

Abstract
Oxidative stress plays a critical role in the progression of pathological cardiac hypertrophy and heart failure. Because crocetin represses oxidative stress in vitro and in vivo, we have suggested that crocetin would repress cardiac hypertrophy by targeting oxidative stress-dependent signalling. We tested this hypothesis using primary cultured cardiac myocytes and fibroblasts and one well-established animal model of cardiac hypertrophy. The results showed that crocetin (1–10 μM) dose-dependently blocked cardiac hypertrophy induced by angiogensin II (Ang II; 1 μM) in vitro. Our data further revealed that crocetin (50 mg/kg/day) both prevented and reversed cardiac hypertrophy induced by aortic banding (AB), as assessed by heart weight/body weight and lung weight/body weight ratios, echocardio-graphic parameters and gene expression of hypertrophic markers. The inhibitory effect of crocetin on cardiac hypertrophy is mediated by blocking the reactive oxygen species (ROS)-dependent mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase-1/2 (MEK/ERK1/2) pathway and GATA binding protein 4 (GATA-4) activation. Further investigation demonstrated that crocetin inhibited inflammation by blocking nuclear factor kappa B (NF-κB) signalling and attenuated fibrosis and collagen synthesis by abrogating MEK-ERK1/2 signalling. Overall, our results indicate that crocetin, which is a potentially safe and inexpensive therapy for clinical use, has protective potential in targeting cardiac hypertrophy and fibrosis by suppression of ROS-dependent signalling pathways.
Keywords: crocetin, reactive oxygen species, cardiac remodelling, fibrosis, NF-κB, ERK1/2
Introduction
Heart failure is one of most prevalent diseases in the world and frequently results from sustained biomechanical overload [1]. After a prolonged period of compensatory adaptation of cardiac hypertrophy, the myocardium undergoes functional and histological deterioration [2, 3]. Increasing evidence suggests that mechanical left ventricular wall stress induces cardiac hypertrophy and failure, in part, by induction of reactive oxygen species (ROS) [4, 5]. ROS can mediate the hypertrophic signals induced by phenylephrine (PE) and angiogensin II (Ang II). Many of the signalling events stimulated by ROS are mediated by members of the mitogen-activated protein kinase (MAPK) family, including the extracellular signal-regulated kinase (ERK1/2), p38, and the c-Jun NH2-terminal kinase (JNK) [6, 7]. Among the MAPKs, ERK1/2 has been considered as the essential regulator of a hypertrophic response, although JNK and p38 were recently examined in regulating cardiac hypertrophy [8]. The activation of the ERK1/2 is triggered by MAPK/ERK kinase-1/2 (MEK1/2) via phosphorylation of serine/threonine residues [9]. Therefore, blocking ROS will attenuate MAPK signalling and then inhibit the development of cardiac hypertrophy. Consistent with this notion, recent studies demonstrated that treatment with antioxidants inhibits the hypertrophic response of cardiac myocytes [10–13]. Natural antioxidants, such as polyphenols from grapes and grape products, have recently attracted considerable attention for the prevention of oxidative stress-related diseases [14, 15]. The fruits and flowers of Crocus sativus (saffron) are important dietary ingredients in traditional India and China. Pistils of saffron are always used in traditional Indian medicine as analgesics and cardioprotective agents. Crocetin is a kind of carotenoid present in saffron, and accumulating evidence demonstrated that it possesses a number of pharmacological activities [16–18]. Crocetin can show strong antioxidant effect including scavenging oxygen free radicals, suppressing lipid per-oxidation, protecting myocardial cell and modulating intracellular Ca 2+ flux. Crocetin has also been reported to inhibit tumour cell proliferation, protect against hepatotoxicity, prevent atherosclerosis in hyperlipidaemic rabbits and improve insulin resistance in rats [19, 20]. Recently, Shen et al.[21, 22] demonstrated that crocetin could prevent cardiac hypertrophy induced by overloading pressure and norepinephrine in rats. However, the effects of crocetin on cardiac hypertrophy and the related signalling mechanisms still remain unclear. The aims of this study were, therefore, to determine whether crocetin can attenuate cardiac hypertrophy induced by Ang II in cultured neonatal rat cardiac myocytes in vitro and pressure overload-induced cardiac hypertrophy in mice in vivo, as well as to identify the molecular mechanisms that may be responsible for its putative effects.
Methods and materials
Materials
The antibodies used to recognize total phosphorylation of ERK1/2, P38 and JNK1/2 as well as of phospho-Smad2, lκB kinase alpha (IKKα), lκB kinase beta (IKKβ), phospho-IκBα and inhibitor of κB alpha (IκBα) were purchased from Cell Signaling Technology (Danvers, MA, USA). [3H]-Leucine and [3H]-proline were purchased from Amersham. The BCA protein assay kit was purchased from Pierce (Rockford, IL, USA), and the IKK activity kit was obtained from B&D Bioscience (San Jose, CA, USA). All other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Transforming growth factor (TGF)-β1 was purchased from R&D Systems (Minneapolis, MN, USA). Foetal calf serum (FCS) was obtained from Hyclone (Logan, UT, USA). NF-κB-, connective tissue growth factor (CTGF)- and collagen 1A2 (COL1A2)-luc report constructs were described previously [23]. Crocetin was obtained from MP Biomedicals (Solon, OH, USA), and cell culture reagents and all other reagents were obtained from Sigma (Oakville, ON, Canada). Crocetin was dissolved in dimethylsulphoxide (DMSO) medium for all in vitro studies.
Cultured neonatal rat cardiac myocytes and fibroblasts
Primary cultures of cardiac myocytes were prepared as described previously [23, 24]. Cells from the hearts of 1- to 2-day-old Sprague-Dawley rats were seeded at a density of 1 × 106/well into 6-well culture plates in a plating medium consisting of F10 medium supplemented with 10% FCS and penicillin/streptomycin. After 48 hrs, the culture medium was replaced with F10 medium containing 0.1% FCS and 5-Bromo-2′-deoxyuridine (BrdU) (100 μM). After 24 hrs of serum starvation, crocetin alone or crocetin followed by Ang II (1 μM) was added to the medium and the cultures were incubated for the indicated time. Viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay.
Cultures of neonatal rat cardiac fibroblasts were described previously [23]. Briefly, the hearts obtained from neonatal rats were enzyme-digested, as described above for myocytes. The adherent non-myocyte fractions obtained during pre-plating were grown in Dulbecco's Modified Eagle Medium (DMEM) containing 10% FCS until confluent and passaged with trypsin-ethylenediaminetetraacetic acid (EDTA). All experiments were performed on cells from the first or second passages that were placed in DMEM containing 0.1% FCS for 24 hrs before the experiment. The purity of these cultures was greater than 95% cardiac fibroblasts, as determined by positive staining for vimentin and negative staining for smooth muscle actin and von Willebrand factor. For cell infection, 1×106/well cardiac myocytes or cardiac fibroblasts were cultured in 6-well plates and exposed to 2 × 108 pfu of each virus in 1 ml of serum-free medium for 24 hrs. The cells were then washed and incubated in serum-containing media for 24 hrs.
[3H]-Leucine incorporation, surface area and collagen synthesis assay
[3H]-Leucine incorporation was measured as described previously [23, 24]. Briefly, cardiac myocytes were pre-treated with crocetin for 60 min., subsequently stimulated with Ang II (1 μM), endotelin-1 (ET-1; 0.1 μM), phenylephrine (100 μM) and insulin-like growth factor-1 (IGF-1; 0.1 μM) and coincubated with [3H]-leucine (1 μCi/ml) for the indicated time. At the end of the experiment, the cells were washed with Hanks’ solution, scraped off the well and then treated with 10% trichloroacetic acid (TCA) at 4°C for 60 min. The precipitates were then dissolved in NaOH (1 N) and subsequently counted in a scintillation counter. For surface areas, the cells were fixed with 3.7% formaldehyde in PBS, permeabilized in 0.1% Triton X-100 in PBS and stained with α-actinin (Sigma) at a dilution of 1:100 by standard immunocytochemical techniques. Collagen synthesis was evaluated by measuring [3H]-proline incorporation, as described previously [23]. In brief, cardiac fibroblasts were made quiescent by culturing in 0.1% FCS DMEM for 24 hrs, pre-treating with crocetin for 60 min. and subsequently incubating with Ang II and 2 μCi/ml [3H]-proline for the indicated time. The cells were washed with PBS twice, treated with ice-cold 5% TCA for 1 hr and washed with distilled water twice. They were then lysed with 1 N NaOH solutions and counted in a liquid scintillation counter. The count representing the amount of newly synthesized collagen was normalized to the cell number.
Reporter assays, Western blotting and quantitative real-time RT-PCR
Cardiac fibroblasts were seeded in triplicate in 6-well plates. They were transfected with 0.5 μg of luciferase reporter constructs and internal control plasmid DNA using 10 μl of LipofectAMINE reagent (Invitrogen) according to the manufacturer's instructions. After 6 hrs of exposure to the DNA–LipofectAMINE complex, the cells were cultured in a medium containing 10% serum for 24 hrs and then incubated with serum-free medium for 12 hrs. The cells were pre-treated with crocetin for 60 min. and then treated with Ang II. They were harvested using a passive lysis buffer (Promega, Madison, WI, USA), according to the manufacturer's protocol. The luciferase activity was normalized by control plasmid. All experiments were done in triplicate and repeated at least three times. For Western blot, cardiac tissue and cultured cardiac myocytes or fibroblasts were lysed in RIPA lysis buffer. Fifty micrograms of cell lysate were used for SDS-PAGE, and the proteins were then transferred to an Immobilon-P membrane (Millipore). Specific protein expression levels were normalized to either the glyceraldehyde-3-phosphate dehy-drogenase (GAPDH) protein for total cell lysate and cytosolic protein or the lamin-B1 protein for nuclear protein signal on the same nitrocellulose membrane. For real-time PCR, total RNA was extracted from frozen, pulverized mouse tissues using TRIzol (Invitrogen) and from synthesized cDNA using oligo (dT) primers with the Advantage RT-for-PCR kit (BD Biosciences). We quantified PCR amplifications using SYBR Green PCR Master Mix (Applied Biosystems) and normalized results against GAPDH gene expression.
Electrophoretic mobility shift assay and IKK assay
To examine the DNA-binding activities of NF-κB and GATA-4, electrophoretic mobility shift assays (EMSA) were performed, according to the manufacturer's instructions (Gel Shift Assay System E3300; Promega). Nuclear proteins were isolated, as described previously [11, 12]. Protein concentrations were measured by the BCA protein assay reagents (Pierce) using bovine serum albumin (BSA) as a standard. To determine the effect of crocetin on IKK activation, the IKK assay was performed, as described previously [11].
Animal models, echocardiography and blood pressure
All protocols were approved by institutional guidelines. All surgeries and subsequent analyses were performed in a blinded fashion for all groups. Adult male C57/B6 mice (8- to 10-week-old) were used in the current study, which were purchased from the Jackson Laboratory and acclimatized for 1 week prior to experimental use. Aortic banding (AB) was performed as described previously [23]. Doppler analysis was performed to ensure that physiological constriction of the aorta was induced. Crocetin suspension was prepared using 0.5% carboxymethylcellulose solution for animal experiments. Suspensions were freshly prepared and administered at a constant volume of 1 ml/100 g body weight by oral gavage three times a day. The control group of these animal experiments was given the same volume of liquid but comprising solely of the vehicle solution (0.5% carboxymethylcellulose). The internal diameter and wall thickness of the left ventricle were assessed by echocardiography in the indicated time after surgery or infusion. The hearts and lungs of the killed mice were dissected and weighed to compare heart weight/body weight (HW/BW; mg/g) and lung weight/body weight (LW/BW; mg/g) ratios in the crocetin-treated and vehicle-treated mice. Echocardiography was performed by SONOS 5500 ultrasound (Philips Electronics, Amsterdam, The Netherlands) with a 15-MHz linear array ultrasound transducer. The left ventricle was assessed in both parasternal long-axis and short-axis views at a frame rate of 120 Hz. End-systole or end-diastole was defined as the phase in which the smallest or the largest area of the left ventricle, respectively, was obtained. Left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD) were measured from the left ventricular M-mode tracing, with a sweep speed of 50 mm/s at the mid-papillary muscle level. Blood pressure was recorded by a microtip catheter transducer (SPR-839; Millar Instruments, Houston, TX, USA) inserted into the right carotid artery and advanced into the left ventricle for haemodynamical measurements.
Histological analysis
The hearts were excised, washed with saline solution and placed in 10% formalin. They were cut transversely close to the apex to visualize the left and right ventricles. Several sections of the heart (4- to 5-μm-thick) were prepared and stained with haematoxylin and eosin and wheat germ agglutinin (WGA) for histopathology or Picrosirius red (PSR) for collagen deposition and then visualized by light microscopy. For myocyte cross-sectional area, the sections were stained with haematoxylin and eosin. A single myocyte was measured with an image quantitative digital analysis system (Image Pro-Plus 4.5, Media Cybernetics, Silver Spring, MD, USA). The outline of 100–200 myocytes was traced in each section.
Measurements of ROS in vitro and in vivo
Cardiac myocytes were cultured on coverslips in 35-mm dishes and then pre-treated with crocetin and subsequently stimulated with 1 μM Ang II for the indicated time. Intracellular generation of ROS was quantified using 2′,7′-dichlorofluorescein dilacerate (DCFH-DA). The cells were incubated with 5 μM DCFH-DA in the dark for 60 min., and immunofluorescence was visualized using laser scanning confocal microscope (488 nm, 200 mW). ROS in the heart tissue were quantified using electron spin resonance (ESR) spectroscopy with 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (hydroxy-TEMPO), as described previously [11].
Statistical analysis
Data are expressed as means ± S.E.M. Differences among groups were tested by one-way anova. Comparisons between two groups were performed by an unpaired Student's t-test. A value of P < 0.05 was considered statistically different.
Results
Crocetin attenuates cardiac hypertrophy in vitro
To rule out the possibility of cytotoxicity, we determined the number of viable cells using MTT assay. Crocetin was determined to be non-cytotoxic for cardiac myocytes at all tested concentrations as the cells were observed to be healthy even in the presence of 10 μM crocetin at the end of 48 hrs (Fig. 1A). In the current study, crocetin was dissolved in DMSO medium for the in vitro studies. There were also no observable adverse effects by the administration of DMSO and Ang II on cellular viability for any of the treatment conditions (data not shown). DMSO alone, without crocetin, served as a control and did not show any effect on cell viability, cardiac hypertrophy and collagen synthesis (data not shown). To examine the effects of crocetin on cardiac hypertrophy cardiac myocytes were incubated with crocetin at the indicated concentrations for 60 min. and subsequently treated with Ang II (1 μM) for 48 hrs. Pre-treatment with crocetin demonstrated a dose-dependent reduction in Ang II-induced increases of [3H]-leucine incorporation that showed maximal effects at 10 μM (Fig. 1B). Additionally, the increase in cardiac myocyte size seen after 48 hrs of culture in the presence of Ang II was also markedly attenuated after treatment with crocetin (Fig. 1C). Further studies demonstrated that crocetin significantly decreased atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) protein expression levels and promoter activities induced by Ang II (Fig. 1D and E). However, crocetin alone had no significant difference compared with control in [3H]-leucine incorporation, cardiac myocyte size and the expression of ANP and BNP genes. We also tested whether crocetin could block cardiac hypertrophy induced by ET-1, PE and IGF-1. The result revealed that crocetin also significantly attenuated cardiac hypertrophy induced by ET-1, PE and IGF-1 (Fig. 1F). Therefore, these data clearly demonstrate that crocetin inhibits cardiac hypertrophy in vitro.
1.
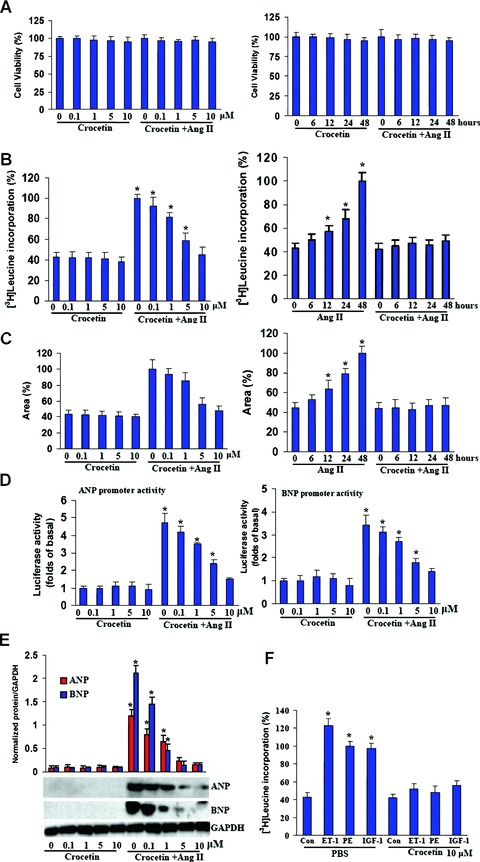
Crocetin inhibits cardiac hypertrophy in vitro. (A) Effect of crocetin and/or Ang II on cell viability in cardiac myocyte. (B) Crocetin inhibited Ang II-induced [3H]-leucine incorporation. (C) Quantification of cell cross-sectional area by measuring 100 random cells. (D, E) Crocetin blunted Ang II-induced ANP and BNP promoter activities and protein expression levels. Cardiac myocytes were incubated with different doses of crocetin (1–10 μM) for 48 hrs or pre-treated with 10 μM crocetin for 60 min. and then incubated with 1 μM Ang II for the indicated time. (F) Crocetin inhibited protein synthesis induced by endotelin-1 (ET-1; 0.1 μM), phenylephrine (PE; 100 μM) and insulin-like growth factor-1 (IGF-1; 0.1 μM) in cardiac myocytes. Cell viability [3H]-leucine incorporation, reporter assay and Western blot were measured as described under Methods and materials. The results were reproducible in three separate experiments. *P< 0.05 versus exposed to control.
Crocetin attenuates cardiac hypertrophy in vivo
To investigate whether our in vitro findings have any physiological relevance, we examined the effects of crocetin on cardiac hypertrophy induced by chronic pressure overload. To evaluate the dose-response relationship, we administered three different doses of crocetin (10, 25 and 50 mg/kg/day) by oral gavage three times a day for 1 week and then subjected the mice to either chronic pressure overload generated by AB or to sham surgery as the control group. Our results demonstrated that the effects of 50 mg/kg/day of crocetin were much more effective than those of 25 mg/kg/day of crocetin in suppressing cardiac hypertrophy, suggesting that maximal efficacy had been achieved at 50 mg/kg/day (Table 1). Moreover, no apparent effect on cell toxicity was observed with either dose of crocetin (data not shown), and 50 mg/kg/day was therefore chosen as the experimental dose.
1.
Echocardiographic data showed the dose-dependent effects of crocetin on cardiac hypertrophy induced by AB model
| Sham | AB 4 weeks | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group (mg) | Saline | 10 | 25 | 50 | Saline | 10 | 25 | 50 | ||||||
| Number | n = 8 | n = 9 | n = 9 | n = 8 | n = 13 | n = 10 | n = 10 | n = 11 | ||||||
| BW, g | 26.6 ± 1.3 | 26.7 ± 1.2 | 27.3 ± 1.4 | 27.5 ± 1.8 | 26.7 ± 1.3 | 27.1 ± 1.2 | 26.8 ± 1.5 | 27.2 ± 1.6 | ||||||
| HW/BW (mg/g) | 5.25 ± 0.04 | 5.18 ± 0.06 | 5.14 ± 0.06 | 5.14 ± 0.07 | 6.88 ± 0.05 | 6.25 ± 0.06 | 6.11 ± 0.05 | 5.45 ± 0.03* | ||||||
| LW/BW (mg/g) | 5.11 ± 0.03 | 5.17 ± 0.04 | 5.13 ± 0.04 | 5.21 ± 0.05 | 5.79 ± 0.03 | 5.67 ± 0.04 | 5.56 ± 0.03 | 5.24 ± 0.03* | ||||||
| SBP, mmHg | 115.2 ± 4.4 | 115.5 ± 3.1 | 113.5 ± 2.7 | 118.5 ± 1.3 | 146.4 ± 2.2 | 145.5 ± 4.1 | 143.5 ± 2.7 | 147.1 ± 3.1 | ||||||
| HR, beats/min | 456 ± 21 | 457 ± 22 | 467 ± 20 | 456 ± 24 | 473 ± 31 | 468 ± 17 | 475 ± 19 | 473 ± 32 | ||||||
| PWT (mm) | 1.25 ± 0.03 | 1.24 ± 0.06 | 1.26 ± 0.07 | 1.22 ± 0.04 | 1.98 ± 0.04 | 1.89 ± 0.04 | 1.72 ± 0.03 | 1.41 ± 0.03* | ||||||
| LVEDD (mm) | 3.65 ± 0.03 | 3.66 ± 0.03 | 3.59 ± 0.03 | 3.52 ± 0.02 | 4.47 ± 0.03 | 4.26 ± 0.03 | 3.97 ± 0.04 | 3.75 ± 0.01* | ||||||
| LVESD (mm) | 2.51 ± 0.04 | 2.52 ± 0.01 | 2.54 ± 0.03 | 2.50 ± 0.02 | 3.22 ± 0.02 | 3.13 ± 0.02 | 2.97 ± 0.02 | 2.71 ± 0.02* | ||||||
| IVSd (mm) | 0.75 ± 0.02 | 0.75 ± 0.02 | 0.73 ± 0.01 | 0.74 ± 0.03 | 1.26 ± 0.01 | 1.19 ± 0.02 | 0.98 ± 0.02 | 0.81 ± 0.02* | ||||||
| LVPWd (mm) | 0.64 ± 0.03 | 0.64 ± 0.02 | 0.62 ± 0.05 | 0.65 ± 0.03 | 1.16 ± 0.03 | 1.11 ± 0.02 | 0.92 ± 0.03 | 0.73 ± 0.01* | ||||||
| FS (%) | 54.5 ± 3.1 | 53.74 ± 2.1 | 55.0 ± 1.7 | 54.2 ± 1.4 | 37.4 ± 3.4 | 40.1 ± 1.2 | 44.2 ± 1.3* | 49.2 ± 1.2* | ||||||
SBP, systolic blood pressure; HR, heart rate; BW, body weight; HW, heart weight; PWT, posterior wall thickness; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; IVSd, left ventricular septum, diastolic; LVPWd, left ventricular posterior wall, diastolic; FS, fractional shortening.
All values are mean ± S.E.M.
P < 0.05 versus AB/saline group after AB.
To further assess the effect of crocetin on cardiac hypertrophy, the mice were randomly assigned into four groups. Pre-treatment with vehicle or 50 mg/kg/day of crocetin for 1 week prior to AB surgery or sham operation allowed for a critical evaluation. As expected, all vehicle-treated AB mice 8 weeks after surgery demonstrated the classical increase in heart size and dilatation of ventricular chambers as compared with the sham control group. Crocetin treatment of the AB mice, on the other hand, resulted in marked reduction in hypertrophic growth, as measured by the ratios of HW/BW and LW/BW, and the cardiomyocyte cross-sectional area (Fig. 2A). No significant changes were observed in the sham-operated mice treated with crocetin or vehicle. Subsequent assessment of chamber size and wall thickness using M-mode echocardiography confirmed these findings. Crocetin treatment prevented the development of adverse cardiac remodelling and ventricular dysfunction, as demonstrated by decreased LVESD, LVEDD and percent fractional shortening (%FS) (Fig. 2B). Haematoxylin and eosin and WGA staining of histological sections further confirmed the inhibitory effect of crocetin on cardiac remodelling in AB hearts (Fig. 2C). To determine whether crocetin affected the mRNA expression levels of markers of cardiac hypertrophy, real-time PCR analysis of ANP, BNP, myosin heavy chain 6 (Myh6) and myosin heavy chain 7 (Myh7) was performed. The results revealed a significant attenuation of the observed increase in the expression level from the vehicle-treated AB group when these animals were treated with crocetin (Fig. 2D). These results indicate that crocetin inhibits the expression of cardiac hypertrophy markers ANP, BNP and Myh7 in the heart and results in an attenuated cardiac hypertrophic response induced by pressure overload. Taken together, our findings indicate that crocetin prevents the development of cardiac hypertrophy in vivo.
2.
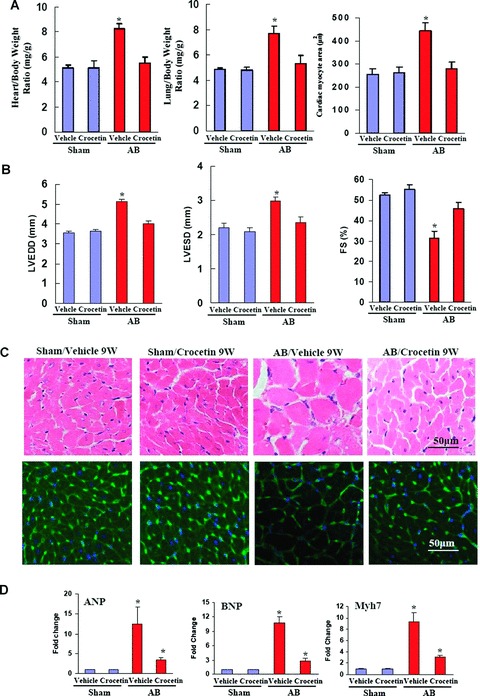
Crocetin blunts cardiac hypertrophy in vivo. (A) Statistical results of HW/BW ratio, LW/BW ratio and myocyte cross-sectional areas (n = 300 cells per section) at 8 weeks after AB surgery (n = 8). (B) Echocardiography results from four groups of mice at 8 weeks after AB or sham surgery. (C) Histology: top, representative of haematoxylin and eosin staining; bottom, WGA-FITC staining at 8 weeks after AB surgery. (D) Analysis of hypertrophic markers (n= 4). Total RNA was isolated from the hearts of mice of the indicated groups, and the expression of transcripts for ANP, BNP and Myh7 induced by AB was determined by RT-PCR analysis. *P < 0.05 for difference from vehicle/sham values in the AB model.
Crocetin inhibits ROS-dependent MEK-ERK1/2 signalling in response to hypertrophic stimuli
Cardiac myocytes were pre-treated for 60 min. with different concentrations of crocetin and then stimulated with 1 μM Ang II for 120 min. As shown in Fig. 3A, Ang II significantly increased the levels of ROS, and such increase was markedly attenuated by crocetin. To further validate these in vitro findings, we evaluated the levels of ROS in the murine heart receiving crocetin treatment. Myocardial production of ROS was evaluated by ESR spectroscopy with hydroxy-TEMPO as a spin probe. The intensity of ESR signals declined more rapidly in banded mice than in the sham-operated controls, and a linear relation was observed in the semi-logarithmic plot of peak signal intensity versus time (data not shown). The rate of signal decay has been shown to reflect the concentration of ROS in the reaction mixture. As shown in Fig. 3B, the rate of signal decay was significantly higher in banded mice than in the sham-operated animals, which was markedly reduced by treatment with crocetin. Treatment by crocetin alone had no significant effects.
3.
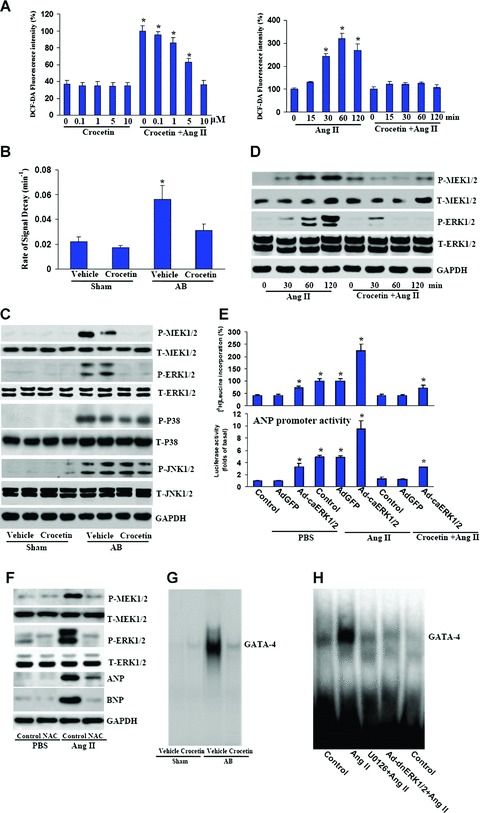
Crocetin inhibits MEK-ERK1/2 signalling in vitro and in vivo. (A) The dose and time courses of crocetin in the generation of ROS induced by Ang II. Cardiac myocytes were pre-treated with different concentrations of crocetin for 30 min. and subsequently incubated with 1 μM Ang II for 60 min. Four parallel experiments were indicated. *P < 0.05 versus exposed to control. (B) Crocetin inhibits pressure overload-induced increase of signal decay rate. *P < 0.05 versus sham/ vehicle. (C) Representative blots of MEK1/2, ERK1/2, p38 and JNK phos-phorylation and their total protein expression in the indicated group's mice. (D) Representative blots of MEK1/2 and ERK1/2 phosphorylation and their total protein expression in cultured cardiac myocytes. (E) The effect of ERK1/2 activation on [3H]-leucine incorporation and ANP promoter activity induced by Ang II. Cardiac myocytes were pre-treated with crocetin for 60 min. and treated with Ang II for 48 hrs. The results were reproducible in three separate experiments as mean ± S.E.M. *P < 0.01 was obtained for the PBS-treated control group. (F) The effect of NAC on MEK1/2 and ERK1/2 activation as well as on ANP and BNP protein expressions induced by Ang II in cultured myocytes. Cardiac myocytes were pre-treated with 10 mM NAC for 30 min. and incubated with Ang II for 120 min. (G) Crocetin inhibits pressure overload-induced GATA-4 DNA-binding activity. (H) Inhibition of ERK1/2 by treatment of U0126 (5 μM) or infection with Ad-dnERK1/2-blocked GATA-4 activation induced by Ang II.
To explore the molecular mechanisms through which crocetin impairs the cardiac hypertrophic response, we further examined the effects of crocetin on MAPK signalling pathway. We found that MEK1, ERK1/2, p38 and JNK1/2 were significantly phosphorylated in AB mice. However, the phosphorylation of MEK1/2 and ERK1/2 was almost completely blocked by crocetin, whereas p38 and JNK1/2 were not significantly affected (Fig. 3C). Collectively, these data suggest that crocetin blunts the activation of MEK-ERK1/2 signalling, although it has no effect on p38 or JNK activation in the hearts subjected to AB. To further test this, we exposed cultured neonatal rat cardiac myocytes to 1 μM Ang II with or without crocetin. Ang II induced a significant increase in the phosphorylated levels of MEK1/2 and ERK1/2; however, crocetin treatment markedly blocked the activation MEK1 and ERK1/2 and sustained it for all tested points in time (Fig. 3D). More importantly, constitutively active ERK1/2 (Ad-caERK1/2) reversed the inhibitory effect of crocetin on cardiac hypertrophy (Fig. 3E). In addition, N-acetylcysteine (NAC; 10 mM), a typical antioxidant, also markedly inhibited ANP and BNP protein expression and blocked MEK1-ERK1/2 signalling induced by Ang II (Fig. 3F). Our findings suggest that crocetin inhibits ROS-dependent MEK-ERK1/2 signalling in vitroand in vivo in response to hypertrophic stimuli. ERK1/2 activation has also been shown to activate hypertrophy-responsive transcription factors including GATA-4. GATA-4 is required for the transcriptional activation of cardiac genes whose expression is up-regulated during cardiac hypertrophy. Therefore, the effects of crocetin on GATA-4 DNA-binding activity induced by AB were further examined. The results showed that crocetin markedly blocked GATA-4 activation (Fig. 3G). Blocking MEK-ERK1/2 signalling by MEK inhibitor U0126 or Ad-dnERK1/2 significantly attenuated Ang II-induced GATA-4 activation, indicating GATA-4 activation is dependent on MEK-ERK1/2 signalling (Fig. 3H).
Crocetin blunts inflammatory response induced by pressure overload
Increasing number of studies suggest that inflammation plays an important role in the development of cardiac and vascular diseases [25, 26]. To determine whether crocetin can suppress the inflammatory responses in the heart, we examined the expression of inflammatory mediators MCP-1, interleukin (IL)-1β and tumour necrosis factor (TNF)-α in cardiac tissue. Our results showed that crocetin significantly decreased the levels of monocyte chemoat-tractant protein 1 (MCP-1), IL-1β and TNF-α mRNA and protein expression compared with the vehicle-treated AB mice (Fig. 4A and B). To evaluate the underlying mechanisms accounting for the potential aetiologies of the inhibitory effects of crocetin on inflammatory responses, we evaluated NF-κB signalling in the mice. Treatment with crocetin abolished the increased activation of NF-κB observed in the myocardium of the vehicle-treated mice 8 weeks after AB (Fig. 4C). To further determine the molecular mechanisms through which crocetin blocks NF-κB activation in vivo, we analysed IκBα phosphorylation and IKK activation processing. Heart lysates from samples obtained from mice 8 weeks after AB were prepared and Western blot analysis was performed. The detection of IκBα phosphorylation and IκBα degradation in the vehicle-treated AB mice was significantly impaired after treatment with crocetin (Fig. 4D). Because the phosphorylation of IκBα is mediated by IKKβ, these findings suggested to us that crocetin might have an inhibitory role in IKKβ activation. Indeed, we observed that the activation, but not the protein expression level, of IKKβ was significantly attenuated by the administration of crocetin (Fig. 4E). Importantly, our further study demonstrated that NAC also blocked Ang II-induced NF-κB activation in cardiac myocytes (Fig. 4F). These results indicate that crocetin inhibits inflammation by blocking NF-κB signalling in response to chronic pressure overload.
4.
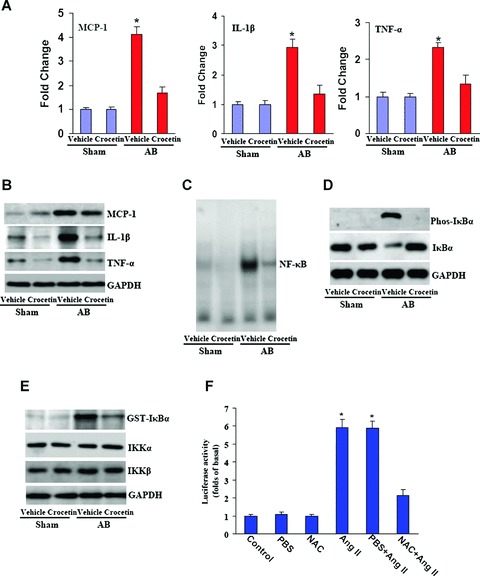
Crocetin inhibits inflammation induced by pressure overload. (A, B) Real-time PCR and Western blot analysis of TNF-α, IL-1β and MCP-1 mRNA and protein expression in the myocardium obtained from the indicated groups (n = 6). (C) The DNA-binding activity of NF-κB in the left ventricle of the indicated groups (n = 5). (D) Western blot analysis of IκBα degradation and IκBα phosphorylation of the myocardium was obtained from the indicated animals at 8 weeks after AB (n= 4). (E) IKKβ activity was shown in the indicated group (n = 4). Each assay was performed in triplicate. *P < 0.01 for difference from vehicle/sham values. (F) 10 mM NAC treatment blocked Ang II-induced NF-κB activity. Luciferase assay were performed as described in Methods and materials. *P < 0.01 for difference from control group.
Crocetin inhibits collagen synthesis induced by Ang II in vitro
To further investigate the mechanism by which crocetin inhibits cardiac hypertrophy, we examined the ability of crocetin to inhibit collagen synthesis stimulated by Ang II in cardiac fibroblasts. We initially assessed the toxicity of crocetin on cardiac fibroblasts using MTT assay. Crocetin was shown to be non-cytotoxic for cardiac fibroblasts at all tested concentrations and time points (data not shown). The cells were serum-starved for 24 hrs in 0.5% FCS and then treated with 1 μM Ang II for the indicated time. Our results revealed that Ang II markedly stimulated [3H]-proline incorporation, which was effectively blocked by pre-treatment with crocetin in a dose-dependent manner (Fig. 5A). To confirm that the observed effects of crocetin on [3H]-proline incorporation were specific to the synthesis of collagen, immunoblot analyses and luciferase assay were employed to assess the protein expression levels and promoter activities of COL1A2 or connective tissue growth factor (CTGF). As shown in Fig. 5B and C, treatment with 1 μM Ang II for up to 48 hrs resulted in increased protein expression and promoter activities of COL1A2 and CTGF, whereas pre-treatment with crocetin for 60 min. significantly blocked Ang II-induced protein expression and promoter activities at all tested time periods.
5.
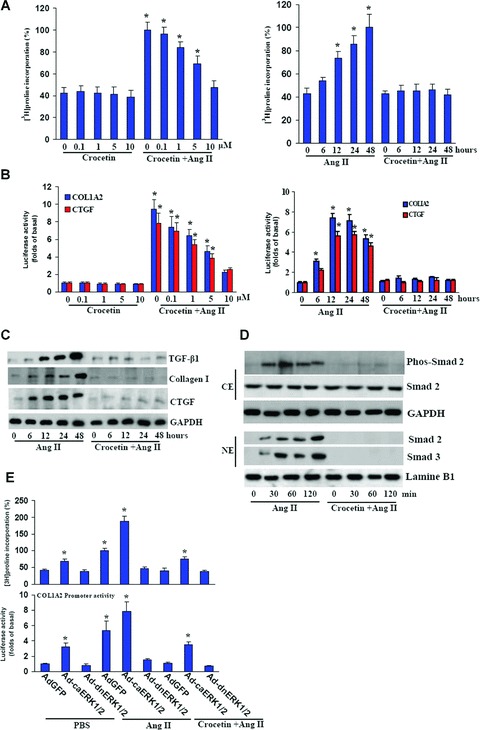
Crocetin blocks collagen synthesis induced by Ang II in vitro. (A) The dose and time course of crocetin in Ang II-induced [3H]-proline incorporation. In these experiments, cells were pre-treated with different doses of crocetin for 60 min. and then incubated with 1 μM Ang II for 48 hrs. [3H]-proline incorporation was measured as described in Methods and materials. (B, C) Crocetin was shown to inhibit the promoter activities of COL1A2 and CTGF as well as protein expression of collagen I and CTGF. Cells were pre-treated with different doses of crocetin or 10 μM crocetin for 60 min. and then incubated with 1 μM Ang II for up to 48 hrs. Luciferase assay and Western blot analysis were performed as described in Methods and materials. Each assay was performed in triplicate. *P < 0.05 versus exposed to control. (D) Representative blots of Smad-2 phosphorylation and Smad-2/3 translocation induced by Ang II in cardiac fibroblasts after treatment with crocetin. (E) The effect of ERK1/2 activation on collagen synthesis along with promoter activity of COL1A2. Cells were infected with or without indicated adenovirus for 24 hrs and then incubated with 1 μM Ang II for up to 48 hrs. [3H]-proline incorporation and luciferase assay were performed as described in Methods and materials. The results were reproducible in three separate experiments.
To examine the molecular mechanisms of crocetin in collagen synthesis, we initially tested whether Ang II induces the phosphorylation and expression of Smad-2 and the expression of Smad 2/3. To accomplish this, we treated cardiac fibroblasts with Ang II for specified time periods and performed Western blot analyses. Our results revealed significant phosphorylation of Smad 2 and translocation of Smad 2/3 without any significant alterations in Smad 2 after 30 min. of Ang II treatment (Fig. 5D). Crocetin almost completely suppressed Smad 2 phosphorylation as well as Smad 2/3 nuclear translocation but had negligible effects on Smad 2 protein expression (Fig. 5D). To further examine the mechanisms involved, we used confluent cardiac fibroblasts infected with Ad-GFP, Ad-caERK1/2 or Ad-dnERK1/2. Activation of ERK1/2 by infection with Ad-caERK1/2 revealed a significant increase in collagen synthesis and COL1A2 promoter activity in response to Ang II, whereas blocking ERK1/2 activity by Ad-dnERK1/2 infection almost completely abrogated them (Fig. 5E). Interestingly, Ad-caERK1/2 infection obviously reversed the inhibitory effects of crocetin on collagen synthesis and COL1A2 promoter activity (Fig. 5E). These findings suggest that crocetin blocks collagen synthesis by disrupting MEK-ERK1/ 2-dependent Smad2/3 signalling.
Crocetin blocks fibrosis induced by pressure overload
To further confirm our in vitro findings, we examined the potential anti-fibrotic effect of crocetin in vivo. To determine the extent of fibrosis in the heart 8 weeks after AB, paraffin-embedded slides were stained with PSR and examined under a light microscope. In contrast to normal hearts with little fibrotic tissue, marked interstitial fibrosis was detected in the vehicle-treated AB mice. Crocetin treatment, however, significantly reduced the extent of cardiac fibrosis in vivo (Fig. 6A and B). Subsequent analysis of mRNA expression levels of known mediators of fibrosis including TGF-β1, collagen I and CTGF demonstrated a blunted response following crocetin administration compared with the vehicle-treated group (Fig. 6B and C). To further elucidate the cellular mechanisms underlying the anti-fibrotic effects of crocetin, we assessed its regulatory role in Smad cascade activation. Upon analysis of the crocetin-treated group, we observed suppressed Smad-2 phosphorylation and Smad 2/3 nuclear translocation in mice (Fig. 6D).
6.
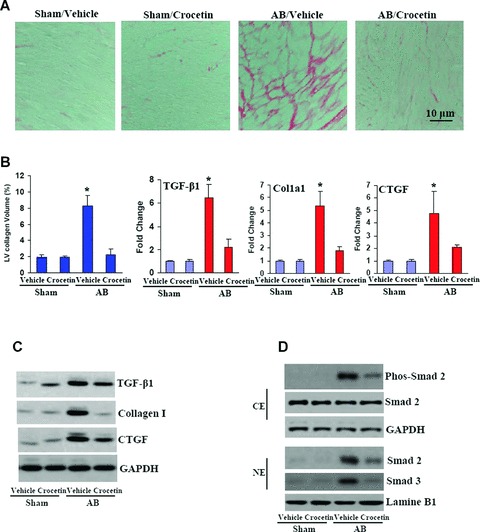
Crocetin inhibits fibrosis induced by pressure overload. (A) PSR staining on histological sections of the left ventricle was performed on each group 8 weeks after AB. (B, C) Fibrotic areas from histological sections were quantified using an image-analyzing system (n = 6). RT-PCR and Western blot analyses of TGF-β1, collagen I and CTGF were performed to determine mRNA and protein. The expression levels in each group 8 weeks after AB (n = 4). GAPDH was used as the sample loading control. *P < 0.05 was obtained for the vehicle/sham values. (D) Representative blots of Smad-2 phosphorylation and Smad-2/3 translocation from the indicated groups 8 weeks after AB (n = 3).
Crocetin ameliorates established cardiac hypertrophy in vivo
We next examined a more clinically relevant model by assessing whether crocetin is able to reverse established cardiac hypertrophy. For these studies, we subjected mice to AB surgery and sham-operated controls. As expected, cardiac hypertrophy was confirmed, as measured by the increase in HW/BW ratio, by direct observation of the gross morphology of the heart and from echocardiographic analyses including increases in LVEDD, LVESD, and posterior wall thickness (PWT) after a 2-week period. Continuation of AB for a subsequent 6 weeks resulted in the transition to heart failure. The cardiac hypertrophy was progressive, as demonstrated by a further decline in %FS, increase in LVEDD and LVESD, as well as increase in HW/BW and LW/BW ratios (Fig. 7A and B). Interestingly, crocetin treatment starting after the initial 2 weeks of AB for a period of 6 weeks was able to reverse the remodelling, contractile dysfunction and cardiac ANP, BNP and Myh7 mRNA expression levels towards normal control values, ultimately preventing the transition to heart failure (Fig. 7A–D).
7.
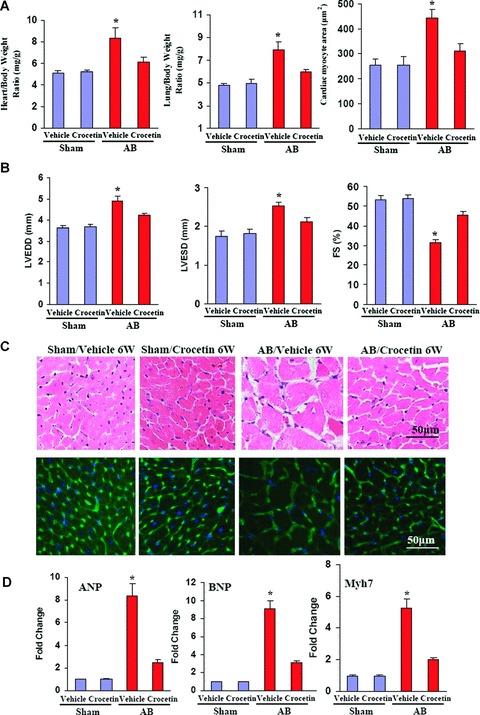
Crocetin ameliorates established cardiac hypertrophy. (A) Statistical results of HW/BW ratio, LW/BW ratio and myocyte cross-sectional areas (n = 300 cells per section) at 8 weeks after AB surgery (n = 8). Mice began treatment with crocetin at 2 weeks after AB or sham surgery and then were killed 8 weeks later. The heart and lung tissues were freshly isolated from each group and HW/BW and LW/BW ratios were determined. (B) Echocardiography results from mice treated with or without crocetin in response to AB. (C) Histology: top, haematoxylin and eosin staining; bottom, WGA-FITC staining at 8 weeks after AB surgery. (D) Analysis of hypertrophic markers (n = 6). Total RNA was isolated from the hearts of mice of the indicated groups, and the expression of transcripts for ANP, BNP and Myh7 induced by AB was determined by RT-PCR analysis. *P < 0.05 for difference from vehicle/sham values.
Discussion
The results from our study demonstrate that crocetin protects against cardiac hypertrophy both in vitro and in vivo. The protective role of crocetin in cardiac hypertrophy is mediated by direct interruption of ROS-dependent MEK-ERK1/2 signalling. This results in the protection of the host from the combined deleterious effects of cardiac hypertrophy, inflammation and fibrosis (Fig. 8). The biological effects of crocetin are also robust enough to reverse established cardiac hypertrophy induced by chronic pressure overload. To our knowledge, this is the first report of the inhibitory effects of crocetin on cardiac hypertrophy in vitro and in vivo. These findings support the concept that crocetin could be an effective preventive and therapeutical candidate against cardiac hypertrophy and heart failure.
8.
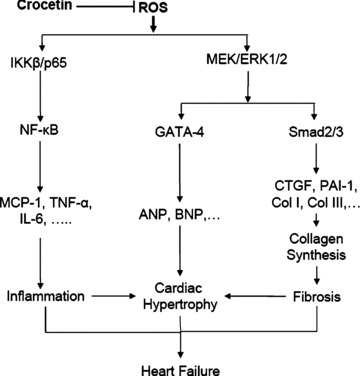
Proposed model of the effect of crocetin on cardiac hypertrophy, inflammation and fibrosis. Crocetin blocks the production of ROS induced by hypertrophic stimulant and leads to the inhibition of cardiac hypertrophy and heart failure. In this study, we have shown that the inhibitory effects of crocetin are achieved by blocking ROS-dependent three proposed signalling pathways. First, increased level of ROS leads to signalling through the IKKβ/p65/NF-κB pathway, promoting cytokine expression and leading to inflammation. Second, increased ROS level may also activate MEK-ERK1/2-dependent GATA-4 activation, activate ANP and BNP expression and subsequently lead to cardiac hypertrophy. Third, ROS is shown to promote the Smad signalling pathway by activation of MEK-ERK1/2 signalling. Subsequent expression of fibrosis markers CTGF, collagen (Col) I and Col II leads to collagen synthesis and fibrosis. Crocetin blocks all of the proposed ROS-dependent signalling pathways and then protects against cardiac hypertrophy, inflammation and fibrosis and the progression of heart failure.
Crocetin is one of about 600 naturally occurring carotenoids and exists mainly in the stigma of saffron and the fruit of Cardenia jasminoides[27]. Recent studies provide additional evidence that crocetin could also exert protective effects against the cardiovascular system, including blocking excessive proliferation of vascular smooth cells [28], lowering serum cholesterol levels, increasing high-density lipoprotein and reducing evolving atherosclerosis [29]. In the current study, we have demonstrated that crocetin not only attenuated cardiac hypertrophy in vitro and in vivo in response to hypertrophic stimuli but also improved cardiac performance and reduced chamber dimensions. Moreover, despite significantly increased blood pressure in our banding model, crocetin treatment did not decrease blood pressure. This indicates that the primary target of crocetin action is heart protection, rather than lowering of blood pressure. Another key finding of this study is that crocetin could reverse pre-established cardiac hypertrophy and dysfunction induced by chronic pressure overload, which is of significant clinical relevance.
The mechanism by which crocetin mediates its anti-hypertrophic effects remains largely unclear. There is increasing evidence for the involvement of ROS generation in pathological cardiac hypertrophy and heart failure [30, 31]. Nakamura et al.[32] first reported that Ang II induced ROS generation in cultured cardiac myocytes, and that pre-treatment with antioxidants led to the abolishment of Ang II-induced cardiac hypertrophy. Shih et al.[33] reported that Ang II-induced expression of the hypertrophic marker β-MHC (myosin heavy chain) was mediated by ROS-dependent activation of MAPK pathway. Date et al.[34] showed that treatment with the antioxidant N-2-mercaptopropionylglycine attenuated myocardial hypertrophy caused by transaortic constriction in mice, indicating that ROS play a key role in pressure overload-induced hypertrophy. In the present report, we investigated whether the anti-hypertrophic effect of crocetin is mediated by the inhibition of ROS generation. Intriguingly, the results indicate that inhibition of ROS in vitro and in vivo was a key mechanism for the anti-hypertrophic activity of crocetin. Although ROS play an important role in the pathogenesis of cardiac hypertrophy, some studies found that hypertrophied hearts exhibited a reduction in oxidative stress. The differences in antioxidant reserve among experimental modes may explain the discrepancy regarding the effect of antioxidants on cardiac hypertrophy. These findings suggest that both ROS-dependent and -independent signalling pathways are present in the signal transduction system in cardiac hypertrophy [11]. Although ROS play a key role in mediating myocardial hypertrophy, the precise molecular targets by which ROS regulate growth signalling are not well defined. To explore the molecular mechanisms through which crocetin impairs the cardiac hypertrophic response, we examined the effects of crocetin on MAPK signalling pathway. Our results clearly demonstrated that crocetin markedly blocked MEK-ERK1/2 signalling pathway in vivo and in vitro in response to hypertrophic stimulus. In accordance with our finding, two recent reports also found that crocetin blocks the proliferation of vascular smooth cells by inhibiting ERK1/2 signalling [28, 35]. Importantly, the classical antioxidant NAC simulated the effects of crocetin on the activation of MEK-ERK1/2, suggesting that the inhibitory effect of crocetin on MEK-ERK1/2 signalling is mainly via inhibition of ROS.
Increasing evidence showed that ERK1/2 activation leads to the activation of the transcription factor, GATA-4, which resulted in cardiac hypertrophy [36, 37]. Compelling evidence has also accumulated to show that GATA-4 is a zinc finger-containing transcription factor that plays an essential role in promoting cardiac development and differentiation of the myocardium, as well as in regulating hypertrophic growth of the heart [38, 39]. GATA-4 functions as a key transcriptional regulator of numerous cardiac genes including ANP, BNP and β-MHC [40]. It also mediates inducible gene expression in response to hypertrophic stimuli, including pressure overload, isoproterenol, PE and ET-1. Therefore, the inhibitory mechanisms of crocetin in cardiac hypertrophy were examined for its effects on the GATA-4 activation. Our data clearly revealed that crocetin blocked GATA-4 activation induced by pressure overload, and that such effect is dependent on ERK1/2 signalling.
Mounting evidence has strongly suggested that inflammation plays a key role in the development of cardiac hypertrophy and heart failure [41, 42]. In line with the growing evidence, we observed a marked induction of cytokines TNF-α, IL-6, MCP-1 and IL-1β in the hypertrophic hearts. When crocetin was administered, cytokine production was attenuated, minimizing the progression of cardiac hypertrophy and heart failure. It is well documented that NF-κB signalling molecules critically regulate inflammation by controlling expression of several families of cytokine gene [43, 44]. Thus, we investigated the status of NF-κB signalling to find out the mechanism by which crocetin blocked inflammation in the heart. Our present data, using both in vivo and in vitro models, suggest that crocetin abrogates NF-κB activation by disrupting DNA-binding and transcriptional activity by blocking the phosphorylation and degradation of IκB as well as IKKβ activation. By blocking NF-κB activation, crocetin may inhibit the early steps of inflammation and modulate the amplification of multiple cytokine signalling cascades. Therefore, treatment by specifically targeting NF-κB seems a conceptually superior approach to blocking each respective cytokine individually.
Fibrosis is another classical feature of pathological hypertrophy, which is characterized by the expansion of the extracellular matrix due to the accumulation of collagen [45, 46]. Thus, it is important to understand the mechanisms that stimulate collagen deposition in the heart and define approaches to limit these processes. In this study, we analysed the anti-fibrotic properties of crocetin in the heart. This study, for the first time, demonstrates that crocetin attenuates cardiac fibrosis and inhibits the expression of several fibrotic mediators induced by pressure overload. Furthermore, this anti-fibrotic effect was further examined in vitro by determining its effectiveness on cultured cardiac fibroblasts. Our findings demonstrate that crocetin blocks Ang II-induced collagen synthesis. Our study is the first to report inhibition of collagen synthesis in cardiac fibroblasts by crocetin. In an attempt to elucidate the mechanisms underlying the inhibitory effect of crocetin on fibrosis, we examined Smad signalling, which plays an important role in the progression of fibrosis. Phosphorylation of Smad 2 and its subsequent translocation to the nucleus are critical steps in the modulation of Smad signalling pathway [47, 48]. Our data suggest, for the first time, that crocetin abrogates Smad 2 phosphorylation and Smad 2/3 translocation in both cardiac fibroblasts culture and hypertrophied hearts and subsequently inhibits collagen synthesis and fibrosis. Recent studies indicate that Smad signalling can be regulated by ERK1/2 signalling [49]. We therefore examined the effects of MEK-ERK1/2 activation on fibrotic signalling and found that blocking MEK-ERK1/2 activation led to significant inhibition, whereas activation of MEK-ERK1/2 resulted in up-regulation of collagen synthesis and Smad 2/3 signalling, indicating that croctin attenuates fibrosis by blocking MEK-ERK1/2-dependent Smad signalling.
In summary, our present work demonstrates for the first time that crocetin inhibits cardiac hypertrophy in vitro and in vivo. We have shown that crocetin can not only prevent the development of cardiac hypertrophy but also reverse established cardiac hypertrophy by blocking ROS-MEK-ERK1/2-dependent hypertrophy, inflammation and fibrosis. Our data confirm that MEK-ERK1/2 is a target of crocetin's inhibitory actions. This study is highly relevant to the understanding of the inhibitory effect of crocetin on cardiac hypertrophy and related molecular mechanisms. It also serves to elucidate the dominant signalling pathways leading to cardiac hypertrophy, inflammation and fibrosis in response to hypertrophic stimuli. Future studies should examine the hypothesis that crocetin may be an effective approach to treating cardiac hypertrophy and preventing the transition to heart failure.
Acknowledgments
The authors thank Dr. Michael Raher (Cardiovascular Research Center, Massachusetts General Hospital, Harvard Medical School) for review of the manuscript and helpful comments. This work was supported by grants from the National Natural Science Foundation of China (No. 30600337 and No. 30770875) to Dr. Yang Xin-Chun.
References
- 1.Liehn EA, Merx MW, Postea O, Becher S, Djalali-Talab Y, Shagdarsuren E, Kelm M, Zernecke A, Weber C. Ccr1 deficiency reduces inflammatory remodeling and preserves left ventricular function after myocardial infarction. J Cell Mol Med. 2008;12:496–506. doi: 10.1111/j.1582-4934.2007.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–50. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 3.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- 4.Neri M, Cerretani D, Fiaschi AI, Laghi PF, Lazzerini PE, Maffione AB, Micheli L, Bruni G, Nencini C, Giorgi G, D’Errico S, Fiore C, Pomara C, Riezzo I, Turillazzi E, Fineschi V. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. J Cell Mol Med. 2007;11:156–70. doi: 10.1111/j.1582-4934.2007.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin II signaling in the heart. J Cell Mol Med. 2004;8:144–52. doi: 10.1111/j.1582-4934.2004.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das M, Das S, Das DK. Caveolin and MAP kinase interaction in angiotensin II preconditioning of the myocardium. J Cell Mol Med. 2007;11:788–97. doi: 10.1111/j.1582-4934.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Das S, Otani H, Maulik N, Das DK. Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling. J Mol Cell Cardiol. 2006;41:248–55. doi: 10.1016/j.yjmcc.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Petrich BG, Wang Y. Stress-activated MAP kinases in cardiac remodeling and heart failure; new insights from transgenic studies. Trends Cardiovasc Med. 2004;14:50–5. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, Hikoso S, Hirotani S, Sadamitsu C, Ichijo H, Baccarini M, Hori M, Otsu K. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest. 2004;114:937–43. doi: 10.1172/JCI20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S, Das DK. Resveratrol: a therapeutic promise for cardiovascular diseases. Recent Patents Cardiovasc Drug Discov. 2007;2:133–8. doi: 10.2174/157489007780832560. [DOI] [PubMed] [Google Scholar]
- 11.Li HL, Huang Y, Zhang CN, Liu G, Wei YS, Wang AB, Liu YQ, Hui RT, Wei C, Williams GM, Liu DP, Liang CC. Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and -independent signal pathways. Free Radic Biol Med. 2006;40:1756–75. doi: 10.1016/j.freeradbiomed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Li HL, Wang AB, Huang Y, Liu DP, Wei C, Williams GM, Zhang CN, Liu G, Liu YQ, Hao DL, Hui RT, Lin M, Liang CC. Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic Biol Med. 2005;38:243–57. doi: 10.1016/j.freeradbiomed.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 13.Tappia PS, Dent MR, Dhalla NS. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic Biol Med. 2006;41:349–61. doi: 10.1016/j.freeradbiomed.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 14.Saini HK, Xu YJ, Arneja AS, Tappia PS, Dhalla NS. Pharmacological basis of different targets for the treatment of atherosclerosis. J Cell Mol Med. 2005;9:818–39. doi: 10.1111/j.1582-4934.2005.tb00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makazan Z, Saini HK, Dhalla NS. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol. 2007;292:H1986–94. doi: 10.1152/ajpheart.01214.2006. [DOI] [PubMed] [Google Scholar]
- 16.Meng L, Cui L. Inhibitory effects of crocetin on high glucose-induced apoptosis in cultured human umbilical vein endothelial cells and its mechanism. Arch Pharm Res. 2008;31:357–63. doi: 10.1007/s12272-001-1164-y. [DOI] [PubMed] [Google Scholar]
- 17.Xi L, Qian Z, Xu G, Zhou C, Sun S. Crocetin attenuates palmitate-induced insulin insensitivity and disordered tumor necrosis factor-alpha and adiponectin expression in rat adipocytes. Br J Pharmacol. 2007;151:610–7. doi: 10.1038/sj.bjp.0707276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanakis CD, Tarantilis PA, Tajmir-Riahi HA, Polissiou MG. Crocetin, dimethylcrocetin, and safranal bind human serum albumin: stability and antioxidative properties. J Agric Food Chem. 2007;55:970–7. doi: 10.1021/jf062638l. [DOI] [PubMed] [Google Scholar]
- 19.He SY, Qian ZY, Wen N, Tang FT, Xu GL, Zhou CH. Influence of crocetin on experimental atherosclerosis in hyperlipidamic-diet quails. Eur J Pharmacol. 2007;554:191–5. doi: 10.1016/j.ejphar.2006.09.071. [DOI] [PubMed] [Google Scholar]
- 20.Xi L, Qian Z, Xu G, Zheng S, Sun S, Wen N, Sheng L, Shi Y, Zhang Y. Beneficial impact of crocetin, a carotenoid from saffron, on insulin sensitivity in fructose-fed rats. J Nutr Biochem. 2007;18:64–72. doi: 10.1016/j.jnutbio.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Shen XC, Qian ZY. Effect of crocetin on cardiac hypertrophy induced by overloading pressure in rats. Yao Xue Xue Bao. 2004;39:172–5. [PubMed] [Google Scholar]
- 22.Shen XC, Qian ZY. Effects of crocetin on antioxidant enzymatic activities in cardiac hypertrophy induced by norepi-nephrine in rats. Pharmazie. 2006;61:348–52. [PubMed] [Google Scholar]
- 23.Li HL, Liu C, De Couto G, Ouzounian M, Sun M, Wang AB, Huang Y, He CW, Shi Y, Chen X, Nghiem MP, Liu Y, Chen M, Dawood F, Fukuoka M, Maekawa Y, Zhang L, Leask A, Ghosh AK, Kirshenbaum LA, Liu PP. Curcumin prevents and reverses murine cardiac hypertrophy. J Clin Invest. 2008;118:879–93. doi: 10.1172/JCI32865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Li HL, She ZG, Li TB, Wang AB, Yang Q, Wei YS, Wang YG, Liu DP. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension. 2007;49:1399–408. doi: 10.1161/HYPERTENSIONAHA.106.085399. [DOI] [PubMed] [Google Scholar]
- 25.Liehn EA, Schober A, Weber C. Blockade of keratinocyte-derived chemokine inhibits endothelial recovery and enhances plaque formation after arterial injury in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:1891–6. doi: 10.1161/01.ATV.0000143135.71440.75. [DOI] [PubMed] [Google Scholar]
- 26.Liehn EA, Zernecke A, Postea O, Weber C. Chemokines: inflammatory mediators of atherosclerosis. Arch Physiol Biochem. 2006;112:229–38. doi: 10.1080/13813450601093583. [DOI] [PubMed] [Google Scholar]
- 27.Magesh V, Singh JP, Selvendiran K, Ekambaram G, Sakthisekaran D. Antitumour activity of crocetin in accordance to tumor incidence, antioxidant status, drug metabolizing enzymes and histopathological studies. Mol Cell Biochem. 2006;287:127–35. doi: 10.1007/s11010-005-9088-0. [DOI] [PubMed] [Google Scholar]
- 28.Zhou CH, Qian ZY, Zheng SG, Xiang M. ERK1/2 pathway is involved in the inhibitory effect of crocetin on angiotensin II-induced vascular smooth muscle cell proliferation. Eur J Pharmacol. 2006;535:61–8. doi: 10.1016/j.ejphar.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Zheng S, Qian Z, Sheng L, Wen N. Crocetin attenuates atherosclerosis in hyperlipidemic rabbits through inhibition of LDL oxidation. J Cardiovasc Pharmacol. 2006;47:70–6. doi: 10.1097/01.fjc.0000194686.11712.02. [DOI] [PubMed] [Google Scholar]
- 30.Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest. 2005;115:2784–92. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–8. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98:794–9. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 33.Shih NL, Cheng TH, Loh SH, Cheng PY, Wang DL, Chen YS, Liu SH, Liew CC, Chen JJ. Reactive oxygen species modulate angiotensin II-induced beta-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2001;283:143–8. doi: 10.1006/bbrc.2001.4744. [DOI] [PubMed] [Google Scholar]
- 34.Date MO, Morita T, Yamashita N, Nishida K, Yamaguchi O, Higuchi Y, Hirotani S, Matsumura Y, Hori M, Tada M, Otsu K. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J Am Coll Cardiol. 2002;39:907–12. doi: 10.1016/s0735-1097(01)01826-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhou CH, Qian ZY, Xiang M, He SY. Involvement of Ca2+ in the inhibition by crocetin of angiotensin II-induced ERK1/2 activation in vascular smooth muscle cells. Eur J Pharmacol. 2007;554:85–91. doi: 10.1016/j.ejphar.2006.09.069. [DOI] [PubMed] [Google Scholar]
- 36.Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol. 2001;21:7460–9. doi: 10.1128/MCB.21.21.7460-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bisping E, Ikeda S, Kong SW, Tarnavski O, Bodyak N, McMullen JR, Rajagopal S, Son JK, Ma Q, Springer Z, Kang PM, Izumo S, Pu WT. GATA4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci USA. 2006;103:14471–6. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrino C, Rockman HA. GATA4 and the two sides of gene expression reprogram-ming. Circ Res. 2006;98:715–6. doi: 10.1161/01.RES.0000217593.07196.af. [DOI] [PubMed] [Google Scholar]
- 39.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of GATA4 reveals its requirement for hypertrophy compensation, and myocyte viability. Circ Res. 2006;98:837–45. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 40.Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovasc Res. 2004;63:196–207. doi: 10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Navab M, Gharavi N, Watson AD. Inflammation and metabolic disorders. Curr Opin Clin Nutr Metab Care. 2008;11:459–64. doi: 10.1097/MCO.0b013e32830460c2. [DOI] [PubMed] [Google Scholar]
- 42.Sadoshima J, Montagne O, Wang Q, Yang G, Warden J, Liu J, Takagi G, Karoor V, Hong C, Johnson GL, Vatner DE, Vatner SF. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J Clin Invest. 2002;110:271–9. doi: 10.1172/JCI14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 44.Baeuerle PA. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell. 1998;95:729–31. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz-Ortega M, Rodrìguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–75. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–92. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 48.Polyakova V, Miyagawa S, Szalay Z, Risteli J, Kostin S. Atrial extracellular matrix remodelling in patients with atrial fibrillation. J Cell Mol Med. 2008;12:189–208. doi: 10.1111/j.1582-4934.2008.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, Sun SQ, Hassid A, Ostrom RS. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol. 2006;70:1992–2003. doi: 10.1124/mol.106.028951. [DOI] [PubMed] [Google Scholar]