. However, various conditions can interfere with the ER function leading to ER stress. Stress is the response of any system to perturbations of its normal state. Thus, ER stress can arise from a disturbance in protein folding which results in an accumulation of unfolded or misfolded proteins within the organelle [36]. During such disturbances, in order to carry out the correct folding of proteins, the ER has evolved as a specialized protein-folding machine with cellular mechanisms that promote proper folding of aberrant protein, thus preventing its aggregation. Therefore, when ER homeostasis is altered by misfolded proteins, the ER responds by inducing the expression of specific genes in an attempt to restore normal ER function to and maintain stability [37]. The principle mechanisms of conformational disorders contained within the four pillars of ER stress: (i) protein degradation, (ii) endoplasmic overload response (EOR), (iii) unfolded protein response (UPR) and (iv) cellular death pathway. This four-stage model of ER stress toxicity helps explain its role in the onset of clinical manifestations. Two ER stress-induced signal transduction pathways have been described: the UPR [38] and the EOR [39]. The function of these pathways is to adapt to the disturbance and attempt to reestablish normal ER function [40]. However, excessive or prolonged ER stress may overwhelm the cell and elicit the cell death programme or apoptosis [41].
ER stress has been implicated as a critical component in diabetes [42], neurodegeneration [43, 44] and cancer [45].
The evidence linking HATs/HDACs to ER Stress is not as well established as that for inflammation, with most studies utilising histone deacetylase inhibitors (HDi). However, in a recent study in hepatocytes on Mallory body (cytokeratin aggresomes) formation, decreased lysine acetyltransferase and increased histone deacetylase activity was observed [46]. In a similar model of oxidative stress induced inclusion formation, treatment of cells with 4-phenylbutyrate was found to alleviate formation of these inclusions [47].
Direct physical evidence for the association of HATs and HDACs with critical regulatory elements within the ER stress pathway is emerging. CHOP (C/EBP homologous protein) an ER stress-inducible protein which plays a critical role in regulating programmed cell death in stressed cells has recently been shown to directly associate with the lysine acetyltransferase KAT3B, and inhibition of HDACs prevents the degradation of CHOP [48].
Using chromatin immunoprecipitation strategies, KAT3B has been shown to bind to the promoter for the GRP78/BiP a prosurvival ER chaperone gene under conditions of ER stress [49]. In similar studies examining the promoters of ER-stress responsive genes, histone H4 acetylation was observed to show a promoter-specific increase following induction of stress [50].
B lymphocyte-induced maturation protein-1 (BLIMP-1) has been shown to be associated with cellular stress and is rapidly up-regulated during the UPR in some cellular models [51]. Of interest though is that fact that this repressor protein has also been shown to directly associate with histone deacetylases to repress transcription [52], indicating that histone deacetylases may utilize BLIMP-1 to down-regulate important genes during ER stress.
Initial in vitro data emerged demonstrating the efficacy of the HDi 4-phenylbutyrate in relieving ER stress in cell line models of cystic fibrosis [53], and also the liver disease mutant α1-anti-trypsin Z (α1-ATZ) [54]. Since these initial observations several studies have shown that 4-phenylbutyrate may act as a chemical chaperone to relieve ER stress induced in models of ischaemia [55, 56]. Similar data have emerged for models of cataract formation [57], Parkinson's disease [58], retinitis pigmentosa [59], glaucoma [60] and confirmation of its effects in cystic fibrosis [61]. 4-Phenybutyrate has also been shown to relieve the ER stress observed in diabetes, where it has been shown to reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes, by the restoration of systemic insulin sensitivity, resolution of fatty liver disease, and enhancement of insulin action in liver, muscle and adipose tissues [62]. In hepatocytes, ER stress in liver induced ischaemia is ameliorated by treatments with 4-phenylbutyrate [56]. This compound has also been shown to have profound effects in alleviating ER tress induced by oxidative stress in cultured hepatocytes and hepatoma cells [47], and preventing ER stress mediated aggregate formation in a model of hereditary haemochromatosis [63].
Another HDi, valproate/valproic acid (VPA), has also been shown to have potential in the treatment of ER stress. The first indications that this drug may be of use in the treatment of ER stress came from studies which showed that treatment of cells with valproate caused the up-regulation of the expression of GRP78/BiP, a key ER-mediated chaperone [64]. Follow-up studies subsequently confirmed that valproate could increase the expression of additional important ER stress proteins, GRP94 and cal-reticulin [65–67]. In the same model system it was subsequently shown that valproate protected against oxidative stress induced protein damage and had neuroprotective capability [68–70].
The potential role of histone deacetylase inhibitors in the treatment of cancer
Non-small cell cancer (NSCLC)
Aberrant epigenetic modifications are a frequent hallmark of cancer and include both DNA CpG methylation [71], and histone post-translational modifications [72]. In NSCLC, altered patterns of DNA CpG methylation [73] and histone modifications [74] are found. In one study, no direct association between polymorphisms in histone deacetylase genes and risk of developing lung cancer was found [75]. Strong evidence is emerging, however, linking HDACs and their activities to NSCLC pathogenesis and prognosis.
Chronic obstructive pulmonary disease (COPD) is a systemic inflammatory condition of the lung frequently associated with a higher risk of developing lung cancer [76]. A link between histone deacetylase activities and COPD was identified when studies revealed that in lung tissue of patients with increasing clinical stages of COPD, graded reductions in HDAC activity, reduced levels of HDAC2 protein and lower mRNA levels for HDACs 2, 5 and 8 were observed [77]. The decreased levels of HDAC2 have also been observed in separate studies of the effects of cigarette smoking in patients with COPD [78, 79]. Of note, in contrast, elevated levels of HDAC1 have been observed in higher stage (stage III or IV) NSCLC [80]. A more recent study has also observed high expression of all class I HDACs in lung [81]. Using antibody arrays, HDAC3 protein was found to be elevated HDAC3 in 92% of squamous cell carcinomas of the lung [82]. It has, however, also been observed that decreased levels of class II HDACs have poor prognosis in NSCLC [83]. Another histone deacetylase SIRT1 has also recently been shown to be down-regulated in patients with COPD [84]. Most recently aberrant histone post-translational modifications have been shown to have prognostic value in NSCLC [74].
Decreased expression of mSin3A, a member of a multiple component corepressor complex that contains histone deacetylases, has also been observed in NSCLC [85]. Metastasis-associated protein 1 (MTA-1) has recently been shown to be significantly elevated in NSCLC and is strongly associated with both invasiveness and metastasis [86]. As MTA-1 is known to associate with histone deacetylases [87–92], this would suggest that HDAC activity associated with MTA-1 is involved with NSCLC invasiveness and metastasis.
Mutations within the lysine acetyltransferase KAT3A have also been identified in a small subset of NSCLC [93]. The importance of lysine acetyltransferase activity in lung development has been shown in mouse studies, where HATs were shown to be highly expressed in the developing lung [94–96], and confirmation of their importance in lung development came from KAT3B knockout studies [97]. Evidence is also emerging that histone deacetylases play important roles in the developing lung. Recently a homeodomain protein (HOP), which in the cardiac system associates with HDAC2 to repress cardiac-specific genes, was observed to be present in airway epithelium along with HDAC2. HOP was shown to represses lung-specific gene expression in an HDAC-dependent manner, and loss of HOP expression in vivo resulted in defective type 2 pneumocyte development [98].
The data emerging clearly show that HDACs and their activities play important roles in lung, and may also be important in the development of lung disease. As such they may also prove to be important therapeutic targets.
In vitro evidence for targeting HDACs in NSCLC
The potential for therapeutic targeting of HDACs in NSCLC has been extensively studied in vitro using HDi. Some of the earliest reports examining the effects of sodium butyrate on lung cell lines demonstrated both cell growth effects and altered DNA hyperme-thylation [99, 100]. In a Lewis lung carcinoma cells study, sodium butyrate was found to enhance the lung-colonizing ability of these cells [101]. Despite having adverse effects on growth and lung colonization, a subsequent study found that sodium butyrate potentiated DNA radiation sensitivity.
In contrast to the data obtained in 1986, a later study on transformed human lung fibroblasts found a dose-dependent induction of apoptosis and reduction in cell numbers after exposure to sodium butyrate [102]. Sodium butyrate was also shown to greatly enhance the antiproliferative effect in vitro and in vivo of interferon-α (IFN-α) on several human lung adenocarcinoma cell lines [103].
With the development of more sophisticated HDi, multiple studies have set out to examine the potential of HDi either alone or as combinatorial agents for the treatment of NCSLC. One of the first such studies, examined the effect of trapoxin on lung cancer cells, and found significant cell cycle arrest and apoptosis following treatment [104]. Further evidence for the potential of HDi in the treatment of lung cancer came from studies using suberoylanilide hydroxamic acid, SAHA (vorinostat), which was found to have chemopreventative efficacy against a tobacco-specific carcinogen model of lung tumourigenesis [105], and has also been shown to have growth inhibitory activity against NSCLC cell lines [106].
Trichostatin A (TSA) is another HDi that has been extensively studied as a potential therapeutic agent in the treatment on NSCLC. When the response of this drug against four NSCLC cell lines was compared against a normal lung fibroblast tenfold greater growth inhibition was found in the cancer cells, along with significant up-regulation of p21 [107]. TSA was shown to increase the expression of RECK and attenuate matrix metalloproteinase expression in lung cancer cells indicating HDi may help to prevent lung cancer cell invasion [108]. Death-associated protein kinase (DAPK) is a pro-apoptotic serine/threonine kinase involved in apoptosis, which is frequently down-regulated in NSCLC, and TSA has been shown to restore expression of DAPK in several NSCLC cell lines [109]. Loss of E-cadherin expression in NSCLC is also associated with de-differentiation, invasion and metastasis. WNT7a has been shown to regulate E-cadherin expression via a β-catenin specific mechanism or via a positive feedback loop, and loss of expression of WNT7a is a frequent event in NSCLC. In NSCLC cells E-cadherin and WNT7a expression was restored following treatment of cells with TSA [110]. RhoB is a small GTPase that is frequently down-regulated in NSCLC, and trapoxin was shown to significantly up-regulate the expression of RhoB in NSCLC cells [111], a finding subsequently confirmed using TSA [112]. Apoptosis induced by TSA in NSCLC cells has been shown to be associated with inhibition of cyclooxygenase 2 [113]. At the same time HDAC inhibitors such as sodium butyrate, scriptaid, apicidin and oxamflatin have also been shown to up-regulate the expression of 15-hydroxyprostaglandin dehy-drogenase (15-PGDH), a potential cyclooxygenase-2 (COX-2) antagonist in a time and concentration dependent manner in NSCLC cells [114].
A study testing the HDi (SAHA, TSA and sodium butyrate) demonstrated that the apoptotic response seen in NSCLC cells in response to HDi occurs via induction of caspase-3 activity [115, 116]. Critically, activation of the NF-κB pathway via tumour necrosis factor-α (TNF-a) has been shown to be abrogated in NSCLC cell lines by TSA by down-regulating the mRNA and protein of its cognate receptor [117].
Another HDi romidepsin (FK228, FR901228, depsipeptide) was shown to affect NSCLC cell line tumour growth [118], and affect matrix metalloproteinase (MMP) expression in a manner similar to that observed for TSA. When administered to NSCLC cell lines romidepsin significantly decreased the expression of MMP-2 and MMP-9 in cells [119]. Treatment of NSCLC cells with romidepsin has also been shown to result in increased p21 and phosphorylated retinoblastoma protein (pRb) [120]. NVP-LAQ824 is another histone deacetylase that has been shown to inhibit cellular proliferation in lung cancer cells [121]. The oral histone deacetylase compound CI-994 (N-acetyldinaline) has also been studied in NSCLC cell lines, and found to have cytostatic properties [122]. Another synthetic HDi (SK-7041) has now been shown to have cell growth inhibitory properties greater than SAHA (vorinostat) in human lung cancer cell lines with lesser effects on normal human bronchial epithelial cells [123].
Combinatorial therapies involving HDi in NSCLC
Synergy between inhibitors of DNA methyltransferases and histone deacetylases in the re-expression of genes silenced in colorectal cancer cell lines was established in 1989 [124]. Similar studies demonstrated the same synergies between Hdi and DNA methyl-transferase inhibitors in NSCLC cell lines [125–127], and also have been found to suppress tumorigenicity in NSCLC xenograft models but only when the tumours were small, and did not affect large well-established tumours. This may have important consequences for the use of HDi within the clinical setting [128]. Combined treatment using the HDi 4-phenylbutyrate and the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine, was shown to prevent tobacco carcinogen-induced lung cancer in wild-type mice [129].
A retinoid compound with the properties of retinoic acid (RA) and sodium butyrate (HDi) called (4-BPRE) was found to have greater cytotoxicity in A549 lung adenocarcinoma cells that RA alone indicating that this might be a good dual therapy for the treatment of NSCLC [130]. A further combinatorial treatment, which appears to have efficacy in NSCLC lung cancer cell lines, is a combination of a proteasome inhibitor (bortezomib) and an HDi. Initial studies using sodium butyrate as the HDi found that this combination could increase apoptosis 3- to 4- fold in NSCLC cell lines [131]. A follow up study using SAHA/vorinostat as the HDi, demonstrated that the increased efficacy of this combination (botezomib/SAHA) was due to a synergistic increase in reactive oxygen species [132].
Using a compound that targets the activation of the NF-κB pathway has also been examined in combination with the HDi SAHA. In this study it was found that the combination of these two compounds significantly induced more apoptosis and cell death than either drug alone [133], while a combination of a protein kinase C inhibitor (calphostin C) and a HDI (TSA) found that approximately 90% to 96% of NSCLC cells under-went apoptosis after exposure to this combination [134].
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is another compound that initially showed promise as a potential treatment for cancer. However, it has proven ineffective at inducing cell death when applied as a single agent. In one study, A549 lung carcinoma cells co-exposed to TRAIL and SAHA, sodium butyrate or TSA underwent substantial apoptosis [135]. VPA is another compound which also has HDi activity and a combination of this and TRAIL resulted in increased sensitization of cultured NSCLC cells to Apo2L/TRAIL, resulting in a 4- to >20-fold reduction of Apo2L/TRAIL IC50 values in combination-treated cells. As stand alone agents, VPA or Apo2L/TRAIL induced less than 20% cell death, whereas in combination they caused 60% to 90% apoptosis of NSCLC cells.
Using a combination of SAHA and gemcitabine in NSCLC cell lines sequential treatment offered no improvement over concurrent treatment, whereas combined treatment showed enhanced apoptosis [136]. Similar results have also been observed for a combination utilising phenylbutyrate [137].
The efficacy of a combination of HDi (sodium butyrate) and a phosphatidylinositol 3-kinase/Akt inhibitor (LY294002) in NSCLC has also been demonstrated both in vitro and in vivo, where it was found that this combination was tumoristatic [138]. Similar results were observed when using a combination of a multiple receptor tyrosine kinase inhibitor AEE788 and HDi. When NSCLC cells were treated with AEE788 and HDAC inhibitors (LBH589, NVP-LAQ824 and TSA) in combination synergistic induction of apoptosis was observed which correlated with increased reactive oxygen species accumulation [139]. Similar studies using Romidepsin in combination with the kinase inhibitors AG1478, AG825, PD98059 and LY294002 found markedly enhanced FK228-induced apoptosis in NSCLC cells [140]. Romidepsin was also found to enhance the antitumour effects of an adenoviral telomerase-specific replication-selective agent (OBP-301) on NSCLC cell lines [141].
The bombesin/gastrin-releasing peptide receptor antagonist PD176252 was found to have enhanced inhibitory activity in lung cancer cell lines when combined with the HDi MS-275 [142].
Increased radiation sensitivity has been observed for a novel HDi (LBH589) targeting the classes I and II HDACs. Clonogenic survival showed that there was a greater than additive effect when LBH589 was administered prior to irradiation compared with irradiation alone. Subsequent in vivo tumour volume studies showed a growth delay of 20 days with combined treatment compared with 4 (radiation) and 2 days (LBH589) [143]. A similar increase in radiation sensitivity has also been observed in lung cancer cells treated with the HDi (NVP-LAQ824) [144].
Activating mutations of the epidermal growth factor receptor (EGFR) play critical roles in NSCLC survival, and have led to the development of targeted therapies [145]. A study examining a combination of the EGFR tyrosine kinase inhibitor erlotinib in combination with the HDi LBH589 found synergistic effects on lung cancer cells dependent on EGFR for growth and/or survival, and triggered apoptosis only in lung cancer cells which harboured EGFR mutations indicating that HDi treatments in NSCLC may prove to be of benefit to those patients which harbour EGFR mutations [146].
Another combinatorial treatment which shows efficacy in NSCLC cell lines involves TSA and etoposide, where co-treatment with these drugs induced apoptotic cell death in drug-resistant NSCLC cells, extending the notion that HDAC inhibitors in combination with conventional chemotherapeutic drugs could be a valuable therapeutic option in the treatment of NSCLC cancer [147].
Finally, a combination of an anti-inflammatory drug Sulindac, and SAHA has been shown to significantly enhance growth suppression and apoptosis in a NSCLC cell line, primarily through an enhancement of mitochondrial membrane potential collapse, release of cytochrome c and caspase activation [148].
Hepatocellular carcinoma (HCC)
Another cancer for which strong evidence exists linking the activities of lysine acetyltransferases and histone deacetylases to disease is HCC. In agreement with NSCLC, altered patterns of both DNA CpG methylation [149], and histone modifications [150] have also been observed in HCC.
We initially described the overexpression of histone deacetylases in the paediatric liver tumour hepatoblastoma [151]. Recent studies have shown that in patients with HCC high expression of HDAC1 was correlated with a higher incidence of cancer cell invasion into the portal vein, a poorer histological differentiation, a more advanced tumour node metastasis stage and had poorer prognosis [152]. In a gene microarray analysis of HCCs poor prognosis was observed for a subset of patients, which had elevated levels of various histone, modifying enzymes including HDAC2 [153]. Further evidence for the importance of histone deacetylases in the liver comes from transgenic mice overex-pressing HDAC1. These mice exhibit a high incidence of hepatic steatosis [154]. As non-alcoholic steatohepatitis frequently develop HCC [155], this would indicate that overexpression of HDAC expression may play a critical role in HCC pathogenesis. Other histone modifying complexes have been shown to be strongly associated with HCC including histone methyltransferases (SMYD3) [156, 157], while a subunit of the TFTC/STAGA histone acetyltransferase complex has also been shown to be deregulated in HCC [158]. Finally, metastatic tumour antigen 1 MTA1 is another gene frequently overexpressed in HCC [159–161]. This protein has been shown to associate with various histone deacetylase complexes and further links aberrant chromatin remodelling activities to HCC [88, 91, 92].
In vitro evidence for the use of HDi in liver cancer
We initially indicated that TSA might have use as a therapeutic modality in the treatment of HCC. Treatment of HCC cell lines with TSA was found to dramatically increase the expression of cyclin dependent kinase inhibitors, insulin-like growth factor binding protein-3 and transforming growth factor β (TGF-β), the overexpression of which are commonly associated with cell cycle arrest and/or apoptosis [162, 163]. TSA and sodium butyrate were subsequently shown to decrease telomerase activity in hepatoma cell lines in addition to decreasing cellular proliferation [164]. TSA was shown to increase the activation of caspase-3 and promote apoptosis in hepatoma cells [165, 166]. Using gene microarray profiling, we and others have examined the global gene expression changes for several HDi in hepatoma cells and identified many genes functionally altered following treatments of HDi [167–170]. Proteomic analysis of hepatoma cells treated with SAHA identified 55 differentially expressed proteins of which 34 were subsequently identified using mass spectrometry analysis [171]. We also demonstrated the efficacy of 4-phenylbutyrate in inducing apoptosis and tumour remission in hepatoma tumour xenografts [172]. Similar results were obtained for the HDi HA-But. This is a butyric acid coupled to hyaluronic acid via an esterification, and which has strong affinity for the CD44 membrane receptor. Most importantly, this compound had strong uptake into liver and was able to prevent hepatic metastases in a xenograft model in vivo[173]. Recently a new phenylbutyrate-derived HDi has been developed which shows strong antitumour activity in hepatoma models both in vitro (cell line) and in vivo (xenograft) even in xenografts with high tumour burden (>500 mm3) [174].
Treating hepatoma cells with sodium butyrate researchers demonstrated that MMP-1 was being down-regulated, which correlated with a decreased ability to invade a matrigel assay [175]. Other HDi have also been shown to induce apoptosis in hepatoma cells including VPA and ITF2357 [176], while in onco-genically Ras-transformed rat liver epithelial (WB-Ras) cells, treatment of the cells with sodium butyrate caused the cells to undergo apoptosis and correlated with down-regulation of Ras-specific proteins [177]. Hepatoma cells pre-treated with VPA induce the expression of ligands that activate NK cell receptors and consequently mediate natural killer cell-mediated lysis of hepatoma cells [178].
Combinatorial treatments involving HDi in hepatoma
Similarly to NSCLC, epigenetic targeting using a combination of HDi and demethylating agents have shown promise in an in vivo xenograft hepatoma model [179]. For instance, a triple combination of SAHA, irinotecan and 5-flurouracil (5-FU) led to a significant induction of apoptosis and cell death in hepatoma cells (92% after 72 hrs). Significantly, the double combination of irinotecan and 5-FU only led to a moderate increase in apoptosis and proliferation inhibition [180].
VPA has also been used to sensitize hepatoma cells to apoptosis in combination with the chemotherapy drug epirubicin [181]. Another study using VPA or another HDi (ITF2357) found that a combination of TRAIL and VPA or ITF2357 was able to selectively overcome the resistance of HCC cells toward TRAIL-mediated apoptosis [182]. Similarly to the effects seen for NSCLC cell lines, combinations of SAHA and bortezomib synergistically enhance apoptosis and cell death in hepatoma cells [183].
Potential role of histone deacetylase inhibitors in the treatment of diabetes
Within diabetes pathogenesis, HATs and HDACs can be envisioned as affecting the expression of critical subsets of genes via three central mechanisms. At a basic level, the activities of these enzymes can affect chromatin at target genes themselves, and any alterations or disruptions of their activities could consequently lead to aberrant transcription of such genes. Alternatively, several proteins, which have been identified as the causative factors in monogenic autosomal dominant forms of type two diabetes (MODY, maturity onset diabetes of the young), have also been shown to associate with HATs/HDACs [184]. Indeed some of the mutations identified in these proteins result in loss of association with HATs/HDACs, or lead to a loss in HAT enzymatic activity [184]. There is an intimate network of associations between MODY proteins and consequently these mutant autosomal dominant conditions have aberrant regulation of critical downstream genes, which ultimately leads to the development of diabetes. Finally, the activities of HATs and HDACs are not limited to histones as they often directly modify transcription factors and regulatory proteins [7]. Alterations to the activities of HATs/HDACs may functionally result in the aberrant regulation of transcription of sets of target genes in diabetes pathogenesis.
Disease models, knockouts and assays
Currently, no direct models exist to test the hypothesis that histone deacetylases are directly involved in diabetes pathogenesis. Some evidence has emerged for a role of HATs/HDACs in this disease comes from a mutant mouse model of a lysine acetyltransferase KAT3A. In this model, mice heterozygous for mutated KAT3A demonstrate increased insulin sensitivity and glucose tolerance even while demonstrating marked lipodystrophy of white adipose tissue [185]. Other indications that HATs/HDACs may be important therapeutic targets for diabetes come from studies on their roles in the TGF-β signalling pathway [184]. The TGF-β signalling pathway has well-established associations with HATs/HDACs and many of its downstream signalling processes including the signal transducer and activator of transcription (STAT) proteins have been linked to diabetes and links between TGF-β, STATs, HATs and HDACs well established [184].
Additional important evidence for the role of HATs and HDACs in diabetes comes from studies in both adipocyte and pancreas development, and due to space constraints the reader is directed to the following review on this topic [184]. Finally, a recent patent filed by Biovitrum AB has shown that in mouse models of insulin resistance, an interaction between insulin receptor substrate (IRS)-1 and HDAC2 occurs, which is absent in mouse models that have insulin sensitivity [186].
Pancreatic islet protection using histone deacetylase inhibitors
Several recent publications have demonstrated the ability of Hdi to protect pancreatic p cell apoptosis. In various diabetes models, nicotinamide has frequently been observed to both ameliorate and/or, accelerate the reversal of diabetes and prevent irreversible B-cell damage [187–190]. However, the European Nicotinamide Diabetes Intervention Trial (ENDIT) clinical trial assessing whether the pre-treatment with nicotinamide of non-diabetic individuals predisposed to the development of diabetes could prevent or delay clinical onset of diabetes was ineffective at the dose used [191], but did however reduce high secretion of IFN-gamma in high-risk individuals [192]. Insulin-secreting cells exposed long-term to either nicotinamide or sodium butyrate were found to have reduced viability and insulin sensitivity, yet enhanced insulin secretory responsiveness to a wide range of β cell stimulators [193]. Most recently, both TSA and SAHA were shown to prevent cytokine-induced toxicity in pancreatic β cells [194].
Additional in vitro evidence
Cell culture experimental systems are generating further evidence that Hdi may play important roles in targeting diabetes pathogenesis. A recent European patent has demonstrated that HDAC2 functionally associates with IRS-1, and that insulin sensitivity could be restored to a cell culture model of insulin resistance through the use of TSA [186]. Another example involves the restoration of mutant low-density lipoprotein receptor functionality using 4-phenylbutyrate [195]. As previously discussed the HDi 4-phenylbutyrate has been shown to have profound effects on relieving ER stress in a mouse model of diabetes, which resulted in improved glucose homeostasis [62].
Stem cells, HDACs and histone deacetylase inhibitors
A currently hotly pursued therapeutic avenue for diabetes centres on embryonic stem (ES) cell technology [196]. Evidence is emerging indicating that histone deacetylases may be an important consideration in the development of this technology. Indeed histone deacetylase activity has been shown to be required for ES cell differentiation [197]. The importance of HDAC inhibitors in differentiating ES cells in general has been reviewed recently elsewhere and the reader is directed to the following reviews [198, 199].
Nicotinamide, a SIRT-specific inhibitor, was also used to differentiate ES cells into structures resembling pancreatic islets and which secreted insulin [200]. More recently, under appropriate culture conditions, Bone marrow stem cells (BMSC) cultured in the presence of the HDi TSA differentiated into islet-like clusters similar to the cells of the islets of the pancreas and capable of secreting insulin [201]. Using BMSCs derived from diabetic patients, similar results were obtained using Nicotinamide as one of the final steps in the differentiation process [202].
Two recent articles have utilized the HDi sodium butyrate to (i) stimulate early pancreatic development in ES cells [203] and (ii) generate Islet-like clusters from human ES cells grown under feeder-free conditions [204]. Finally, Scharfmann and colleagues have used various HDi to demonstrate that these enzymes are responsible for the timing and determination of pancreatic cell fate, by promoting the NGN3 pro-endocrine lineage leading to an increased pool of endocrine progenitors and modified endocrine sub-type lineage choices. Treatment of cells with TSA or sodium butyrate also enhanced the pool of β cells [205]. These results clearly demonstrate the potential of HDi in expanding pancreatic β cells from a stem cell population.
The potential role of histone deacetylase inhibitors in the treatment of neurodegenerative conditions
Neuronal traits are modulated by HDAC/REST complexes
A study carried out in 1975 examined the acetylation status of histones in neuronal fractions [206]. Most recently it has now been demonstrated that HDACs play important roles in both neuron differentiation [207], expression of neuron-specific genes [208], and indeed regulate diverse cues such as maternal grooming [209], and addiction [210].
One of the best-established mechanisms in neurons involving HDACs concerns genes that are controlled by a specific protein repressor, neuron restrictive silencing transcription factor (NRSF, also known as REST) [211]. REST contains two distinct repressor domains, one located at the N-terminus and the other at the C-terminus of the protein and several distinct neuronal repressor complexes have now been isolated containing both REST and HDACs [211]. REST also associates with a novel protein called CoREST that interacts with HDACs to actively repress genes essential for neuronal phenotype [212]. The ATP-dependent remodelling complex SWI/SNF also plays a role in REST-mediated neuronal gene regulation, as it has recently emerged that CoREST recruits several SWI/SNF members, indicating that active chromatin remodelling is an element in REST-mediated repression [213, 214]. CoREST complexes have also been shown to contain lysine methyltransferases, and recently an LSD1-CoREST-CtBP corepres-sor complex was shown to be required for late cell-lineage determination and differentiation during pituitary organogenesis [215].
Class II and Class III histone deacetylases also play pivotal roles in the proliferation and differentiation of neurons. SIRT1 has also been shown to protect primary cultures of cerebral granule neurons from FOXO induced cell death, while inactivation of a MEF2D/HDAC5 complex by depolarization-mediated calcium influx protects cerebellar granule neuron survival [211].
E2F, HDACs and neuronal survival mechanisms
An essential feature for neuronal survival has also been linked to constitutive repression of E2F1 transcriptional activity through HDAC proteins. Elevated levels of E2f1 lead to neuronal apoptosis and enhanced immune cell proliferation, factors that could be deleterious in MS [211]. Using microarray analysis enhanced E2F pathway transcription was observed in the peripheral blood mononuclear cells from multiple sclerosis (MS) patients [216]. Subsequently, we demonstrated that HDAC inhibitors reduce levels of E2f class I proteins in vivo[217]. These observations may therefore help to explain why HDAC inhibitors, block immune cell proliferation [218] and enhance neuronal survival [219].
HDACs play important roles in stem cell neuronal differentiation
HDACs have also been shown to play important roles in neuronal stem cell (NSC) differentiation. Using ‘dominant negative’ stem cell lines expressing mutant, Flag-tagged HDACs with reduced enzymatic activity, Howard and colleagues found that mutant HDAC1 reduced differentiation to neurons by 50%[220]. The importance of HDACs in neuronal differentiation has also been demonstrated using HDAC inhibitors. In an in vitro study on the effects of the HDAC inhibitor TSA on the differentiation pattern of embryonic mouse NSCs during culture in a minimal, serum-free medium, it was found that under these conditions TSA treatment increased neuronal differentiation of the NSCs and decreased astrocyte differentiation [221]. In lineage-committed oligodendrocyte precursor cells inhibition of HDAC activity in these cells acted as a priming event in the induction of developmental plasticity [222]. A similar study examining the ability of oligodendrocyte progenitors to acquire the identity of myelin-expressing cells or choose alternative fates found that the activity of histone deacetylases was critical to these processes [223]. Emphasizing this finding, the transcription factor Yin Yang 1 (YY1) was shown to be a critical regulator of oligodendrocyte progenitor differentiation, acting as a lineage-specific repressor of transcriptional inhibitors of myelin gene expression (Tcf4 and Id4), through the recruitment of histone deacetylase-1 to their promoters during oligodendrocyte differentiation [224]. These studies underline the importance of HDACs in neuronal differentiation.
A direct role for histone deacetylases in the regulation of neural stem cell proliferation has been shown where the orphan nuclear receptor TLX, a critical regulator of stem cell proliferation was found to associate with HDAC3 and HDAC5. Inhibition of HDAC activity or knockdown of HDAC expression led to marked induction of TLX target gene expression and dramatically reduced neural stem cell proliferation [225]. REST is also critically involved with neural stem cell differentiation. Activation of REST is sufficient to cause neuronal differentiation [226]. REST complexes are able to both silence and repress neuronal genes in embryonic neural stem cells through the creation of chromatin environments that contain both repressive and active local epigenetic signatures [212, 227, 228].
It is now well established that several neuronal conditions can be linked to aberrant activities of lysine acetyltransferases and deacetylases including Huntington's disease, Rubinstein-Taybi syndrome, spinocerebellar ataxia type 1, spinocerebellar ataxia type 3, Alzheimer's, Parkinson's, spinal muscular atrophy, and amyotrophic lateral sclerosis [211, 229–239].
Histone deacetylase inhibitors
SAHA has been the first directed HDi which has been FDA approved for the treatment of advanced primary cutaneous T-cell lymphoma [240]. In the following sections we will discuss the pharmacological and clinical data emerging from clinical trials using HDi (Table 2). Furthermore we will also discuss one of the issues emerging within the literature on whether the therapeutic efficiency of HDi is via transcriptional mechanisms, or through their ability to enhance chaperone activity.
2.
Clinical parameters from clinical trials using HDi as single agents
| Drug | HDACs targeted | Phase | Method of delivery | Dosage range | Adverse reactions |
|---|---|---|---|---|---|
| Panobinostat (LBH589) | Class I, II | I | Oral | 20 mg/m2 | Grade 3 diarrhoea |
| MGCD0103 | Class I | I | Oral | 60 mg/m2 | Fatigue, nausea, vomiting and diarrhoea |
| Vorinostat | Class I, II | FDA approved | Oral | 200–600 mg/m2 | Fatigue, dehydration, nausea and vomiting |
| MS-275 | Class I | II | Oral | 2–6 mg/m2 | Hypophosphatemia, hyponatremia and hypoalbuminemia |
| VPA | Class I | II | Oral | 60 mg/kg/day | Grade 3, 4 neurocognitive impairment |
| Phenylbutyrate | Class I,II | I/II | Oral and intravenous (i.v.) infusion | 60–360 mg/kg/day | Reversible neurocortical toxicity characterized by somnolence and confusion, short-term memory loss, sedation, confusion, nausea and vomiting |
| Pivanex (AN-9) | II | i.v. infusion | 2.34 g/m2/day | Nausea, vomiting, hyperglycaemia, fatigue, diarrhoea and visual complaints | |
| CI-994 | Class I | I/II | Oral | 4–6 mg/m2/day | Thrombocytopenia, fatigue, nausea, vomiting, diarrhoea, constipation and mucositis |
| Belinostat (PXD101) | Class I, II | I/II | i.v. infusion | 1000 mg/m2/day | Nausea, vomiting, fatigue and flushing atrial fibrillation |
| Two cases of grade 4 renal failure | |||||
| Romidepsin | Class I | I/II | i.v. infusion | 10–26 mg/m2 | Reversible cardiac dysrhythmias and non-specific ECG abnormalities thromocytopenia |
-
•
Initial pharmacological and clinical data
Several studies have attempted to examine the pharmacological clearance of HDi. Initial studies on TSA showed that it undergoes intensive phase I biotransformation in rat hepatocytes, which has important consequences for its potential development as a drug, as this will lead to poor in vivo bioavailability of this drug [241]. These results were subsequently confirmed in a mouse model using intraperitoneal administration of TSA [242].
In human liver microsomes, the metabolism of FK228 (romidepsin) has also been investigated. This compound gets metabolized rapidly into at least 10 metabolites at a Vmax 561.9 pmol/min./mg protein [243]. Hepatic clearance of apicidin has also been examined and an intrinsic Vmax of 927.0 ng/min/mg determined for human hepatic microsomes. In contrast another HDi MS-275 shows markedly little metabolism indicating that this is a minor of elimination for this drug [244].
-
•
Current trials
Several HDi have undergone both phase I/II clinical trials as anticancer agents [245], and as previously mentioned SAHA (vorinostat) has been FDA approved for the treatment of cutaneous T-cell lymphoma. As these trials proceed, greater understanding of the potential side effects and dose-limiting toxicities (DLTs) of these drugs begins to emerge. For the most part these first generation inhibitors have shown well-tolerated safety profiles.
SAHA (vorinostat)
Several phase I clinical trials of vorinostat in solid and haematological tumours have been completed. In initial trials this drug was introduced intravenously and the most significant DLTs observed were leukopenia and thrombocytopenia [246]. An oral version of vorinostat was subsequently developed and phase I trials were found to have linear pharmacokinetics from 200 to 600 mg, with an apparent half-life ranging from 91 to 127 min. and 43% oral bioavailability [247]. Following the development of this oral form, a phase I trial was conducted in patients with mesothelioma. Similar toxicities to the original phase I trials were observed primarily fatigue, dehydration, nausea, and vomiting. Of four patients who completed greater than six cycles of therapy, two showed partial responses, which have led to a placebo-controlled, randomized phase III study of oral vorinostat for mesothelioma patients for whom treatment with pemetrexed has failed [248]. A phase II trial of vorinostat for refractory cutaneous T-cell lymphoma (CTCL) demonstrated both partial responses and ruritus relief, with limited toxicities of fatigue, thrombocytopenia, diarrhoea and nausea [249], and went on for further development. Following the completion of a single-arm, open-label, multi-centre pivotal trial and 11 other trials, clinical efficacy was assessed. Vorinostat showed activity in CTCL, and skin responses were a clinical benefit, and vorinostat was subsequently approved for treatment of cutaneous manifestations of CTCL [250]. A phase II trial of vorinostat has also been completed in patients with recurrent and/or metastatic head and neck cancer. In this trial, 12 patients received 400 mg once daily. Three patients had stable disease ranging from 9 to 26 weeks. Nine patients discontinued due to progressive disease, two withdrew consent, and one discontinued therapy for grade 3 anorexia. Overall, vorinostat was generally well tolerated but did not demonstrate efficacy as defined by tumour response [251]. In an open centre early phase II trial of oral vorinostat, patients with measurable, relapsed or refractory breast or non-small cell lung cancer who had received >/= 1 prior therapy or colorectal cancer who had received >/= 2 prior therapies were enrolled. Oral vorinostat (200, 300 or 400 mg) was taken twice daily for 14 days, followed by a 7-day rest until disease progression or intolerable toxicity was conducted, and the response rate, safety and tolerability were evaluated. Of the 16 patients recruited no DLTs were observed at 200 mg. Disease stabilization was observed in eight patients, but there were no confirmed responses [252].
Phenylbutyrate
Phenylbutyrate is another HDi for which several clinical trials have been carried out. When taken orally, the most common toxicities observed are grade 1–2 dyspepsia and fatigue [253]. Two phase I studies found that the DLTs included reversible neurocortical toxicity characterized by somnolence and confusion [254–256]. In a phase I study of sodium phenylbutyrate on patients with Huntington's disease, toxicities observed at the higher doses included vomiting, light-headedness, confusion and gait instability, but with no significant laboratory or electrocardiographic abnormalities [257].
Several trials testing phenylbutyrate on spinal muscular atrophy (SMA) have been carried out. The results of these studies found that the major side effect was a temporary stomach ache [258, 259]. Unfortunately, in a randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy no significant improvements were observed [260]. However, it must be noted that in this trial, SMA patients were treated for only 13 weeks, and this may be too short a treatment period to identify any clinical benefit from PB. However, other trials continue to show further evidence that phenylbutyrate may have efficacy in the treatment of disease. In a phase I dose-escalation study in cystic fibrosis, a statistically significant induction of chloride transport was observed, with minimal adverse reactions [261].
Phenylbutyrate has also been used in phase I clinical trials for solid tumours. In this trial common adverse effects included grade I nausea/vomiting, fatigue and light-headedness, and the DLTs observed were short-term memory loss, sedation, confusion, nausea and vomiting. The maximum tolerated dose (MTD) was 300 mg/kg/day, and three of the twenty-one patients enrolled achieved stable disease [262]. A dose escalation study of oral phenylbutyrate has also been carried out in patients with recurrent malignant glioma. In agreement with the trials presented above, fatigue and somnolence were the worst toxicities observed. Of the twenty-three patients enrolled one patient had a complete response for 5 years [263].
Valproic acid
VPA has also entered clinical trials as a stand-alone agent in patients with advanced refractory cancer. Dose escalation was carried out in three-patient cohorts on twenty-six pre-treated patients. The MTD of infused drug was found to be 60 mg/kg/day, and the DLT was found to be grade 3 or 4 neurological side effects occurring in 8 out of 26 patients [264]. A phase II trial of VPA has also been completed in patients with castration-resistant prostate cancer. However, VPA was not found to be well tolerated by this cohort, and could not be administered reliably in order to achieve consistent levels or duration, and it was concluded that oral VPA is not recommended for prostate cancer [265].
In a phase I trial of continuous oral VPA for maintenance treatment in heavily pre-treated paediatric glioma patients, moderate tumour efficacy was observed [266]. VPA also shows potential in the treatment of neurodegenerative conditions. In a phase I trial of VPA on patients with human T-lymphotropic virus type 1 (HTLV-1), which is responsible for HTLV-associated myelopathy/tropical spastic paraparesis, clinical efficacy was observed. 16 patients were recruited and VPA was administrated orally at a maximal dose of 20 mg/kg/day. There was a significant drop in patient viral load from month 0 to month 3, and for the first time provides evidence that VPA leads to depletion of HTLV-1-infected cells in vivo [267]. The Project Cure SMA team has just completed the phase II CARNI-VAL clinical trial examining the effects of a combinatorial treatment of VPA and carnitine on SMA (http://www.fsma.org), but the results of this trial have yet to be disseminated to the greater scientific community.
AN-9
The drug Pivanex (AN-9) has completed both phase I and II clinical trials for advanced solid malignancies. No significant DLTs were observed in the phase I study and mild to moderate reactions included nausea, vomiting, hyperglycaemia, fatigue, diarrhoea and visual complaints [268, 269]. During the phase II trial, similar tolerance was observed, with the worst effects including fatigue and hypokalemia [269].
CI-994
For the inhibitor CI-994 several phase I studies have been completed. The principal DLT observed for all trials was thrombocytopenia, but other side effects observed included nausea, vomiting diarrhoea and mucositis [270–273].
Romidepsin
The HDi romidepsin has completed several phase I clinical trials on cancer and has also entered/completed phase II trials. In the phase I trials the major DLTs for this drug was again thromocytopenia. However, for this drug a significant increase in reversible cardiac dysrhythmias and non-specific ECG abnormalities were observed [274–276]. In a phase I dose-escalation trial of romidepsin in paediatric solid tumours romidepsin was administered as a 4-hr infusion weekly for three consecutive weeks every 28 days at dose levels of 10, 13, 17 and 22 mg/m2. Of the 24 patients enrolled, 18 could be assessed for toxicity. In agreement with the other phase I trials non-specific reversible ECG abnormalities were observed. Of the 18 patients three achieved long-term stable disease. For phase II trails the recommended dose was determined to be the recommended phase II dose in children with solid tumours is 17 mg/m2[277].
A second phase I trial in patients with acute myelogenous leukaemia or advanced myelodysplastic syndromes used 18 mg/m2 intravenous on days 1 and 5 every 3 weeks. The most common grade 3/4 toxicities were febrile neutropenia/infection, neutropenia/thrombocytopenia, nausea and asymptomatic hypophosphatemia, with no clinically significant cardiac toxicity. The best responses of 11 assessed patients was one complete remission (CR) in a patient with acute myeloid leukaemia (AML), stable disease in six patients. Notably however, histone H3 and H4 acetylation levels evaluated in five patients showed no consistent changes [278].
Two phase II trials of romidepsin have been completed. In a trial on twenty-nine patients with refractory metastatic renal cell cancer, the most common serious toxicities were fatigue, nausea, vomiting and anaemia. However, one patient developed a grade 3 atrial fibrillation, one patient developed tachycardia, and there was 1 sudden death. Within the study group itself two patients achieved an objective response, which was insufficient to bring this agent forward for further study in the treatment of renal cell cancer [279]. In a phase II trial on T-cell lymphoma, cardiac monitoring of 42 patients was carried out. The data obtained as part of this study indicate that the administration of romidepsin is not associated with myocardial damage or impaired cardiac function [280]. However, in a phase II study of romidepsin in neuroendocrine tumours, the study was terminated prematurely due to an unexpected high number of serious cardiac adverse events [281].
MS-275
MS-275 has just completed two phase I trials in solid tumours. The MTD of this drug was found to be 6 mg/m2, with a mean terminal half-life of 33.9 ± 26.2 and a T(max) ranging from 0.5 to 24 hrs. Dose-limiting grade 3 toxicities were found to be reversible and included hypophosphatemia, hypoasthenia, hyponatremia and hypoalbuminemia [282, 283] . In an earlier phase I study, the MTD was found to be 10 mg/m2 and DLTs were nausea, vomiting, anorexia and fatigue. In addition the half-life was found to be within the range 39–80 hrs [284]. In a phase I trial of MS-275 in patients with advanced acute leukaemias, the MTD was found to be 8 mg/m2 weekly for 4 weeks every 6 weeks. DLTs included infections and neurological toxicity manifesting as unsteady gait and somnolence. Other frequent toxicities observed were fatigue, anorexia, nausea, vomiting, hypoalbuminemia and hypocalcaemia [285]. In a trial to determine pharmacokinetic data for oral MS-275 in 64 adult patients receiving MS-275 orally (dose range, 2 to 12 mg/m2), no metabolites could be detected after incubation of MS-275 in human liver microsomes, suggesting that hepatic metabolism is a minor pathway of elimination, while the mean (±S.D.) apparent oral clearance of MS-275 was 38.5 ± 18.7 l/hr [244].
Most recently a phase II trial of MS-275 has been completed in patients with metastatic melanoma. The primary study end-point was objective tumour response, but among 28 patients enrolled, no objective response was detected. Seven patients in showed disease stabilizations [286].
LBH589 (panobinostat)
LBH5985 is currently undergoing an ongoing phase I, open-label, dose-escalation study in patients with solid tumours and non-Hodgkin's lymphoma. From this study, 10 patients with CTCLs were treated with oral panobinostat on a 28-day cycle. From this initial cohort of 10 patients complete responses were observed in 2 patients and partial responses in a further 4 individuals. The major DLT observed was a grade 3 diarrhoea at a dose of 30 mg [287].
MGC D0103
This orally administered HDi has recently completed a phase I clinical trial in patients with leukaemia or myelodysplastic syndromes [288]. This was administered orally three times weekly without interruption, in a dose-escalation study of 20, 40 and 80 mg/m2. The MTD was determined to be 60 mg/m2, with DLTs of fatigue, nausea, vomiting, and diarrhoea observed at the higher doses. Of 29 patients enrolled, 3 achieved a complete bone marrow response (blasts < or = 5%). Pharmacokinetic analyses indicated absorption of MGCD0103 within 1 hr and an elimination half-life in plasma of 9 (±2) hrs [288].
-
•
PXD101 (Belinostat): Two phase I trial of belinostat have been completed, and several phase II clinical trials are in progress. In the first, a DLT study was carried out in 46 patients with confirmed advanced malignancy refractory to standard therapy or for whom no standard therapy existed. The MTD for Belinostat was determined to be 1000 mg/m2/days 1–5 in a 21-d cycle, and of the treated patients, 50% achieved stable disease [289]. The second trial was carried out in patients with advanced haematological neoplasia using the previously determined MTD. No complete or partial remissions were noted in these heavily pre-treated (median of four prior regimens) patients. However, five patients achieved some degree of disease stabilization [290].
-
•
Combinatorial trials using Hdi.
The poor overall response of solid tumours to HDi as single agent therapies has resulted in several trials examining their potential as adjuvant therapies in combination with other drugs.
One HDi valproate, has completed a phase II trial examining its use as a combined epigenetic therapy with hydralazine to overcome chemotherapy resistance in refractory solid tumours. In this trial 17 patients were evaluable for toxicity and 15 for response. Twelve patients were found to respond with four showing a partial response and eight demonstrating stable disease (defined as neither sufficient tumour reduction to qualify for partial response nor sufficient increase to qualify for progressive disease) [291]. A phase I trial using a similar combination (hydralazine/valproate) plus neoadjuvant Doxorubicin Cyclophosphamide was carried out in patients with locally advanced breast cancer. 16 patients were included and received treatment. All were evaluated for clinical response and toxicity and 15 for pathological response. Treatment was well tolerated and the most common toxicity observed was patient drowsiness grades 1–2. Five (31%) patients had clinical response and eight (50%) had a partial response to give an overall response rate of 81%. There was a statistically significant decrease in both global 5-methylcytosine content and HDAC activity. The results obtained in this trial have resulted in the initiation of an ongoing randomized phase III study [292].
A pilot clinical trial involving phenylbutyrate and 5-azacytidine in patients with acute myeloid leukaemia or myelodysplastic syndrome has also been carried out. This combination regimen was well tolerated with common toxicities of injection site skin reaction (90% of the patients) from 5-azacytidine, and somnolence/fatigue from the sodium PB infusion (80% of the patients). Of the 10 patients in this trial, 5 patients (50%) were able to achieve a partial remission or stable disease, and 1 patient was able to proceed to allogeneic stem cell transplantation and was alive without evidence of disease 39 months later [293].
A phase I/II study of the combination of 5-aza-2’-deoxycytidine with VPA in patients with leukaemia was completed. In this study a group of 54 patients was recruited and treated with a fixed dose of decitabine (15 mg/m2 i.v. daily for 10 days) administered concomitantly with escalating doses of VPA orally for 10 days. The MTD of VPA was determined to be 50 mg/kg daily. Twelve (22%) patients had objective response, including 10 (19%) CRs and 2 (3%) CRs with incomplete platelet recovery (CRp). Remission duration was 7.2 months (range, 1.3–12.6+ months), while overall survival was 15.3 months (range, 4.6–20.2+ months) in responders [294].
Several trials involving VPA in combination with all-trans retinoic acid (ATRA) have been carried out. In one study on 26 patients with poor-risk AML VPA (5–10 mg/kg starting dose) and ATRA (45 mg/m2) were administered orally. Of 26 patients recruited, 19 completed at least 4 weeks of VPA/ATRA treatment. Seven patients were withdrawn prematurely because of rapidly progressive disease (n= 3) or unacceptable neurological and cardiovascular toxicity (n= 4). Three patients had partial responses [295]. A larger trial was carried out on 58 patients with advanced AML who were unfit for standard intensive chemotherapy. VPA was administered to reach serum concentrations between 50 and 100 g/ml, the therapeutic range used for VPA in anti-epileptic treatment. There were two different treatment schedules for ATRA, either 80 mg/m2 each day was given in two divided doses, days 1–7, every other week, or 15 mg/m2 was given daily, starting on day 4. Both drugs were administered orally. Treatment was continued as long as neither significant side effects nor disease progression occurred. Overall, treatment was well tolerated, but it was concluded that VPA had beneficial effects but was not sufficiently active to be useful as single-agent therapy for AML [296]. A larger phase II trial involving 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukaemia, found that VPA was only clinically useful in low-risk myelodysplastic syndrome [297]. This was subsequently followed with a phase I/II study combining 5-azacitidine (5-AZA), VPA, and ATRA in patients with acute myeloid leukaemia or high-risk myelodysplastic syndrome. In this trial 53 patients were treated. 5-AZA was administered subcutaneously at a fixed dose of 75 mg/m2 daily for 7 days. VPA was dose-escalated and given orally daily for 7 days concomitantly with 5-AZA. ATRA was given at 45 mg/m2 orally daily for 5 days, starting on day 3. The MTD of VPA in this combination was found to be 50 mg/kg daily for 7 days, and reversible neurotoxicity was the DLT Overall response rate to this treatment was 42%. The median remission duration was 26 weeks, and at the time of publication median survival had not yet been reached [298].
VPA has also undergone a phase I dose-escalation trial in combination with the topoisomerase II inhibitor epirubicin in advanced solid tumours. In this trial forty-eight patients were enrolled, and 44 received at least one cycle of therapy (increasing doses of VPA (days 1 through 3) followed by epirubicin (day 3) in 3-week cycles). DLTs observed were somnolence, confusion, and febrile neutropenia. The maximum-tolerated dose and recommended phase II dose identified was VPA 140 mg/kg/day for 48 hrs followed by epirubicin 100 mg/m2. In the treated patients partial responses were seen across different tumour types in nine patients (22%), and stable disease/minor responses were seen in 16 patients (39%), indicating that this combination may have potential in the treatment of solid tumours [299].
SAHA (vorinostat) has also completed a phase I trial in combination with Carboplatin and Paclitaxel for the treatment of advanced solid malignancies. Twenty-eight patients were enrolled into the study and separated into two arms for vorinostat treatment which was either administered orally once daily for two weeks or given twice daily for 1 week, every 3 weeks. Within each arm, the doses of vorinostat and paclitaxel were dose escalated in sequential cohorts of three patients. The DLT was determined to be 400 mg of vorinostat given once daily. Other non-DLTs included nausea, fatigue, diarrhoea and neuropathy. When patient response was evaluated, 10 of 19 (53%) of patients who had advanced chemo-naive non-small cell lung cancer (NSCLC) experienced a partial response and 4 had stable disease. In comparison, chemo-naive NSCLC patients just receiving carboplatin–paclitaxel generally achieve response rates of approximately (20–30%). These results indicate that vorinostat may enhance this therapy in NSCLC, and a phase II study randomizing advanced NSCLC patients to carboplatin-paclitaxel with either vorinostat or placebo is currently ongoing [300].
A phase I study combining cytotoxic-differentiation therapy with 5-fluorouracil and phenylbutyrate in patients with advanced colorectal cancer has been completed. In this study 5-flurouracil (FUra) was dose escalated (24-hr continuous intravenous infusion (2–2.3 g/m2), in combination with PB (120-hr continuous intravenous infusion at a fixed dose of 410 mg/kg/day × 5), and repeated weekly, in patients with advanced colorectal cancer. Nine patients were recruited and treated of which 8 could be assessed for toxicity. Weekly infusions of FUra followed by PB were fairly well tolerated with dose-dependent, reversible toxicities including somnolence, fatigue, confusion, hearing loss, triglyceridemia and hyperuricaema. Four patients completed eight weeks of treatment. Of these three achieved stable disease [301].
In a phase II randomized, double-blind, placebo-controlled, multi-centre study examining a combination of CI-994 and gemcitabine in patients with advanced pancreatic cancer. A total of 174 patients were recruited and received either a combination of CI-994/gemcitabine (CI-994 6 mg/m2/day days 1–21 plus gemcitabine 1000 mg/m2 days 1, 8 and 15 each 28-day cycle) or placebo/gemcitabine (placebo plus gemcitabine 1000 mg/m days 1, 8 and 15 of each 28-day cycle days 1–21). When the results were assessed, CI-994 offered no advantage over gemcitabine alone in the treatment of these patients, and indeed lowered their quality of life [302].
Caveats
Intriguingly, HDi have been shown to be ineffective in causing apoptosis in non-small cell lung cancer cell line models. This was shown to be due to the transcriptional activation of NF-κB through the Akt pathway, with up-regulation of IL-8, Bcl-XL and MMP-9 transcripts [303]. A follow-up study using vorinostat (SAHA) has shown that this drug stimulates NF-κB transcription through a signalling cascade that involves activation of both the serine/threonine kinase Akt and the KAT3B acetyltransferase [304]. As such these studies provide evidence that HDi, such as SAHA, not only inhibit deacetylase activity but also stimulate active NF-κB transcription and cell survival through signalling pathways involving Akt and increased KAT3B acetyltransferase activity. This may have important implications in the use of histone deacetylases in the treatment of cancer as they may promote cancer cell survival, in contrast they may have important therapeutic implications in both diabetes and neurodegenerative disease as the may promote the survival of pancreatic β cells or protect neuronal cells.
Despite the evidence strongly suggesting that HDi may have an important therapeutic role in the treatment of obesity and diabetes nevertheless there are some caveats that temper this notion. One of the caveats concerns the use of VPA in the treatment of diabetes in that known side effects of this drug in patients are in fact, obesity and insulin resistance [305–311]. However, it must also be noted that in a study on the effects of HDAC inhibitors in adipocytes which included TSA and VPA it was found that VPA reduced leptin mRNA levels while TSA did not, suggesting that VPA therapy may be associated with altered leptin homeostasis contributing to weight gain in vivo, and therefore other HDAC inhibitors may not cause similar effects in relation to obesity and insulin resistance [312].
HDAC inhibitors have also been shown to up-regulate NF-κB driven pro-inflammatory cascades [313, 314], albeit within a neural setting. Nevertheless this may also be true for multiple tissue types. In addition, HDi have also been shown to activate NF-κB, and to sustain the activation of NF-κB by delaying kBa mRNA resynthesis [315, 316].
Within the clinical setting, other studies have shown the ineffectiveness of phenylbutyrate on relieving ER Stress in patients with α-1-antitrypsin deficiency [317].
Do HDAC inhibitors target genes or help chaperone activity as their primary response?
A plethora of studies have clearly shown that HDi can reactivate or alter gene expression. However, several studies now indicate that histone deacetylases and HDi may play important roles in regulating chaperone expression and function. This has important implications in conditions such as diabetes and obesity where aberrant misfolding of proteins can result in ER Stress. Indeed HDAC6 has been recognized as a leading regulator of cellular efforts to counteract the deleterious effects of misfolded protein accumulation [318–321]. Inhibition or depletion of HDAC6 leads to an induction of Hsp90 acetylation inhibiting its chaperone activity and eliciting cellular responses [146,322–326]. It must also be noted that HDAC6 is very resistant to various HDi including VPA and apicidin. The class I HDACs (HDACs 1–3) have been shown to associate with the ATP dependent chaperone Hsp70, and this association enhances deacetylase catalytic activity [327].
Emerging data clearly link the use of HDi in the relief of ER Stress in cell line models. Many of these studies have utilized the chemical 4-phenylbutyrate (PB). In a hepatocyte cell line model of oxidative stress induced ER Stress, treatment of cells with PB was found to alleviate the ER Stress in these cells [47]. General examples of the benefits of PB as a chemical chaperone have been described for relieving apoptosis and/or ER Stress in models of eye disease [59, 60], rescue of defective trafficking of nephrin in kidney [328], rescue of protein trafficking in the lysosomal storage disorder Fabry disease [329], correction of autodominant hypoparathyroidism induced apoptosis [330] and relief of ER stress mediated programmed cell death in arabadopsis [331].
Other evidence for the potential of HDi influencing chaperone-like activities to relieve ER Stress have come from studies in neuroprotection lung, and liver disease. Within the neuronal setting, initial studies on mood stabilizing drugs such as VPA demonstrated increased expression of ER stress proteins in cerebral cortex, hippocampus, neuronal and glial cells [64–67, 332, 333]. Neuroprotective effects of these HDi have been observed to protect against ischaemia [55, 334, 335], malonate toxicity [336], rotenone [337, 338], thapsigargin [339] and relieved ER Stress marker induction in a model of autosomal recessive juvenile Parkinsonism [58].
Studies on cystic fibrosis have shown that phenylbutyrate can restore CFTR trafficking and function [53, 236, 261, 340–345], through the induction of Hsp90 [345]. Other lung conditions for which beneficial responses have been observed using HDi as chemical chaperones include respiratory distress syndrome [346], and emphysema [54].
Benefits accruing to the use of HDi as chemical chaperones in the liver have also been described. In one instance PB was used to protect liver cells from ER stress mediated apoptosis induced by liver ischaemia [56], enhances the cell surface expression and transport capacity of mutated bile salt export pumps [347], and reduces ER Stress induced formation of Mallory bodies in hepatocytes [47].
Increasing evidence is linking the activities of HDi to ER Stress in obesity. For instance, in a cell line model, phenylbutyrate has been shown to restore functionality to misfolded low-density lipoprotein receptors, and shuttle them to the cell surface. The authors concluded that their results indicate that phenylbutyrate did not just solely mediate this response by its ability to induce gene expression of proteins involved in intracellular transport, but could also mediate this effect via a direct chemical chaperone activity [195]. VPA was also shown to protect cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3 [348]. In a mouse model of type 2 diabetes, phenylbutyrate was found to reduce ER Stress and restore glucose homeostasis in the mutant mice [62]. This critical result underlines the potential importance of HDi as both regulators of gene expression and as chemical chaperones to dampen down inflammation and relieve ER Stress.
Final comments
One critical element that needs to be addressed in future studies examining the efficacy of HDi in combinatorial trials should include studies to study the importance of timing for the scheduling of HDi within the protocol. In vitro studies using a combination of HDi and camptothecin, have shown that in NSCLC, the time of HDi addition is a critical determinant which can either cause cell protection or sensitization to camptothecin [349]. The development of tests that can predict the efficacy of HDi as an antitumour agent will also be an avenue that should be more thoroughly examined. It is interesting to note that a predictive model for HDi antitumour activity in non-small cell lung cancer has recently been developed through gene expression profiling, where a nine-gene classifier set has been found to have predictive value for determining drug sensitivity to HDi [350]. This exciting development raises the possibility of achieving individualized therapy for NSCLC patients, and potentially this could be expanded to individualized patient therapy for any disease.
Another area of critical importance will require the development of isoform-specific HDi, and area currently being actively pursued. The development of such inhibitors will allow better selectivity as tools for probing the biological functions of the isoforms, but also may function as better candidate therapeutic agents with fewer side effects [351, 352]. For instance, a HDAC8-specific HDi PCI-3405 has been developed which has >200-fold selectivity over the other HDAC isoforms, and induces caspase-dependent apoptosis in cell lines derived from T-cell lymphomas or leukaemias, but not in other haematopoietic or solid tumour lines [353].
Finally, one avenue which has only recently begun to emerge concerns the effects of HDi on non-coding RNAs. This is especially important for miRNAs, which have been shown to be regulated via epigenetic mechanisms including histone acetylation and DNA CpG methylation [354–358].
References
- 1.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 4.Lee KK, Workman JL. Histone acetyl-transferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–95. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 5.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 6.Gray SG, Ekström TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 7.Batta K, Das C, Gadad S, Shandilya J, Kundu TK. Reversible acetylation of non histone proteins: role in cellular function and disease. Subcell Biochem. 2007;41:193–212. [PubMed] [Google Scholar]
- 8.Smith CL. A shifting paradigm: histone deacetylases and transcriptional actiation. Bioessays. 2008;30:15–24. doi: 10.1002/bies.20687. [DOI] [PubMed] [Google Scholar]
- 9.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 10.Jones P, Steinkuhler C. From natural products to small molecule ketone histone deacetylase inhibitors: development of new class specific agents. Curr Pharm Des. 2008;14:545–61. doi: 10.2174/138161208783885317. [DOI] [PubMed] [Google Scholar]
- 11.Perwez Hussain S, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–80. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 12.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 13.Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21:1443–55. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- 14.Esiri MM. The interplay between inflammation and neurodegeneration in CNS disease. J Neuroimmunol. 2007;184:4–16. doi: 10.1016/j.jneuroim.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA. 1997;94:2927–32. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–7. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 17.Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J Biol Chem. 1999;274:1879–82. doi: 10.1074/jbc.274.4.1879. [DOI] [PubMed] [Google Scholar]
- 18.Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet. 1998;63:1552–8. doi: 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacety-lase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proin-flammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–76. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 22.Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated dere-pression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–71. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell. 2004;16:245–55. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Park J, Lee JH, La M, Jang MJ, Chae GW, Kim SB, Tak H, Jung Y, Byun B, Ahn JK, Joe CO. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J Mol Biol. 2007;368:388–97. doi: 10.1016/j.jmb.2007.02.047. [DOI] [PubMed] [Google Scholar]
- 25.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci. 2002;115:3319–30. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 26.Kuo HY, Chang CC, Jeng JC, Hu HM, Lin DY, Maul GG, Kwok RP, Shih HM. SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proc Natl Acad Sci USA. 2005;102:16973–8. doi: 10.1073/pnas.0504460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esposito K, Nappo F, Giugliano F, Di Palo C, Ciotola M, Barbieri M, Paolisso G, Giugliano D. Cytokine milieu tends toward inflammation in type 2 diabetes. Diabetes Care. 2003;26:1647. doi: 10.2337/diacare.26.5.1647. [DOI] [PubMed] [Google Scholar]
- 28.Dandona P, Aljada A, Chaudhuri A, Mohanty P. Endothelial dysfunction, inflammation and diabetes. Rev Endocr Metab Disord. 2004;5:189–97. doi: 10.1023/B:REMD.0000032407.88070.0a. [DOI] [PubMed] [Google Scholar]
- 29.Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25:4–7. doi: 10.1016/j.it.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Vanden Berghe W, De Bosscher K, Boone E, Plaisance S, Haegeman G. The nuclear factor-kappa B engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274:32091–8. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–96. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calao M, Burny A, Quivy V, Dekoninck A, Van Lint C. A pervasive role of histone acetyltransferases and deacetylases in an NF-kappaB-signaling code. Trends Biochem Sci. 2008;33:339–49. doi: 10.1016/j.tibs.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 35.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–9. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 36.Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 37.Caruso ME, Chevet E. Systems biology of the endoplasmic reticulum stress response. Subcell Biochem. 2007;43:277–98. doi: 10.1007/978-1-4020-5943-8_13. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 39.Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kappa B. Trends Biochem Sci. 1997;22:63–7. doi: 10.1016/s0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- 40.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–80. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 42.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2007;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 43.Uehara T. Accumulation of misfolded protein through nitrosative stress linked to neurodegenerative disorders. Antioxid Redox Signal. 2007;9:597–601. doi: 10.1089/ars.2006.1517. [DOI] [PubMed] [Google Scholar]
- 44.Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17:469–77. doi: 10.1093/hmg/ddm324. [DOI] [PubMed] [Google Scholar]
- 45.Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007;67:10631–4. doi: 10.1158/0008-5472.CAN-07-1705. [DOI] [PubMed] [Google Scholar]
- 46.Bardag-Gorce F, Dedes J, French BA, Oliva JV, Li J, French SW. Mallory body formation is associated with epigenetic phenotypic change in hepatocytes in vivo. Exp Mol Pathol. 2007;83:160–8. doi: 10.1016/j.yexmp.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanada S, Harada M, Kumemura H, Bishr Omary M, Koga H, Kawaguchi T, Taniguchi E, Yoshida T, Hisamoto T, Yanagimoto C, Maeyama M, Ueno T, Sata M. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol. 2007;47:93–102. doi: 10.1016/j.jhep.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 48.Ohoka N, Hattori T, Kitagawa M, Onozaki K, Hayashi H. Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J Biol Chem. 2007;282:35687–94. doi: 10.1074/jbc.M703735200. [DOI] [PubMed] [Google Scholar]
- 49.Baumeister P, Luo S, Skarnes WC, Sui G, Seto E, Shi Y, Lee AS. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol. 2005;25:4529–40. doi: 10.1128/MCB.25.11.4529-4540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donati G, Imbriano C, Mantovani R. Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters. Nucleic Acids Res. 2006;34:3116–27. doi: 10.1093/nar/gkl304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doody GM, Stephenson S, Tooze RM. BLIMP-1 is a target of cellular stress and downstream of the unfolded protein response. Eur J Immunol. 2006;36:1572–82. doi: 10.1002/eji.200535646. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol. 2000;20:2592–603. doi: 10.1128/mcb.20.7.2592-2603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–67. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- 54.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci USA. 2000;97:1796–801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 56.Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, Eckhoff DE, Contreras JL. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138:342–51. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 57.Mulhern ML, Madson CJ, Kador PF, Randazzo J, Shinohara T. Cellular osmolytes reduce lens epithelial cell death and alleviate cataract formation in galactosemic rats. Mol Vis. 2007;13:1397–405. [PubMed] [Google Scholar]
- 58.Kubota K, Niinuma Y, Kaneko M, Okuma Y, Sugai M, Omura T, Uesugi M, Uehara T, Hosoi T, Nomura Y. Suppressive effects of 4-phenylbutyrate on the aggregation of Pael receptors and endoplasmic reticulum stress. J Neurochem. 2006;97:1259–68. doi: 10.1111/j.1471-4159.2006.03782.x. [DOI] [PubMed] [Google Scholar]
- 59.Bonapace G, Waheed A, Shah GN, Sly WS. Chemical chaperones protect from effects of apoptosis-inducing mutation in carbonic anhydrase IV identified in retinitis pigmentosa 17. Proc Natl Acad Sci USA. 2004;101:12300–5. doi: 10.1073/pnas.0404764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest Ophthalmol Vis Sci. 2007;48:1683–90. doi: 10.1167/iovs.06-0943. [DOI] [PubMed] [Google Scholar]
- 61.Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J Biol Chem. 2006;281:17369–78. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- 62.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azevedo JE, De Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007;282:27905–12. doi: 10.1074/jbc.M702672200. [DOI] [PubMed] [Google Scholar]
- 64.Wang JF, Bown C, Young LT. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. 1999;55:521–7. [PubMed] [Google Scholar]
- 65.Bown CD, Wang JF, Young LT. Increased expression of endoplasmic reticulum stress proteins following chronic valproate treatment of rat C6 glioma cells. Neuropharmacology. 2000;39:2162–9. doi: 10.1016/s0028-3908(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 66.Chen B, Wang JF, Young LT. Chronic valproate treatment increases expression of endoplasmic reticulum stress proteins in the rat cerebral cortex and hippocampus. Biol Psychiatry. 2000;48:658–64. doi: 10.1016/s0006-3223(00)00878-7. [DOI] [PubMed] [Google Scholar]
- 67.Shao L, Sun X, Xu L, Young LT, Wang JF. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci. 2006;78:1317–23. doi: 10.1016/j.lfs.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 68.Wang JF, Azzam JE, Young LT. Valproate inhibits oxidative damage to lipid and protein in primary cultured rat cerebrocortical cells. Neuroscience. 2003;116:485–9. doi: 10.1016/s0306-4522(02)00655-3. [DOI] [PubMed] [Google Scholar]
- 69.Shao L, Young LT, Wang JF. Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry. 2005;58:879–84. doi: 10.1016/j.biopsych.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 70.Cui J, Shao L, Young LT, Wang JF. Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and val-proate. Neuroscience. 2007;144:1447–53. doi: 10.1016/j.neuroscience.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 71.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16:R50–9. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 72.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 73.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 74.Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. 2007;25:4358–64. doi: 10.1200/JCO.2007.11.2599. [DOI] [PubMed] [Google Scholar]
- 75.Park JM, Lee GY, Choi JE, Kang HG, Jang JS, Cha SI, Lee EB, Kim SG, Kim CH, Lee WK, Kam S, Kim DS, Jung TH, Park JY. No association between polymorphisms in the histone deacetylase genes and the risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1841–3. doi: 10.1158/1055-9965.EPI-05-0200. [DOI] [PubMed] [Google Scholar]
- 76.Cazzola M, Ciaprini C, Page CP, Matera MG. Targeting systemic inflammation: novel therapies for the treatment of chronic obstructive pulmonary disease. Expert Opin Ther Targets. 2007;11:1273–86. doi: 10.1517/14728222.11.10.1273. [DOI] [PubMed] [Google Scholar]
- 77.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–76. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 78.Szulakowski P, Crowther AJ, Jimenez LA, Donaldson K, Mayer R, Leonard TB, MacNee W, Drost EM. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:41–50. doi: 10.1164/rccm.200505-725OC. [DOI] [PubMed] [Google Scholar]
- 79.Ito K, Lim S, Caramori G, Chung KF, Barnes PJ, Adcock IM. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001;15:1110–2. [PubMed] [Google Scholar]
- 80.Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Kiriyama M, Fukai I, Yamakawa Y, Fujii Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer. 2004;46:171–8. doi: 10.1016/j.lungcan.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 81.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–74. [PubMed] [Google Scholar]
- 82.Bartling B, Hofmann HS, Boettger T, Hansen G, Burdach S, Silber RE, Simm A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49:145–54. doi: 10.1016/j.lungcan.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 83.Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004;112:26–32. doi: 10.1002/ijc.20395. [DOI] [PubMed] [Google Scholar]
- 84.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and anti-aging protein, is decreased in lungs of patients with COPD. Am J Respir Crit Care Med. 2008;117:861–70. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suzuki H, Ouchida M, Yamamoto H, Yano M, Toyooka S, Aoe M, Shimizu N, Date H, Shimizu K. Decreased expression of the SIN3A gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer. 2008;59:24–31. doi: 10.1016/j.lungcan.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 86.Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Yukiue H, Kaji M, Fukai I, Kiriyama M, Yamakawa Y, Fujii Y. Expression of the MTA1 mRNA in advanced lung cancer. Lung Cancer. 2002;35:149–54. doi: 10.1016/s0169-5002(01)00329-4. [DOI] [PubMed] [Google Scholar]
- 87.Iguchi H, Imura G, Toh Y, Ogata Y. Expression of MTA1, a metastasis-associated gene with histone deacetylase activity in pancreatic cancer. Int J Oncol. 2000;16:1211–4. doi: 10.3892/ijo.16.6.1211. [DOI] [PubMed] [Google Scholar]
- 88.Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, Vadlamudi RK, Kumar R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–7. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 89.Toh Y, Kuninaka S, Endo K, Oshiro T, Ikeda Y, Nakashima H, Baba H, Kohnoe S, Okamura T, Nicolson GL, Sugimachi K. Molecular analysis of a candidate metastasis-associated gene, MTA1: possible interaction with histone deacetylase 1. J Exp Clin Cancer Res. 2000;19:105–11. [PubMed] [Google Scholar]
- 90.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–61. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 91.Yao YL, Yang WM. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J Biol Chem. 2003;278:42560–8. doi: 10.1074/jbc.M302955200. [DOI] [PubMed] [Google Scholar]
- 92.You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–8. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kishimoto M, Kohno T, Okudela K, Otsuka A, Sasaki H, Tanabe C, Sakiyama T, Hirama C, Kitabayashi I, Minna JD, Takenoshita S, Yokota J. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res. 2005;11:512–9. [PubMed] [Google Scholar]
- 94.Li Q, Xiao H, Isobe K. Histone acetyltrans-ferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J Gerontol A Biol Sci Med Sci. 2002;57:B93–8. doi: 10.1093/gerona/57.3.b93. [DOI] [PubMed] [Google Scholar]
- 95.Yamauchi T, Yamauchi J, Kuwata T, Tamura T, Yamashita T, Bae N, Westphal H, Ozato K, Nakatani Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc Natl Acad Sci USA. 2000;97:11303–6. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Naltner A, Wert S, Whitsett JA, Yan C. Temporal/spatial expression of nuclear receptor coactivators in the mouse lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1066–74. doi: 10.1152/ajplung.2000.279.6.L1066. [DOI] [PubMed] [Google Scholar]
- 97.Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, Wittwer J, Scheidweiler A, Eckner R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003;22:5175–85. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yin Z, Gonzales L, Kolla V, Rath N, Zhang Y, Lu MM, Kimura S, Ballard PL, Beers MF, Epstein JA, Morrisey EE. HOP functions downstream of Nkx2.1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. Am J Physiol Lung Cell Mol Physiol. 2006;291:L191–9. doi: 10.1152/ajplung.00385.2005. [DOI] [PubMed] [Google Scholar]
- 99.De Haan JB, Gevers W, Parker MI. Effects of sodium butyrate on the synthesis and methylation of DNA in normal cells and their transformed counterparts. Cancer Res. 1986;46:713–6. [PubMed] [Google Scholar]
- 100.Parker MI, De Haan JB, Gevers W. DNA hypermethylation in sodium butyrate-treated WI-38 fibroblasts. J Biol Chem. 1986;261:2786–90. [PubMed] [Google Scholar]
- 101.Takenaga K. Effect of butyric acid on lung-colonizing ability of cloned low-metastatic Lewis lung carcinoma cells. Cancer Res. 1986;46:1244–9. [PubMed] [Google Scholar]
- 102.Thomas GL, Henley A, Rowland TC, Sahai A, Griffin M, Birckbichler PJ. Enhanced apoptosis in transformed human lung fibroblasts after exposure to sodium butyrate. In Vitro Cell Dev Biol Anim. 1996;32:505–13. doi: 10.1007/BF02723054. [DOI] [PubMed] [Google Scholar]
- 103.Goto I, Yamamoto-Yamaguchi Y, Honma Y. Enhancement of sensitivity of human lung adenocarcinoma cells to growth-inhibitory activity of interferon alpha by differentiation-inducing agents. Br J Cancer. 1996;74:546–54. doi: 10.1038/bjc.1996.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sambucetti LC, Fischer DD, Zabludoff S, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–7. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 105.Desai D, Das A, Cohen L, El-Bayoumy K, Amin S. Chemopreventive efficacy of suberoylanilide hydroxamic acid (SAHA) against 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in female A/J mice. Anticancer Res. 2003;23:499–503. [PubMed] [Google Scholar]
- 106.Komatsu N, Kawamata N, Takeuchi S, Yin D, Chien W, Miller CW, Koeffler HP. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol Rep. 2006;15:187–91. [PubMed] [Google Scholar]
- 107.Mukhopadhyay NK, Weisberg E, Gilchrist D, Bueno R, Sugarbaker DJ, Jaklitsch MT. Effectiveness of trichostatin A as a potential candidate for anticancer therapy in non-small-cell lung cancer. Ann Thorac Surg. 2006;81:1034–42. doi: 10.1016/j.athoracsur.2005.06.059. [DOI] [PubMed] [Google Scholar]
- 108.Liu L-T, Chang H-C, Chiang L-C, Hung W-C. Histone Deacetylase Inhibitor Up-Regulates RECK to Inhibit MMP-2 Activation and Cancer Cell Invasion. Cancer Res. 2003;63:3069–72. [PubMed] [Google Scholar]
- 109.Toyooka S, Toyooka KO, Miyajima K, Reddy JL, Toyota M, Sathyanarayana UG, Padar A, Tockman MS, Lam S, Shivapurkar N, Gazdar AF. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin Cancer Res. 2003;9:3034–41. [PubMed] [Google Scholar]
- 110.Ohira T, Gemmill RM, Ferguson K, Kusy S, Roche J, Brambilla E, Zeng C, Baron A, Bemis L, Erickson P, Wilder E, Rustgi A, Kitajewski J, Gabrielson E, Bremnes R, Franklin W, Drabkin HA. WNT7a induces E-cadherin in lung cancer cells. Proc Natl Acad Sci USA. 2003;100:10429–34. doi: 10.1073/pnas.1734137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang S, Yan-Neale Y, Fischer D, Zeremski M, Cai R, Zhu J, Asselbergs F, Hampton G, Cohen D. Histone deacetylase 1 represses the small GTPase RhoB expression in human nonsmall lung carcinoma cell line. Oncogene. 2003;22:6204–13. doi: 10.1038/sj.onc.1206653. [DOI] [PubMed] [Google Scholar]
- 112.Mazieres J, Tovar D, He B, Nieto-Acosta J, Marty-Detraves C, Clanet C, Pradines A, Jablons DM, Favre G. Epigenetic regulation of RhoB loss of expression in lung cancer. BMC Cancer. 2007;7:220–6. doi: 10.1186/1471-2407-7-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choi YH. Induction of apoptosis by trichostatin A, a histone deacetylase inhibitor, is associated with inhibition of cyclooxygenase-2 activity in human non-small cell lung cancer cells. Int J Oncol. 2005;27:473–9. [PubMed] [Google Scholar]
- 114.Tong M, Ding Y, Tai HH. Histone deacetylase inhibitors and transforming growth factor-beta induce 15-hydroxyprostaglandin dehydrogenase expression in human lung adenocarcinoma cells. Biochem Pharmacol. 2006;72:701–9. doi: 10.1016/j.bcp.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 115.Sonnemann J, Hartwig M, Plath A, Saravana Kumar K, Muller C, Beck JF. Histone deacetylase inhibitors require cas-pase activity to induce apoptosis in lung and prostate carcinoma cells. Cancer Lett. 2006;232:148–60. doi: 10.1016/j.canlet.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 116.Kim HR, Kim EJ, Yang SH, Jeong ET, Park C, Lee JH, Youn MJ, So HS, Park R. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp Mol Med. 2006;38:616–24. doi: 10.1038/emm.2006.73. [DOI] [PubMed] [Google Scholar]
- 117.Imre G, Gekeler V, Leja A, Beckers T, Boehm M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer Res. 2006;66:5409–18. doi: 10.1158/0008-5472.CAN-05-4225. [DOI] [PubMed] [Google Scholar]
- 118.Sasakawa Y, Naoe Y, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Effects of FK228, a novel histone deacetylase inhibitor, on tumor growth and expression of p21 and c-myc genes in vivo. Cancer Lett. 2003;195:161–8. doi: 10.1016/s0304-3835(03)00184-8. [DOI] [PubMed] [Google Scholar]
- 119.Vinodhkumar R, Song YS, Ravikumar V, Ramakrishnan G, Devaki T. Depsipeptide a histone deacetlyase inhibitor down regulates levels of matrix metalloproteinases 2 and 9 mRNA and protein expressions in lung cancer cells (A549) Chem Biol Interact. 2007;165:220–9. doi: 10.1016/j.cbi.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 120.Radhakrishnan V, Song YS, Thiruvengadam D. Romidepsin (depsipeptide) induced cell cycle arrest, apop-tosis and histone hyperacetylation in lung carcinoma cells (A549) are associated with increase in p21 and hypophosphorylated retinoblastoma proteins expression. Biomed Pharmacother. 2008;62:85–93. doi: 10.1016/j.biopha.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 121.Remiszewski SW, Sambucetti LC, Bair KW, Bontempo J, Cesarz D, Chandramouli N, Chen R, Cheung M, Cornell-Kennon S, Dean K, Diamantidis G, France D, Green MA, Howell KL, Kashi R, Kwon P, Lassota P, Martin MS, Mou Y, Perez LB, Sharma S, Smith T, Sorensen E, Taplin F, Trogani N, Versace R, Walker H, Weltchek-Engler S, Wood A, Wu A, Atadja P. N-hydroxy-3-phenyl-2-propenamides as novel inhibitors of human histone deacetylase with in vivo antitumor activity: discovery of (2E)-N-hydroxy-3-[4-[[(2-hydroxyethyl)[2-(1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide (NVPLAQ824) J Med Chem. 2003;46:4609–24. doi: 10.1021/jm030235w. [DOI] [PubMed] [Google Scholar]
- 122.Loprevite M, Tiseo M, Grossi F, Scolaro T, Semino C, Pandolfi A, Favoni R, Ardizzoni A. In vitro study of CI-994, a histone deacetylase inhibitor, in non-small cell lung cancer cell lines. Oncol Res. 2005;15:39–48. doi: 10.3727/096504005775082066. [DOI] [PubMed] [Google Scholar]
- 123.Lee KW, Kim JH, Park JH, Kim HP, Song SH, Kim SG, Kim TY, Jong HS, Jung KH, Im SA, Kim TY, Kim NK, Bang YJ. Antitumor activity of SK-7041, a novel histone deacetylase inhibitor, in human lung and breast cancer cells. Anticancer Res. 2006;26:3429–38. [PubMed] [Google Scholar]
- 124.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 125.Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2001;61:1327–33. [PubMed] [Google Scholar]
- 126.Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, Fischette MR, Yu X, Chen GA, Hong JA, Stewart JH, Nguyen DM, Rosenberg SA, Schrump DS. Sequential 5-Aza-2 deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother. 2001;24:151–61. doi: 10.1097/00002371-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 127.Boivin AJ, Momparler LF, Hurtubise A, Momparler RL. Antineoplastic action of 5-aza-2’-deoxycytidine and phenylbutyrate on human lung carcinoma cells. Anticancer Drugs. 2002;13:869–74. doi: 10.1097/00001813-200209000-00013. [DOI] [PubMed] [Google Scholar]
- 128.Cantor JP, Lliopoulos D, Rao AS, Druck T, Semba S, Han SY, McCorkell KA, Lakshman TV, Collins JE, Wachsberger P, Friedberg JS, Huebner K. Epigenetic modulation of endogenous tumor suppressor expression in lung cancer xenografts suppresses tumorigenicity. Int J Cancer. 2007;120:24–31. doi: 10.1002/ijc.22073. [DOI] [PubMed] [Google Scholar]
- 129.Belinsky SA, Klinge DM, Stidley CA, Issa JP, Herman JG, March TH, Baylin SB. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–93. [PubMed] [Google Scholar]
- 130.Um SJ, Han HS, Kwon YJ, Park SH, Bae TS, Rho YS, Sin HS. In vitro antitumor potential of 4-BPRE, a butyryl aminophenyl ester of retinoic acid: role of the butyryl group. Oncol Rep. 2004;11:719–26. [PubMed] [Google Scholar]
- 131.Denlinger CE, Keller MD, Mayo MW, Broad RM, Jones DR. Combined proteasome and histone deacetylase inhibition in non-small cell lung cancer. J Thorac Cardiovasc Surg. 2004;127:1078–86. doi: 10.1016/s0022-5223(03)01321-7. [DOI] [PubMed] [Google Scholar]
- 132.Denlinger CE, Rundall BK, Jones DR. Proteasome inhibition sensitizes non-small cell lung cancer to histone deacetylase inhibitor-induced apoptosis through the generation of reactive oxygen species. J Thorac Cardiovasc Surg. 2004;128:740–8. doi: 10.1016/j.jtcvs.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 133.Rundall BK, Denlinger CE, Jones DR. Combined histone deacetylase and NF-kappaB inhibition sensitizes non-small cell lung cancer to cell death. Surgery. 2004;136:416–25. doi: 10.1016/j.surg.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 134.Maxhimer JB, Reddy RM, Zuo J, Cole GW, Schrump DS, Nguyen DM. Induction of apoptosis of lung and esophageal cancer cells treated with the combination of histone deacetylase inhibitor (trichostatin A) and protein kinase C inhibitor (calphostin C) J Thorac Cardiovasc Surg. 2005;129:53–63. doi: 10.1016/j.jtcvs.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 135.Sonnemann J, Gange J, Kumar KS, Muller C, Bader P, Beck JF. Histone deacetylase inhibitors interact synergisti-cally with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to induce apoptosis in carcinoma cell lines. Invest New Drugs. 2005;23:99–109. doi: 10.1007/s10637-005-5854-9. [DOI] [PubMed] [Google Scholar]
- 136.Rundall BK, Denlinger CE, Jones DR. Suberoylanilide hydroxamic acid combined with gemcitabine enhances apopto-sis in non-small cell lung cancer. Surgery. 2005;138:360–7. doi: 10.1016/j.surg.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 137.Schniewind B, Heintz K, Kurdow R, Ammerpohl O, Trauzold A, Emme D, Dohrmann P, Kalthoff H. Combination phenylbutyrate/gemcitabine therapy effectively inhibits in vitro and in vivo growth of NSCLC by intrinsic apoptotic pathways. J Carcinog. 2006;5:25–35. doi: 10.1186/1477-3163-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Denlinger CE, Rundall BK, Jones DR. Inhibition of phosphatidylinositol 3-kinase/Akt and histone deacetylase activity induces apoptosis in non-small cell lung cancer in vitro and in vivo. J Thorac Cardiovasc Surg. 2005;130:1422–9. doi: 10.1016/j.jtcvs.2005.06.051. [DOI] [PubMed] [Google Scholar]
- 139.Yu C, Friday BB, Lai JP, McCollum A, Atadja P, Roberts LR, Adjei AA. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res. 2007;13:1140–8. doi: 10.1158/1078-0432.CCR-06-1751. [DOI] [PubMed] [Google Scholar]
- 140.Yu XD, Wang SY, Chen GA, Hou CM, Zhao M, Hong JA, Nguyen DM, Schrump DS. Apoptosis induced by depsipeptide FK228 coincides with inhibition of survival signaling in lung cancer cells. Cancer J. 2007;13:105–13. doi: 10.1097/PPO.0b013e318046eedc. [DOI] [PubMed] [Google Scholar]
- 141.Watanabe T, Hioki M, Fujiwara T, Nishizaki M, Kagawa S, Taki M, Kishimoto H, Endo Y, Urata Y, Tanaka N, Fujiwara T. Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp Cell Res. 2006;312:256–65. doi: 10.1016/j.yexcr.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 142.Moody TW, Nakagawa T, Kang Y, Jakowlew S, Chan D, Jensen RT. Bombesin/gastrin-releasing peptide receptor antagonists increase the ability of histone deacetylase inhibitors to reduce lung cancer proliferation. J Mol Neurosci. 2006;28:231–8. doi: 10.1385/JMN:28:3:231. [DOI] [PubMed] [Google Scholar]
- 143.Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- 144.Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs. 2007;18:793–800. doi: 10.1097/CAD.0b013e3280b10d57. [DOI] [PubMed] [Google Scholar]
- 145.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 146.Edwards A, Li J, Atadja P, Bhalla K, Haura EB. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol Cancer Ther. 2007;6:2515–24. doi: 10.1158/1535-7163.MCT-06-0761. [DOI] [PubMed] [Google Scholar]
- 147.Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene. 2008;27:3134–44. doi: 10.1038/sj.onc.1210976. [DOI] [PubMed] [Google Scholar]
- 148.Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, Yoo DH, Lee SJ, An S, Rhee CH, Hong SI, Choe TB, Park IC. Combined effects of sulindac and suberoylanilide hydroxamic acid on apoptosis induction in human lung cancer cells. Mol Pharmacol. 2008;73:1005–12. doi: 10.1124/mol.107.041293. [DOI] [PubMed] [Google Scholar]
- 149.Gray SG, Eriksson T, Ekstrom TJ. Methylation, gene expression and the chromatin connection in cancer (review) Int J Mol Med. 1999;4:333–50. doi: 10.3892/ijmm.4.4.333. [DOI] [PubMed] [Google Scholar]
- 150.Sistayanarain A, Tsuneyama K, Zheng H, Takahashi H, Nomoto K, Cheng C, Murai Y, Tanaka A, Takano Y. Expression of Aurora-B kinase and phosphorylated histone H3 in hepatocellular carcinoma. Anticancer Res. 2006;26:3585–93. [PubMed] [Google Scholar]
- 151.Gray SG, Hartmann W, Eriksson T, Ekstrom C, Holm S, Kytola S, Von Schweinitz D, Pietsch T, Larsson C, Kogner P, Sandstedt B, Ekstrom TJ. Expression of genes involved with cell cycle control, cell growth and chromatin modification are altered in hepatoblastomas. Int J Mol Med. 2000;6:161–9. [PubMed] [Google Scholar]
- 152.Rikimaru T, Taketomi A, Yamashita Y, Yamashita Y, Shirabe K, Hamatsu T, Shimada M, Maehara Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- 153.Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–76. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 154.Wang AG, Seo SB, Moon HB, Shin HJ, Kim DH, Kim JM, Lee TH, Kwon HJ, Yu DY, Lee DS. Hepatic steatosis in transgenic mice overexpressing human histone deacetylase 1. Biochem Biophys Res Commun. 2005;330:461–6. doi: 10.1016/j.bbrc.2005.02.179. [DOI] [PubMed] [Google Scholar]
- 155.Nugent C, Younossi ZM. Evaluation and management of obesity-related nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4:432–41. doi: 10.1038/ncpgasthep0879. [DOI] [PubMed] [Google Scholar]
- 156.Tsuge M, Hamamoto R, Silva FP, Ohnishi Y, Chayama K, Kamatani N, Furukawa Y, Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5’ flanking region of SMYD3 is a risk factor for human cancers. Nat Genet. 2005;37:1104–7. doi: 10.1038/ng1638. [DOI] [PubMed] [Google Scholar]
- 157.Hamamoto R, Furukawa Y, Morita M, Limura Y, Silva FP, Li M, Yagyu R, Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004;6:731–40. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 158.Kurabe N, Katagiri K, Komiya Y, Ito R, Sugiyama A, Kawasaki Y, Tashiro F. Deregulated expression of a novel component of TFTC/STAGA histone acetyltransferase complexes, rat SGF29, in hepatocellular carcinoma: possible implication for the oncogenic potential of c-Myc. Oncogene. 2007;26:5626–34. doi: 10.1038/sj.onc.1210349. [DOI] [PubMed] [Google Scholar]
- 159.Hamatsu T, Rikimaru T, Yamashita Y, Aishima S, Tanaka S, Shirabe K, Shimada M, Toh Y, Sugimachi K. The role of MTA1 gene expression in human hepatocellular carcinoma. Oncol Rep. 2003;10:599–604. [PubMed] [Google Scholar]
- 160.Moon WS, Chang K, Tarnawski AS. Overexpression of metastatic tumor antigen 1 in hepatocellular carcinoma: relationship to vascular invasion and estrogen receptor-alpha. Hum Pathol. 2004;35:424–9. doi: 10.1016/j.humpath.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 161.Moon HE, Cheon H, Lee MS. Metastasis-associated protein 1 inhibits p53-induced apoptosis. Oncol Rep. 2007;18:1311–4. [PubMed] [Google Scholar]
- 162.Gray SG, Kytola S, Lui WO, Larsson C, Ekstrom TJ. Modulating IGFBP-3 expression by trichostatin A: potential therapeutic role in the treatment of hepatocellular carcinoma. Int J Mol Med. 2000;5:33–41. doi: 10.3892/ijmm.5.1.33. [DOI] [PubMed] [Google Scholar]
- 163.Gray SG, Yakovleva T, Hartmann W, Tally M, Bakalin G, Ekström TJ. IGF-II enhances trichostatin A-induced TGFβ1 and p21Waf1,Cip1,Sdi1 expression in Hep3B cells. Exp Cell Res. 1999;253:618–28. doi: 10.1006/excr.1999.4661. [DOI] [PubMed] [Google Scholar]
- 164.Nakamura M, Saito H, Ebinuma H, Wakabayashi K, Saito Y, Takagi T, Nakamoto N, Ishii H. Reduction of telom-erase activity in human liver cancer cells by a histone deacetylase inhibitor. J Cell Physiol. 2001;187:392–401. doi: 10.1002/jcp.1087. [DOI] [PubMed] [Google Scholar]
- 165.Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG, Schuppan D. The histone-deacetylase inhibitor trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol. 2002;36:233–40. doi: 10.1016/s0168-8278(01)00257-4. [DOI] [PubMed] [Google Scholar]
- 166.Yamashita Y, Shimada M, Harimoto N, Rikimaru T, Shirabe K, Tanaka S, Sugimachi K. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. 2003;103:572–6. doi: 10.1002/ijc.10699. [DOI] [PubMed] [Google Scholar]
- 167.Gray SG, Qian CN, Furge K, Guo X, Teh BT. Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int J Oncol. 2004;24:773–95. doi: 10.3892/ijo.24.4.773. [DOI] [PubMed] [Google Scholar]
- 168.Chiba T, Yokosuka O, Arai M, Tada M, Fukai K, Imazeki F, Kato M, Seki N, Saisho H. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J Hepatol. 2004;41:436–45. doi: 10.1016/j.jhep.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 169.Chiba T, Yokosuka O, Fukai K, Kojima H, Tada M, Arai M, Imazeki F, Saisho H. Cell growth inhibition and gene expression induced by the histone deacetylase inhibitor, trichostatin A, on human hepatoma cells. Oncology. 2004;66:481–91. doi: 10.1159/000079503. [DOI] [PubMed] [Google Scholar]
- 170.Wakabayashi K, Saito H, Kaneko F, Nakamoto N, Tada S, Hibi T. Gene expression associated with the decrease in malignant phenotype of human liver cancer cells following stimulation with a histone deacetylase inhibitor. Int J Oncol. 2005;26:233–9. [PubMed] [Google Scholar]
- 171.Tong A, Zhang H, Li Z, Gou L, Wang Z, Wei H, Tang M, Liang S, Chen L, Huang C, Wei Y. Proteomic analysis of liver cancer cells treated with suberonylanilide hydroxamic acid. Cancer Chemother Pharmacol. 2008;61:791–802. doi: 10.1007/s00280-007-0536-2. [DOI] [PubMed] [Google Scholar]
- 172.Svechnikova I, Gray SG, Kundrotiene J, Ponthan F, Kogner P, Ekstrom TJ. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int J Oncol. 2003;22:579–88. [PubMed] [Google Scholar]
- 173.Coradini D, Zorzet S, Rossin R, Scarlata I, Pellizzaro C, Turrin C, Bello M, Cantoni S, Speranza A, Sava G, Mazzi U, Perbellini A. Inhibition of hepatocellular carcinomas in vitro and hepatic metastases in vivo in mice by the histone deacetylase inhibitor HA-But. Clin Cancer Res. 2004;10:4822–30. doi: 10.1158/1078-0432.CCR-04-0349. [DOI] [PubMed] [Google Scholar]
- 174.Lu YS, Kashida Y, Kulp SK, Wang YC, Wang D, Hung JH, Tang M, Lin ZZ, Chen TJ, Cheng AL, Chen CS. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology. 2007;46:1119–30. doi: 10.1002/hep.21804. [DOI] [PubMed] [Google Scholar]
- 175.Kaneko F, Saito H, Saito Y, Wakabayashi K, Nakamoto N, Tada S, Suzuki H, Tsunematsu S, Kumagai N, Ishii H. Down-regulation of matrix-invasive potential of human liver cancer cells by type I interferon and a histone deacetylase inhibitor sodium butyrate. Int J Oncol. 2004;24:837–45. [PubMed] [Google Scholar]
- 176.Armeanu S, Pathil A, Venturelli S, Mascagni P, Weiss TS, Gottlicher M, Gregor M, Lauer UM, Bitzer M. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol. 2005;42:210–7. doi: 10.1016/j.jhep.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 177.Jung JW, Cho SD, Ahn NS, Yang SR, Park JS, Jo EH, Hwang JW, Jung JY, Kim SH, Kang KS, Lee YS. Ras/MAP kinase pathways are involved in Ras specific apoptosis induced by sodium butyrate. Cancer Lett. 2005;225:199–206. doi: 10.1016/j.canlet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 178.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, Steinle A, Salih HR. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- 179.Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, Wehrmann M, Gregor M, Lauer UM, Bitzer M. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer. 2007;109:2132–41. doi: 10.1002/cncr.22652. [DOI] [PubMed] [Google Scholar]
- 180.Ocker M, Alajati A, Ganslmayer M, Zopf S, Luders M, Neureiter D, Hahn EG, Schuppan D, Herold C. The histone-deacetylase inhibitor SAHA potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cells. J Cancer Res Clin Oncol. 2005;131:385–94. doi: 10.1007/s00432-004-0664-6. [DOI] [PubMed] [Google Scholar]
- 181.Schuchmann M, Schulze-Bergkamen H, Fleischer B, Schattenberg JM, Siebler J, Weinmann A, Teufel A, Worns M, Fischer T, Strand S, Lohse AW, Galle PR. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol Rep. 2006;15:227–30. doi: 10.3892/or.15.1.227. [DOI] [PubMed] [Google Scholar]
- 182.Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, Lauer UM, Bitzer M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43:425–34. doi: 10.1002/hep.21054. [DOI] [PubMed] [Google Scholar]
- 183.Emanuele S, Lauricella M, Carlisi D, Vassallo B, D’Anneo A, Di Fazio P, Vento R, Tesoriere G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor bortezomib. Apoptosis. 2007;12:1327–38. doi: 10.1007/s10495-007-0063-y. [DOI] [PubMed] [Google Scholar]
- 184.Gray SG, De Meyts P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab Res Rev. 2005;21:416–33. doi: 10.1002/dmrr.559. [DOI] [PubMed] [Google Scholar]
- 185.Yamauchi T, Oike Y, Kamon J, Waki H, Komeda K, Tsuchida A, Date Y, Li MX, Miki H, Akanuma Y, Nagai R, Kimura S, Saheki T, Nakazato M, Naitoh T, Yamamura K, Kadowaki T. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet. 2002;30:221–6. doi: 10.1038/ng829. [DOI] [PubMed] [Google Scholar]
- 186.James S, Kaiser C. Methods for identification of compounds modulating insulin resistance. In: KC JS, JS KC, editors. BIOVITRUM AB (SE) 2003. [Google Scholar]
- 187.Schein PS, Cooney DA, Vernon ML. The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer Res. 1967;27:2324–32. [PubMed] [Google Scholar]
- 188.Stauffacher W, Burr I, Gutzeit A, Beaven D, Veleminsky J, Renold AE. Streptozotocin diabetes: time course of irreversible B-cell damage; further observations on prevention by nicotinamide. Proc Soc Exp Biol Med. 1970;133:194–200. doi: 10.3181/00379727-133-34439. [DOI] [PubMed] [Google Scholar]
- 189.Sandler S, Andersson A, Korsgren O, Tollemar J, Petersson B, Groth CG, Hellerstrom C. Tissue culture of human fetal pancreas. Effects of nicotinamide on insulin production and formation of isletlike cell clusters. Diabetes. 1989;38:168–71. doi: 10.2337/diab.38.1.s168. [DOI] [PubMed] [Google Scholar]
- 190.Korsgren O, Andersson A, Sandler S. Pretreatment of fetal porcine pancreas in culture with nicotinamide accelerates reversal of diabetes after transplantation to nude mice. Surgery. 1993;113:205–14. [PubMed] [Google Scholar]
- 191.Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925–31. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 192.Hedman M, Ludvigsson J, Faresjo MK. Nicotinamide reduces high secretion of IFN-gamma in high-risk relatives even though it does not prevent type 1 diabetes. J Interferon Cytokine Res. 2006;26:207–13. doi: 10.1089/jir.2006.26.207. [DOI] [PubMed] [Google Scholar]
- 193.Liu HK, Green BD, Flatt PR, McClenaghan NH, McCluskey JT. Effects of long-term exposure to nicotinamide and sodium butyrate on growth, viability, and the function of clonal insulin secreting cells. Endocr Res. 2004;30:61–8. doi: 10.1081/erc-120028485. [DOI] [PubMed] [Google Scholar]
- 194.Larsen L, Tonnesen M, Ronn SG, Storling J, Jorgensen S, Mascagni P, Dinarello CA, Billestrup N, Mandrup-Poulsen T. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia. 2007;50:779–89. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 195.Tveten K, Holla OL, Ranheim T, Berge KE, Leren TP, Kulseth MA. 4-Phenylbutyrate restores the functionality of a misfolded mutant low-density lipoprotein receptor. FEBS J. 2007;274:1881–93. doi: 10.1111/j.1742-4658.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- 196.Gangaram-Panday ST, Faas MM, De Vos P. Towards stem-cell therapy in the endocrine pancreas. Trends Mol Med. 2007;13:164–73. doi: 10.1016/j.molmed.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 197.Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32–8. doi: 10.1002/gene.10250. [DOI] [PubMed] [Google Scholar]
- 198.Romagnani P, Lasagni L, Mazzinghi B, Lazzeri E, Romagnani S. Pharmacological modulation of stem cell function. Curr Med Chem. 2007;14:1129–39. doi: 10.2174/092986707780362880. [DOI] [PubMed] [Google Scholar]
- 199.Okazaki K, Maltepe E. Oxygen, epigenetics and stem cell fate. Regen Med. 2006;1:71–83. doi: 10.2217/17460751.1.1.71. [DOI] [PubMed] [Google Scholar]
- 200.Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science. 2001;292:1389–94. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 201.Tayaramma T, Ma B, Rohde M, Mayer H. Chromatin-remodeling factors allow differentiation of bone marrow cells into insulin-producing cells. Stem Cells. 2006;24:2858–67. doi: 10.1634/stemcells.2006-0109. [DOI] [PubMed] [Google Scholar]
- 202.Sun Y, Chen L, Hou XG, Hou WK, Dong JJ, Sun L, Tang KX, Wang B, Song J, Li H, Wang KX. Differentiation of bone marrow-derived mesenchymal stem cells from diabetic patients into insulin-producing cells in vitro. Chin Med J. 2007;120:771–6. [PubMed] [Google Scholar]
- 203.Goicoa S, Alvarez S, Ricordi C, Inverardi L, Dominguez-Bendala J. Sodium butyrate activates genes of early pancreatic development in embryonic stem cells. Cloning Stem Cells. 2006 Fall;8:140–9. doi: 10.1089/clo.2006.8.140. [DOI] [PubMed] [Google Scholar]
- 204.Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G, Majumdar AS. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25:1940–53. doi: 10.1634/stemcells.2006-0761. [DOI] [PubMed] [Google Scholar]
- 205.Haumaitre C, Lenoir O, Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol Cell Biol. 2008;28:6373–83. doi: 10.1128/MCB.00413-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sarkander HI, Fleischer-Lambropoulos H, Brade WP. A comparative study of histone acetylation in neuronal and glial nuclei enriched rat brain fractions. FEBS Lett. 1975;52:40–3. doi: 10.1016/0014-5793(75)80633-8. [DOI] [PubMed] [Google Scholar]
- 207.Liu H, Hu Q, Kaufman A, D’Ercole AJ, Ye P. Developmental expression of histone deacetylase 11 in the murine brain. J Neurosci Res. 2008;86:537–43. doi: 10.1002/jnr.21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lakowski B, Roelens I, Jacob S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci. 2006;29:227–39. doi: 10.1385/JMN:29:3:227. [DOI] [PubMed] [Google Scholar]
- 209.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 210.Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–29. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 211.Gray SG, Dangond F. Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics. 2006;1:67–75. doi: 10.4161/epi.1.2.2678. [DOI] [PubMed] [Google Scholar]
- 212.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chro-matin throughout neurogenesis. Cell. 2005;121:645–57. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 213.Battaglioli E, Andres ME, Rose DW, Chenoweth JG, Rosenfeld MG, Anderson ME, Mandel G. REST repression of neuronal genes requires components of the hSWI.SNF complex. J Biol Chem. 2002;277:41038–45. doi: 10.1074/jbc.M205691200. [DOI] [PubMed] [Google Scholar]
- 214.Watanabe H, Mizutani T, Haraguchi T, Yamamichi N, Minoguchi S, Yamamichi-Nishina M, Mori N, Kameda T, Sugiyama T, Iba H. SWI/SNF complex is essential for NRSF-mediated suppression of neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene. 2006;25:470–9. doi: 10.1038/sj.onc.1209068. [DOI] [PubMed] [Google Scholar]
- 215.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–7. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 216.Iglesias AH, Camelo S, Hwang D, Villanueva R, Stephanopoulos G, Dangond F. Microarray detection of E2F pathway activation and other targets in multiple sclerosis peripheral blood mononuclear cells. J Neuroimmunol. 2004;150:163–77. doi: 10.1016/j.jneuroim.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 217.Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 218.Dangond F, Hafler DA, Tong JK, Randall J, Kojima R, Utku N, Gullans SR. Differential display cloning of a novel human histone deacetylase (HDAC3) cDNA from PHA-activated immune cells. Biochem Biophys Res Commun. 1998;242:648–52. doi: 10.1006/bbrc.1997.8033. [DOI] [PubMed] [Google Scholar]
- 219.Ryu H, Lee J, Olofsson BA, Mwidau A, Deodeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci USA. 2003;100:4281–6. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Humphrey GW, Wang YH, Hirai T, Padmanabhan R, Panchision DM, Newell LF, McKay RD, Howard BH. Complementary roles for histone deacetylases 1, 2, and 3 in differentiation of pluripotent stem cells. Differentiation. 2008;76:348–56. doi: 10.1111/j.1432-0436.2007.00232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Balasubramaniyan V, Boddeke E, Bakels R, Kust B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006;143:939–51. doi: 10.1016/j.neuroscience.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 222.Lyssiotis CA, Walker J, Wu C, Kondo T, Schultz PG, Wu X. Inhibition of histone deacetylase activity induces developmental plasticity in oligodendrocyte precursor cells. Proc Natl Acad Sci USA. 2007;104:14982–7. doi: 10.1073/pnas.0707044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Liu A, Han YR, Li J, Sun D, Ouyang M, Plummer MR, Casaccia-Bonnefil P. The glial or neuronal fate choice of oligodendrocyte progenitors is modulated by their ability to acquire an epigenetic memory. J Neurosci. 2007;27:7339–43. doi: 10.1523/JNEUROSCI.1226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–30. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sun G, Yu RT, Evans RM, Shi Y. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc Natl Acad Sci USA. 2007;104:15282–7. doi: 10.1073/pnas.0704089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Su X, Kameoka S, Lentz S, Majumder S. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol Cell Biol. 2004;24:8018–25. doi: 10.1128/MCB.24.18.8018-8025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Greenway DJ, Street M, Jeffries A, Buckley NJ. RE1 silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells. 2007;25:354–63. doi: 10.1634/stemcells.2006-0207. [DOI] [PubMed] [Google Scholar]
- 228.Sun YM, Greenway DJ, Johnson R, Street M, Belyaev ND, Deuchars J, Bee T, Wilde S, Buckley NJ. Distinct profiles of REST interactions with its target genes at different stages of neuronal development. Mol Biol Cell. 2005;16:5630–8. doi: 10.1091/mbc.E05-07-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gray SG, Iglesias AH, Teh BT, Dangond F. Modulation of splicing events in histone deacetylase 3 by various extracellular and signal transduction pathways. Gene Expr. 2003;11:13–21. doi: 10.3727/000000003783992342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 231.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–74. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 232.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amy-otrophic lateral sclerosis. EMBO J. 2007;26:3169–79. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–95. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–98. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 235.Gardian G, Yang L, Cleren C, Calingasan NY, Klivenyi P, Beal MF. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromolecular Med. 2004;5:235–41. doi: 10.1385/NMM:5:3:235. [DOI] [PubMed] [Google Scholar]
- 236.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–23. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 237.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–9. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 238.Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a his-tone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–9. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 239.Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amy-otrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–45. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 241.Elaut G, Torok G, Vinken M, Laus G, Papeleu P, Tourwe D, Rogiers V. Major phase I biotransformation pathways of Trichostatin a in rat hepatocytes and in rat and human liver microsomes. Drug Metab Dispos. 2002;30:1320–8. doi: 10.1124/dmd.30.12.1320. [DOI] [PubMed] [Google Scholar]
- 242.Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, Vigushin DM. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab Dispos. 2004;32:1132–8. doi: 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- 243.Shiraga T, Tozuka Z, Ishimura R, Kawamura A, Kagayama A. Identification of cytochrome P450 enzymes involved in the metabolism of FK228, a potent histone deacetylase inhibitor, in human liver micro-somes. Biol Pharm Bull. 2005;28:124–9. doi: 10.1248/bpb.28.124. [DOI] [PubMed] [Google Scholar]
- 244.Acharya MR, Karp JE, Sausville EA, Hwang K, Ryan Q, Gojo I, Venitz J, Figg WD, Sparreboom A. Factors affecting the pharmacokinetic profile of MS-275, a novel histone deacetylase inhibitor, in patients with cancer. Invest New Drugs. 2006;24:367–75. doi: 10.1007/s10637-005-5707-6. [DOI] [PubMed] [Google Scholar]
- 245.Kim TY, Bang YJ, Robertson KD. Histone deacetylase inhibitors for cancer therapy. Epigenetics. 2006;1:14–23. doi: 10.4161/epi.1.1.2644. [DOI] [PubMed] [Google Scholar]
- 246.Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–88. [PubMed] [Google Scholar]
- 247.Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Krug LM, Curley T, Schwartz L, Richardson S, Marks P, Chiao J, Kelly WK. Potential role of histone deacetylase inhibitors in mesothelioma: clinical experience with suberoylanilide hydroxamic acid. Clin Lung Cancer. 2006;7:257–61. doi: 10.3816/CLC.2006.n.003. [DOI] [PubMed] [Google Scholar]
- 249.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, Verbois L, Morse DE, Jee JM, Pope S, Harapanhalli RS, Dagher R, Farrell A, Justice R, Pazdur R. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. 2007;13:2318–22. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
- 251.Blumenschein GR, Jr, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinzatrade mark, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–7. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 252.Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26:483–8. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 253.Gilbert J, Baker SD, Bowling MK, Grochow L, Figg WD, Zabelina Y, Donehower RC, Carducci MA. A phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin Cancer Res. 2001;7:2292–300. [PubMed] [Google Scholar]
- 254.Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, Zabelina Y, Chen TL, Grochow LB, Donehower RC. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res. 2001;7:3047–55. [PubMed] [Google Scholar]
- 255.Gore SD, Weng LJ, Figg WD, Zhai S, Donehower RC, Dover G, Grever MR, Griffin C, Grochow LB, Hawkins A, Burks K, Zabelena Y, Miller CB. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res. 2002;8:963–70. [PubMed] [Google Scholar]
- 256.Gore SD, Weng LJ, Zhai S, Figg WD, Donehower RC, Dover GJ, Grever M, Griffin CA, Grochow LB, Rowinsky EK, Zabalena Y, Hawkins AL, Burks K, Miller CB. Impact of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res. 2001;7:2330–9. [PubMed] [Google Scholar]
- 257.Hogarth P, Lovrecic L, Krainc D. Sodium phenylbutyrate in Huntington's disease: a dose-finding study. Mov Disord. 2007;22:1962–4. doi: 10.1002/mds.21632. [DOI] [PubMed] [Google Scholar]
- 258.Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13:256–9. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 259.Mercuri E, Bertini E, Messina S, Pelliccioni M, D’Amico A, Colitto F, Mirabella M, Tiziano FD, Vitali T, Angelozzi C, Kinali M, Main M, Brahe C. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul Disord. 2004;14:130–5. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 260.Mercuri E, Bertini E, Messina S, Solari A, D’Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68:51–5. doi: 10.1212/01.wnl.0000249142.82285.d6. [DOI] [PubMed] [Google Scholar]
- 261.Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee CK, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther. 2002;6:119–26. doi: 10.1006/mthe.2002.0639. [DOI] [PubMed] [Google Scholar]
- 262.Camacho LH, Olson J, Tong WP, Young CW, Spriggs DR, Malkin MG. Phase I dose escalation clinical trial of phenylbu-tyrate sodium administered twice daily to patients with advanced solid tumors. Invest New Drugs. 2007;25:131–8. doi: 10.1007/s10637-006-9017-4. [DOI] [PubMed] [Google Scholar]
- 263.Phuphanich S, Baker SD, Grossman SA, Carson KA, Gilbert MR, Fisher JD, Carducci MA. Oral sodium phenylbutyrate in patients with recurrent malignant gliomas: a dose escalation and pharmacologic study. Neuro Oncol. 2005;7:177–82. doi: 10.1215/S1152851704000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Atmaca A, Al-Batran SE, Maurer A, Neumann A, Heinzel T, Hentsch B, Schwarz SE, Hovelmann S, Gottlicher M, Knuth A, Jager E. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97:177–82. doi: 10.1038/sj.bjc.6603851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Sharma S, Symanowski J, Wong B, Dino P, Manno P, Vogelzang N. A phase II clinical trial of oral valproic acid in patients with castration-resistant prostate cancers using an intensive biomarker sampling Strategy. Transl Oncol. 2008;1:141–7. doi: 10.1593/tlo.08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wolff JE, Kramm C, Kortmann RD, Pietsch T, Rutkowski S, Jorch N, Gnekow A, Driever PH. Valproic acid was well tolerated in heavily pretreated pediatric patients with high-grade glioma. J Neurooncol. 2008;90:309–14. doi: 10.1007/s11060-008-9662-x. [DOI] [PubMed] [Google Scholar]
- 267.Lezin A, Gillet N, Olindo S, Signate A, Grandvaux N, Verlaeten O, Belrose G, De Carvalho Bittencourt M, Hiscott J, Asquith B, Burny A, Smadja D, Cesaire R, Willems L. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood. 2007;110:3722–8. doi: 10.1182/blood-2007-04-085076. [DOI] [PubMed] [Google Scholar]
- 268.Patnaik A, Rowinsky EK, Villalona MA, Hammond LA, Britten CD, Siu LL, Goetz A, Felton SA, Burton S, Valone FH, Eckhardt SG. A phase I study of pivaloyloxymethyl butyrate, a prodrug of the differentiating agent butyric acid, in patients with advanced solid malignancies. Clin Cancer Res. 2002;8:2142–8. [PubMed] [Google Scholar]
- 269.Reid T, Valone F, Lipera W, Irwin D, Paroly W, Natale R, Sreedharan S, Keer H, Lum B, Scappaticci F, Bhatnagar A. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer. 2004;45:381–6. doi: 10.1016/j.lungcan.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 270.Prakash S, Foster BJ, Meyer M, Wozniak A, Heilbrun LK, Flaherty L, Zalupski M, Radulovic L, Valdivieso M, LoRusso PM. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs. 2001;19:1–11. doi: 10.1023/a:1006489328324. [DOI] [PubMed] [Google Scholar]
- 271.Nemunaitis JJ, Orr D, Eager R, Cunningham CC, Williams A, Mennel R, Grove W, Olson S. Phase I study of oral CI-994 in combination with gemcitabine in treatment of patients with advanced cancer. Cancer J. 2003;9:58–66. doi: 10.1097/00130404-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 272.Undevia SD, Kindler HL, Janisch L, Olson SC, Schilsky RL, Vogelzang NJ, Kimmel KA, Macek TA, Ratain MJ. A phase I study of the oral combination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Ann Oncol. 2004;15:1705–11. doi: 10.1093/annonc/mdh438. [DOI] [PubMed] [Google Scholar]
- 273.Pauer LR, Olivares J, Cunningham C, Williams A, Grove W, Kraker A, Olson S, Nemunaitis J. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Invest. 2004;22:886–96. doi: 10.1081/cnv-200039852. [DOI] [PubMed] [Google Scholar]
- 274.Marshall JL, Rizvi N, Kauh J, Dahut W, Figuera M, Kang MH, Figg WD, Wainer I, Chaissang C, Li MZ, Hawkins MJ. A phase I trial of depsipeptide (FR901228) in patients with advanced cancer. J Exp Ther Oncol. 2002;2:325–32. doi: 10.1046/j.1359-4117.2002.01039.x. [DOI] [PubMed] [Google Scholar]
- 275.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8:718–28. [PubMed] [Google Scholar]
- 276.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, Chan KK, Grever MR. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–67. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 277.Fouladi M, Furman WL, Chin T, Freeman BB, 3rd, Dudkin L, Stewart CF, Krailo MD, Speights R, Ingle AM, Houghton PJ, Wright J, Adamson PC, Blaney SM. Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children's Oncology Group report. J Clin Oncol. 2006;24:3678–85. doi: 10.1200/JCO.2006.06.4964. [DOI] [PubMed] [Google Scholar]
- 278.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, Panageas K, Wright JJ, Pandolfi PP, Nimer SD. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826–32. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 279.Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin Genitourin Cancer. 2006;5:57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 280.Piekarz RL, Frye AR, Wright JJ, Steinberg SM, Liewehr DJ, Rosing DR, Sachdev V, Fojo T, Bates SE. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin Cancer Res. 2006;12:3762–73. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- 281.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]
- 282.Kummar S, Gutierrez M, Gardner ER, Donovan E, Hwang K, Chung EJ, Lee MJ, Maynard K, Kalnitskiy M, Chen A, Melillo G, Ryan QC, Conley B, Figg WD, Trepel JB, Zwiebel J, Doroshow JH, Murgo AJ. Phase I trial of MS-275, a histone deacety-lase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res. 2007;13:5411–7. doi: 10.1158/1078-0432.CCR-07-0791. [DOI] [PubMed] [Google Scholar]
- 283.Gore L, Rothenberg ML, O’Bryant CL, Schultz MK, Sandler AB, Coffin D, McCoy C, Schott A, Scholz C, Eckhardt SG. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14:4517–25. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. Phase I and pharmacoki-netic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–22. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 285.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, Karp JE. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–90. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Hauschild A, Trefzer U, Garbe C, Kaehler KC, Ugurel S, Kiecker F, Eigentler T, Krissel H, Schott A, Schadendorf D. Multicenter phase II trial of the histone deacetylase inhibitor pyridylmethyl-N-{4-[(2-aminophenyl)-carbamoyl]-benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Res. 2008;18:274–8. doi: 10.1097/CMR.0b013e328307c248. [DOI] [PubMed] [Google Scholar]
- 287.Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, Mita M, Beck J, Burris H, Ryan G, Atadja P, Butterfoss D, Dugan M, Culver K, Johnstone RW, Prince HM. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14:4500–10. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 288.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH, Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–9. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Steele NL, Plumb JA, Vidal L, Tjornelund J, Knoblauch P, Rasmussen A, Ooi CE, Buhl-Jensen P, Brown R, Evans TR, DeBono JS. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res. 2008;14:804–10. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 290.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81:170–6. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 291.Candelaria M, Gallardo-Rincon D, Arce C, Cetina L, Aguilar-Ponce JL, Arrieta O, Gonzalez-Fierro A, Chavez-Blanco A, De La Cruz-Hernandez E, Camargo MF, Trejo-Becerril C, Perez-Cardenas E, Perez-Plasencia C, Taja-Chayeb L, Wegman-Ostrosky T, Revilla-Vazquez A, Duenas-Gonzalez A. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18:1529–38. doi: 10.1093/annonc/mdm204. [DOI] [PubMed] [Google Scholar]
- 292.Arce C, Perez-Plasencia C, Gonzalez-Fierro A, De La Cruz-Hernandez E, Revilla-Vazquez A, Chavez-Blanco A, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Bargallo E, Villarreal P, Ramirez T, Vela T, Candelaria M, Camargo MF, Robles E, Duenas-Gonzalez A. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS ONE. 2006;1:e98. doi: 10.1371/journal.pone.0000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Maslak P, Chanel S, Camacho LH, Soignet S, Pandolfi PP, Guernah I, Warrell R, Nimer S. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20:212–7. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 294.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–9. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104:2717–25. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- 296.Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106:112–9. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- 297.Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, Germing U, Haas R, Gattermann N. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol. 2005;84:61–6. doi: 10.1007/s00277-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 298.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–8. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 299.Minister P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, Simon G, Fishman M, Minton S, Garrett C, Chiappori A, Lush R, Sullivan D, Daud A. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. 2007;25:1979–85. doi: 10.1200/JCO.2006.08.6165. [DOI] [PubMed] [Google Scholar]
- 300.Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007;13:3605–10. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 301.Sung MW, Waxman S. Combination of cytotoxic-differentiation therapy with 5-fluorouracil and phenylbutyrate in patients with advanced colorectal cancer. Anticancer Res. 2007;27:995–1001. [PubMed] [Google Scholar]
- 302.Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, Grove W, Macdonald K, Gulyas S, Clark M, Dasse KD. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multi-center study. Ann Oncol. 2006;17:1096–102. doi: 10.1093/annonc/mdl081. [DOI] [PubMed] [Google Scholar]
- 303.Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, Jones DR. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the Akt pathway. J Biol Chem. 2003;278:18980–9. doi: 10.1074/jbc.M211695200. [DOI] [PubMed] [Google Scholar]
- 304.Liu Y, Denlinger CE, Rundall BK, Smith PW, Jones DR. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J Biol Chem. 2006;281:31359–68. doi: 10.1074/jbc.M604478200. [DOI] [PubMed] [Google Scholar]
- 305.Greco R, Latini G, Chiarelli F, Iannetti P, Verrotti A. Leptin, ghrelin, and adiponectin in epileptic patients treated with valproic acid. Neurology. 2005;65:1808–9. doi: 10.1212/01.wnl.0000187074.27586.d1. [DOI] [PubMed] [Google Scholar]
- 306.Pylvanen V, Knip M, Pakarinen AJ, Turkka J, Kotila M, Rattya J, Myllyla VV, Isojarvi JI. Fasting serum insulin and lipid levels in men with epilepsy. Neurology. 2003;60:571–4. doi: 10.1212/01.wnl.0000048209.07526.86. [DOI] [PubMed] [Google Scholar]
- 307.Pylvanen V, Pakarinen A, Knip M, Isojarvi J. Characterization of insulin secretion in Valproate-treated patients with epilepsy. Epilepsia. 2006;47:1460–4. doi: 10.1111/j.1528-1167.2006.00546.x. [DOI] [PubMed] [Google Scholar]
- 308.Pylvanen V, Pakarinen A, Knip M, Isojarvi J. Insulin-related metabolic changes during treatment with valproate in patients with epilepsy. Epilepsy Behav. 2006;8:643–8. doi: 10.1016/j.yebeh.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 309.Qiao L, Schaack J, Shao J. Suppression of adiponectin gene expression by histone deacetylase inhibitor valproic acid. Endocrinology. 2006;147:865–74. doi: 10.1210/en.2005-1030. [DOI] [PubMed] [Google Scholar]
- 310.Tan H, Orbak Z, Kantarci M, Kocak N, Karaca L. Valproate-induced insulin resistance in prepubertal girls with epilepsy. J Pediatr Endocrinol Metab. 2005;18:985–9. doi: 10.1515/JPEM.2005.18.10.985. [DOI] [PubMed] [Google Scholar]
- 311.Verrotti A, Di Corcia G, Salladini C, Trotta D, Morgese G, Chiarelli F. Serum insulin and leptin levels and valproate-associated obesity. Epilepsia. 2003;44:1606. doi: 10.1111/j.0013-9580.2003.t01-2-33003.x. [DOI] [PubMed] [Google Scholar]
- 312.Lagace DC, Nachtigal MW. Inhibition of histone deacetylase activity by valproic acid blocks adipogenesis. J Biol Chem. 2004;279:18851–60. doi: 10.1074/jbc.M312795200. [DOI] [PubMed] [Google Scholar]
- 313.Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61:203–6. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- 314.Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem. 2003;87:407–16. doi: 10.1046/j.1471-4159.2003.02004.x. [DOI] [PubMed] [Google Scholar]
- 315.Haggarty SJ, Koeller KM, Wong JC, Butcher RA, Schreiber SL. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem Biol. 2003;10:383–96. doi: 10.1016/s1074-5521(03)00095-4. [DOI] [PubMed] [Google Scholar]
- 316.Kim YK, Lee EK, Kang JK, Kim JA, You JS, Park JH, Seo DW, Hwang JW, Kim SN, Lee HY, Lee HW, Han JW. Activation of NF-kappaB by HDAC inhibitor apicidin through Sp1-dependent de novo protein synthesis: its implication for resistance to apoptosis. Cell Death Differ. 2006;13:2033–41. doi: 10.1038/sj.cdd.4401915. [DOI] [PubMed] [Google Scholar]
- 317.Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antit-rypsin in patients with alpha-1-antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr. 2004;39:34–7. doi: 10.1097/00005176-200407000-00007. [DOI] [PubMed] [Google Scholar]
- 318.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 319.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–66. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc’h C, Matthias P, Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, Yao TP. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol. 2007;27:8637–47. doi: 10.1128/MCB.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Fiskus W, Ren Y, Mohapatra A, Bali P, Mandawat A, Rao R, Herger B, Yang Y, Atadja P, Wu J, Bhalla K. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13:4882–90. doi: 10.1158/1078-0432.CCR-06-3093. [DOI] [PubMed] [Google Scholar]
- 323.Wang Y, Wang SY, Zhang XH, Zhao M, Hou CM, Xu YJ, Du ZY, Yu XD. FK228 inhibits Hsp90 chaperone function in K562 cells via hyperacetylation of Hsp70. Biochem Biophys Res Commun. 2007;356:998–1003. doi: 10.1016/j.bbrc.2007.03.076. [DOI] [PubMed] [Google Scholar]
- 324.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. 2007;25:151–9. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 326.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Johnson CA, White DA, Lavender JS, O’Neill LP, Turner BM. Human Class I Histone Deacetylase Complexes Show Enhanced Catalytic Activity in the Presence of ATP and Co-immunoprecipitate with the ATP-dependent Chaperone Protein Hsp70. J Biol Chem. 2002;277:9590–7. doi: 10.1074/jbc.M107942200. [DOI] [PubMed] [Google Scholar]
- 328.Liu XL, Done SC, Yan K, Kilpelainen P, Pikkarainen T, Tryggvason K. Defective trafficking of nephrin missense mutants rescued by a chemical chaperone. J Am Soc Nephrol. 2004;15:1731–8. doi: 10.1097/01.asn.0000129826.28932.fd. [DOI] [PubMed] [Google Scholar]
- 329.Yam GH, Roth J, Zuber C. 4-Phenylbutyrate rescues trafficking incompetent mutant alpha-galactosidase A without restoring its functionality. Biochem Biophys Res Commun. 2007;360:375–80. doi: 10.1016/j.bbrc.2007.06.048. [DOI] [PubMed] [Google Scholar]
- 330.Datta R, Waheed A, Shah GN, Sly WS. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci USA. 2007;104:19989–94. doi: 10.1073/pnas.0708725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Watanabe N, Lam E. BAX inhibitor-1 modulates endoplasmic reticulum stress-mediated programmed cell death in Arabidopsis. J Biol Chem. 2008;283:3200–10. doi: 10.1074/jbc.M706659200. [DOI] [PubMed] [Google Scholar]
- 332.Corson TW, Woo KK, Li PP, Warsh JJ. Cell-type specific regulation of calreticulin and Bcl-2 expression by mood stabilizer drugs. Eur Neuropsychopharmacol. 2004;14:143–50. doi: 10.1016/S0924-977X(03)00102-0. [DOI] [PubMed] [Google Scholar]
- 333.Wang JF, Bown CD, Chen B, Young LT. Identification of mood stabilizer-regulated genes by differential-display PCR. Int J Neuropsychopharmacol. 2001;4:65–74. doi: 10.1017/S1461145701002231. [DOI] [PubMed] [Google Scholar]
- 334.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 335.Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–67. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 336.Morland C, Boldingh KA, Iversen EG, Hassel B. Valproate is neuroprotective against malonate toxicity in rat striatum: an association with augmentation of high-affinity glutamate uptake. J Cereb Blood Flow Metab. 2004;24:1226–34. doi: 10.1097/01.WCB.0000138666.25305.A7. [DOI] [PubMed] [Google Scholar]
- 337.Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, Taira T, Ariga H, Shimohama S. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem. 2007;101:1491–504. doi: 10.1111/j.1471-4159.2006.04440.x. [DOI] [PubMed] [Google Scholar]
- 338.Pan T, Li X, Xie W, Jankovic J, Le W. Valproic acid-mediated Hsp70 induction and anti-apoptotic neuroprotection in SH-SY5Y cells. FEBS Lett. 2005;579:6716–20. doi: 10.1016/j.febslet.2005.10.067. [DOI] [PubMed] [Google Scholar]
- 339.Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium, GRP78 and Bcl-2. Pharmacogenomics J. 2005;5:102–11. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- 340.Singh OV, Vij N, Mogayzel PJ, Jr, Jozwik C, Pollard HB, Zeitlin PL. Pharmacoproteomics of 4-phenylbutyrate-treated IB3–1 cystic fibrosis bronchial epithelial cells. J Proteome Res. 2006;5:562–71. doi: 10.1021/pr050319o. [DOI] [PubMed] [Google Scholar]
- 341.Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–90. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- 342.Rubenstein RC, Lyons BM. Sodium 4-phenylbutyrate downregulates HSC70 expression by facilitating mRNA degradation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L43–51. doi: 10.1152/ajplung.2001.281.1.L43. [DOI] [PubMed] [Google Scholar]
- 343.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–65. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Lim M, McKenzie K, Floyd AD, Kwon E, Zeitlin PL. Modulation of deltaF508 cystic fibrosis transmembrane regulator trafficking and function with 4-phenylbutyrate and flavonoids. Am J Respir Cell Mol Biol. 2004;31:351–7. doi: 10.1165/rcmb.2002-0086OC. [DOI] [PubMed] [Google Scholar]
- 345.Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol. 2001;281:L58–68. doi: 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- 346.Cheong N, Madesh M, Gonzales LW, Zhao M, Yu K, Ballard PL, Shuman H. Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J Biol Chem. 2006;281:9791–800. doi: 10.1074/jbc.M507515200. [DOI] [PubMed] [Google Scholar]
- 347.Hayashi H, Sugiyama Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology. 2007;45:1506–16. doi: 10.1002/hep.21630. [DOI] [PubMed] [Google Scholar]
- 348.Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- 349.Bevins RL, Zimmer SG. It's about time: scheduling alters effect of histone deacetylase inhibitors on camptothecin-treated cells. Cancer Res. 2005;65:6957–66. doi: 10.1158/0008-5472.CAN-05-0836. [DOI] [PubMed] [Google Scholar]
- 350.Miyanaga A, Gemma A, Noro R, Kataoka K, Matsuda K, Nara M, Okano T, Seike M, Yoshimura A, Kawakami A, Uesaka H, Nakae H, Kudoh S. Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol Cancer Ther. 2008;7:1923–30. doi: 10.1158/1535-7163.MCT-07-2140. [DOI] [PubMed] [Google Scholar]
- 351.Itoh Y, Suzuki T, Miyata N. Isoform-selective histone deacetylase inhibitors. Curr Pharm Des. 2008;14:529–44. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- 352.Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–28. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- 353.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–34. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 354.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the protooncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 355.Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66:1277–81. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- 356.Lujambio A, Esteller M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle. 2007;6:1455–9. [PubMed] [Google Scholar]
- 357.Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, Liu CG, Volinia S, Croce CM, Schmittgen TD, Ghoshal K, Jacob ST. Methylation mediated silencing of MicroRNA-1 gene and its role in hepato-cellular carcinogenesis. Cancer Res. 2008;68:5049–58. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 358.Nasser MW, Datta J, Nuovo G, Kutay H, Motiwala T, Majumder S, Wang B, Suster S, Jacob ST, Ghoshal K. Downregulation of microRNA-1 (miR-1) in lung cancer: Suppression of tumorigenic property of lung cancer cells and their sensitization to doxorubicin induced apoptosis bymiR-1. J Biol Chem. 2008:M804788200. doi: 10.1074/jbc.M804788200. [Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 359.Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–67. doi: 10.1038/sj.onc.1210613. [DOI] [PubMed] [Google Scholar]
- 360.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]